

Abstract
Cockayne Syndrome (CS) is a rare human genetic disorder characterized by progressive multisystem degeneration and segmental premature aging. The CS complementation group B (CSB) protein is engaged in transcription coupled and global nucleotide excision repair, base excision repair and general transcription. However, the precise molecular function of the CSB protein is still unclear. In the current review we discuss the involvement of CSB in some of these processes, with focus on the role of CSB in repair of oxidative damage, as deficiencies in the repair of these lesions may be an important aspect of the premature aging phenotype of CS.
1. Premature aging syndromes as model systems for studying human aging
Genome instability is an important component of aging in all eukaryotes; but how age-related genome instability develops, remains an important question. Most human progeroid disorders are linked to defects in genome maintenance (Kyng and Bohr, 2005). Altered transcription seems to be involved in the pathology of most if not all progeroid syndromes and is thought to play a causative role in aging (Kyng and Bohr, 2005). Almost all cases of progeroid syndromes are caused by mutation of a single gene (Martin, 1978). Most of the genes are cloned, and their function can be investigated in isogenic cell lines, where the cellular defects in deficient cell lines can be complemented by transfection with the functional or a mutated version of the otherwise deficient gene. Thus, segmental premature aging syndromes are valuable model systems for studying human aging. CS is such a genetic disease, which in most cases is due to a mutated CSB gene (Licht et al., 2003).
2.1 Clinical manifestations of CS
CS is a rare inherited autosomal recessive disease with diverse clinical symptoms including severe impairment of physical development, cachectic dwarfism, progressive neurological degeneration, white matter hypomyelination, central nervous system (CNS) calcification, sensorineural hearing loss, lack of subcutaneous fat, cataracts and hypersensitivity to sunlight. Interestingly, CS patients appear to lack cancer predisposition, however variant forms of the CSB gene have been associated with increased risk of lung cancer (Lin et al., 2008) and mice disrupted in the CSB gene (also called ERCC6) have increased susceptibility to skin cancer (van der Horst et al., 1997). The life expectancy of patients with CS is approximately 12 years (for extensive reviews on clinical characteristics of CS see (Nance and Berry, 1992) and (Licht et al., 2003). Several of the traits are reminiscent of normal aging, and CS has therefore been classified as a segmental premature aging syndrome. Interestingly, some of the clinical features of CS such as hearing loss, neuronal demyelination and myopathy are reminiscent of syndromes that involve mitochondrial dysfunction (Wallace et al., 1998).
2.2 Mutations causing CS
Eighty percent of the CS cases are caused by a defect in the CSB gene (Mallery et al., 1998), while most of the remaining cases are caused by mutations in the CSA gene (also called CNK1 or ERCC8). Symptoms of CS-A are indistinguishable from CS-B. In addition, rare examples of Xeroderma Pigmentosum (XP) complementation group B (XP-B) patients and certain XP-D and XP-G patients display features of CS in addition to the XP phenotype (Vermeulen et al., 1994). The CSA gene (cloned in 1995) is located on chromosome 5q12—q13, and the gene product, which consists of 396 amino acids, belongs to the “WD repeat” family of structural and regulatory proteins that lack enzymatic activity by itself (Henning et al., 1995). The CSA protein is part of a multisubunit ubiquitin ligase complex, containing Cullin 4A, Roc 1 (Rbx1) and DNA damage binding protein 1 (DDB1) (Groisman et al., 2003). However, detailed literature on CSA is still relatively sparse. The CSB gene, which is located on chromosome 10q11, was cloned in 1990 and described in greater detail in 1992 (Troelstra et al., 1992a; Troelstra et al., 1992b). The severity of the disease does not seem to correlate with the site or nature of the CSB mutation, suggesting that the genetic background and/or environmental factors or downstream targets of a CSB protein-dependent regulation may be involved in determining the specific pathological phenotype of CS (Colella et al., 1999; Mallery et al., 1998). A report from Tanaka and coworkers (Horibata et al., 2004), which describes a patient suffering from UV-sensitivity Syndrome, shows that complete absence of the CSB gene product can give rise to UV-sensitivity but not Cockayne Syndrome. This suggests that presence of truncated or non-functional CSB gene products may prevent the completion of (a) certain process(es). In a recent report from Dolfuss and coworkers this hypothesis is questioned, though, as they describe two patients with no detectable CSB mRNA or CSB protein but displaying classical CS characteristics (Laugel et al., 2008).
2.3 Biochemical characteristics of the CSB protein
The CSB gene product consists of 1493 amino acids and has an estimated molecular weight of 168 KDa. It contains several specific domains including an acidic domain, a glycine rich region and two putative nuclear localization signal (NLS) sequences. In addition, CSB is a member of the SWI2/SNF2-family of DNA dependent ATPases that contain seven characteristic ATPase motifs, which are also present in DNA and RNA helicases (Eisen et al., 1995; Troelstra et al., 1992b) (Figure 1). Like other members of this family, no activity has yet been demonstrated for CSB when using the conventional strand displacement assay (Pazin and Kadonaga, 1997; Selby and Sancar, 1997b). Instead, Vermeulen and coworkers showed that CSB has ATP-dependent chromatin remodeling activity (Citterio et al., 2000). Furthermore, it was recently discovered that CSB also has strand annealing and exchange activities (Muftuoglu et al., 2006). The SNF-2 like ATPase domain is critical for CSB-catalyzed ATPase activity, but the ATPase activity is differentially affected by mutations in the different domains. Thus, a motif II mutant is more affected than motif V and VI mutants, indicating that motif II is the most important one for the catalytic activity of CSB among the motifs (Christiansen et al., 2003).
Fig. 1.
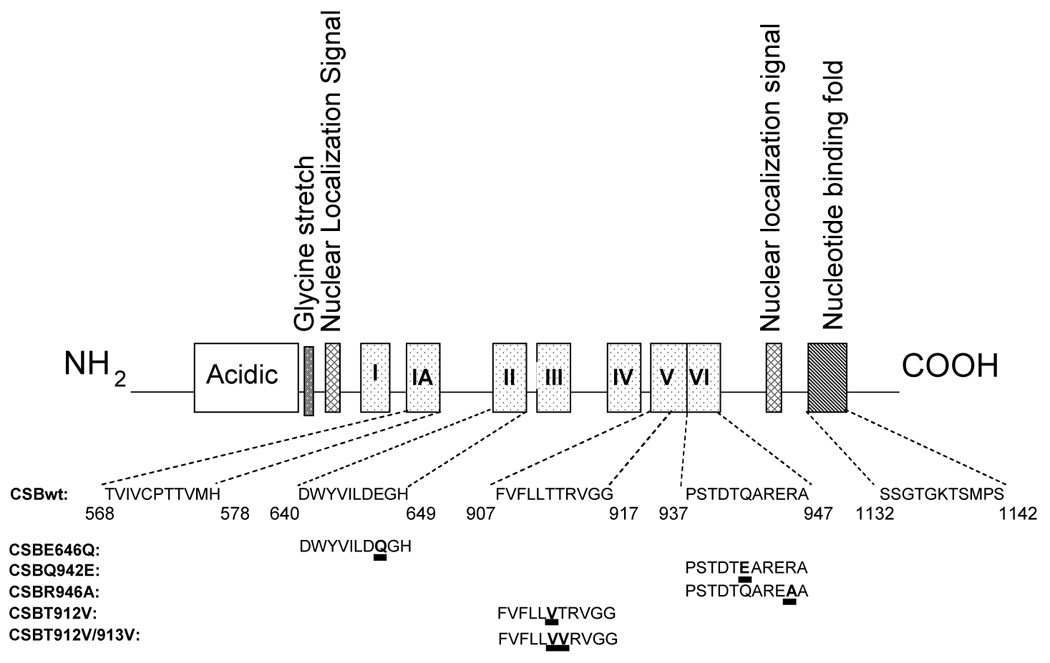
Characteristic motifs of the CSB protein. Mutants that are discussed in the review are indicated.
The enzymatically active CSB protein functions as a homodimer, and the dimerization occurs through the central ATPase domain of the protein (Christiansen et al., 2005). Using scanning force microscopy for analysis of CSB-DNA complexes, Beerens and co-workers demonstrated that DNA wraps around dimers of CSB (Beerens et al., 2005). DNA wrapping and unwrapping may allow CSB to actively alter the DNA double helix conformation, which could influence nucleosomes and other protein-DNA interactions, as discussed below. This may partly explain the chromatin remodeling effect observed by Vermeulen and coworkers (Citterio et al., 2000).
3. The role of CSB in various cellular processes
A hallmark of the CS phenotype is the sensitivity to UV light. UV-light mainly leads to induction of cyclobutane pyrimidine dimers (CPD) and 6-pyrimidine-4-pyrimidone products. Normally, UV-lesions are preferentially removed from the transcribed strand of actively transcribed regions of the genome by the so-called transcription coupled repair (TCR), which is a sub-pathway of nucleotide excision repair (NER). This pathway does not function in CSB deficient cells, indicating that CSB plays an important role in TCR (Licht et al., 2003; Venema et al., 1990). TCR is initiated when elongating RNA polymerase II (RNAPII) progression is blocked by damage in the transcribed strand. RNAPII complex must be displaced and/or degraded in order for efficient repair because it shields the DNA lesion and prevents accessibility of NER proteins (reviewed in Licht et al., 2003; Fousteri et al., 2008). Several models have been proposed regarding how CSB rescues RNAPII complexes that are stalled at DNA lesions (Selby et al., 1997; Tornaletti et al., 1999), but the details of this process remains unclear. It has been suggested that CSB is involved in initiation of TCR through recognition of blocked RNAPII and binding to this complex, followed by recruitment of other NER proteins to the damaged site. As summarized in Figure 2, CSB interacts with several proteins involved in TCR/NER pathway. Recent studies also indicate that TCR can be accomplished without dissociation of RNAPII, and in this model CSB might help remodeling the RNAPII complex without release, allowing repair and the resumption of transcription (Laine et al., 2006a; Laine et al., 2006b; Tremeau-Bravard et al., 2004).
Fig. 2.

Pathways in which CSB might participate - based on proteins which have been demonstrated to interact with CSB.
It has also been suggested that CSB has additional functions outside of TCR, possibly in base excision repair (BER) of some types of oxidative DNA damage in nuclei and mitochondria as described below in detail.
CSB also appears to be implicated in other cellular processes, including transcription by RNA pol I, RNA pol II and possibly also RNA pol III (Licht et al., 2003; Yuan et al., 2007). Besides the defect in coping with many types of DNA damaging agents, CSB cells have markedly reduced transcription (Balajee et al., 1997; Dianov et al., 1997). CSB stimulates transcription by stimulating elongation of actively transcribing RNA polymerase bound to DNA and nascent RNA (Selby and Sancar, 1997a; Tantin et al., 1997; van Gool et al., 1997). Furthermore, using array analysis, Kyng et al. (Kyng et al., 2003) demonstrated that the transcriptional response after oxidative stress is defective in CSB cells. Some of the major defects were found to be in the transcription of genes involved in DNA repair, signal transduction and ribosomal functions. A study by Egly and co-workers (Proietti-De-Santis et al., 2006) supports a role for CSB in regulating the transcription pattern after UV damage as they found a function for CSB in transcription from some but not other promoters. Another comparative gene expression study established that CSB is involved in transcription of genes involved in chromatin maintenance and remodeling (Newman et al., 2006).
CS cells display a stronger apoptotic response to DNA damaging agents than normal cells (Balajee et al., 2000; Laposa et al., 2007; Liu et al., 2006), which in part could explain why no cancer is seen in the patients. This CSB dependent apoptosis response seems independent of p53 (Balajee et al., 2000; Lu et al., 2001).
4. The role of CSB in repair of oxidative DNA damage
Several studies investigated the role of the CSB protein in the cellular repair of oxidative DNA damage, which results from endogenous and exogenous sources. Oxidatively induced DNA damage is mainly repaired by BER, which involves lesion-specific DNA glycosylases in the first step of the repair process. The repair then proceeds with apurinic/apyrimidinic endonuclease 1 (APE1) leaving a single-stranded gap, which is filled by DNA polymerase β (pol β) and ligated by a DNA ligase. A number of studies indicate that BER undergoes age-related changes in several tissues. These include decrease in the activity of the DNA glycosylases or pol β and reduction in the protein levels or expression of pol β or APE1 with age (reviewed in (Cabelof et al., 2006; Gorbunova et al., 2007)). Thus, a decline of BER function with age might contribute to the accumulation of oxidative DNA damage and mutations, and the onset of aging (reviewed in (Gorbunova et al., 2007)). Although there is limited evidence linking the decrease in the amount and activities of these core BER proteins with premature aging, defects in several other factors that have auxiliary, nonessential functions in BER could also contribute significantly to premature aging characteristics.
Cellular studies demonstrated accumulation of oxidative DNA damage in CSB deficient cells after oxidative stress (Tuo et al., 2003), supporting an important potential role for CSB in BER. To dissect the molecular mechanisms underlying the role of CSB in the repair of oxidative lesions, several different biochemical approaches have been used. Dianov and coworkers made the first demonstration that CSB mutant cells are defective in the incision of 8-oxoG (Dianov et al., 1999). The reduced repair of 8-oxoG is associated with a down-regulation of human 8-oxoguanine DNA glycosylase (hOGG1) gene expression and protein level in CSB mutant cells (Dianov et al., 1999; Tuo et al., 2002b). This deficiency is complemented by transfection of CSB mutant cells with the normal CSB gene (Dianov et al., 1999). It was shown that CSB status does not affect the incision activities of two other DNA glycosylases, thymine glycol DNA glycosylase (hNTH) and uracil DNA glycosylase (hUDG) (Dianov et al., 1999). As described below, CSB mutant cells are also defective in the repair of 8-oxoA, another abundant lesion in oxidatively damaged DNA, indicating that CSB might be one of the factors important for the repair of 8-oxoA (Tuo et al., 2003; Tuo et al., 2002b). The glycosylase involved specifically in the repair of this lesion has, however, not yet been identified (Jensen et al., 2003). Furthermore, cell extracts from stably transformed human cell lines with site-directed CSB mutations in various ATPase domains have shown that ATPase domains V and VI of CSB are important for the role of the protein in the processing of 8-oxoG lesions (Tuo et al., 2001), as discussed below, whereas only domain VI appears to be involved in the repair of 8-oxoA (Tuo et al., 2002b). It is possible that CSB plays an important role in the repair of oxidatively modified bases via its interaction with lesion-specific DNA glycosylases.
In humans, OGG1 exists in both nuclear and mitochondrial (mtOGG1) isoforms, which are generated by alternative splicing. The mtOGG1 protein level is low in CSB deficient cells (Stevnsner et al., 2002). We showed a reduced 8-oxoG incision activity in both the mitochondrial extracts of CSB deficient cells and that of CSB knockout mouse liver cells, indicating a potential role for CSB also in the mitochondrial repair of oxidative base damage (Stevnsner et al., 2002). This activity of the CSB protein in mitochondria is specific for the repair of 8-oxoG, since CSB deficient cells have normal levels of uracil, thymine glycol and hypoxanthine incision activities (Stevnsner et al., 2002). Mitochondria possess an independent BER machinery, the components of which are coded by nuclear genes, resembling nuclear BER in the major molecular steps (described above). Mitochondrial BER plays a crucial role in protecting the integrity of mitochondrial DNA (mtDNA). Since mitochondria are the primary source of endogenous ROS, the accumulation of oxidative DNA damage and mutations in mtDNA may lead to mitochondrial dysfunction and consequently to cell loss (Ames et al., 1995). Certain aspects of the clinical features of CSB overlap with the phenotypes associated with mitochondrial dysfunction, including severe neurological deficiencies, dysfunction in skeletal muscle and heart, and premature aging symptoms. Thus, we have previously suggested that CSB deficient cells accumulate mutations in mtDNA and develop mitochondrial dysfunction that contributes to the phenotype and progression of disease in CSB patients (Stevnsner et al., 2002).
Apart from the in vitro incision assays, the role of CSB in the repair of oxidative DNA damage has also been examined in vivo by the removal of formamidopyrimidine DNA glycosylase (Fpg)-sensitive sites in the genome of CSB deficient cells after DNA damage induction with light-activated photosensitizers, such as acridine orange, methylene blue and RO19-8022. These agents introduce mostly 8-oxoG lesions. Fpg is an E. coli glycosylase that excises 8-oxoG and formamidopyrimidine lesions, followed by DNA strand cleavage. We demonstrated that expression of the wild type or ATPase-domain II mutated CSB gene (CSBE646Q) increased the repair rate of Fpg-sensitive sites in mtDNA of CSB deficient cells (Stevnsner et al., 2002). Furthermore, mitochondrial extracts from these ATPase deficient CSB mutant cells stimulated the repair of Fpg-sensitive sites or the incision of 8-oxoG to the same extent as wild-type CSB extracts, indicating that this domain or ATPase activity of the CSB protein is not required for the mitochondrial repair of oxidative base lesions.
The above observations suggest that CSB is involved in the incision process of oxidative base damage. However, the precise mechanism by which CSB participates in the repair of 8-oxoG in both nuclear DNA and mtDNA is not yet known. Although CSB affects the function of hOGG1 as described above, there is no direct physical interaction between CSB and hOGG1; however, the proteins are present in the same protein complex (Tuo et al., 2002a). CSB also stimulates the expression of mtOGG1, but it has not yet been possible to demonstrate that the CSB protein is actually present in mitochondria. Finally, data from Epe and coworkers suggests that CSB may play a role in an OGG1 independent removal of 8-oxoG from the general genome (Osterod et al., 2002).
Recently, CSB has been shown to interact physically and/or functionally with more proteins involved in the BER pathway including poly(ADP-ribose) polymerase 1 (PARP-1) (Thorslund et al., 2005) and APE1 (Wong et al., 2007). PARP-1 is an abundant nuclear DNA damage surveillance protein that can be characterized as a “molecular nick sensor”. PARP-1 binds with high affinity to and is activated by DNA SSBs. When activated, PARP-1 adds polymers of ADP-ribose to various proteins using NAD+ as a substrate (reviewed in (Herceg and Wang, 2001)). CSB interacts with PARP-1 in vivo both in absence and presence of oxidative stress, but interestingly, the CSB/PARP-1 complex relocates to sites of DNA damage in the cell after oxidative stress (Thorslund et al., 2005). It has been suggested that PARP-1 stimulation of BER depends on CSB (Flohr et al., 2003). CSB-deficient cells are significantly more sensitive to PARP inhibitors than CSB-complemented cells and this sensitivity can not be rescued by complementing with CSB protein containing site-directed mutations in the ATPase domain (Thorslund et al., 2005).
CSB also interacts with APE1, stimulating the AP site incision activity of APE1 in an ATP independent manner (Wong et al., 2007). It was suggested that the CSB–APE1 interaction is most critical to regions of the genome where complex DNA structures are formed, such as during transcription or replication, or at sites of recombination, where the local, relative concentrations of these proteins may also be higher. Thus, in addition to coordination with DNA glycosylases in endogenous base damage repair and PARP-1 in DNA damage responses, the interaction with APE1 suggests a more general function of CSB in modulating BER processes (Wong et al., 2007).
It has also been hypothesized that oxidative DNA damage may be repaired by TCR. It is argued that the TCR deficiency of oxidative damage could be responsible for the progressive neurological disorders seen in CS patients (de Waard et al., 2003; Laposa et al., 2007; Osterod et al., 2002; Pastoriza-Gallego et al., 2007). This area of study has been filled with controversy, but one model proposes that the TCR of oxidative damage would be due to the arrest of RNA polymerases at oxidative DNA base lesions. For the initiation of TCR, RNA pol II is thought to be arrested when it encounters a lesion located in the transcribed strand of active gene. However, there are some discrepancies among studies showing the ability of oxidative DNA lesions (e.g. 8-oxoG, thymine glycol) to block RNA pol II during transcription. Some studies show that RNA pol II can bypass an 8-oxoG lesion (Kathe et al., 2004; Larsen et al., 2004; Tornaletti et al., 2004; Viswanathan and Doetsch, 1998), which is a non-bulky lesion repaired by BER, while some other groups could see a partial stalling of RNA pol II (Kuraoka et al., 2003). Sarasin and coworkers have suggested that the repair of 8-oxoG on a transcribed strand results in a competition between BER and TCR components, dependent upon which enzymes arrive first to the lesion (Pastoriza-Gallego et al., 2007). There is a particular class of endogenous oxidative DNA lesions that can block RNA Pol II transcription and are repaired by NER, 8,5'-cyclopurine-2'-deoxynucleosides (Brooks, 2007; Brooks et al., 2000). These lesions are not repaired by BER because of the presence of the covalent 8,5' bond. It has been suggested that the accumulation of these lesions can cause neuronal death by blocking transcription, and that this is a possible source of neurodegeneration in XP (Brooks, 2007). CSB deficient cells might also be defective in the repair of these oxidative lesions and their accumulation could contribute to the CS phenotype. Additional studies on the molecular mechanism of oxidative DNA damage repair in the brain will be important in order to address these possibilities.
5. Sensitivities of CSB deficient cells to various genotoxins
CSB deficient cells are hypersensitive to several types of DNA damaging agents including 4-nitroquinoline-1-oxide (4-NQO), N-acetoxy-2-acetylaminofluorene (NA-AAF), ionizing radiation (IR), paraquat, methyl methanesulfonate (MMS) and 5-hydroxymethyl-2’-doxyuridine. These findings have been obtained by studying different CSB deficient cell types and biological systems. Primary and SV-40 transformed CSB deficient fibroblasts are hypersensitive to 4-NQO, which induces alkali labile single-strand DNA breaks (SSBs) and bulky adducts that are repaired without strand bias. In addition, these fibroblasts are also found to be sensitive to the agent NA-AAF, which also induces adducts that are repaired by global genome repair (GGR), suggesting that CSB might participate in this repair pathway.
Importantly, after exposure to IR, CSB deficient transformed fibroblasts, mouse embryonic fibroblasts (MEF), embryonic stem (ES) cells and keratinocytes from CSB knockout mice all show a marked reduction in survival (de Waard et al., 2004; de Waard et al., 2003; Leadon and Cooper, 1993; Tuo et al., 2001). While IR induces a variety of DNA lesions including single stranded DNA breaks (SSBs), double-strand DNA breaks (DSBs) and oxidative base damage, the observed hypersensitivity has been ascribed to oxidative DNA modifications (de Waard et al., 2004; de Waard et al., 2003), which normally are repaired by BER. Indeed, after IR treatment, CSB deficient primary and transformed fibroblasts, and cells with mutated ATPase domains V (CSBT912V and CSBT912/913V) and VI (CSBQ942E and CSBR946A) accumulated significant amounts of the oxidative base modifications 8-hydroxyguanine (8-oxoG) and 8-hydroxyadenine (8-oxoA) (Tuo et al., 2003; Tuo et al., 2002b). This indicates that CSB could be involved in the repair of oxidized purines through its ATPase domains V and VI, since the IR doses used in these studies mainly cause oxidative DNA base modifications.
Another oxidative DNA base product, 5-hydroxymethyl-2'-deoxyuridine (HmdU), is formed in DNA after exposure to IR or formed by attack of endogenous ROS (Boorstein et al., 1992). Recently, CSB deficient transformed fibroblasts were found to be hypersensitive to HmdU, which generates cytotoxic BER intermediates/products after incorporation. Hence, it was suggested that CSB deficient cells are defective in the removal of these cytotoxic BER intermediates, likely AP sites and/or DNA SSBs (Wong et al., 2007). Paraquat is a widely used broad spectrum herbicide toxin. Enzymatic reduction of paraquat produces paraquat radicals (PQ+), which react with molecular oxygen to generate reactive oxygen species (ROS) in several tissues inflicting oxidative stress to cells and thereby causes oxidative DNA lesions (Ali et al., 1996). CSB knockout MEFs were found also to be hypersensitive to paraquat (de Waard et al., 2004; de Waard et al., 2003). Potassium bromate (KBrO3) generally acts mostly as a one-electron oxidant that leads almost exclusively to the formation of 8-oxoG and formamidopyrimidine derivatives. The effect of KBrO3 on 8-oxoguanine glycosylase (OGG1)-initiated BER, which generates direct DNA strand breaks in CSB mutant fibroblasts, was investigated by the comet assay. Elevated levels of DNA breakage after KBrO3 have been observed in CSB mutant fibroblasts suggesting an inability to process 8-oxoG in these cells (Mosesso et al., 2004). Recently, it was also shown that CSB deficient fibroblasts display hypersensitivity to elevated concentrations of KBrO3 (Ropolo et al., 2007). Altogether, it is evident that CSB deficient cells have limited capacity to cope with elevated levels of oxidatively damaged DNA.
Human fibroblasts defective in CSB are also hypersensitive to methyl methanesulfonate (MMS) treatment (Wong et al., 2007). MMS introduces BER substrates/intermediates. The most critical biological lesion caused by MMS is presumed to be N-methylation base products, which frequently give rise to AP sites via enhanced hydrolysis of the N-glycosylic bond or DNA glycosylase-mediated base release (Wyatt and Pittman, 2006). CSB deficient cells expressing an ATPase domain II mutant CSB protein (CSBE646Q) display intermediate sensitivity to MMS challenge. These findings provide indication for a direct role of CSB in the repair of BER substrates/intermediates.
6. Regulation of CSB activity – and relation to DNA repair
The observation that CSB protein is dephosphorylated at serine and/or threonine in vivo in cells which have been UV-irradiated and that dephosphorylation of CSB stimulates CSB-catalyzed ATPase activity in vitro (Christiansen et al., 2003) suggests that phosphorylation of CSB may regulate its activity in cells with DNA damage. Furthermore, the CSB catalyzed annealing activity is also sensitive to the phosphorylation state of CSB as the strand annealing reaction is stimulated by dephosphorylation of CSB (Muftuoglu et al., 2006). Furthermore, Imam et al. found that c-Abl-dependent phosphorylation of CSB was increased in cells treated with hydrogen peroxide and decreased in cells pre-treated with a c-Abl-specific protein kinase inhibitor, STI-571 (Imam et al., 2007). The activity of c-Abl is normally activated in response to genotoxic or oxidative stress, but in CSB null cells this response is absent. This suggests that c-Abl and CSB may regulate each other in a reciprocal manner in response to oxidative stress. It may be speculated that the hydrogen peroxide induced phosphorylation leads to changes in cellular localization, interactions or stability of CSB. It remains to be investigated whether other types of posttranslational modifications also affect the activity or localization of the CSB protein.
7. CSB and chromatin structure
There are two generally different ways to regulate protein accessibility to a certain area of chromatinized DNA. The histones can either be chemically modified by acetylation, methylation, phosphorylation or ubiquitination, which can serve as a signal to loosen the grip on the DNA, or the nucleosomes can slide or be evicted from the DNA (reviewed in (Osley et al., 2007)). As described above the CSB protein belongs to the SWI2/SNF-2 like family. The family contains members involved in transcription regulation, chromosome stability, DNA repair and recombination (Eisen et al., 1995). A common feature of SWI/SNF-2 proteins is destabilization of protein-DNA interactions (reviewed in (Christiansen et al., 2003)). As described above, several studies indicate involvement of CSB in transcription regulation, TC-NER and BER of some oxidative lesions. All three processes are affected by chromatin structure.
A biochemical in vitro study reported ATPase independent topological changes of a plasmid as a consequence of CSB binding (Citterio et al., 2000). This change in topology could be caused by a wrapping of DNA around CSB. This hypothesis is supported by a study reporting that CSB induces negative supercoils in plasmid DNA and shortening of the contour length of a plasmid by approximately 125 bp. Both effects were dependent on ATP binding, but not on ATP hydrolysis (Beerens et al., 2005). Together, the shortening and the topological change argue that CSB, upon ATP binding, wraps around DNA, in a manner similar to a nucleosome. DNA binding and ATPase assays indicate that binding and hydrolysis of ATP result in DNA binding and release (Beerens et al., 2005; Christiansen et al., 2003), although it is not clear whether it is ATP binding or hydrolysis that results in DNA binding. This mechanism could imply that CSB tracks along DNA and alters its topology as a result of the wrapping. In accord with this, CSB have been reported to mediate ATPase dependent nucleosome remodeling of a mononucleosome, which did not result in core histone release (Citterio et al., 2000). Release of histones is, however, not needed for chromatin remodeling to have an effect on DNA repair, since chromatin remodelers that do not release histones but only open up the chromatin structure have a positive effect on BER (Menoni et al., 2007).
In vivo transcription in CSB deficient cells have been reported to be 50% of wild type cells and in vitro transcription studies on chromatin from CSB deficient cells found that transcription elongation was significantly decreased in the absence of CSB (Selby and Sancar, 1997a). When compared to CSB deficient cells, the transcription from chromatin in wild type cells was more resistant to the detergent Triton X-100, indicating that the transcription machinery is less tightly associated with chromatin in CSB deficient cells compared to wild type cells (Balajee et al., 1997). A study by Weiner and coworkers found that the transcription pattern of CSB deficient cells had a significant overlap with the transcription pattern of cells treated with the histone deacetylase (HDAC) inhibitor trichostatin A (TSA), indicating that CSB could play a role in chromatin maintenance (Newman et al., 2006). The similarity of HDAC inhibited cells to CSB deficient cells indicates that a role of CSB is to decrease the amount of histone acetylation. In contrast, the acetylation of histone H4 near certain promoters is decreased in CSB deficient cells before and/or after UV irradiation, indicating CSB dependent histone acetylase (HAT) activity, as argued by Egly and coworkers (Proietti-De-Santis et al., 2006). Hence, studies indicate that the chromatin pattern of CSB deficient cells is affected, but it is not clear whether CSB is involved in increasing or decreasing the histone acetylation.
8. The role of CSB in DNA repair – and consequences for the aging process
A major theory of aging suggests that free radicals produced as a consequence of the oxidative phosphorylation processes in the mitochondria lead to damage of the mitochondrial genome. This results in dysfunction of the mitochondria and will again lead to production of more free radicals. Thus, a vicious cycle is initiated where oxidative DNA damage accumulates. The accumulation however, must necessarily depend on the efficiency of the repair mechanisms. There has been considerable discussion about whether the complex clinical phenotype of CS is due to a primary defect in DNA repair or transcription. Much data including array analysis by Kyng et al. (Kyng et al., 2003) would suggest that a major defect in this disease lies in the transcriptional response to oxidative stress. This is compatible with the substantial amounts of data suggesting that progeria in CS is linked to accumulated endogenous DNA damage (Andressoo et al., 2006). The observation that a defect in repair of some types of oxidative damage, such as 8-oxoG, can be observed in whole cell, nuclear and mitochondrial extracts from CS cells (Dianov et al., 1999; Stevnsner et al., 2002; Tuo et al., 2002b) suggests that the CSB protein either plays a direct role in the repair process, or that it regulates expression, activity or localization of one of the involved repair proteins. It has not yet been determined whether CSB is present in mitochondria, and it is therefore possible that CSB plays an indirect role in the repair of oxidative damage in these organelles.
Studies of retinal degeneration in a mouse model of CSB supports the oxidative DNA damage theory since expression of established oxidative stress marker genes were found to be up regulated in Csbm/m retina, where the mutation results in a premature stop codon (Gorgels et al., 2007). The neurological deficiencies observed in CS and in several age associated diseases are likely (in part) to be due to insufficient BER of oxidative DNA lesions. However, TC-NER may also play a role in the CS-phenotype and the normal aging process since DNA lesions such as 8,5’-cyclopurine-2’-deoxynucleosides, which can be induced by free radicals, are capable of blocking RNA pol II and are repaired by NER (Brooks et al., 2000).
Within the last few years it has become evident, that the mtBER capacity in the brain is relatively low compared to other organs of the body and that the repair capacity varies between different brain regions (Imam et al., 2006). There also seems to be region specific differences in how dramatically the DNA repair capacity changes with age. It is therefore possible that some regions in CS brains also are more affected by decreased mtBER capacity than others, which could partly explain the neuropathology seen for the CS phenotype, which includes multifocal patchy demyelination in the cerebral and cerebellar cortex and calcium deposits in basal ganglia and cerebral cortex (Itoh et al., 1999).
9. Concluding remarks
CS is a very significant human disorder that has a strong premature aging phenotype. The precise function(s) of the CSB protein still remains unclear. The CSB protein functions in many DNA metabolic pathways and appears to have diverse significant functions in transcription, signaling, chromatin structure and DNA repair (Figure 2). It is not clear whether the BER deficiency in CS cells is due primarily to reduced transcription of BER proteins or to a more direct role of the CSB protein in the BER processes. As a result of the ongoing leakage of ROS from the mitochondria, the genome is continuously damaged by various oxygen species. As we have described in this review it seems conceivable that the defective repair of oxidatively damaged DNA contributes to the CS phenotype – especially the component that has to do with the central nervous system, because of the potential association between mitochondrial dysfunction and neurodegeneration.
Acknowledgements
This work was supported by funds from the intramural program of the National Institute on Aging, National Institutes of Health, and by grants from the Danish Research Councils (22-03-0253), The Velux Foundation, The Danish Cancer Society (DP05118) and EU (LSHM-CT-2004-512020).
Abbreviations
- APE1
apurinic/apyrimidinic endonuclease 1
- BER
base excision repair
- CNS
central nervous system
- CPD
cyclobutane pyrimidine dimers
- CS
Cockayne Syndrome
- CSA
Cockayne Syndrome complementation group A
- CSB
Cockayne syndrome complementation group B
- DDB1
DNA damage binding protein 1
- DSBs
double strand breaks
- Fpg
formamido pyrimidine DNA glycosylase
- GGR
global genome repair
- HAT
histone acetylase
- HDAC
histone deacetylase
- HmdU
5-hydroxymethyl-2’-deoxyuridine
- hNTH
thymine glycol DNA glycosylase
- hUDG
human uracil DNA glycosylase
- IR
ionizing radiation
- KBrO3
potassium bromate
- MMS
methyl methanesulfonate
- mtDNA
mitochondrial DNA
- NA-AAF
N-acetoxy-2-acetylaminofluorene
- NER
nucleotide excision repair
- OGG1
8-oxoguanine glycosylase
- PARP1
poly(ADP-ribose)polymerase
- polβ
DNA polymerase β
- PQ+
paraquat radicals
- ROS
reactive oxygen species
- SSBs
single strand breaks
- TCR
transcription coupled repair
- TC-NER
transcription coupled NER
- TSA
trichostatin A
- 4-NQO
4-nitroquinoline-1-oxide
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Ali S, Jain SK, Abdulla M, Athar M. Paraquat induced DNA damage by reactive oxygen species. Biochem Mol Biol Int. 1996;39:63–67. doi: 10.1080/15216549600201061. [DOI] [PubMed] [Google Scholar]
- Ames BN, Shigenaga MK, Hagen TM. Mitochondrial decay in aging. Biochim Biophys Acta. 1995;1271:165–170. doi: 10.1016/0925-4439(95)00024-x. [DOI] [PubMed] [Google Scholar]
- Andressoo JO, Hoeijmakers JH, Mitchell JR. Nucleotide excision repair disorders and the balance between cancer and aging. Cell Cycle. 2006;5:2886–2888. doi: 10.4161/cc.5.24.3565. [DOI] [PubMed] [Google Scholar]
- Balajee AS, May A, Dianov GL, Friedberg EC, Bohr VA. Reduced RNA polymerase II transcription in intact and permeabilized Cockayne syndrome group B cells. Proc Natl Acad Sci U S A. 1997;94:4306–4311. doi: 10.1073/pnas.94.9.4306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balajee AS, Proietti De Santis L, Brosh RM, Jr., Selzer R, Bohr VA. Role of the ATPase domain of the Cockayne syndrome group B protein in UV induced apoptosis. Oncogene. 2000;19:477–489. doi: 10.1038/sj.onc.1203372. [DOI] [PubMed] [Google Scholar]
- Beerens N, Hoeijmakers JH, Kanaar R, Vermeulen W, Wyman C. The CSB protein actively wraps DNA. J Biol Chem. 2005;280:4722–4729. doi: 10.1074/jbc.M409147200. [DOI] [PubMed] [Google Scholar]
- Boorstein RJ, Chiu LN, Teebor GW. A mammalian cell line deficient in activity of the DNA repair enzyme 5-hydroxymethyluracil-DNA glycosylase is resistant to the toxic effects of the thymidine analog 5-hydroxymethyl-2'-deoxyuridine. Mol Cell Biol. 1992;12:5536–5540. doi: 10.1128/mcb.12.12.5536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooks PJ. The case for 8,5'-cyclopurine-2'-deoxynucleosides as endogenous DNA lesions that cause neurodegeneration in xeroderma pigmentosum. Neuroscience. 2007;145:1407–1417. doi: 10.1016/j.neuroscience.2006.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooks PJ, Wise DS, Berry DA, Kosmoski JV, Smerdon MJ, Somers RL, Mackie H, Spoonde AY, Ackerman EJ, Coleman K, Tarone RE, Robbins JH. The oxidative DNA lesion 8,5'-(S)-cyclo-2'-deoxyadenosine is repaired by the nucleotide excision repair pathway and blocks gene expression in mammalian cells. J Biol Chem. 2000;275:22355–22362. doi: 10.1074/jbc.M002259200. [DOI] [PubMed] [Google Scholar]
- Cabelof DC, Raffoul JJ, Ge Y, Van Remmen H, Matherly LH, Heydari AR. Age-related loss of the DNA repair response following exposure to oxidative stress. J Gerontol A Biol Sci Med Sci. 2006;61:427–434. doi: 10.1093/gerona/61.5.427. [DOI] [PubMed] [Google Scholar]
- Christiansen M, Stevnsner T, Modin C, Martensen PM, Brosh RM, Jr., Bohr VA. Functional consequences of mutations in the conserved SF2 motifs and post-translational phosphorylation of the CSB protein. Nucleic Acids Res. 2003;31:963–973. doi: 10.1093/nar/gkg164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christiansen M, Thorslund T, Jochimsen B, Bohr VA, Stevnsner T. The Cockayne syndrome group B protein is a functional dimer. Febs J. 2005;272:4306–4314. doi: 10.1111/j.1742-4658.2005.04844.x. [DOI] [PubMed] [Google Scholar]
- Citterio E, Van Den Boom V, Schnitzler G, Kanaar R, Bonte E, Kingston RE, Hoeijmakers JH, Vermeulen W. ATP-dependent chromatin remodeling by the Cockayne syndrome B DNA repair-transcription-coupling factor. Mol Cell Biol. 2000;20:7643–7653. doi: 10.1128/mcb.20.20.7643-7653.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colella S, Nardo T, Mallery D, Borrone C, Ricci R, Ruffa G, Lehmann AR, Stefanini M. Alterations in the CSB gene in three Italian patients with the severe form of Cockayne syndrome (CS) but without clinical photosensitivity. Hum Mol Genet. 1999;8:935–941. doi: 10.1093/hmg/8.5.935. [DOI] [PubMed] [Google Scholar]
- de Waard H, de Wit J, Andressoo JO, van Oostrom CT, Riis B, Weimann A, Poulsen HE, van Steeg H, Hoeijmakers JH, van der Horst GT. Different effects of CSA and CSB deficiency on sensitivity to oxidative DNA damage. Mol Cell Biol. 2004;24:7941–7948. doi: 10.1128/MCB.24.18.7941-7948.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Waard H, de Wit J, Gorgels TG, van den Aardweg G, Andressoo JO, Vermeij M, van Steeg H, Hoeijmakers JH, van der Horst GT. Cell type-specific hypersensitivity to oxidative damage in CSB and XPA mice. DNA Repair (Amst) 2003;2:13–25. doi: 10.1016/s1568-7864(02)00188-x. [DOI] [PubMed] [Google Scholar]
- Dianov G, Bischoff C, Sunesen M, Bohr VA. Repair of 8-oxoguanine in DNA is deficient in Cockayne syndrome group B cells. Nucleic Acids Res. 1999;27:1365–1368. doi: 10.1093/nar/27.5.1365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dianov GL, Houle JF, Iyer N, Bohr VA, Friedberg EC. Reduced RNA polymerase II transcription in extracts of cockayne syndrome and xeroderma pigmentosum/Cockayne syndrome cells. Nucleic Acids Res. 1997;25:3636–3642. doi: 10.1093/nar/25.18.3636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisen JA, Sweder KS, Hanawalt PC. Evolution of the SNF2 family of proteins: subfamilies with distinct sequences and functions. Nucleic Acids Res. 1995;23:2715–2723. doi: 10.1093/nar/23.14.2715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flohr C, Burkle A, Radicella JP, Epe B. Poly(ADP-ribosyl)ation accelerates DNA repair in a pathway dependent on Cockayne syndrome B protein. Nucleic Acids Res. 2003;31:5332–5337. doi: 10.1093/nar/gkg715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fousteri M, Mullenders LHF. Transcription-coupled nucleotide excision repair in mammalian cells: molecular mechanisms and biological effects. Cell Res. 2008;18:73–84. doi: 10.1038/cr.2008.6. [DOI] [PubMed] [Google Scholar]
- Gorbunova V, Seluanov A, Mao Z, Hine C. Changes in DNA repair during aging. Nucleic Acids Res. 2007;35:7466–7474. doi: 10.1093/nar/gkm756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorgels TG, van der Pluijm I, Brandt RM, Garinis GA, van Steeg H, van den Aardweg G, Jansen GH, Ruijter JM, Bergen AA, van Norren D, Hoeijmakers JH, van der Horst GT. Retinal degeneration and ionizing radiation hypersensitivity in a mouse model for Cockayne syndrome. Mol Cell Biol. 2007;27:1433–1441. doi: 10.1128/MCB.01037-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groisman R, Polanowska J, Kuraoka I, Sawada J, Saijo M, Drapkin R, Kisselev AF, Tanaka K, Nakatani Y. The ubiquitin ligase activity in the DDB2 and CSA complexes is differentially regulated by the COP9 signalosome in response to DNA damage. Cell. 2003;113:357–367. doi: 10.1016/s0092-8674(03)00316-7. [DOI] [PubMed] [Google Scholar]
- Henning KA, Li L, Iyer N, McDaniel LD, Reagan MS, Legerski R, Schultz RA, Stefanini M, Lehmann AR, Mayne LV, Friedberg EC. The Cockayne syndrome group A gene encodes a WD repeat protein that interacts with CSB protein and a subunit of RNA polymerase II TFIIH. Cell. 1995;82:555–564. doi: 10.1016/0092-8674(95)90028-4. [DOI] [PubMed] [Google Scholar]
- Herceg Z, Wang ZQ. Functions of poly(ADP-ribose) polymerase (PARP) in DNA repair, genomic integrity and cell death. Mutat Res. 2001;477:97–110. doi: 10.1016/s0027-5107(01)00111-7. [DOI] [PubMed] [Google Scholar]
- Horibata K, Iwamoto Y, Kuraoka I, Jaspers NG, Kurimasa A, Oshimura M, Ichihashi M, Tanaka K. Complete absence of Cockayne syndrome group B gene product gives rise to UV-sensitive syndrome but not Cockayne syndrome. Proc Natl Acad Sci U S A. 2004;101:15410–15415. doi: 10.1073/pnas.0404587101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imam SZ, Indig FE, Cheng WH, Saxena SP, Stevnsner T, Kufe D, Bohr VA. Cockayne syndrome protein B interacts with and is phosphorylated by c-Abl tyrosine kinase. Nucleic Acids Res. 2007;35:4941–4951. doi: 10.1093/nar/gkm386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imam SZ, Karahalil B, Hogue BA, Souza-Pinto NC, Bohr VA. Mitochondrial and nuclear DNA-repair capacity of various brain regions in mouse is altered in an age-dependent manner. Neurobiol Aging. 2006;27:1129–1136. doi: 10.1016/j.neurobiolaging.2005.06.002. [DOI] [PubMed] [Google Scholar]
- Itoh M, Hayashi M, Shioda K, Minagawa M, Isa F, Tamagawa K, Morimatsu Y, Oda M. Neurodegeneration in hereditary nucleotide repair disorders. Brain Dev. 1999;21:326–333. doi: 10.1016/s0387-7604(99)00033-9. [DOI] [PubMed] [Google Scholar]
- Jensen A, Calvayrac G, Karahalil B, Bohr VA, Stevnsner T. Mammalian 8-oxoguanine DNA glycosylase 1 incises 8-oxoadenine opposite cytosine in nuclei and mitochondria, while a different glycosylase incises 8-oxoadenine opposite guanine in nuclei. J Biol Chem. 2003;278:19541–19548. doi: 10.1074/jbc.M301504200. [DOI] [PubMed] [Google Scholar]
- Kathe SD, Shen GP, Wallace SS. Single-stranded breaks in DNA but not oxidative DNA base damages block transcriptional elongation by RNA polymerase II in HeLa cell nuclear extracts. J Biol Chem. 2004;279:18511–18520. doi: 10.1074/jbc.M313598200. [DOI] [PubMed] [Google Scholar]
- Kuraoka I, Endou M, Yamaguchi Y, Wada T, Handa H, Tanaka K. Effects of endogenous DNA base lesions on transcription elongation by mammalian RNA polymerase II. Implications for transcription-coupled DNA repair and transcriptional mutagenesis. J Biol Chem. 2003;278:7294–7299. doi: 10.1074/jbc.M208102200. [DOI] [PubMed] [Google Scholar]
- Kyng KJ, Bohr VA. Gene expression and DNA repair in progeroid syndromes and human aging. Ageing Res Rev. 2005;4:579–602. doi: 10.1016/j.arr.2005.06.008. [DOI] [PubMed] [Google Scholar]
- Kyng KJ, May A, Brosh RM, Jr., Cheng WH, Chen C, Becker KG, Bohr VA. The transcriptional response after oxidative stress is defective in Cockayne syndrome group B cells. Oncogene. 2003;22:1135–1149. doi: 10.1038/sj.onc.1206187. [DOI] [PubMed] [Google Scholar]
- Laine JP, Egly JM. Initiation of DNA repair mediated by a stalled RNA polymerase IIo. EMBO J. 2006a;2:387–397. doi: 10.1038/sj.emboj.7600933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laine JP, Egly JM. When transcription and repair meet: a complex system. Trends Genet. 2006b;8:430. doi: 10.1016/j.tig.2006.06.006. [DOI] [PubMed] [Google Scholar]
- Laposa RR, Huang EJ, Cleaver JE. Increased apoptosis, p53 up-regulation, and cerebellar neuronal degeneration in repair-deficient Cockayne syndrome mice. Proc Natl Acad Sci U S A. 2007;104:1389–1394. doi: 10.1073/pnas.0610619104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsen E, Kwon K, Coin F, Egly JM, Klungland A. Transcription activities at 8-oxoG lesions in DNA. DNA Repair (Amst) 2004;3:1457–1468. doi: 10.1016/j.dnarep.2004.06.008. [DOI] [PubMed] [Google Scholar]
- Laugel V, Dalloz C, Stary A, Cormier-Daire V, Desguerre I, Renouil M, Fourmaintraux A, Velez-Cruz R, Egly JM, Sarasin A, Dollfus H. Deletion of 5' sequences of the CSB gene provides insight into the pathophysiology of Cockayne syndrome. Eur J Hum Genet. 2008;16:320–327. doi: 10.1038/sj.ejhg.5201991. [DOI] [PubMed] [Google Scholar]
- Leadon SA, Cooper PK. Preferential repair of ionizing radiation-induced damage in the transcribed strand of an active human gene is defective in Cockayne syndrome. Proc Natl Acad Sci U S A. 1993;90:10499–10503. doi: 10.1073/pnas.90.22.10499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Licht CL, Stevnsner T, Bohr VA. Cockayne syndrome group B cellular and biochemical functions. Am J Hum Genet. 2003;73:1217–1239. doi: 10.1086/380399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin Z, Zhang X, Tuo J, Guo Y, Green B, Chan CC, Tan W, Huang Y, Ling W, Kadlubar FF, Lin D, Ning B. A variant of the Cockayne syndrome B gene ERCC6 confers risk of lung cancer. Hum Mutat. 2008;29:113–122. doi: 10.1002/humu.20610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu F, Yu ZJ, Sui JL, Bai B, Zhou PK. siRNA-mediated silencing of Cockayne Cyndrome group B gene potentiates radiation-induced apoptosis and antiproliferative effect in HeLa cells. Chin Med J (Engl) 2006;119:731–739. [PubMed] [Google Scholar]
- Lu Y, Lian H, Sharma P, Schreiber-Agus N, Russell RG, Chin L, van der Horst GT, Bregman DB. Disruption of the Cockayne syndrome B gene impairs spontaneous tumorigenesis in cancer-predisposed Ink4a/ARF knockout mice. Mol Cell Biol. 2001;21:1810–1818. doi: 10.1128/MCB.21.5.1810-1818.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mallery DL, Tanganelli B, Colella S, Steingrimsdottir H, van Gool AJ, Troelstra C, Stefanini M, Lehmann AR. Molecular analysis of mutations in the CSB (ERCC6) gene in patients with Cockayne syndrome. Am J Hum Genet. 1998;62:77–85. doi: 10.1086/301686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin GM. Genetic syndromes in man with potential relevance to the pathobiology of aging. Birth Defects Orig Artic Ser. 1978;14:5–39. [PubMed] [Google Scholar]
- Menoni H, Gasparutto D, Hamiche A, Cadet J, Dimitrov S, Bouvet P, Angelov D. ATP-dependent chromatin remodeling is required for base excision repair in conventional but not in variant H2A.Bbd nucleosomes. Mol Cell Biol. 2007;27:5949–5956. doi: 10.1128/MCB.00376-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mosesso P, Penna S, Pepe G, Lorenti-Garcia C, Palitti F. Potassium bromate but not X-rays cause unexpectedly elevated levels of DNA breakage similar to those induced by ultraviolet light in Cockayne syndrome (CS-B) fibroblasts. Cytogenet Genome Res. 2004;104:178–181. doi: 10.1159/000077485. [DOI] [PubMed] [Google Scholar]
- Muftuoglu M, Selzer R, Tuo J, Brosh RM, Jr., Bohr VA. Phenotypic consequences of mutations in the conserved motifs of the putative helicase domain of the human Cockayne syndrome group B gene. Gene. 2002;283:27–40. doi: 10.1016/s0378-1119(01)00870-8. [DOI] [PubMed] [Google Scholar]
- Muftuoglu M, Sharma S, Thorslund T, Stevnsner T, Soerensen MM, Brosh RM, Jr., Bohr VA. Cockayne syndrome group B protein has novel strand annealing and exchange activities. Nucleic Acids Res. 2006;34:295–304. doi: 10.1093/nar/gkj410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nance MA, Berry SA. Cockayne syndrome: review of 140 cases. Am J Med Genet. 1992;42:68–84. doi: 10.1002/ajmg.1320420115. [DOI] [PubMed] [Google Scholar]
- Newman JC, Bailey AD, Weiner AM. Cockayne syndrome group B protein (CSB) plays a general role in chromatin maintenance and remodeling. Proc Natl Acad Sci U S A. 2006;103:9613–9618. doi: 10.1073/pnas.0510909103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osley MA, Tsukuda T, Nickoloff JA. ATP-dependent chromatin remodeling factors and DNA damage repair. Mutat Res. 2007;618:65–80. doi: 10.1016/j.mrfmmm.2006.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osterod M, Larsen E, Le Page F, Hengstler JG, Van Der Horst GT, Boiteux S, Klungland A, Epe B. A global DNA repair mechanism involving the Cockayne syndrome B (CSB) gene product can prevent the in vivo accumulation of endogenous oxidative DNA base damage. Oncogene. 2002;21:8232–8239. doi: 10.1038/sj.onc.1206027. [DOI] [PubMed] [Google Scholar]
- Pastoriza-Gallego M, Armier J, Sarasin A. Transcription through 8-oxoguanine in DNA repair-proficient and Csb(-)/Ogg1(-) DNA repair-deficient mouse embryonic fibroblasts is dependent upon promoter strength and sequence context. Mutagenesis. 2007;22:343–351. doi: 10.1093/mutage/gem024. [DOI] [PubMed] [Google Scholar]
- Pazin MJ, Kadonaga JT. SWI2/SNF2 and related proteins: ATP-driven motors that disrupt protein-DNA interactions? Cell. 1997;88:737–740. doi: 10.1016/s0092-8674(00)81918-2. [DOI] [PubMed] [Google Scholar]
- Proietti-De-Santis L, Drane P, Egly JM. Cockayne syndrome B protein regulates the transcriptional program after UV irradiation. Embo J. 2006;25:1915–1923. doi: 10.1038/sj.emboj.7601071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ropolo M, Degan P, Foresta M, D'Errico M, Lasiglie D, Dogliotti E, Casartelli G, Zupo S, Poggi A, Frosina G. Complementation of the oxidatively damaged DNA repair defect in Cockayne syndrome A and B cells by Escherichia coli formamidopyrimidine DNA glycosylase. Free Radic Biol Med. 2007;42:1807–1817. doi: 10.1016/j.freeradbiomed.2007.03.008. [DOI] [PubMed] [Google Scholar]
- Selby CP, Drapkin R, Reinberg D, Sancar A. RNA polymerase II stalled at a thymine dimer: footprint and effect on excision repair. Nucleic Acids Res. 1997;25:787–793. doi: 10.1093/nar/25.4.787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selby CP, Sancar A. Cockayne syndrome group B protein enhances elongation by RNA polymerase II. Proc Natl Acad Sci U S A. 1997a;94:11205–11209. doi: 10.1073/pnas.94.21.11205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selby CP, Sancar A. Human transcription-repair coupling factor CSB/ERCC6 is a DNA-stimulated ATPase but is not a helicase and does not disrupt the ternary transcription complex of stalled RNA polymerase II. J Biol Chem. 1997b;272:1885–1890. doi: 10.1074/jbc.272.3.1885. [DOI] [PubMed] [Google Scholar]
- Stevnsner T, Nyaga S, de Souza-Pinto NC, van der Horst GT, Gorgels TG, Hogue BA, Thorslund T, Bohr VA. Mitochondrial repair of 8-oxoguanine is deficient in Cockayne syndrome group B. Oncogene. 2002;21:8675–8682. doi: 10.1038/sj.onc.1205994. [DOI] [PubMed] [Google Scholar]
- Sunesen M, Selzer RR, Brosh RM, Jr., Balajee AS, Stevnsner T, Bohr VA. Molecular characterization of an acidic region deletion mutant of Cockayne syndrome group B protein. Nucleic Acids Res. 2000;28:3151–3159. doi: 10.1093/nar/28.16.3151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tantin D, Kansal A, Carey M. Recruitment of the putative transcription-repair coupling factor CSB/ERCC6 to RNA polymerase II elongation complexes. Mol Cell Biol. 1997;17:6803–6814. doi: 10.1128/mcb.17.12.6803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorslund T, von Kobbe C, Harrigan JA, Indig FE, Christiansen M, Stevnsner T, Bohr VA. Cooperation of the Cockayne syndrome group B protein and poly(ADP-ribose) polymerase 1 in the response to oxidative stress. Mol Cell Biol. 2005;25:7625–7636. doi: 10.1128/MCB.25.17.7625-7636.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tornaletti S, Maeda LS, Kolodner RD, Hanawalt PC. Effect of 8-oxoguanine on transcription elongation by T7 RNA polymerase and mammalian RNA polymerase II. DNA Repair (Amst) 2004;3:483–494. doi: 10.1016/j.dnarep.2004.01.003. [DOI] [PubMed] [Google Scholar]
- Tornaletti S, Reines D, Hanawalt PC. Structural characterization of RNA polymerase II complexes arrested by a cyclobutane pyrimidine dimer in the transcribed strand of template DNA. J Biol Chem. 1999;274:24124–24130. doi: 10.1074/jbc.274.34.24124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tremeau-Bravard A, Riedl T, Egly JM, Dahmus ME. Fate of RNA polymerase II stalled at a cisplatin lesion. J Biol Chem. 2004;279:7751–7759. doi: 10.1074/jbc.M309853200. [DOI] [PubMed] [Google Scholar]
- Troelstra C, Landsvater RM, Wiegant J, van der Ploeg M, Viel G, Buys CH, Hoeijmakers JH. Localization of the nucleotide excision repair gene ERCC6 to human chromosome 10q11–q21. Genomics. 1992a;12:745–749. doi: 10.1016/0888-7543(92)90304-b. [DOI] [PubMed] [Google Scholar]
- Troelstra C, van Gool A, de Wit J, Vermeulen W, Bootsma D, Hoeijmakers JH. ERCC6, a member of a subfamily of putative helicases, is involved in Cockayne's syndrome and preferential repair of active genes. Cell. 1992b;71:939–953. doi: 10.1016/0092-8674(92)90390-x. [DOI] [PubMed] [Google Scholar]
- Tuo J, Chen C, Zeng X, Christiansen M, Bohr VA. Functional crosstalk between hOgg1 and the helicase domain of Cockayne syndrome group B protein. DNA Repair (Amst) 2002a;1:913–927. doi: 10.1016/s1568-7864(02)00116-7. [DOI] [PubMed] [Google Scholar]
- Tuo J, Jaruga P, Rodriguez H, Bohr VA, Dizdaroglu M. Primary fibroblasts of Cockayne syndrome patients are defective in cellular repair of 8-hydroxyguanine and 8-hydroxyadenine resulting from oxidative stress. Faseb J. 2003;17:668–674. doi: 10.1096/fj.02-0851com. [DOI] [PubMed] [Google Scholar]
- Tuo J, Jaruga P, Rodriguez H, Dizdaroglu M, Bohr VA. The cockayne syndrome group B gene product is involved in cellular repair of 8-hydroxyadenine in DNA. J Biol Chem. 2002b;277:30832–30837. doi: 10.1074/jbc.M204814200. [DOI] [PubMed] [Google Scholar]
- Tuo J, Muftuoglu M, Chen C, Jaruga P, Selzer RR, Brosh RM, Jr., Rodriguez H, Dizdaroglu M, Bohr VA. The Cockayne Syndrome group B gene product is involved in general genome base excision repair of 8-hydroxyguanine in DNA. J Biol Chem. 2001;276:45772–45779. doi: 10.1074/jbc.M107888200. [DOI] [PubMed] [Google Scholar]
- van der Horst GT, van Steeg H, Berg RJ, van Gool AJ, de Wit J, Weeda G, Morreau H, Beems RB, van Kreijl CF, de Gruijl FR, Bootsma D, Hoeijmakers JH. Defective transcription-coupled repair in Cockayne syndrome B mice is associated with skin cancer predisposition. Cell. 1997;89:425–435. doi: 10.1016/s0092-8674(00)80223-8. [DOI] [PubMed] [Google Scholar]
- van Gool AJ, Citterio E, Rademakers S, van Os R, Vermeulen W, Constantinou A, Egly JM, Bootsma D, Hoeijmakers JH. The Cockayne syndrome B protein, involved in transcription-coupled DNA repair, resides in an RNA polymerase II-containing complex. Embo J. 1997;16:5955–5965. doi: 10.1093/emboj/16.19.5955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venema J, Mullenders LH, Natarajan AT, van Zeeland AA, Mayne LV. The genetic defect in Cockayne syndrome is associated with a defect in repair of UV-induced DNA damage in transcriptionally active DNA. Proc Natl Acad Sci U S A. 1990;87:4707–4711. doi: 10.1073/pnas.87.12.4707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vermeulen W, van Vuuren AJ, Chipoulet M, Schaeffer L, Appeldoorn E, Weeda G, Jaspers NG, Priestley A, Arlett CF, Lehmann AR, et al. Three unusual repair deficiencies associated with transcription factor BTF2(TFIIH): evidence for the existence of a transcription syndrome. Cold Spring Harb Symp Quant Biol. 1994;59:317–329. doi: 10.1101/sqb.1994.059.01.036. [DOI] [PubMed] [Google Scholar]
- Viswanathan A, Doetsch PW. Effects of nonbulky DNA base damages on Escherichia coli RNA polymerase-mediated elongation and promoter clearance. J Biol Chem. 1998;273:21276–21281. doi: 10.1074/jbc.273.33.21276. [DOI] [PubMed] [Google Scholar]
- Wade MH, Chu EH. Effects of DNA damaging agents on cultured fibroblasts derived from patients with Cockayne syndrome. Mutat Res. 1979;59:49–60. doi: 10.1016/0027-5107(79)90194-5. [DOI] [PubMed] [Google Scholar]
- Wallace DC, Brown MD, Melov S, Graham B, Lott M. Mitochondrial biology, degenerative diseases and aging. Biofactors. 1998;7:187–190. doi: 10.1002/biof.5520070303. [DOI] [PubMed] [Google Scholar]
- Wong HK, Muftuoglu M, Beck G, Imam SZ, Bohr VA, Wilson DM., 3rd Cockayne syndrome B protein stimulates apurinic endonuclease 1 activity and protects against agents that introduce base excision repair intermediates. Nucleic Acids Res. 2007;35:4103–4113. doi: 10.1093/nar/gkm404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyatt MD, Pittman DL. Methylating agents and DNA repair responses: Methylated bases and sources of strand breaks. Chem Res Toxicol. 2006;19:1580–1594. doi: 10.1021/tx060164e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan X, Feng W, Imhof A, Grummt I, Zhou Y. Activation of RNA polymerase I transcription by cockayne syndrome group B protein and histone methyltransferase G9a. Mol Cell. 2007;27:585–595. doi: 10.1016/j.molcel.2007.06.021. [DOI] [PubMed] [Google Scholar]