

Abstract
Understanding the molecular mechanisms that regulate cellular proliferation and differentiation is a central theme of developmental biology. MicroRNAs (miRNAs) are a class of regulatory RNAs of ~22 nucleotides that post-transcriptionally regulate gene expression1,2. Increasing evidence points to the potential role of miRNAs in various biological processes3–8. Here we show that miRNA-1 (miR-1) and miRNA-133 (miR-133), which are clustered on the same chromosomal loci, are transcribed together in a tissue-specific manner during development. miR-1 and miR-133 have distinct roles in modulating skeletal muscle proliferation and differentiation in cultured myoblasts in vitro and in Xenopus laevis embryos in vivo. miR-1 promotes myogenesis by targeting histone deacetylase 4 (HDAC4), a transcriptional repressor of muscle gene expression. By contrast, miR-133 enhances myoblast proliferation by repressing serum response factor (SRF). Our results show that two mature miRNAs, derived from the same miRNA polycistron and transcribed together, can carry out distinct biological functions. Together, our studies suggest a molecular mechanism in which miRNAs participate in transcriptional circuits that control skeletal muscle gene expression and embryonic development.
To understand the potential involvement of miRNAs in skeletal muscle proliferation and differentiation, we analyzed the expression of miRNAs during skeletal muscle differentiation using an established microarray analysis9. We used C2C12 myoblasts because these cells faithfully mimic skeletal muscle differentiation in vitro, as shown by their induction to terminally differentiated myotubes when serum is withdrawn from the culture medium10–12. We found that the expression of a few of the miRNAs examined was upregulated in differentiated C2C12 myoblasts or myotubes (Fig. 1a and Supplementary Fig. 1 online). This increase in expression of miR-1 and miR-133 in differentiated myoblasts was confirmed by RNA blot analysis (Fig. 1b and Supplementary Fig. 2 online).
Figure 1.

Expression of miR-1 and miR-133 in cardiac and skeletal muscle during development. (a) miRNA array expression data from C2C12 myoblasts cultured in growth medium (GM) or in differentiation medium (DM) for 0, 1, 3 or 5 d. Normalized log2 data were hierarchically clustered by gene and are plotted as a heat map. The signal ranged from a fourfold decrease to a fourfold increase. Yellow denotes high expression and blue denotes low expression relative to the median; only the miRNA nodes that were upregulated in differentiation medium are shown. (b) Northern blot analysis of miR-1 and miR-133 expression using total RNA isolated from C2C12 myoblasts cultured in growth medium or in differentiation medium DM for 0, 1, 3 or 5 d. tRNAs were used as a loading control. (c) Northern blot analysis of miR-1 and miR-133 expression in adult mouse tissues. (d) Northern blot analysis of miR-1 and miR-133 expression in E13.5 and E16.5 mouse tissues. (e) Northern blot analysis of miR-1 and miR-133 expression in neonatal mouse tissues. The same amount of total RNAs from adult heart and skeletal muscle was loaded on the blots to compare embryonic and neonate RNA (d,e).
miR-1 and miR-133 are specifically expressed in adult cardiac and skeletal muscle tissues, but not in other tissues tested13–15 (Fig. 1c and Supplementary Fig. 3 online). Little is known, however, about the spatio-temporal distribution of specific miRNAs during mammalian development. We therefore examined the expression of miR-1 and miR-133 in mouse embryos and neonates. miR-1 and miR-133 are expressed in very small amounts in the developing hearts and skeletal muscle of embryonic day 13.5 (E13.5) and E16.5 mice (Fig. 1d and Supplementary Fig. 3). Increasing expression of miR-1 and miR-133 was found in neonatal hearts and skeletal muscle, although it was still substantially lower than that in adult tissues (Fig. 1e and Supplementary Fig. 3). These data are consistent with findings in zebrafish showing that most miRNAs are expressed relatively late during embryogenesis16.
miR-1 and miR-133 are clustered together both on mouse chromosome 2, where they are separated by 9.3 kb, and on mouse chromosome 18, where they are separated by 2.5 kb (Supplementary Fig. 4 online and ref. 14). We carried out northern blot analysis using genomic probes of ~300 bp including the miR-1 or miR-133 sequence (Supplementary Fig. 4). The probes for miR-1 and miR-133 on chromosome 18 detected a single primary transcript of ~6 kb in total RNAs isolated from heart and skeletal muscle (Supplementary Fig. 4), indicating that miR-1 and miR-133 are indeed transcribed together. Whereas the probes for miR-1 and miR-133 on chromosome 2 both detected a transcript of ~10 kb in the heart and skeletal muscle, the miR-133 probe hybridized to two additional transcripts of ~4.5 kb and ~2.2 kb, and the miR-1 probe detected an additional major transcript of ~6 kb (Supplementary Fig. 4), suggesting that post-transcriptional processing may be involved in the production of miR-1 and miR-133. Together, our data indicate that cardiac-specific and skeletal muscle–specific expression of miR-1 and miR-133 is dictated at the primary transcription step.
We reasoned that the regulatory elements that control the transcription of both the chromosome 2 and the chromosome 18 miR-1 and miR-133 clusters are probably conserved. We therefore used sequence analysis to identify a highly conserved region (~2 kb) that lies about 50 kb upstream of the miR-1–miR-133 clusters on both chromosome 2 and chromosome 18 (Supplementary Fig. 5 online). When this genomic fragment from chromosome 2 was used to drive the expression of a dsRed reporter gene in transgenic X. laevis, we found cardiac-specific and skeletal muscle–specific expression of the transgene (Supplementary Fig. 5).
To assess the function of miR-1 and miR-133 in skeletal muscle, we first attempted to overexpress miR-1 and miR-133 in mammalian cells. We tested and validated the expression and activity of both miRNAs by RNA blot analysis, as well as by using miR-1 and miR-133 ‘sensors’17, in which the complementary sequences of miR-1 and miR-133 are cloned downstream of a dsRed coding sequence (Supplementary Fig. 6 online and data not shown). We transfected C2C12 myoblasts with miR-1 or miR-133 and then either maintained cells in growth medium or transferred them to differentiation medium after transfection. miR-1 strongly enhanced myogenesis, as indicated by an increase in expression of the respective early and late myogenic markers myogenin and myosin heavy chain (MHC), as well as other myogenic markers including MyoD, MEF2 and skeletal α-actin (Fig. 2 and Table 1). miR-1 induced the expression of myogenic marker genes in cells maintained in the log-phase growth condition (Fig. 2c) and in the differentiation condition (Fig. 2d,e).
Figure 2.
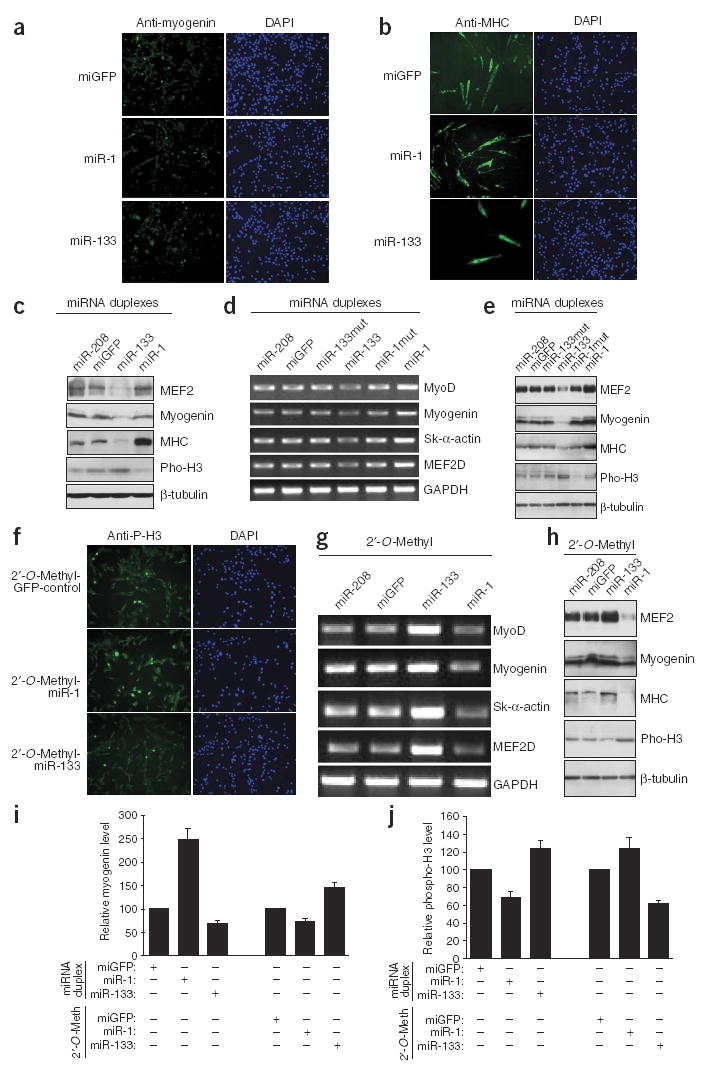
Regulation of myoblast proliferation and differentiation by miR-1 and miR-133. C2C12 myoblasts cultured in growth medium were electroporated with double-stranded miR-1, miR-133 or control miGFP. (a,b) Cells were continuously cultured in growth medium for 24 h after transfection and then transferred to differentiation medium for either 12 h before immunostaining for myogenin (a) or 36 h before immunostaining for MHC (b). (c–e) C2C12 myoblasts cultured in GM were electroporated with double-stranded miR-1, miR-133 (or their mutants as indicated) or control miR-208 or miGFP and cultured for 24 h before being either subjected to immunoblotting with the indicated antibodies (c), or transferred to differentiation medium for 24 h and subjected to RT-PCR for the indicated genes (d) or to immunoblotting with the indicated antibodies (e). (f–h) C2C12 myoblasts cultured in GM were electroporated with a 2′-O-Methyl antisense oligonucleotide inhibitors of miR-1, miR-133, miR-208 or miGFP as controls. Cells were cultured in growth medium for 24 h after transfection and then transferred into differentiation medium for 12 h before immunostaining for phospho–histone H3 (f), 24 h before RT-PCR for the indicated genes (g) or 24 h before immunoblotting with the indicated antibodies (h). (i,j) C2C12 myoblasts cultured in growth medium were electroporated with miRNA duplexes or with 2′-O-Methyl antisense oligonucleotide inhibitors as indicated. Cells were cultured in growth medium for 24 h after transfection and then transferred into DM for 12 h before immunostaining for myogenin (i) or phospho–histone H3 (j). Positive stained cells were counted and data are presented as the expression level relative to a miGFP control (100%).
Table 1.
Effect on myogenic proliferation and differentiation of miR-1 and miR-133 overexpression and knock down
| Differentiation medium (8 h)
|
Differentiation medium (12 h)
|
Differentiation medium (24 h)
|
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Myogenin-positive cells
|
Myogenin-positive cells
|
Phospho-H3–positive cells
|
Myogenin-positive cells
|
Phospho-H3–positive cells
|
MHC-positive cells
|
|||||||
| Treatment | Total number | Relative to control (%) | Total number | Relative to control (%) | Total number | Relative to control (%) | Total number | Relative to control (%) | Total number | Relative to control (%) | Total number | Relative to control (%) |
| MiGFP | 172 | 100 | 93 | 100 | 135 | 100 | 118 | 100 | 137 | 100 | 22 | 100 |
| miR-1 | 206 | 121 | 230 | 247.3 | 93 | 68.9 | 251 | 212.7 | 76 | 55.5 | 56 | 254.5 |
| miR-133 | 89 | 51.7 | 68 | 73.1 | 168 | 124.4 | 93 | 78.8 | 201 | 146.7 | 12 | 54.5 |
| 2′-O-Methyl-GFP | 146 | 100 | 145 | 100 | 172 | 100 | 348 | 100 | 207 | 100 | 22 | 100 |
| 2′-O-Methyl-miR-1 | 120 | 82.2 | 98 | 67.6 | 214 | 124.4 | 299 | 85.9 | 283 | 136.7 | 18 | 81.8 |
| 2′-O-Methyl-miR-133 | 205 | 140.4 | 211 | 145.5 | 107 | 62.2 | 498 | 143.1 | 191 | 92.3 | 44 | 200 |
C2C12 myoblasts cultured in growth medium were electroporated with a double-stranded miRNA duplex or 2′-O-Methyl antisense oligoribonucleotides targeted towards miR-1, miR-133 or miGFP as a negative control. After 36 h, growth medium was replaced with differentiation medium for 8, 12 or 24 h and cells were fixed for immunohistochemical analysis using antibodies to myogenin, to phospho–histone H3 and to MHC. Positive cells were counted out of 5,000 DAPI-positive cells in a randomly chosen field. Three independent assays were done with comparable results.
Accelerated myogenic differentiation induced by miR-1 was accompanied by a decrease in cell proliferation, as marked by a significant decrease in expression of phosphorylated histone H3 (phospho–histone H3; Fig. 2c,e and Table 1). We found that miR-1-induced myogenesis is specific, because overexpression of a green fluorescent protein control RNA duplex (miGFP) or miR-208, which is not endogenously expressed in skeletal muscle cells, showed no effect (Fig. 2a–e). In addition, mutations introduced into miR-1 ‘seed’ sequences abolished its ability to activate myogenic gene expression (Fig. 2d,e). By contrast, overexpression of miR-133 repressed the expression of myogenin and MHC (Fig. 2a–e and Table 1) and promoted myoblast proliferation (Fig. 2c,e and Table 1). Again, we found that the effect of miR-133 on myoblasts proliferation is specific, because controls showed no effect and the mutation introduced abolished the function of miR-133 (Fig. 2a–e,j).
We carried out a reciprocal experiment wherein we transfected C2C12 myoblasts with 2′-O-methyl antisense inhibitory oligoribonucleotides, which have been shown to inhibit the function of miRNAs18,19, targeted towards miR-1 or miR-133 (or control miGFP and miR-208). Cells transfected with the miR-1 inhibitor showed inhibition of myogenesis and promotion of myoblast proliferation, as indicated by a decrease in myogenic markers and an increase in phospho–histone H3 (Fig. 2f–i and Table 1). Consistent with the role of miR-133 in promoting myoblast proliferation and repressing differentiation, inhibition of miR-133 caused an opposing effect, whereby myogenesis was enhanced and cell proliferation was repressed (Fig. 2f–j and Table 1). By contrast, the control 2′-O-methyl inhibitors showed no effects (Fig. 2f–j). We conclude that miR-1 and miR-133 have distinct roles in skeletal muscle proliferation and differentiation: miR-1 promotes myoblast differentiation, whereas miR-133 stimulates myoblast proliferation.
Both miR-1 and miR-133 have been found in most animal species, from Drosophila to human, suggesting that they are evolutionary conserved. To test the effects of miR-1 and miR-133 on skeletal muscle and heart development in vivo, we identified copies of miR-1 and miR-133 in X. laevis and tested their function through misexpression. Introduction of miR-1 at the one-cell stage led to a markedly shortened axis with an accompanying reduction in anterior structures and an increase in body size along the dorsal-ventral axis, as compared with either uninjected or miGFP-injected controls (n > 50, two independent experiments; Fig. 3). Although somites formed in embryos injected with miR-1 (Fig. 3), whole-mount antibody staining and serial sectioning showed that the tissue was highly disorganized and did not develop into segmented structures (Fig. 3e,f,j). Cardiac tissue was completely absent, as judged by histology, staining for tropomyosin (Fig. 3f,j) and staining for cardiac actin (data not shown). In addition to these defects, there was a marked decrease in phospho–histone H3 staining (Fig. 3i–k), consistent with the notion that miR-1 is essential in regulating muscle cell proliferation and differentiation.
Figure 3.
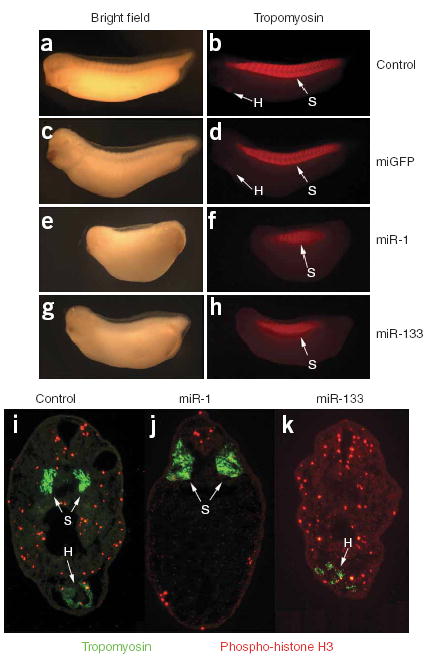
Control of cardiac and skeletal muscle development by miR-1 and miR-133 in vivo. (a–h) Images of uninjected (a,b), control miGFP-injected (c,d), miR-1-injected (e,f) and miR-133-injected (g,h) X. laevis embryos stained with anti-tropomyosin and shown at stage 32 under bright-field (a,c,e,g) or fluorescence (b,d,f,h) microscopy. Note the lack of staining for heart tissue (H, arrows) and the disruption of segmented somites (S, arrows) in f and h. (i–k) Transverse sections corresponding to the position of the heart at stage 32 in uninjected (i), miR-1-injected (j) and miR-133-injected (k) X. laevis embryos stained with anti-tropomyosin to visualize somites (S, arrows) and cardiac tissue (H, arrows), and antibody to phospho–histone H3 (red) to visualize cells in S phase. Each set of injections was conducted at least twice independently, and the phenotype was observed in at least 90% of a minimum of 50 embryos scored by whole-mount immunostaining.
Misexpression of miR-133 also led to a reduction in anterior structures and defects in somite development but, in contrast to misexpression of miR-1, there was only a modest reduction in anterior-posterior length and somitic defects were most severe in the more anterior or posterior aspects of the embryo where somites failed to form (Fig. 3g,h). In addition, cardiac tissue frequently formed in miR-133-injected embryos, although it was highly disorganized and did not undergo cardiac looping or chamber formation (Fig. 3g,h,k and data not shown). Collectively, these data suggest that correct temporal expression and amounts of both miR-1 and miR-133 are required for proper skeletal muscle and heart development.
To identify target genes that might mediate the observed effects of miR-1 and miR-133 on skeletal muscle proliferation and differentiation, we next examined potential targets of these two miRNAs. Many computational and/or bioinformatics-based approaches have been used to predict numerous potential targets of miRNAs20–22. Strikingly, many transcription factors have been suggested to be targets of miRNAs, raising the possibility that miRNAs might be involved in transcriptional regulation of gene expression. Among the predicted targets of miR-1, HDAC4 has been shown to inhibit muscle differentiation and skeletal muscle gene expression, mainly by repressing MEF2C, an essential muscle-related transcription factor12,23. HDAC4 contains two naturally occurring putative miR-1-binding sites at its 3′ UTR, which are evolutionarily conserved among vertebrate species (Supplementary Fig. 7 online). Similarly, two conserved miR-133-binding sites are found in the 3′ UTR of the mammalian gene encoding SRF (Supplementary Fig. 7), which has been shown to be important in muscle proliferation and differentiation in vitro and in vivo11,24,25.
We fused the 3′ UTRs of mouse SRF and HDAC4 to a luciferase reporter gene and transfected these constructs, along with transfection controls, into mammalian cells. Ectopic overexpression of miR-1 strongly repressed the HDAC4 3′ UTR luciferase reporter gene, whereas over-expression of miR-133 inhibited the SRF 3′ UTR luciferase reporter gene (Fig. 4a). By contrast, mutations introduced into miR-1 or miR-133 seed sequences abolished this repression, indicating the specificity of the action (Fig. 4a).
Figure 4.
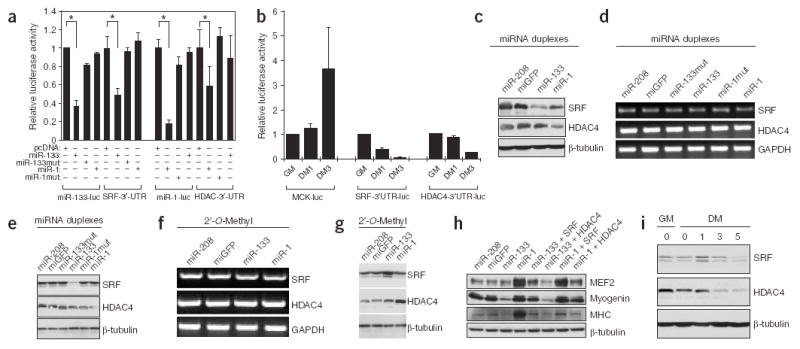
Identification of miR-1 and miR-133 target genes in skeletal muscle. (a) Repression of SRF and HDAC4 3′ UTRs by miR-133 and miR-1. Luciferase reporters containing either miR-133 complementary sites from mouse SRF 3′ UTR (SRF-3′-UTR), miR-1 complementary sites from mouse HDAC4 3′ UTR (HDAC4-3′-UTR) or the perfect antisense sequences of miR-133 (miR-133-luc) or miR-1 (miR-1-luc) were cotransfected with the indicated miRNA expression vectors. Luciferase activity was determined 48 h after transfection. Data represent the mean ± s.d. from at least three independent experiments done in duplicate (*P < 0.05). (b) SRF-3′-UTR, HDAC4-3′-UTR and MCK-luc luciferase reporters were transfected into C2C12 myoblasts. Cells were maintained in growth medium for 24 h (GM) or transferred into differentiation medium for 1 d (DM1) or 3 d (DM3) before luciferase activity was determined. (c–e) C2C12 myoblasts cultured in growth medium were electroporated with the indicated miRNA duplexes (or their mutants), or miR-208 and miGFP as controls. Cells were cultured in growth medium for 24 h after transfection before being either subjected to immunoblotting with anti-SRF and anti-HDAC4 antibodies (c), or transferred into differentiation medium for 24 h and subjected to RT-PCR for the indicated genes (d) or to immunoblotting with the indicated antibodies (e). (f,g) C2C12 myoblasts cultured in growth medium were electroporated with the indicated 2′-O-Methyl antisense oligonucleotide inhibitors. Cells were cultured in growth medium for 24 h after transfection and transferred into differentiation medium for 24 h before being subjected to RT-PCR for the indicated genes (f) or to immunoblotting with indicated antibodies (g). (h) C2C12 myoblasts cultured in growth medium were electroporated with the indicated miRNA duplexes and/or expression plasmids for SRF or HDAC4, as indicated. Cells were cultured in growth medium for 24 h after transfection. Immunoblotting with the indicated antibodies was done 24 h after transfer into differentiation medium. (i) C2C12 myoblasts were cultured in growth medium or differentiation medium for 0, 1, 3 or 5 d and subjected to immunoblotting with the indicated antibodies.
When the above reporters were transfected into C2C12 myoblasts and luciferase activity was measured before and after the induction of cell differentiation, reporter gene activity was markedly repressed in differentiated cells (Fig. 4b), indicating that the increase in endogenous miR-1 and miR-133 inhibited the reporter gene. The effects and specificity of endogenous miR-1 and miR-133 were monitored by the miRNA sensor (Supplementary Fig. 6). By contrast, the activity of a luciferase reporter gene for MCK, an indicator of muscle differentiation, was increased in differentiated muscle cells (Fig. 4b). In addition, overexpression of miR-1 led to the downregulation of endogenous HDAC4 protein in C2C12 cells in both the growth condition (Fig. 4c) and the differentiation condition (Fig. 4e), whereas overexpression of miR-133 repressed the expression of endogenous SRF proteins (Fig. 4c,e). By contrast, the mRNA levels of SRF and HDAC4 were not altered by these miRNAs (Fig. 4d), supporting the notion that miRNAs repress the function of their target genes mainly by inhibiting translation. The application of 2′-O-methyl-antisense oligoribonucleotides targeted towards miR-1 or miR-133 relieved the repression of HDAC4 or SRF protein, respectively (Fig. 4g), but had no effect on their mRNA levels (Fig. 4f).
To verify that HDAC4 and SRF are cognate targets of miR-1 and miR-133 in regulating skeletal muscle gene expression, we tested whether cotransfecting expression plasmids encoding SRF and HDAC4 could ‘suppress’ miRNA-mediated myogenesis. Indeed, over-expression of SRF partially reversed the myogenic gene repression induced by miR-133 (Fig. 4h), whereas overexpression of HDAC4 counteracted the effects of miR-1 on skeletal muscle gene expression (Fig. 4h). Consistent with the potential involvement of HDAC4 and SRF in miR-1- and miR-133-dependent skeletal muscle proliferation and differentiation, endogenous HDAC4 and SRF protein was down-regulated in differentiated C2C12 cells, coupled with a concomitant increase in expression of myogenic differentiation markers and a decrease in expression of the mitotic index marker phospho–histone H3 (Fig. 4i and Supplementary Fig. 2 online). The decrease in expression of SRF and HDAC4 protein was accompanied by an increase in expression of miR-1 and miR-133 (compare Fig. 4i with Fig. 1b). Together, these data show that miR-1 and miR-133 specifically repress HDAC4 and SRF protein, respectively, which in turn contributes to (at least in part) the regulatory effects of these miRNAs on myoblast proliferation and differentiation.
In summary, we have characterized cardiac-specific and skeletal muscle–specific miR-1 and miR-133 and have shown their essential functions in controlling skeletal muscle proliferation and differentiation. Notably, we have shown that miR-1 and miR-133, which are clustered on the same chromosomal loci and transcribed together as a single transcript, become two independent, mature miRNAs with distinct biological functions achieved by inhibiting different target genes. This finding implicates miRNAs in complex molecular mechanisms. Although the tissue-specific expression of miR-1 and miR-133 is controlled by MyoD and SRF8, expression of SRF is repressed by miR-133. Thus, these findings identify a negative regulatory loop in which miRNAs participate to control cellular proliferation and differentiation (Fig. 5). In the future, it will be interesting to determine whether miR-1 and miR-133 are involved in cardiac-related and skeletal muscle–related human diseases.
Figure 5.
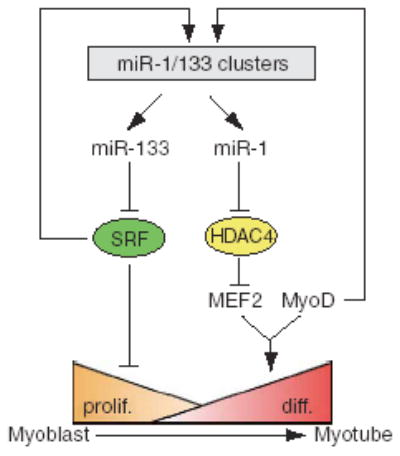
Model of miR-1 and miR-133-mediated regulation of skeletal muscle proliferation and differentiation. Tissue-specific expression of miR-1 and miR-133 clusters is regulated by SRF and myogenic transcription factor MyoD. miR-1 and miR-133 modulate muscle proliferation and differentiation, in part, by targeting HDAC4 and SRF, respectively.
METHODS
Analysis of microRNA expression by microarray
Total RNA was isolated from C2C12 cells cultured in growth medium comprising DMEM (Sigma) plus 10% fetal bovine serum (Sigma) and 1% penicillin or streptomycin (Invitrogen) or differentiation medium comprising DMEM (Sigma) plus 2% horse serum (Sigma) at different time points (days 0, 1, 3 and 5, where day 0 was the first day after transfer into differentiation medium). Microarray hybridization and data analysis were done as described9. In brief, 2.5 μg of isolated RNA was labeled with 5′-phosphate-cytidyl-uridyl-Cy3-3′ (Dharmacon) using RNA ligase and hybridized with a 0.5-mM mixture of oligoribonucleotide probes for 124 miRNAs labeled with Alexa 647 (Cy5) in SLF-0601 disposable chambers (MJ Research). Normalized log2 data were hierarchically clustered by gene and plotted as a heat map.
Northern blot analysis
Total RNA was extracted from C2C12 cells, mouse embryonic tissue or adult tissue with Trizol Reagent (Invitrogen). To aid analysis of miRNA, polyethylene glycol (PEG) was applied to remove large-sized RNAs. In brief, 30 μg of each total RNA sample was mixed 1:1 with 5× PEG solution and placed on ice for 10 min. After 10 min of centrifugation at maximum speed at 4 °C, the supernatant was transferred to a fresh tube. RNAs were then precipitated by adding 2.5 volumes of 100% ethanol, followed by centrifugation for 30 min at maximum speed. Northern blot analysis of miRNAs was done as described13. The miR-1 and miR-133 oligonucleotide sequences used as probes are listed in Supplementary Table 1 online. Northern blot analysis to detect primary transcripts of miRNAs was done as described26 using 20 μg of total RNA from each sample. Genomic fragments of miR-1 and miR-133 were cloned by PCR and used as probes.
Cloning and expression of miR-1 and miR-133
Genomic fragments of miR-1 and miR-133 precursors from mouse chromosomes 2 and 18 were amplified by PCR using mouse genomic DNA as a template (for PCR primers, see Supplementary Table 1). The PCR products were cloned into the pDNA(+)3.1 vector (Invitrogen) and miRNA expression was detected by RNA blot analysis after transfecting the expression vectors into mammalian cells (COS7, 10T1/2 or C2C12).
Cell culture, in vitro myogenesis differentiation and luciferase reporter assay
C2C12 myoblast cells were cultured and myogenesis was induced as described12. Transient transfection luciferase reporter assays were done as described12,26. miRNA duplexes and 2′-O-methyl antisense oligoribo-nucleotides targeted towards miR-1, miR-133, miR-208 and miGFP were purchased from Dharmacon (see Supplementary Table 1 for sequences) and introduced into mammalian cells by either Lipofectamine (Invitrogen) transfection (200 nM) or electroporation with the Nucleofector (Amaxa) system (5 μg).
For construction of the 3′-UTR-luciferase reporter, the multiple cloning site of the pGL3-Control vector (Promega) was removed and placed downstream of the luciferase gene. We amplified the 3′ UTRs of mouse HDAC4 and SRF by PCR and cloned them into the modified pGL3-Control vector, resulting in the constructs SRF-3′-UTR and HDAC4-3′-UTR (see Supplementary Table 1 for PCR primer sequences). Luciferase reporter assays were done as described26.
Immunoblotting and immunostaining
Immunoblotting was done as described27 using antibodies to myogenin, SRF, MEF2, HDAC4 and β-tubulin (Santa Cruz Biotechnology), and to phospho–histone H3 (Upstate Biotechnology). The MF20 antibody, which recognizes striated muscle-specific MHC, was obtained from the DSHB (University of Iowa). For immunostaining, C2C12 cells treated in 12-well plates were fixed with 4% formaldehyde for 5 min at 37 °C and washed in 0.1% Nonidet P40 (NP40) in PBS for 15 min at room temperature. Primary antibodies were incubated in 0.1% NP40 in PBS plus 3% bovine serum albumin (BSA) for 2 h in the following concentrations: anti-myogenin, 1:20 dilution; anti–phospho–histone H3, 1:100 dilution; MF20, 1:10 dilution. The secondary antibodies, fluorescein-conjugated anti-mouse and anti-rabbit (1:100 dilution, Vector Laboratories), were added in 0.1% NP40 plus 3% BSA in PBS for 1 h at 37 °C. 4′,6-Diamidino-2-phenylindole dihydrochloride (DAPI) was then added for 5 min at room temperature. After several washes with PBS, cells were subjected to fluorescence microscopy. For each well, ten fields covering the whole well were picked and green fluorescence–positive cells and total cells with DAPI staining were counted.
RT-PCR analysis
RT-PCR was done essentially as described27. Total RNA was extracted from C2C12 cells with Trizol Reagent (Invitrogen), and 2.0-μg samples were reverse-transcribed to cDNA by using random hexamers and MMLV reverse transcriptase (Invitrogen). In each analysis, 2.5% of the cDNA pool was used for amplification and PCR was done for 24–28 cycles. The sequences of the PCR primers are given in Supplementary Table 1.
X. laevis embryo injections and transgenesis
Standard methods were used to obtain and culture X. laevis embryos. DNA constructs were linearized with KpnI and transgenic embryos were generated as described28. Expression of the transgene was analyzed under an MZFLIII microscope (Leica). Preparation and injection of X. laevis with miRNAs was done essentially as described29, except that RNA was not capped before injection. Whole-mount immunohistological analysis was done as described30.
Acknowledgments
We thank members of our laboratories for discussion and support; T. McKinsey for advice on the HDAC4 antibodies; E. Amaya for the dsRed construct; S. Smyth use of electroporation apparatus; M. von Drehl for histology; M. Majesky and C. Patterson for discussion and critically reading the manuscript. E.M.M. is funded by a National Science Foundation graduate research fellowship. J.M.T. is a Frederick Gardner Cottrell Postdoctoral Fellow. S.M.H. is a General Motors Cancer Research Foundation Scholar. F.L.C. was supported by the US National Institutes of Health (NIH) and the American Heart Association. D.-Z.W. is a Basil O’Connor Scholar of the March of Dimes Birth Defects Foundation and was supported by the NIH, the Muscular Dystrophy Association and an American Heart Association Grant-in-Aid.
Footnotes
COMPETING INTERESTS STATEMENT
The authors declare that they have no competing financial interests.
References
- 1.Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116:281–297. doi: 10.1016/s0092-8674(04)00045-5. [DOI] [PubMed] [Google Scholar]
- 2.Ambros V. The functions of animal microRNAs. Nature. 2004;431:350–355. doi: 10.1038/nature02871. [DOI] [PubMed] [Google Scholar]
- 3.Lee RC, Feinbaum RL, Ambros V. The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell. 1993;75:843–854. doi: 10.1016/0092-8674(93)90529-y. [DOI] [PubMed] [Google Scholar]
- 4.Wightman B, Ha I, Ruvkun G. Posttranscriptional regulation of the heterochronic gene lin-14 by lin-4 mediates temporal pattern formation in C. elegans Cell. 1993;75:855–862. doi: 10.1016/0092-8674(93)90530-4. [DOI] [PubMed] [Google Scholar]
- 5.Chen CZ, Li L, Lodish HF, Bartel DP. MicroRNAs modulate hematopoietic lineage differentiation. Science. 2004;303:83–86. doi: 10.1126/science.1091903. [DOI] [PubMed] [Google Scholar]
- 6.He L, et al. A microRNA polycistron as a potential human oncogene. Nature. 2005;435:828–833. doi: 10.1038/nature03552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Giraldez AJ, et al. MicroRNAs regulate brain morphogenesis in zebrafish. Science. 2005;308:833–838. doi: 10.1126/science.1109020. [DOI] [PubMed] [Google Scholar]
- 8.Zhao Y, Samal E, Srivastava D. Serum response factor regulates a muscle-specific microRNA that targets Hand2 during cardiogenesis. Nature. 2005;436:214–220. doi: 10.1038/nature03817. [DOI] [PubMed] [Google Scholar]
- 9.Thomson JM, Parker J, Perou CM, Hammond SM. A custom microarray platform for analysis of microRNA gene expression. Nat Methods. 2004;1:47–53. doi: 10.1038/nmeth704. [DOI] [PubMed] [Google Scholar]
- 10.Blau HM, et al. Plasticity of the differentiated state. Science. 1985;230:758–766. doi: 10.1126/science.2414846. [DOI] [PubMed] [Google Scholar]
- 11.Soulez M, et al. Growth and differentiation of C2 myogenic cells are dependent on serum response factor. Mol Cell Biol. 1996;16:6065–6074. doi: 10.1128/mcb.16.11.6065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lu J, McKinsey TA, Zhang CL, Olson EN. Regulation of skeletal myogenesis by association of the MEF2 transcription factor with class II histone deacetylases. Mol Cell. 2000;6:233–244. doi: 10.1016/s1097-2765(00)00025-3. [DOI] [PubMed] [Google Scholar]
- 13.Lee RC, Ambros V. An extensive class of small RNAs in Caenorhabditis elegans. Science. 2001;294:862–864. doi: 10.1126/science.1065329. [DOI] [PubMed] [Google Scholar]
- 14.Lagos-Quintana M, et al. Identification of tissue-specific microRNAs from mouse. Curr Biol. 2002;12:735–739. doi: 10.1016/s0960-9822(02)00809-6. [DOI] [PubMed] [Google Scholar]
- 15.Sempere LF, et al. Expression profiling of mammalian microRNAs uncovers a subset of brain-expressed microRNAs with possible roles in murine and human neuronal differentiation. Genome Biol. 2004;5:R13. doi: 10.1186/gb-2004-5-3-r13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wienholds E, et al. MicroRNA expression in zebrafish embryonic development. Science. 2005;309:310–311. doi: 10.1126/science.1114519. [DOI] [PubMed] [Google Scholar]
- 17.Mansfield JH, et al. MicroRNA-responsive ‘sensor’ transgenes uncover Hox-like and other developmentally regulated patterns of vertebrate microRNA expression. Nat Genet. 2004;36:1079–1083. doi: 10.1038/ng1421. [DOI] [PubMed] [Google Scholar]
- 18.Hutvagner G, Simard MJ, Mello CC, Zamore PD. Sequence-specific inhibition of small RNA function. PLoS Biol. 2004;2:E98. doi: 10.1371/journal.pbio.0020098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Meister G, Landthaler M, Dorsett Y, Tuschl T. Sequence-specific inhibition of microRNA- and siRNA-induced RNA silencing. RNA. 2004;10:544–550. doi: 10.1261/rna.5235104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lewis BP, Shih IH, Jones-Rhoades MW, Bartel DP, Burge CB. Prediction of mammalian microRNA targets. Cell. 2003;115:787–798. doi: 10.1016/s0092-8674(03)01018-3. [DOI] [PubMed] [Google Scholar]
- 21.Kiriakidou M, et al. A combined computational-experimental approach predicts human microRNA targets. Genes Dev. 2004;18:1165–1178. doi: 10.1101/gad.1184704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Krek A, et al. Combinatorial microRNA target predictions. Nat Genet. 2005;37:495–500. doi: 10.1038/ng1536. [DOI] [PubMed] [Google Scholar]
- 23.McKinsey TA, Zhang CL, Lu J, Olson EN. Signal-dependent nuclear export of a histone deacetylase regulates muscle differentiation. Nature. 2000;408:106–111. doi: 10.1038/35040593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang D, et al. Regulation of cardiac growth and development by SRF and its cofactors. Cold Spring Harb Symp Quant Biol. 2002;67:97–105. doi: 10.1101/sqb.2002.67.97. [DOI] [PubMed] [Google Scholar]
- 25.Li S, et al. Requirement for serum response factor for skeletal muscle growth and maturation revealed by tissue-specific gene deletion in mice. Proc Natl Acad Sci USA. 2005;102:1082–1087. doi: 10.1073/pnas.0409103102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang D, et al. Activation of cardiac gene expression by myocardin, a transcriptional cofactor for serum response factor. Cell. 2001;105:851–862. doi: 10.1016/s0092-8674(01)00404-4. [DOI] [PubMed] [Google Scholar]
- 27.Cao D, et al. Modulation of smooth muscle gene expression by association of histone acetyltransferases and deacetylases with myocardin. Mol Cell Biol. 2005;25:364–376. doi: 10.1128/MCB.25.1.364-376.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kroll KL, Amaya E. Transgenic Xenopus embryos from sperm nuclear transplantations reveal FGF signaling requirements during gastrulation. Development. 1996;122:3173–3183. doi: 10.1242/dev.122.10.3173. [DOI] [PubMed] [Google Scholar]
- 29.Conlon FL, Sedgwick SG, Weston KM, Smith JC. Inhibition of Xbra transcription activation causes defects in mesodermal patterning and reveals autoregulation of Xbra in dorsal mesoderm. Development. 1996;122:2427–2435. doi: 10.1242/dev.122.8.2427. [DOI] [PubMed] [Google Scholar]
- 30.Brown DD, et al. Tbx5 and Tbx20 act synergistically to control vertebrate heart morphogenesis. Development. 2005;132:553–563. doi: 10.1242/dev.01596. [DOI] [PMC free article] [PubMed] [Google Scholar]