

Abstract
Background: Ethnic differences may affect the phenotypic expression of genetic disorders. However, data regarding the effect of ethnicity on outcome in patients with genetic cardiac disorders are limited. We compared the clinical course of Caucasian and Japanese long QT type‐1 (LQT1) patients who were matched for mutations in the KCNQ1 gene.
Methods: The study population comprised 62 Caucasian and 38 Japanese LQT1 patients from the International LQTS Registry who were identified as having six identical KCNQ1 mutations. The biophysical function of the mutations was categorized into dominant‐negative (>50%) or haploinsufficiency (≤50%) reduction in cardiac repolarizing IKs potassium channel current. The primary end point of the study was the occurrence of a first cardiac event from birth through age 40 years.
Results: Japanese patients had a significantly higher cumulative rate of cardiac events (67%) than Caucasian patients (39%; P = 0.01). The respective frequencies of dominant negative mutations in the two ethnic groups were 63% and 28% (P < 0.001). In multivariate analysis, Japanese patients had an 81% increase in the risk of cardiac events (P = 0.06) as compared with Caucasians. However, when the biophysical function of the mutations was included in the multivariate model, the risk associated with Japanese ethnicity was no longer evident (HR = 1.05; P = 0.89). Harboring a dominant negative mutation was shown to be the most powerful and significant predictor of outcome (HR = 3.78; P < 0.001).
Conclusions: Our data indicate that ethnic differences in the clinical expression of LQTS can be attributed to the differences in frequencies of the specific mutations within the two populations.
Keywords: long‐QT syndrome, genetics, ethnicity
The congenital Long QT Syndrome (LQTS) is caused by mutations in eleven defined genes that encode channels that regulate sodium, potassium, and calcium currents, and by mutations that affect trafficking of ion channels, resulting in prolonged ventricular repolarization and an increased risk for sustained ventricular tachyarrhythmias. 1 , 2 , 3 , 4 Mutations in the KCNQ1 gene, also known as KvLQT1, cause the more common long QT type‐1 (LQT1) genotype by creating faulty alpha subunits that are part of the delayed rectifier repolarizing potassium (IKs) channel. 5 LQT1 mutations are associated with two distinct biophysical mechanisms: (1) coassembly or cellular trafficking defects of the mutant subunits that allow only normal, wild‐type, subunits to be successfully transported to the cell membrane, resulting in reduced channel function by ≤50%; (haploinsufficiency); and (2) formation of defective channels involving mutant subunits with the altered channel protein transported to the cell membrane, resulting in a dysfunctional channel having >50% reduction in channel current (dominant‐negative effect). 6 We have recently shown that the degree of ion channel dysfunction caused by the mutations is a significant predictor of outcome in LQT1 patients. 7
Studies reporting the detection of single nucleotide polymorphisms (SNPs) within different populations have shown that there is a large variability in the LQTS genes across countries and ethnic groups. 8 , 9 , 10 , 11 , 12 , 13 , 14 , 15 However, the effect of ethnicity on the phenotypic expression of LQTS has not been studied. To assess possible mechanisms by which ethnicity influences the risk of cardiac events in LQTS patients, we compared the clinical course of Japanese and Caucasian LQT1 patients from the International LQTS Registry who shared identical mutations in the KCNQ1 gene.
METHODS
Study Population
Study patients were drawn from a population of 600 subjects with genetically confirmed KCNQ1 mutations derived from 101 proband‐identified LQT1 families. Patients were enrolled in the U.S. portion of the International LQTS Registry (n = 425), The Netherlands' LQTS Registry (n = 93), and the Japanese LQTS Registry (n = 82), ethnicity was self reported. In our comparison study, Caucasian subjects from the U.S. and The Netherlands' LQTS Registries were matched with Japanese subjects harboring the same mutations. This analysis identified six identical LQT1 mutations in the two ethnic groups, resulting in a final study population of 100 patients (62 Caucasians and 38 Japanese). Patients with homozygous KCNQ1 mutations characteristic of the Jervell‐Lange Nielsen syndrome, or with a second mutation in another LQTS ion‐channel gene were excluded from the study. All subjects or their guardians provided informed consent for the genetic and clinical studies.
Data Collection and Management
For each patient, data on personal and family history, cardiac events, and therapy were systematically recorded at each visit or medical contact on prospectively designed forms. Clinical data included patient and family history, demographics, electrocardiographic (ECG), therapeutic, and cardiac event information. Upon enrollment in the LQTS Registry, a 12‐lead ECG was obtained from each patient. From this first recorded ECG, the duration of the QT interval was assessed from lead II (or lead I or III if the QT interval could not be measured from lead II) and corrected for heart rate (QTc) using Bazett's formula. Data common to all three LQTS registries involving genetically identified LQT1 patients were electronically merged into a common database.
Genotype Characterization
The KCNQ1 mutations were identified using standard genetic tests performed in academic molecular‐genetic laboratories. Genetic alterations of the amino acid sequence were characterized by location in the channel protein, the type of mutation (missense, splice site, in‐frame insertions/deletions, nonsense [stop codon], and frameshift), and the biophysical functional effect of the mutation (dominant negative or haploinsufficiency; Table 1). The biophysical function was classified based on studies reported in literature; 12 , 16 , 17 , 18 mutations that have not yet been characterized were assumed haploinsufficiency if identified as splice site, nonsense, in‐frame deletion, or frame‐shift mutations. The transmembrane region of the KCNQ1‐encoded channel included 6 membrane‐spanning segments [S1–S6], and was defined as the coding sequence involving amino acid residues from 120 through 355 (comprising two cytoplasmic loops between S2–S3 and S4–S5, and the S5‐pore–S6 region), with the N‐terminus region defined before residue 120, and the C‐terminus region after residue 355.
Table 1.
KCNQ1 Mutations in Study Patients
| Mutation | Caucasian N = 62 | Japanese N = 38 | Position | Exon | Type | Biophysical Functional Effect |
|---|---|---|---|---|---|---|
| A341V | 4 (6%) | 16 (42%)* | S6 | 7 | Missense | Dominant negative 12 |
| G314S | 4 (6%) | 4 (11%) | Pore loop | 7 | Missense | Dominant negative 16 |
| R243C | 9 (15%) | 4 (11%) | S4/S5 | 5 | Missense | Dominant negative 17 |
| delF340 | 4 (6%) | 3 (8%) | S6 | 7 | In‐frame deletion | Haploinsufficiency† |
| A344A/sp | 20 (32%) | 7 (18%) | S6 | 7 | Splice site | Haploinsufficiency† |
| G269S | 21 (34%) | 4 (11%)* | S5 | 6 | Missense | Loss of function 18 |
*P value for the comparison between Caucasian and Japanese patients < 0.05.
†Splice site and in‐frame deletion mutation‐types were assumed haploinsufficiency.
End Points
The end point of the study was the first occurrence of an LQTS‐related cardiac event (syncope, aborted cardiac arrest [ACA] or LQTS‐related death). In the primary analysis we compared the outcome by ethnicity, with and without adjustment for biophysical function (e.g., dominant negative vs haploinsufficiency). In a secondary exploratory analysis, the ethnic group comparison was further stratified by gender.
Statistical Analysis
The clinical characteristics of Caucasian and Japanese subjects were compared using the chi‐square test, and the Fisher's exact test, as appropriate. The Kaplan‐Meier life‐table method was used to assess the time to a first LQTS‐related cardiac event and the cumulative event rates for each ethnic group and mutation‐type. The results were compared using the log‐rank statistic.
Multivariate Cox proportional hazards regression modeling was carried out to determine the significant and independent contribution of each factor as a predictor for a first cardiac event during follow‐up. Prespecified factors included gender, ethnicity, QTc interval duration >500 ms, the biophysical functional effect of the mutation (e.g., dominant negative vs haploinsufficiency), and time‐dependent beta‐blocker therapy. Data from the International LQTS Registry have shown that an implanted cardioverter defibrillator (ICD) is very effective in preventing fatal LQTS‐related cardiac events. 19 Therefore, to validate the consistency of the results the non‐ICD population, all analyses were repeated with censoring of patients upon implantation of an ICD.
The statistical software used for the analyses was SAS version 9.13 (SAS, Cary, NC, USA). A 2‐sided probability value < 0.05 was used for declaring statistical significance.
RESULTS
Clinical and ECG characteristics of Caucasian and Japanese study patients are shown in Table 2, panel A. Gender distribution and baseline QTc duration were similar between the two ethnic groups. However, when the baseline QTc was analyzed by gender, Japanese male patients exhibited a significantly longer QTc duration as compared with their Caucasian counterparts. β‐blocker therapy was administered to a similar proportion of Japanese and Caucasian patients, whereas ICD implantation occurred only in Caucasian LQT1 subjects.
Table 2.
Characteristics of Caucasian and Japanese LQT1 Patients
| Caucasian (n = 62) | Japanese (n = 38) | P‐Value | |
|---|---|---|---|
| A. Characteristics | |||
| Female, n (%) | 38 (61) | 21 (55) | 0.55 |
| QTc, ms | 487 ± 53 | 497 ± 35 | 0.30 |
| Female, ms | 498 ± 59 | 496 ± 39 | 0.92 |
| Male, ms | 468 ± 38 | 499 ± 32 | 0.02 |
| Beta‐blocker use, n (%) | 28 (45) | 14 (36) | 0.41 |
| ICD, n | 10 | 0 | |
| Pacemaker, n (%) | 5 (8) | 1 (3) | 0.27 |
| Sympathectomy, n | 0 | 0 | |
| Mutation characteristics | |||
| Missense mutation, n (%) | 38 (61) | 28 (74) | 0.20 |
| Dominant negative, n (%) | 17 (28) | 24 (63) | <0.001 |
| B. Cardiac Events | |||
| Any cardiac event, n (%)* | 25 (40) | 22 (58) | 0.09 |
| Syncope, n (%) | 21 (34) | 17 (45) | 0.28 |
| Aborted cardiac arrest, n (%) | 4 (7) | 6 (16) | 0.13 |
| Sudden cardiac death, n | 5 (8) | 2 (5) | 0.59 |
| Other Death, n (%) | 3 (5) | 0 (0) | 0.17 |
| Age at first cardiac event, y | 11 | 5 | 0.12 |
| Syncope as a first cardiac event | 21 (34) | 16 (42) | 0.41 |
| Aborted cardiac arrest as a first cardiac event | 1 (2) | 5 (13) | 0.02 |
| Sudden cardiac death as first a cardiac event | 3 (5) | 1 (3) | 0.59 |
Plus‐minus values are means ± SD.
*Denotes the number of patients who experienced at least one cardiac event during follow‐up. Mean QTc values include 87 cases; 13 cases were missing: 6 males and 7 females.
ICD = implantable cardioverter defibrillator.
The two ethnic groups exhibited significant differences in the frequencies of individual mutations (Table 1) and their biophysical functional effects, with the higher‐risk dominant negative mutations being significantly more frequent among Japanese patients than among Caucasians (Table 2, panel A).
Cardiac Events During Follow‐Up
A comparison of the frequency of cardiac events experienced between the two populations revealed a higher event rate among Japanese patients (Table 2, panel B). Notably, the frequency of presentation with ACA as a first cardiac event was significantly higher in Japanese patients as compared with Caucasians. Accordingly, the cumulative probability of a first cardiac event from birth through age 40 years was significantly higher among Japanese patients (67%) than among Caucasians (39%, P = 0.007; Fig. 1). Furthermore, cardiac events occurred at a significantly earlier age in the former ethnic group. Thus, at age 10 years Japanese and Caucasians exhibited a respective cardiac event rate of 43% and 18% (Fig. 1).
Figure 1.
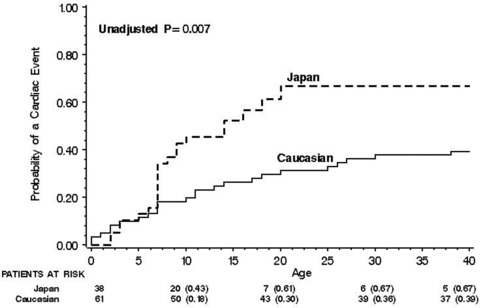
Kaplan‐Meier estimates of the probability of a first cardiac event from birth through age 40 years by ethnicity.
Comparison of event rates for the individual mutations showed a lower rate for mutations resulting in haploinsufficiency (G269S, A344A/sp, and delF340), and an increasing event rate for mutations with dominant negative ion current effects (R243C, G314S, and A341V; Fig. 2). Accordingly, dominant negative mutation carriers exhibited a significantly greater probability of a first cardiac event than those with haploinsufficiency (Fig. 3), and this finding was consistent in both the Japanese (82% vs 38%, respectively; P = 0.001) and Caucasian (71% vs 29%, respectively; P = 0.001) groups.
Figure 2.
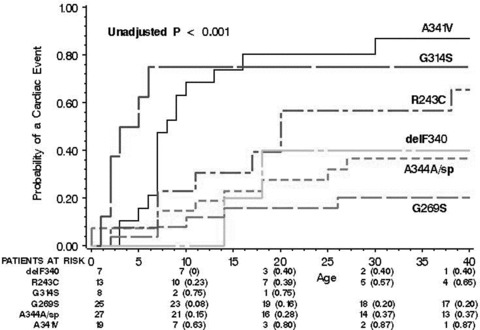
Kaplan‐Meier estimates of the probability of a first cardiac event from birth through age 40 years for the six individual KCNQ1 mutations included in the study.
Figure 3.
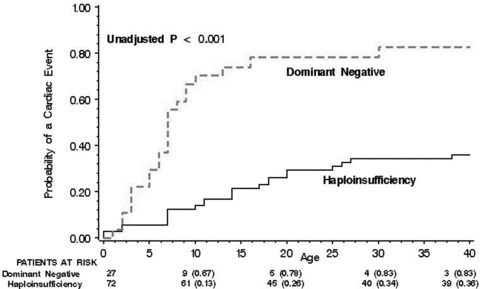
Kaplan‐Meier estimates of the probability of a first cardiac event from birth through age 40 years by the biophysical functional effect of the mutations.
Multivariate Analysis
Cox proportional hazards regression modeling demonstrated that Japanese ethnicity was associated with a marginally significant 81% increase in the risk of cardiac events as compared to Caucasian ethnicity, after adjustment for gender, QTc duration, and time‐dependent beta‐blocker therapy (Table 3, panel A). However, the risk associated with ethnicity was no longer evident when the biophysical functional effect of the mutation was added to the multivariate model (Table 3, panel B). In this model, a dominant negative mutation effect was shown to be the most powerful and significant predictor of outcome in study patients, and was associated with nearly a fourfold increase in the risk of cardiac events during follow‐up. The risk associated with dominant negative mutations was consistent for both Japanese and Caucasian patients (P value for ethnicity × mutation effect interaction = 0.54).
Table 3.
Multivariate Analysis*
| Variable | Hazard Ratio | 95% Confidence Interval | P Value |
|---|---|---|---|
| A. Risk of a first cardiac event by | |||
| ethnicity after adjustment for, gender, | |||
| QTc and time‐dependent beta‐blocker therapy | |||
| Japan:Caucasian | 1.81 | 0.96–3.40 | 0.06 |
| Female:Male | 0.72 | 0.37–1.38 | 0.32 |
| QTc>500:QTc≤500 ms | 1.71 | 0.90–3.24 | 0.10 |
| Beta‐blockers no beta‐blockers† | 0.31 | 0.07–1.32 | 0.11 |
| B. Risk of a first cardiac event by | |||
| ethnicity after further adjustment for | |||
| the biophysical function of the mutations | |||
| Japan:Caucasian | 1.05 | 0.53–2.10 | 0.89 |
| Dominant negative:haploinsufficiency | 3.78 | 1.90–7.51 | <0.001 |
| Female:Male | 0.76 | 0.39–1.49 | 0.43 |
| QTc>500:QTc≤500 ms | 1.50 | 0.78–2.90 | 0.22 |
| Beta‐blockers No beta‐blockers† | 0.33 | 0.08–1.40 | 0.13 |
*The analysis involved 87 cases; 13 cases were omitted due to missing QTc values; virtually identical results were obtained when analysis were repeating with censoring of patients upon implantation of an ICD.
†Beta‐blocker therapy was analyzed as a time‐dependent variable in the multivariate model.
Gender Differences within Ethnic Groups
We have previously shown that in LQT1 patients the risk of cardiac events is affected by age and gender. 20 We have therefore carried out a further exploratory analysis, in which the clinical course of males and females was compared and related to the biophysical functional effect of the mutations in the 2 ethnic groups. Overall, gender did not contribute significantly to outcome in study patients after adjustment for ethnicity or mutation effect (Table 3, panel A and panel B, respectively). However, when gender was related to the biophysical functional effect of the LQT1 mutations, dominant negative mutations were shown to exhibit a different gender effect in the two ethnic groups. The risk of cardiac events among Japanese males with dominant negative mutations was significantly higher than among Japanese females with the same mutations (HR = 3.46 [95% CI 1.06–11.27]; P = 0.03), whereas male and female Caucasians with dominant negative mutations exhibited a similar risk of cardiac events during follow‐up (HR = 0.79 [95% CI 0.13–4.65]; P = 0.80).
DISCUSSION
In the present study, we sought to explore the extent of phenotypic variation that occurs across two ethnic groups that possess the same LQT1 mutations. Our findings suggest that the clinical severity of LQTS in different ethnic groups is largely determined by the frequency distribution of the LQT1 mutations in the population.
Ethnicity and Single Nucleotide Polymorphisms
Genotype‐phenotype relationships and risk factors for cardiac events in LQTS patients have been the topic of much investigation. 20 , 21 , 22 , 23 , 24 , 25 However, previous studies that have analyzed the variability in disease expression and phenotype severity did not assess the effect of ethnicity on outcome in LQTS patients. Modifier genes and/or environmental factors may differ among ethnic groups and may explain why a given genotype confers variable susceptibility to cardiac arrhythmias in different ethnic groups. Thus, the identification of risk factors among LQTS populations with similar mutations may be important for risk‐stratification in this genetic disorder. Ethnic‐specific polymorphisms on the LQTS‐related genes have been described in previous reports. 8 , 9 , 10 , 11 , 12 , 13 , 14 , 15 , 25 , 26 , 27 Notably, Ackerman et al. 8 studied 744 healthy black, white, Asian and Hispanic subjects and reported 49 distinct amino acid‐altering variants of four LQTS‐related potassium channel genes. These findings reflect the genetic diversity that may be a source of phenotype variation among different ethnic populations.
The evidence that certain polymorphisms on the KCNQ1 gene may increase susceptibility to cardiac events has given credence to the concept of modifier genes. Kubota et al. 13 identified six patients that exhibited mild signs of LQTS as carriers of the G643S polymorphism, which is found in approximately 11% of the Japanese population. Through patch‐clamp tests, it was found that this polymorphism, coexpressed with wild‐type KCNQ1 and KCNE1, conferred approximately a 30% reduction in current. This same polymorphism has also been associated with QT intervals that vary with gender and age in Japanese subjects. 26 In addition, ethnic‐specific SNPs have been recognized as important in the correct diagnosis and clinical management of LQTS suspected patients. In one report, a previously established KCNQ1 mutation, P488R, was found to be a common polymorphism in 14% of healthy Chinese volunteers after a healthy child of a proband was mistakenly diagnosed with LQTS while his undiagnosed brother was later found to have a novel HERG mutation. 27 Therefore, diagnosis based on genotyping requires correct discrimination of genetic polymorphisms from pathologic mutations, which may be confounded by ethnic genetic variability.
It has been previously suggested that the observed phenotypic differences between Caucasian and Japanese LQTS patients may be attributed to the effect of ethnic‐specific common polymorphisms. 25 Evidence from the current study suggests that the clinical course of individual patients is dependent on the biophysical function of their mutations and is independent of ethnic origin. Japanese patients possessed a larger proportion of dominant negative mutations that are powerful and significant predictors of outcome in both ethnic groups, whereas the frequency of the lower‐risk, haploinsufficiency mutations, was significantly higher among Caucasians. Accordingly, the risk of cardiac events among the Japanese patients was higher than among the Caucasians patients without adjustment for mutation effect, whereas a similar ethnic risk was shown when the biophysical functional effect of the mutations was incorporated into the multivariate model.
Notably, 42% of Japanese patients harbored the dominant negative A341V mutation compared with only 6% of Caucasian patients. The A341V mutation has been associated with a severe clinical course and early onset disease. 12 Consistently, in the current study we have shown that Japanese and Caucasian patients who carried the A341V mutation experienced the highest rate of cardiac events, and at an earlier age, compared to the other five shared mutations (Fig. 2). Eighty percent of the patients who were identified as carriers of the A341V mutation had a cardiac event prior to age 20 years. Therefore, our findings suggest that identification of the functional effect of KCNQ1 mutations should comprise a primary component in the risk stratification and clinical management of LQT1 patients, regardless of their ethnic origin.
Possible Gender‐Related Phenotype Variability in Japanese and Caucasian Patients
A previous study from the International LQTS Registry demonstrated an age‐gender interaction in LQT1 patients, in which the risk of cardiac events is higher in males than females before adolescence, with risk‐reversal in the postadolescence period. 20 Possibly due to sample size limitations, we did not show an age‐gender interaction in the two ethnic groups in the current study population. However, our secondary exploratory analysis suggests a possible gender‐related difference in the functional effect of LQT1 mutation between the two ethnic groups. Japanese males who possessed dominant negative mutations had a significantly higher risk of cardiac events than Japanese females with the same mutations, whereas the effect of the dominant negative mutations did not show a significant gender‐related difference in the Caucasian population. These findings may be due to the fact that Japanese patients with dominant negative mutations experienced a relatively high rate of cardiac events during childhood, a time‐period that has been shown to be associated with a higher risk among LQT1 males. 20 It is also possible that modifier genes affect gender differences in risk in the two ethnic groups.
Limitations
The Japanese, United States, and the Netherlands' LQTS Registries have similar enrollment criteria. However, it is possible that higher risk patients were preferentially enrolled in the Japanese registry, leading to an unbalanced mutation distribution in the two ethnic groups. Therefore, the frequencies of individual KCNQ1 mutations in different ethnic groups need to be further assessed in large population studies. Nevertheless, our findings suggest that the risk associated with the biophysical function of a mutation in an individual is more important than the risk associated with his/her ethnic origin.
The findings regarding gender differences in the risk between the two ethnic groups should be regarded as secondary and preliminary due to sample size limitations, and therefore need to be further validated in future studies.
Conclusions and Clinical Implications
The growing data regarding identification of gene defects in congenital LQTS has revolutionized our understanding of the basic mechanisms underlying QT prolongation and cardiac arrhythmias, and raises the possibility of mutation‐specific therapeutic intervention. The LQT1 genotype accounts for approximately 50% of genotyped LQTS patients, and has shown to be associated with variable clinical expression, incomplete penetrance, 1 and ethnic‐specific phenotypic variation. 25 Our findings suggest that, regardless of ethnic origin, risk‐assessment in LQT1 patients should consider the biophysical functional effect of the mutation in an individual as an important determinant of outcome.
Financial support: This study was supported in part by research grants HL‐33843 and HL‐51618 from the National Institutes of Health, Bethesda, Maryland.
REFERENCES
- 1. Splawski I, Shen J, Timothy KW, et al Spectrum of mutations in long‐QT syndrome genes KVLQT1, HERG, SCN5A, KCNE1, and KCNE2. Circulation 2000;102:1178–1185. [DOI] [PubMed] [Google Scholar]
- 2. Wang Q, Shen J, Splawski I, et al SCN5A mutations associated with an inherited cardiac arrhythmia, long QT syndrome. Cell 1995;80:805–811. [DOI] [PubMed] [Google Scholar]
- 3. Sanguinetti MC, Jiang C, Curran ME, et al A mechanistic link between an inherited and an acquired cardiac arrhythmia: HERG encodes the IKr potassium channel. Cell 1995;81:299–307. [DOI] [PubMed] [Google Scholar]
- 4. Cronk LB, Ye B, Kaku T, et al Novel mechanism for sudden infant death syndrome: Persistent late sodium current secondary to mutations in caveolin‐3. Heart Rhythm 2007;4:161–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Wang Q, Curran ME, Splawski I, et al Positional cloning of a novel potassium channel gene: KVLQT1 mutations cause cardiac arrhythmias. Nat Genet 1996;12:17–23. [DOI] [PubMed] [Google Scholar]
- 6. Wang Z, Tristani‐Firouzi M, Xu Q, et al Functional effects of mutations in KvLQT1 that cause long QT syndrome. J Cardiovasc Electrophysiol 1999;10:817–826. [DOI] [PubMed] [Google Scholar]
- 7. Moss AJ, Shimizu W, Wilde AA, et al Clinical aspects of type‐1 long‐QT syndrome by location, coding type, and biophysical function of mutations involving the KCNQ1 gene. Circulation 2007;115:2481–2489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Ackerman MJ, Tester DJ, Jones GS, et al Ethnic differences in cardiac potassium channel variants: Implications for genetic susceptibility to sudden cardiac death and genetic testing for congenital long QT syndrome. Mayo Clin Proc 2003;78:1479–1487. [DOI] [PubMed] [Google Scholar]
- 9. Modell SM, Lehmann MH. The long QT syndrome family of cardiac ion channelopathies: A HuGE review. Genet Med 2006;8:143–155. [DOI] [PubMed] [Google Scholar]
- 10. Niu DM, Hwang B, Hwang HW, et al A common SCN5A polymorphism attenuates a severe cardiac phenotype caused by a nonsense SCN5A mutation in a Chinese family with an inherited cardiac conduction defect. J Med Genet 2006;43:817–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Chen J, Xie X, Zhu J, et al Single‐nucleotide polymorphisms in SCN5A gene in Chinese Han population and their correlation with cardiac arrhythmias. Genet Med 2004;6:159. [DOI] [PubMed] [Google Scholar]
- 12. Brink PA, Crotti L, Corfield V, et al Phenotypic variability and unusual clinical severity of congenital long‐QT syndrome in a founder population. Circulation 2005;112:2602–2610. [DOI] [PubMed] [Google Scholar]
- 13. Kubota T, Horie M, Takano M, et al Evidence for a single nucleotide polymorphism in the KCNQ1 potassium channel that underlies susceptibility to life‐threatening arrhythmias. J Cardiovasc Electrophysiol 2001;12:1223–1229. [DOI] [PubMed] [Google Scholar]
- 14. Hiraoka M. Inherited arrhythmic disorders in Japan. J Cardiovasc Electrophysiol 2003;14:431–434. [DOI] [PubMed] [Google Scholar]
- 15. Fodstad H, Bendahhou S, Rougier JS, et al Molecular characterization of two founder mutations causing long QT syndrome and identification of compound heterozygous patients. Ann Med 2006;38:294–304. [DOI] [PubMed] [Google Scholar]
- 16. Kobori A, Sarai N, Shimizu W, et al Additional gene variants reduce effectiveness of beta‐blockers in the LQT1 form of long QT syndrome. J Cardiovasc Electrophysiol 2004;15:190–199. [DOI] [PubMed] [Google Scholar]
- 17. Franqueza L, Lin M, Shen J, et al Long QT syndrome‐associated mutations in the S4‐S5 linker of KvLQT1 potassium channels modify gating and interaction with minK subunits. J Biol Chem 1999;274:21063–21070. [DOI] [PubMed] [Google Scholar]
- 18. Murray A, Potet F, Bellocq C, et al Mutation in KCNQ1 that has both recessive and dominant characteristics. J Med Genet 2002;39:681–685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Zareba W, Moss AJ, Daubert JP, et al Implantable cardioverter defibrillator in high‐risk long QT syndrome patients. J Cardiovasc Electrophysiol 2003;14:337–341. [DOI] [PubMed] [Google Scholar]
- 20. Zareba W, Moss AJ, Locati EH, et al International Long QT Syndrome Registry. Modulating effects of age and gender on the clinical course of long QT syndrome by genotype. J Am Coll Cardiol 2003;42:103–109. [DOI] [PubMed] [Google Scholar]
- 21. Moss AJ, Schwartz PJ, Crampton RS, et al The long QT syndrome. Prospective longitudinal study of 328 families. Circulation 1991;84:1136–1144. [DOI] [PubMed] [Google Scholar]
- 22. Priori SG, Napolitano C, Schwartz PJ, et al Association of long QT syndrome loci and cardiac events among patients treated with beta‐blockers. JAMA 2004;292:1341–1344. [DOI] [PubMed] [Google Scholar]
- 23. Zareba W, Moss AJ, Schwartz PJ, et al Influence of genotype on the clinical course of the long‐QT syndrome. International Long‐QT Syndrome Registry Research Group. N Engl J Med 1998;339:960–965. [DOI] [PubMed] [Google Scholar]
- 24. Priori SG, Schwartz PJ, Napolitano C, et al Risk stratification in the long‐QT syndrome. N Engl J Med 2003;348:1866–1874. [DOI] [PubMed] [Google Scholar]
- 25. Shimizu W. The long QT syndrome: Therapeutic implications of a genetic diagnosis. Cardiovasc Res 2005;67:347–56. [DOI] [PubMed] [Google Scholar]
- 26. Ozawa T, Ito M, Tamaki S, et al Gender and age effects on ventricular repolarization abnormality in Japanese general carriers of a G643S common single nucleotide polymorphism for the KCNQ1 gene. Circ J 2006;70:645–650. [DOI] [PubMed] [Google Scholar]
- 27. Sharma D, Glatter KA, Timofeyev V, et al Characterization of a KCNQ1/KVLQT1 polymorphism in Asian families with LQT2: Implications for genetic testing. J Mol Cell Cardiol 2004;37:79–89. [DOI] [PubMed] [Google Scholar]