

Abstract
BACKGROUND
Human embryonic stem cell (hESC) lines derived from poor quality embryos usually have either normal or abnormal karyotypes. However, it is still unclear whether their biological characteristics are similar.
METHODS
Seven new hESC lines were established using discarded embryos. Five cell lines had normal karyotype, one was with an unbalanced Robertsonian translocation and one had a triploid karyotype. Their biological characteristics, short tandem repeat loci, HLA typing, differentiation capability and imprinted gene, DNA methylation and X chromosome inactivation status were compared between different cell lines.
RESULTS
All seven hESC lines had similar biological characteristics regardless of karyotype (five normal and two abnormal), such as expression of stage-specific embryonic antigen (SSEA)-4, tumor-rejection antigen (TRA)-1-81 and TRA-1-60 proteins, transcription factor octamer binding protein 4 mRNA, no detectable expression of SSEA-1 protein and high levels of alkaline phosphatase activity. All cell lines were able to undergo differentiation. Imprinted gene expression and DNA methylation were also similar among these cell lines. Non-random X chromosome inactivation patterns were found in XX cell lines.
CONCLUSIONS
The present results suggest that hESC lines with abnormal karyotype are also useful experimental materials for cell therapy, developmental biology and genetic research.
Keywords: human embryonic stem cell lines, characterization, karyotype, methylation, X-inactivation
Introduction
Human embryonic stem cell (hESC) research is one of the most rapidly growing areas in cell biology and medicine. Recent evidence has indicated that hESC can be cultured in the laboratory, unlimitedly passed from generation to generation (Thomson et al., 1998; Stojkovic et al., 2004; Oh et al., 2005; Peura et al., 2007) and induced to differentiate into all kinds of somatic cells under appropriate conditions. These differentiated cells can be used to restore damaged tissues and to treat some kinds of diseases (Assady et al., 2001; Kehat et al., 2001; Wang et al., 2005; Lim et al., 2006).
Since the first hESC line was established in 1998 (Thomson et al., 1998), more than 400 hESC lines have been established in 20 countries and some of them have been registered in the National Institutes of Health (http:escr.nih.gov/) (Guhr et al., 2006). To establish new hESC lines, human embryos are required. However, it is difficult to obtain good quality human embryos for research purposes and it is not permitted to use human embryos for research in some countries. Hence, most researchers use discarded human embryos from IVF clinics. Indeed, in IVF clinics, many poor quality human embryos have been discarded because they showed no survival characteristics at the end of culture.
Hardarson et al. (2003) found that 58% of the embryos produced by IVF had chromosomal abnormalities at blastocyst stage. These abnormal embryos can be used to derive hESC lines (Baharvand et al., 2006). However, it is still unknown whether hESC lines with abnormal karyotypes have similar biological characteristics and functions to those with normal karyotypes. Therefore, in the present study, we used the discarded embryos to establish hESC lines and then compare the biological characteristics, imprinted gene expression, DNA methylation and X chromosome between the hESC lines with normal and abnormal karyotypes.
Materials and Methods
Preparation of feeder layers
The feeder layers of murine embryonic fibroblasts (MEF) were prepared from Day 13.5 post-coitum fetuses of Kunming mice as previous described (Li et al., 2004).
Culture of human embryos
This research was approved by the ethics committee of Guangzhou Medical College. Human embryos from IVF centers were donated on Day 3 after the patients signed the consent. The embryos were cultured in G2.3 medium (Vitrolife, Gothenburg, Sweden) until Day 5 (Kim et al., 2005). On Day 5, early blastocysts were cultured for additional 2 days in a blastocyst optimum culture medium, which is G2.3 medium supplemented with 2000 U/ml of human recombinant leukemia inhibitory factor (hLIF; Chemicon, Temecula, CA, USA) and 10 ng/ml of human basic fibroblast growth factor (bFGF; Vitrolife).
Isolation of inner cell mass
Day 7 expanded blastocysts and hatched blastocysts were used to derive the ESC lines. Zona pellucida of expanded blastocyst was removed by treatment with 0.1% pronase (Sigma). The inner cell mass (ICM) of blastocysts were isolated by immunosurgery or mechanical method. Isolated ICMs were then placed on mitomycin C-treated MEF feeder layers for further culture.
Culture of hESCs
After the ICMs were seeded on the feeder layer, the formation of dome structure was examined after 8–9 days of culture. The ICMs were then mechanically broken down into 2–3 small clumps using a small pipette and the ICM clumps were transferred to a freshly prepared feeder layer. These cells were again mechanically dissociated during the initial five passages. After five passages, they were incubated in 1 mg/ml collagenase IV (Invitrogen) for 20–25 min at 37°C before further culture on freshly prepared feeders. The cells were routinely passed every 4–5 days, and the medium was changed every day. The hESC culture medium is knockout-Dulbecco's modified Eagle's medium (Gibco) supplemented with 15% serum replacement (GIBCO), 5% defined fetal bovine serum (Hyclone), 2 mM glutamine, 0.1 mM β-mercaptolethanol, 0.1 mM non-essential amino acids, 100 U/ml penicillin, 100 µg/ml streptomycin, 4 ng/ml bFGF (Invitrogen) and 2000 U/ml hLIF. After 10 passages, hLIF was not added in the culture medium.
Karyotype analysis
For karyotype analysis, ESCs at passages 12, 22 and 32 were incubated in the culture medium with 0.25 µg/ml colcemid (Gibco) for 4 h, then with 0.4% sodium citrate and 0.4% chloratum Kaliumat (1:1, v/v) at 37°C for 5 min, and finally were fixed in methanol:acetic acid (3:1, v/v) solution. After Giemsa staining, at least 20 cells were examined in each group for the karyotype analysis.
Fluorescence in situ hybridization
For fluorescence in situ hybridization (FISH) analysis, ESC suspensions were dropped onto wet slides, dried at 63°C overnight and then dehydrated with ethanol in sequential concentrations of 70%, 85% and 100% before hybridization. FISH was performed using Vysis MultiVysion® PGT Multi-color Probe set (Vysis Inc., No. 32–131080), which includes five probes for chromosomes of 13, 18, 21, X and Y. The samples were stained according to recommended FISH protocols from manufacturer and examined under a fluorescence microscope. At least, 10 cells were examined in each cell line at each time of examination.
Staining for ESC markers
Human ESC marker staining was performed after 20 passages. To detect alkaline phosphatase (AP) activity, ESC colonies were fixed with 90% alcohol for 2 min, washed three times with Tween-BST solution [phosphate-buffered saline (PBS) with 1% bovine serum albumin and 0.2% Tween-20], and then stained with BCIP/NBT (AP substrate solution, Maxim Biotech Inc., USA.) for 30 min. To detect the hESC stage-specific embryonic markers, ESCs were fixed with 4% paraformaldehyde for 30 min and then incubated with 4% goat serum for 1 h before ESC marker staining. Primary antibodies were stage-specific embryonic antigens (SSEA)-4, SSEA-1, tumor-rejection antigen (TRA)-1-81 and TRA-1-60 (Chemicon). All antibodies were diluted 1:50 with PBS and the cells were incubated with antibody solution at room temperature for 1 h. The cells were washed three times with Tween-BST solution for 5 min and then incubated with the secondary antibody [goat anti-mouse immunoglobulin (Ig)G and goat anti-mouse IgM, both 1:100 dilution] conjugated to fluorescein isothiocyanate for 30 min. Negative controls were carried out without the addition of the primary antibodies. Hoechst 33342 was used for nuclear staining. The cells were then washed again and examined under a fluorescence microscope or confocal microscope.
Oct-4 expression
Total RNA was purified using Trizol Kit (Invitrogen) and RT–PCR reaction was carried out using Qiagen One Step for RT–PCR Kit (Qiagen, Germany) according to manufacturer's instructions. Octamer binding protein 4 (Oct-4) primers were used (Table I). RT–PCR was carried out by reverse transcription for 30 min at 50°C, initial PCR activation for 15 min at 95°C, followed by 30 cycles of denaturation for 1 min at 94°C, annealing for 1 min at 54°C and finally extension for 1 min at 72°C. The PCR amplified products were analyzed on 1.5% agarose gel and visualized by ethidium bromide (Invitrogen) staining.
Table I.
RT–PCR and methylation-specific PCR primer sequences.
| Gene | Primer forward 5′-3′ | Primer reverse5′-3′ | Size (bp) |
|---|---|---|---|
| Oct-4 | GTGTTCAGCCAAAAGACCATC | CCCTGAGAAAGGAGACCCA | 387 |
| H19 | CCGGACACAAAACCCTCTAGCT | TGTTCCGATGGTGTCTTTGATG | 142 |
| IGF2 | TCCCCTGATTGCTCTACCCA | GCAGTTTTGCTCACTTCCGATT | 86 |
| SNRPN | TGGCACCTTTAAGGCTTTTG | CCG CTTTTCTTCACGCTCT | 112 |
| GNAS | CAGCACTGCCAGTGGAGATG | TGTCACGGCAGTCGTTGAAC | 101 |
| GAPD | GGAGTCAACGGATTTGGTCG | CCTGGAAGATGGTGATGGG | 218 |
| SNRPN-M | TAAATAAGTACGTTTGCGCGGTC | AACCTTACCCGCTCCATCGCG | 177 |
| SNRPN-P | GTAGGTTGGTGTGTATGTTTAGGT | ACATCAAACATC TCC AACAACCA | 100 |
Oct-4, octamer binding protein 4; IGF2: insulin-like growth factor; SNRPN, small nuclear ribonucleoprotein polypeptide N; GAPD, glyceraldehyde-3-phosphate dehydrogenase; SNRPN-M, used to analyze methylated status; SNRPN-P, used to analyze unmethylated sites.
DNA fingerprinting and HLA typing
Total DNA was extracted using Qiagen DNeasy Tissue Kit (Qiagen) according to manufacturer's instructions. Extracted DNA was amplified for 16 different genetic loci using the Promega PowerPlex 16 System kit (Promega, USA). Capillary electrophoresis was carried out on an automated ABI 3100 Genetic Analyzer (Applied Biosystems). The 16 short tandem repeat (STR) loci were D3S1358, TH01, D21S11, D18S51, Penta E, D5S818, D13S317, D7S820, D16S539, CSF1PO, Penta D, amelogenin, vWA, D8S1179, TPOX and FGA.
HLA typing was performed by PCR with sequence specific primers (Biotest, Landsteinerstr, Dreieich Germany, Biotest HLA SSP Kit http://www.biotest.de). The products were identified using agarose gel electrophoresis followed by the detection of the DNA bands in UV light with the aid of the Biotest SSP typing software to determine the HLA-A, HLA-B and HLA-DR loci. All manipulations were performed according to manufacturer's recommendations.
Differentiation assessment in vitro
The ESC colonies were dissociated with 1 mg/ml collagenase IV and cultured in culture plates to prevent attachment of the cells. After culture for 3 days, the cells were transferred to a new culture plate. Seven days after culture, the formation of embryoid bodies (EBs) was examined. EBs were transferred to 0.1% gelatin-coated culture dish for spontaneous differentiation. The differentiated cells were stained with antibodies against human smooth muscle actin, cardiac troponin I, alpha fetoprotein and nestin (Chemicon). The EB culture medium was the same as hESC culture medium but without bFGF and hLIF.
Differentiation assessment in vivo
The ESC colonies of passage 15 or beyond were harvested and were broken down into 300–400 small ESC colony suspension. The colonies were injected into inguinal groove of 6-week-old male severe combined immunodeficiency (SCID) mice. Twelve weeks later, the resultant tumors were removed, fixed in 4% paraformaldehyde and embedded in paraffin. Sections were prepared, stained with hematoxylin and eosin, and examined for the presence of tissues derived from the three germ layers.
Analysis of imprinted genes in undifferentiated hESCs
In order to identify the imprinted gene expression in undifferentiated hES cells, total RNA was extracted from different hESC lines. Gene expression pattern in undifferentiated cells was profiled using the QIAGENE one step RT–PCR kit. Selected imprinted genes were H19, insulin-like growth factor (IGF)2, small nuclear ribonucleoprotein polypeptide N (SNRPN) and the conditional gene GNAS (Table I). The PCR was performed using 50°C for 30 min, 95°C for 15 min and followed by 94°C for 30 s, 55°C for 30 s and 72°C for 45 s for 45 cycles and 72°C for 5 min (Sun et al., 2006). The PCR products were analyzed by 2% polyacrylamide gel electrophoresis, stained with ethidium bromide and documented using the BioImaging system (UVP, Upland, CA, USA). Glyceraldehyde-3-phosphate dehydrogenase served as a ubiquitously expressed control. Genomic contamination was ruled out by including an RT-negative sample in each PCR set as a control.
DNA methylation analysis
Methylation patterns of the imprint control (IC) region of the human SNRPN-gene (Table I) were studied in the undifferentiated hESCs. Four hESC lines, FY-hESC-1 (46, XY), -5 (unbalanced Robertsonian translocations), -8 (46, XX) and FY-3PN (69, XXX), were analyzed. Prader–Willi syndrome (PWS) and Angelman syndrome (AS) patients as well as normal DNA samples were also analyzed by using methylation-specific PCR (MSP) assay (Kubota et al., 1997). Genomic DNA was extracted according to the manufacturer's instructions (QIAamp DNA Blood Mini Kit). The PCR products were analyzed by 7% polyacrylamide gel electrophoresis.
X chromosome inactivation status
Human androgen receptor gene contains a highly polymorphic trinucleotide repeat in the first exon. It has been found that the methylation of HpaII and HhaI sites <100 bp away from this polymorphic STR correlates with X inactivation. MSP was used to determine the methylation status of the selected sample with XX chromosome (FY-hES-5, -7, -8 and FY-3PN). Genomic DNA was extracted from the XX hESC lines. Two sets of PCR were prepared. One was for methylated X alleles and the other was for unmethylated alleles. MSP primers were Primer ARM-F 5′-GCG AGC GTA GTA TTT TTC GGC-3′, Primer ARM-R 5′-AAC CAA ATA ACC TAT AAA ACC TCT ACG-3′, Primer ARU-F 5′-GTT GTG AGT GTA GTA TTT TTT GGT-3′ and Primer ARU-R 5′-CAA ATA ACC TAT AAA ACC TCT ACA-3′. Amplification and gel analysis were performed as manufacturer's instruction. Bisulfite-converted CpGenome Universal Methylated DNA (Chemicon http://www.chemicon.com) and bisulfite-converted female human blood DNA were used as positive controls. Sample tubes were loaded to Genetic Analyzer 310 for analysis of fragmentation. The size of PCR products of the androgen receptor gene was between 177 and 221 bp (Kubota et al., 1999).
Results
Derivation of hESC lines
In this study, 265 donated embryos were used and 42 (15.8%) developed to early blastocysts on Day 5 (Fig. 1I, A). When these early blastocysts were transferred to blastocyst optimum culture medium for another 2 days, 36 developed to expanded blastocysts and 6 to hatched blastocysts. A total of 42 ICMs (Fig. 1I, B) were isolated using immunosurgery (19 ICM) or mechanical method (23 ICM). All ICMs were seeded on MEF feeder layer (Fig. 1I, C-H).
Figure 1:

Derivation of hESC lines and characteristics.
(I) Human embryos and isolated ICM. (A) A Day 5 blastocyst cultured in G2.3 blastocyst medium. Note the small ICM. (B) A Day 7 blastocyst that has been cultured in the blastocyst optimum medium for another 2 days. The ICM became clearer. (C) An ICM isolated by mechanical method. (D) A round ICM colony surrounded by a group of residual trophectoderm after mechanical isolation. (E) An ICM isolated by immunosurgery. (F) A dome-like structure on the feeder layer formed after 8 days culture of ICM isolated with immunosurgery. Arrows in C to F indicate ICMs. (G) Typical round hESC colonies with very clear boundary at low magnification. (H) hESC morphology in a colony, showing a high nucleus–cytoplasm ratio; note the presence of nucleoli and typical intercellular spaces at high magnification. Bar = 25 µm in A, B, C and D and bar = 100 µm in E, F and G. (II) Immunocytochemical staining of undifferentiated hESC colonies after 32 passages in four cell lines (A) AP, (B) SSEA-1, (C) SSEA-4, (D) TRA-1-60 and (E) TRA-1-81. Bar = 100 µm.
Seven hESC lines have been established in our laboratory (16.7% of the blastocysts or 2.6% of Day 3 embryos). After nine passages, cells at various passages were frozen and thawed to examine the survival status and were found to survive in the subsequent cultures. FY-hES-1 has been in continuous culture for 1 year and 76 passages, whereas FY-3PN, FY-hES-3, -4, -5, -7 and -8 have been in continuous cultures for 44, 38, 30, 27, 20 and 15 passages, respectively (Table II).
Table II.
Human embryonic stem cell (hESC) lines and their characteristics.
| hESC lines | FY-hES-1 | FY-hES-3 | FY-hES-4 | FY-hES-5 | FY-hES-7 | FY-hES-8 | FY-3PN |
|---|---|---|---|---|---|---|---|
| Karyotypes | 46, XY | 46, XY | 46, XY | 46, XX, +13,der (13;13) (q10;q10) | 46, XX | 46, XX | 69,XXX |
| AP activity | + | + | + | + | + | + | + |
| SSEA-4 | + | + | + | + | + | + | + |
| SSEA-1 | − | − | − | − | − | − | − |
| TRA-1-81 | + | + | + | + | + | + | + |
| TRA-1-60 | + | + | + | + | + | + | + |
| STR | + | + | + | + | + | + | + |
| HLA typing | + | + | + | + | + | + | + |
| FISH analysis | + | + | + | + | + | + | + |
| Embryoid in vitro | + | + | + | + | + | + | + |
| Teratomas in vivo | + | + | + | + | + | + | + |
| Imprinted genes | √ | √ | √ | √ | √ | √ | √ |
| DNA methylation | √ | N/A | N/A | √ | √ | N/A | √ |
| X chromosome status | √ | N/A | N/A | √ | √ | √ | √ |
| No. of Passages | 76 | 38 | 30 | 27 | 20 | 15 | 44 |
AP, alkaline phosphatase; SSEA, stage-specific embryonic antigen; TRA, tumor-rejection antigen; STR, short tandem repeat; FISH, fluorescence in-situ hybridization; “√ ” indicates that the samples were analyzed; N/A, not applicable (not analyzed).
Characterization and identification of hESC lines
Cells in all seven lines showed a high level of AP activity and strongly expressed TRA-1-60, TRA-1-81 and SSEA-4 (Fig. 1II, A, D, E and C, respectively) but not SSEA-1 proteins (Fig. 1II, B). Oct-4 mRNA expression was observed in all seven hESC lines (Fig. 2). Sixteen STR loci were analyzed for hESC lines and each cell line showed distinct STR loci indicating that they were derived from different embryos (Fig. 3A–G). HLA typing also showed that the seven lines have different HLA-A and DBR loci (Table III).
Figure 2:
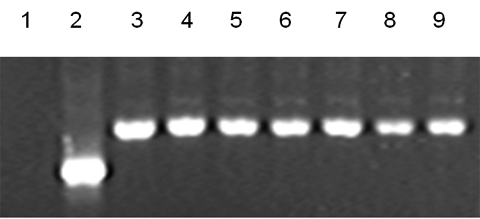
Octamer binding protein 4 expression by RT–PCR.
Lanes 3–9 represent FY-hES-1, -3, -4, -5, -7, -8 and FY-3PN, respectively. Lane 1 is a negative control and lane 2 is human β-actin (265 bp).
Figure 3:

DNA fingerprinting of FY-hES-1 (A), -3 (B), -4 (C), -5 (D), -7 (E), -8 (F) and -3PN (G), respectively.
Table III.
HLA typing of FY-hESC lines.
| HLA type | A | B | DRB |
|---|---|---|---|
| FY-hES-1 | A2/A2 | B46/B60 | DR9/DR16 |
| FY-hES-3 | A11/A11 | B46/B46 | DR9/DR11 |
| FY-hES-4 | A2/A2 | B46/B54 | DR9/DR10 |
| FY-hES-5 | A11/A29 | B7/B54 | DR10/DR13 |
| FY-hES-7 | A11/A24 | B35/B35 | DR14/DR15 |
| FY-hES-8 | A11/A33 | B13/B58 | DR9/DR16 |
| FY-3PN | A11/A33 | B44/B58 | DR17/DR10 |
Karyotypes of the hESC lines
Chromosome analysis and FISH examination showed that FY-hES-1, -3 and -4 had normal 46, XY karyotypes, FY-hES-7 and -8 had normal 46, XX karyotypes, FY-3PN had 69, XXX karyotype (Fig. 4A–F), whereas FY-hES-5 was an unbalanced Robertsonian translocations with 46, XX,+13,der(13;13)(q10;q10) (Fig. 4G) irrespective of the passages 12, 22 and 32. As shown in Fig. 4H, FISH images of FY-hES-5 at the 22 passage showed three chromosome 13 (red signals), 2 chromosome 18 (aqua), 2 chromosome 21 (green), 2 X chromosome (blue) and no Y chromosome after five probe staining, which was the same as examined in other passages. All cells in other cell lines also maintained the same karyotypes as original chromosomal analysis. Embryo donor for FY-hES-5 had normal karyotype.
Figure 4:

Karyotypes of hESC lines.
Karyotype analysis (A–G) and FISH images (H) of hESC. Normal 46, XY karyotypes of FY-hES-1 (A), -3 (B) and -4 (C), normal 46, XX of FY-hES-7 (D) and -8 (E). FY-3PN (F) shows a triploid karyotype of 69, XXX. FY-hES-5 (G) shows unbalanced Robertsonian translocations with 46, XX+13, der (13;13) (q10;q10) karyotype. Blue arrow indicates duplicate chromosome 13. FISH of interphase nuclei from FY-hES-5 cells shows two chromosome 13 (red), two chromosome 21 (green), two chromosome 18 (aqua) and two X chromosomes (blue) (H).
Differentiation of hESC lines
The cells of the hESC lines were cultured in suspension on Petri dishes, simple EBs were formed on Day 3 and cystic EBs on Dasy 6–7. On Day 8, these EBs were transferred to 0.1% gelatin-coated plates for further culture to examine cell differentiation. After 4 days culture, these cells were positively stained by antibodies against human smooth muscle actin (mesoderm), alpha fetoprotein (endoderm) and nestin (ectoderm) (data not shown). Hence, all hESC lines were able to differentiate into three germ layers in vitro. When hESCs were injected into SCID mice, teratomas were first observed at 4–5 weeks and the size of teratomas reached 25 × 30 mm after 12 weeks. After the teratomas were excised and sectioned for examination, three embryonic germ layers including endoderm (gut epithelium), mesoderm (cartilage and muscle) and ectoderm (squamous epithelium, neuroectoderm and neural ganglia) were identified (Fig. 5A–D).
Figure 5:

Histology analysis of teratomas derived from FY-hES-1 and FY-3PN hESC lines.
(A) Squamous epithelium tissue from ectoderm; (B) neural ganglia tissue from ectoderm; (C) cartilage tissue from mesoderm and (D) glandular tissue from endoderm.
Analysis of imprinted genes in undifferentiated hES cells
In order to assess if there are any differences in gene expression between normal and abnormal karyotype hESC lines, we examined maternal expressed imprinted geneH19, paternal expressed imprinted gene IGF2 and SNRPN and conditional gene GNAS as disrupted expression of these genes is associated with human genetic diseases, such as PWS and AS. We found that gene expression patterns in all four of these cell lines were similar. These results indicate that expression of H19, IGF2, SNRPN and GNAS in the abnormal karyotype hESC lines were regulated in a similar way as in the normal ESC lines (Fig. 6).
Figure 6:

RT–PCR analysis of imprinted gene expression (left panel).
Paternally expressed gene IGF2, small nuclear ribonucleoprotein polypeptide N (SNRPN) and maternally expressed gene H19 were analyzed in four hESC lines with different karyotypes. The conditional gene GNAS was used as an additional control. Glyceraldehyde-3-phosphate dehydrogenase (GAPD) was included as mRNA quantitative control. SNRPN gene methylation status (right panel) in FY-hES-1, -5, -7, and -3PN lines, AS, PWS and normal DNA was used as control.
DNA methylation
The SNRPN is a paternally expressed imprinted gene that is located on chromosome 15q11–13, a region related to PWS and AS. The IC-region of the SNRPN-gene showed 23 CpG-sites that are methylated on the maternal chromosome and unmethylated on the paternal chromosome (Zeschnigk et al., 1997). Genomic sequencing of the SNRPN region after bisulfite treatment has revealed that >96% of all CpG dinucleotides are methylated on the maternal chromosome, but none on the paternal chromosome. A normal person or normal hESCs have both methylated and unmethylated SNRPN gene sites. In this study, two pairs of primers were used to analyze SNRPN gene methylation status. Primer SNRPN-M was used to analyze methylated status (174 bp band) and primer SNRPN-P was used to analyze unmethylated sites (100 bp band). Normal and abnormal karyotype hESC lines showed both 100 and 174 bp bands. MSP analysis demonstrated that all of the hESC lines have a normal SNRPN methylation status (Fig. 6).
Determination of X-inactivation pattern
X-inactivation means that one of the X chromosomes is silenced in XX female mammals. Initiation of this process during early development is controlled by the X-inactivation centre, a complex locus that determines how many and which X chromosomes will be inactivated. In order to analyze DNA methylation in XX hESC lines, MSP was used to observe the X-inactivation status in these ESC lines (Table II).
In a random X chromosome inactivation pattern, the XX hESC should have two active alleles and two inactive alleles. The peak area ratio in both active and inactive allele should be 50:50 (the peak area ratio of the small allele to the larger allele). However, in the present study, we found that all of the XX hESC lines have both active and inactive X chromosomes with non-random inactivation patterns of either >80:20 or <20:80 (Fig. 7). FY-hES-5 and -8 had almost complete non-random X chromosome inactivation patterns with only one inactive X chromosome (194 bp in FY-hES-5 and 191 bp in FY-hES-8). The inactivation ratio in the FY-3PN was also non-random (data not shown). In contrast, in the normal female blood DNA samples (as a control), there were two active (194 and 207 bp) and two inactive (194 and 207 bp) X chromosomes with a random X chromosome inactivation pattern of 50:50. The XY hESC line (FY-hES-1) always had one active X chromosome (197 bp) but did not have inactive X chromosome (Fig. 7).
Figure 7:

X chromosome inactivation analysis of the XX hESC line.
Column A represents inactive X chromosome and column B represents active X chromosome. A normal female blood DNA and FY-hES-1 (46, XY) was used as a control (red peak is the marker and blue peak is the product).
Discussion
In the present study, five hESC lines with normal karyotypes and two lines with abnormal karyotypes have been derived from poor quality blastocysts. Successful derivation of hESC lines from poor quality blastocysts has been previously reported by other authors (Hovatta et al., 2003; Mitalipova et al., 2003; Genbacew et al., 2005; Chen et al., 2005; Mateizel et al., 2006; Lerou et al., 2008): the derivation rate from poor quality blastocysts was <10%. In the present study, out of 42 blastocysts, seven ESC lines have been established with a rate of 16.7%. This higher rate may be attributed to the further culture of Day 5 blastocysts in the blastocyst optimum culture medium, which significantly increased the number of cells in the ICM, thus isolation of ICM became much easier. However, if the rate is calculated from Day 3 embryos, it is very low (2.6%) in the present study. This rate is the same as that reported previously (Chen et al., 2005; Genbacew et al., 2005). The low rate is due to poor quality of Day 3 embryos as many of them usually arrest during the subsequent culture due to chromosome abnormalities, such as aneuploidy, mosaicism, haploidy or polyploidy, as these are often found in poor human embryos (Magli et al., 2007; Munne et al., 2007).
Currently, there are standard culture protocols for human ESC culture and there are many poor quality human embryos being discarded from IVF clinics. Thus, if these embryos can be used correctly, it is possible to establish more and more hESC lines.
We also established two ESC lines with abnormal chromosomal constitution (FY-hESC-5) and three pronucleus (FY-3PN) embryos. Some hESC lines with abnormal chromosomal constitution have also been derived previously (Draper et al., 2004; Heins et al., 2004; Munne et al., 2005; Verlinsky et al., 2005; Baharvand et al., 2006). Munne et al. (2005) reported that ESCs derived from trisomic embryos can undergo self-correction, partially or totally, to chromosomally normal cells, thus they observed mosaic in their hESCs. However, we did not observe such self-correction in the present study.
There are two possibilities for the origins of cell lines with normal karyotype. One is that it is derived from normal fertilized oocytes, thus all chromosomes are normal in the subsequent culture, and the other is that it is derived from embryos with chromosomal abnormalities, but the cells undergo a self-correction during subsequent culture, thus the chromosomes are normal in the cell lines. In the present study, because we did not examine the embryo's chromosomal constitutions at Day 3 or 5, we do not know if the cell lines with normal karyotype are derived from embryos with normal chromosomes or embryos with chromosomal abnormalities.
In the present study, self-correction was not observed in a hESC line (FY-3PN) that was derived from a triploid embryo. Triploid embryos used for hESC line derivation may be from a polyspermic oocyte or a diploid oocyte plus a fertilized sperm. In the present study, when we further examined STRs in this cell line, we found that it was a homogeneous triploid cell line (the peak area ratio of each STR locus except D7S820 is almost 1:2.) (Fig. 3G). Therefore, this cell line may result from duplication of the chromosomes in the oocyte. No mosaic in the chromosomal constitutions may indicate that the chromosomes are more stable in the cell lines derived from such triploid embryos than trisomy embryos.
Also, in the unbalanced Robertsonian translocation cell line, the two long arms of 13 chromosomes fused at the centromere and the two short arms were lost. This situation is also different from chromosome separation error in the aneuploid embryo. Thus, it may be difficult to self-correct such an error. From these results, it would appear that self-correction occurs in the ESCs derived from partial chromosomally abnormal embryos, but not in the ESC lines derived from complete triploid embryos or translocation embryos. It is also possible that some cell lines may undergo self-correction, but others may not.
Previous studies indicated that hESC from chromosomally abnormal embryos had all cell markers that hESC should have, and had the ability to differentiate (Heins et al., 2004; Baharvand et al., 2006). In the present study, we examined not only cell markers and ability to differentiate, but also other characteristics, such as imprinted genes, DNA methylation and X chromosome inactivation. Similar to previous studies, we did not find any difference in all characteristics in all seven lines, such as AP activity and cell surface markers, SSEA-4, TRA-1-60 and TRA-1-81. Oct-4, which is an important factor for early embryos and undifferentiated cells (Hay et al., 2004: Lee et al., 2006), was present in all cell lines.
Furthermore, all of these hESC lines are pluripotent. When the cells were injected into immunosuppressed mice to examine the formation of teratomas, we found that they formed cells of all three germ layers including mesoderm (cartilage), ectoderm (epithelium) and endoderm (muscle cells). Also when these cells were spontaneously differentiated in vitro, EBs formed muscular cells, nerve cells and other types of cells.
In order to practically use the established hESCs, the characteristics of each cell line should be clarified. Therefore, we developed a comprehensive database of DNA profiles for each cell line based on STR loci and HLA typing. The exploitation of STR elements in the genome is important in the field of genetic mapping, linkage analysis and human identity testing. STR loci have become the standard for identifying hESC lines (Plaia et al., 2006). STR analysis is also useful in confirming and clarifying some of the anomalies. For example, STR map of FY-3PN showed different peak mapping when compared with others. In order to provide more major histocompatibility complex matched cell lines, HLA typing would be critical for stem cell-based therapies. HLA is a family of cell proteins found on the surface of white blood cells and other nucleated cells in the body. These proteins vary from person to person and are critical for the activation of immune responses. HLA-matched transplantation will minimize the possibility of rejection. Our results from seven cell lines revealed that all of these cells were heterozygous HLA genotype.
Epigenetic stability has profound implications for the use of hESCs in regenerative medicine. Genomic imprinting is erased in the primordial germ cells during development and is reestablished during gametogenesis. Aberrant expression of imprinted genes can cause inherited diseases and induce tumors. For example, the imprinting domain on human chromosome 15q11–13 contains a large cluster of imprinted genes, including paternally expressed SNRPN. Improper regulation of imprinted genes in this cluster results in PWS and AS (Glenn et al., 1997). Loss of imprinting of H19 gene or IGF2 gene, which is normally located at 11p15.5, is related to embryonic cancers, such as BWS [Beckwith–Wiedemann syndrome and Wilm's tumor, Neu-roblastomas and Yolk sac carcinomas (Rainier et al., 1995)]. In our study, no different expression in H19, IGF2, SNRPN and GNAS was found in these cell lines.
DNA methylation is essential for normal mammalian development (Herman et al., 1996). It is not clear whether the potential epigenetic changes occur during long-term ESC culture. Thus, in order to reveal epigenetic stability of imprinted genetic regions of SNRPN in undifferentiated hES cells, we examined DNA methylation status via MSP (Zeschnigk et al., 1997). The SNRPN critical region, such as the maternal allele, is methylated and the paternal allele is not methylated and is transcriptionally active. MSP analysis of these regions demonstrated that all of the hESCs have a normal SNRPN methylation status, indicating that there are no deletions, uniparental disomy or imprinting mutations of SNRPN methylation in these normal and abnormal karyotype hESC lines.
In mice, establishing a stable XX ESC line is not easy due to loss of one X chromosome. The unstable X chrosomosome and DNA methylation have been found in diploid parthenogenetic ESC lines, which results in only one X chromosome (XO genotype) in the cells (Robertson et al., 1983; Zvetkova et al., 2005). Failures of X chromosome inactivation in different hESC lines has also been reported (Dhara and Benvenisty, 2004; Hoffman et al., 2005). Epigenetic variation between hESC lines may also perturb X chromosome inactivation. In order that female embryos express similar levels of X-linked genes to males, one of the two X chromosomes is inactivated at an early embryonic stage. It has been revealed by genetic studies that X chromosome selection is influenced by the X controlling element (Simmler et al., 1993). The X chromosome is randomly inactivated in a 50:50 ratio in most females whereas ∼10% females have a non-random X chromosome inactivation (Kubota et al., 1999). Asymptomatic female carriers of X-linked diseases have the preferential selection of the normal non-mutated X chromosome, which causes extremely non-random inactivation, such as X-linked hyper-IgM syndrome (97:3), Pai syndrome (89:11), multiple congential anomalies (4:96) and unbalanced X autosome translocation (91:9) (Kubota et al., 1999). Therefore, determination of the X-inactivation pattern is important for the detection of carriers of X-linked diseases. Our data showed that all of our XX hESC lines have both active and inactive X chromosomes. Almost extremely non-random X chromosome inactivation patterns (>95:5) were also found in FY-hES-5, FY-hES-8 and FY-3PN. Our data indicate that the patterns of XX hESC with extremely non-random X chromosome inactivation are similar to the patterns of X-linked diseases with skewed X chromosome inactivation. Whether this phenotype means a relationship between XX hESC lines and X-linked disease is unknown, thus further study on the mechanism of non-random X inactivation may be necessary to explain epigenetic states and developmental competence in hESC lines. X chromosome inactivation should be affected in the hESC lines derived from triploidy embryos since there were three X chromosomes. However, we did not find significant differences in X chromosome inactivation in the cell line with XX chromosomes and three X chromosomes. Further studies are necessary to address these issues and hESC lines with abnormal karyotypes are useful materials for these studies.
In conclusion, our results indicate that new hESC lines can be successfully established from poor quality human embryos. All hESC lines established in our laboratory showed all hESC characteristics and could be differentiated into three germ layers, regardless of their karyotype. Through the detailed examination of ESC biological characterizations, gene expression, DNA methylation and X-inactivation, we found that hESC lines with abnormal karyotype are also useful experimental materials for developmental biology and genetic research. Our results also indicate that these ESC lines have potential application in human cell therapy.
Funding
This work was funded by the Guangdong Province Health Department of B30202 and Gangzhou City Science and technology Administration of 2006Z1-E0021.
Acknowledgements
We gratefully acknowledge the support of Dr Y.W. Kan from University of California for critically reading of the manuscript.
References
- Assady S, Maor G, Amit M, Itskovitz-Eldor J, Skorecki KL, Tzukerman M. Insulin production by human embryonic stem cells. Diabetes. 2001;50:1691–1697. doi: 10.2337/diabetes.50.8.1691. [DOI] [PubMed] [Google Scholar]
- Baharvand H, Ashtiani SK, Taee A, Massumi M, Valojerdi MR, Yazdi PE, Moradi SZ, Farrokhi A. Generation of new human embryonic stem cell lines with diploid and triploid karyotypes. Dev Growth Differ. 2006;48:117–128. doi: 10.1111/j.1440-169X.2006.00851.x. [DOI] [PubMed] [Google Scholar]
- Chen H, Qian H, Hu J, Liu D, Lu W, Yang Y, Wang D, Yan H, Zhang S, Zhu G. The derivation of two additional human embryonic stem cell lines from day 3 embryos with low morphological scores. Hum Reprod. 2005;20:2201–2206. doi: 10.1093/humrep/dei010. [DOI] [PubMed] [Google Scholar]
- Dhara SK, Benvenisty N. Gene trap as a tool for genome annotation and analysis of X chromosome inactivation in human embryonic stem cells. Nucleic Acids Res. 2004;32:3995–4002. doi: 10.1093/nar/gkh746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Draper JS, Smith K, Gokhale P, Moore HD, Maltby E, Johnson J, Meisner L, Zwaka TP, Thomson JA, Andrews PW. Recurrent gain of chromosomes 17q and 12 in cultured human embryonic stem cells. Nat Biotechnol. 2004;22:53–54. doi: 10.1038/nbt922. [DOI] [PubMed] [Google Scholar]
- Genbacew O, Krtolica A, Zdravkovic T, Brunette E, Powell S, Nath A, Caceres E, McMaster M, McDonagh S, Li Y, et al. Serum-free derivation of human embryonic stem cell lines on human placental fibroblast feeders. Fertil Steril. 2005;83:1517–1529. doi: 10.1016/j.fertnstert.2005.01.086. [DOI] [PubMed] [Google Scholar]
- Glenn CC, Driscoll DJ, Yang TP, Nicholls RD. Genomic imprinting: potential function and mechanisms revealed by the Prader-Willi and Angelman syndromes. Mol Hum Reprod. 1997;3:321–332. doi: 10.1093/molehr/3.4.321. [DOI] [PubMed] [Google Scholar]
- Guhr A, Kurtz A, Friedgen K, Loser P. Current state of human embryonic stem cell research: an overview of cell lines and their use in experimental work. Stem Cells. 2006;24:2187–2191. doi: 10.1634/stemcells.2006-0053. [DOI] [PubMed] [Google Scholar]
- Hardarson T, Caisander G, Sjogren A, Hanson C, Hamberger L, Lundin K. A morphological and chromosomal study of blastocysts developing from morphologically suboptimal human pre-embryos compared with control blastocysts. Hum Reprod. 2003;18:399–407. doi: 10.1093/humrep/deg092. [DOI] [PubMed] [Google Scholar]
- Hay DC, Sutherland L, Clark J, Burdon T. Oct-4 knockdown induces similar patterns of endoderm and trophoblast differentiation markers in human and mouse embryonic stem cells. Stem cells. 2004;22:225–235. doi: 10.1634/stemcells.22-2-225. [DOI] [PubMed] [Google Scholar]
- Heins N, Englund MC, Sjoblom C, Dahl U, Tonning A, Bergh C, Lindahl A, Hanson C, Semb H. Derivation, characterization, and differentiation of human embryonic stem cells. Stem Cells. 2004;22:367–376. doi: 10.1634/stemcells.22-3-367. [DOI] [PubMed] [Google Scholar]
- Herman JG, Graff JR, Myohanen S, Nelkin BD, Baylin SB. Methylation-specific PCR: a novel PCR assay for methylation status of CpG islands. Proc Natl Acad Sci USA. 1996;93:9821–9826. doi: 10.1073/pnas.93.18.9821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffman LM, Hall L, Batten JL, Young H, Pardasani D, Baetge EE, Lawrence J, Carpenter MK. X-inactivation status varies in human embryonic stem cell lines. Stem cells. 2005;23:1468–1478. doi: 10.1634/stemcells.2004-0371. [DOI] [PubMed] [Google Scholar]
- Hovatta O, Mikkola M, Gertow K, Strömberg AM, Inzunza J, Hreinsson J, Rozell B, Blennow E, Andang M, Ahrlund-Richer L. A culture system using human foreskin fibroblasts as feeder cells allows production of human embryonic stem cells. Hum Reprod. 2003;18:1404–1409. doi: 10.1093/humrep/deg290. [DOI] [PubMed] [Google Scholar]
- Kehat I, Kenyagin-Karsenti D, Snir M, Segev H, Amit M, Gepstein A, Livne E, Binah O, Itskovitz-Eldor J, Gepstein L. Human embryonic stem cells can differentiate into myocytes with structural and functional properties of cardiomyocytes. J Clin Invest. 2001;108:407–414. doi: 10.1172/JCI12131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SJ, Lee JE, Park JH, Lee JB, Kim JM, Yoon BS, Song JM, Roh SI, Kim CG, Yoon HS. Efficient derivation of new human embryonic stem cell lines. Mol Cells. 2005;19:46–53. [PubMed] [Google Scholar]
- Kubota T, Das S, Christian SL, Baylin SB, Herman JG, Ledbetter DH. Methylation-specific PCR simplifies imprinting analysis. Nat Genet. 1997;16:16–17. doi: 10.1038/ng0597-15. [DOI] [PubMed] [Google Scholar]
- Kubota T, Nonoyama S, Tonoki H, Masuno M, Imaizumi K, Kojima M, Wakui K, Shimadzu M, Fukushima Y. A new assay for the analysis of X-chromosome inactivation based on methylation-specific PCR. Hum Genet. 1999;104:49–55. doi: 10.1007/s004390050909. [DOI] [PubMed] [Google Scholar]
- Lee J, Kim HK, Rho JY, Han YM, Kim J. The human OCT-4 isoforms differ in their ability to confer self-renewal. J Biol Chem. 2006;281:33554–33565. doi: 10.1074/jbc.M603937200. [DOI] [PubMed] [Google Scholar]
- Lerou PH, Yabuuchi A, Huo H, Takeuchi A, Shea J, Cimini T, Ince TA, Racowsky C, Daley GO, Ginsbury E. Human embryonic stem cell derivation from poor-quality embryos. Nat Biotech. 2008;26:212–214. doi: 10.1038/nbt1378. [DOI] [PubMed] [Google Scholar]
- Li M, Ma W, Hou Y, Sun XF, Sun QY, Wang WH. Improved isolation and culture of embryonic stem cells from Chinese miniature pig. J Reprod Dev. 2004;50:237–244. doi: 10.1262/jrd.50.237. [DOI] [PubMed] [Google Scholar]
- Lim UM, Sidhu KS, Tuch BE. Derivation of motor neurons from three clonal human embryonic stem cell lines. Curr Neurovasc Res. 2006;3:281–288. doi: 10.2174/156720206778792902. [DOI] [PubMed] [Google Scholar]
- Magli MC, Gianaroli L, Ferraretti AP, Lappi M, Ruberti A, Farfalli V. Embryo morphology and development are dependent on the chromosomal complement. Fertil Steril. 2007;87:534–541. doi: 10.1016/j.fertnstert.2006.07.1512. [DOI] [PubMed] [Google Scholar]
- Mateizel I, De Temmerman N, Ullmann U, Cauffman G, Sermon K, Van de Velde H, De Rycke M, Degreef E, Devroey P, Liebaers I, et al. Derivation of human embryonic stem cell lines from embryos obtained after IVF and after PGD for monogenic disorders. Hum Reprod. 2006;21:503–511. doi: 10.1093/humrep/dei345. [DOI] [PubMed] [Google Scholar]
- Mitalipova M, Calhoun J, Shin S, Wininger D, Schulz T, Noggle S, Vanable A, Lyons I, Robins A, Stice S. Human embryonic stem cell lines derived from discarded embryos. Stem Cell. 2003;21:521–526. doi: 10.1634/stemcells.21-5-521. [DOI] [PubMed] [Google Scholar]
- Munne S, Velilla E, Colls P, Garcia BM, Vemuri MC, Steuerwald N, Garrisi J, Cohen J. Self-correction of chromosomally abnormal embryosin culture and implications for stem cell production. Fertile Steril. 2005;84:1328–1334. doi: 10.1016/j.fertnstert.2005.06.025. [DOI] [PubMed] [Google Scholar]
- Munne S, Chen S, Colls P, Garrisi J, Zheng X, Cekleniak N, Lenzi M, Hughes P, Fischer J, Garrisi M, et al. Maternal age, morphology, development and chromosome abnormalities in over 6000 cleavage-stage embryos. Reprod Biomed Online. 2007;14:628–634. doi: 10.1016/s1472-6483(10)61057-7. [DOI] [PubMed] [Google Scholar]
- Oh SK, Kim HS, Ahn HJ, Seol HW, Kim YY, Park YB, Yoon CJ, Kim DW, Kim SH, Moon SY. Derivation and characterization of new human embryonic stem cell lines: SNUhES1, SNUhES2, and SNUhES3. Stem cells. 2005;23:211–219. doi: 10.1634/stemcells.2004-0122. [DOI] [PubMed] [Google Scholar]
- Peura TT, Bosman A, Stojanov T. Derivation of human embryonic stem cell lines. Theriogenology. 2007;67:32–42. doi: 10.1016/j.theriogenology.2006.09.031. [DOI] [PubMed] [Google Scholar]
- Plaia TW, Josephson R, Liu Y, Zeng X, Ording C, Toumadje A, Brimble SN, Sherrer ES, Uhl EW, Freed WJ, et al. Characterization of a new NIH-registered variant human embryonic stem cell line, BG01V: a tool for human embryonic stem cell research. Stem cells. 2006;24:531–546. doi: 10.1634/stemcells.2005-0315. [DOI] [PubMed] [Google Scholar]
- Rainier S, Dobry CJ, Feinberg AP. Loss of imprinting in hepatoblastoma. Cancer Res. 1995;55:1836–1838. [PubMed] [Google Scholar]
- Robertson EJ, Evans MJ, Kaufman MH. X-chromosome instability in pluripotential stem cell lines derived from parthenogenetic embryos. J Embryol Exp Morphol. 1983;74:297–309. [PubMed] [Google Scholar]
- Simmler MC, Cattanach BM, Rasberry C, Rougeulle C, Avner P. Mapping the murine Xce locus with (CA)n repeats. Mamm Genome. 1993;4:523–530. doi: 10.1007/BF00364788. [DOI] [PubMed] [Google Scholar]
- Stojkovic M, Lako M, Stojkovic P, Stewart R, Przyborski S, Armstrong L, Evans J, Herbert M, Hyslop L, Ahmad S, et al. Derivation of human embryonic stem cells from day-8 blastocysts recovered after three-step in vitro culture. Stem Cells. 2004;22:790–797. doi: 10.1634/stemcells.22-5-790. [DOI] [PubMed] [Google Scholar]
- Sun BW, Yang AC, Feng Y, Sun YJ, Zhu Y, Zhang Y, Jiang H, Li C, Gao FR, Zhang ZH, et al. Temporal and parental-specific expression of imprinted genes in a newly derived Chinese human embryonic stem cell line and embryoid bodies. Hum Mol Genet. 2006;15:65–75. doi: 10.1093/hmg/ddi427. [DOI] [PubMed] [Google Scholar]
- Thomson JA, Itskovitz-Eldor J, Shapiro SS, Waknitz MA, Swiergiel JJ, Marshall VS, Jones JM. Embryonic stem cell lines derived from human blastocysts. Science. 1998;282:1145–1147. doi: 10.1126/science.282.5391.1145. [DOI] [PubMed] [Google Scholar]
- Verlinsky Y, Strelchenko N, Kukharenko V, Rechitsky S, Verlinsky O, Galat V, Kuliev A. Human embryonic stem cell lines with genetic disorders. Reprod Biomed Online. 2005;10:105–110. doi: 10.1016/s1472-6483(10)60810-3. [DOI] [PubMed] [Google Scholar]
- Wang Y, Yates F, Naveiras O, Ernst P, Daley GQ. Embryonic stem cell-derived hematopoietic stem cells. Proc Natl Acad Sci USA. 2005;102:19081–19086. doi: 10.1073/pnas.0506127102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeschnigk M, Schmitz B, Dittrich B, Buiting K, Horsthemke B, Doerfler W. Imprinted segments in the human genome: different DNA methylation patterns in the Prader-Willi/Angelman syndrome region as determined by the genomic sequencing method. Hum Mol Genet. 1997;6:387–395. doi: 10.1093/hmg/6.3.387. [DOI] [PubMed] [Google Scholar]
- Zvetkova I, Apedaile A, Ramsahoye B, Mermoud JE, Crompton LA, John R, Feil R, Brockdorff N. Global hypomethylation of the genome in XX embryonic stem cells. Nat Genet. 2005;37:1274–1279. doi: 10.1038/ng1663. [DOI] [PubMed] [Google Scholar]