

Abstract
Matrix metalloproteinase (MMP) plays a crucial role in periodontal disease and is up-regulated by oral Gram-negative, pathogen-derived LPS. In this study, we reported that simvastatin, a 3-hydroxyl-3-methylglutaryl-CoA reductase inhibitor, effectively inhibited LPS-stimulated MMP-1 as well as MMP-8 and MMP-9 expression by U937 mononuclear cells. Our studies showed that the geranylgeranyl transferase inhibitor inhibited LPS-stimulated MMP-1 expression, and addition of isoprenoid intermediate geranylgeranyl pyrophosphate (GGPP) reduced the inhibitory effect of simvastatin on LPS-stimulated MMP-1 expression. We also demonstrated that simvastatin inhibited the activation of Ras and Rac, and the inhibition was abolished by addition of GGPP. The above results indicate that protein isoprenylation is involved in the regulation of MMP-1 expression by LPS and simvastatin. Moreover, we showed that simvastatin inhibited LPS-stimulated nuclear AP-1, but not NF-κB activity, and the inhibition was reversed by addition of GGPP. Simvastatin also inhibited LPS-stimulated ERK but not p38 MAPK and JNK. Finally, we showed that the inhibition of LPS-stimulated ERK activation by simvastatin was reversed by GGPP. Taken together, this study showed that simvastatin suppresses LPS-induced MMP-1 expression in U937 mononuclear cells by targeting protein isoprenylation-mediated ERK activation.
Keywords: matrix metalloproteinases, statin, lipopolysaccharide, mitogen-activated protein kinase, gene expression
INTRODUCTION
Matrix metalloproteinases (MMPs), a group of zinc-dependent proteases responsible for degradation of collagen and other extracellular matrix components, play a central role in the connective tissue destruction in periodontal disease [1,2,3]. Studies have shown that patients with periodontal disease have increased MMP gene expression and decreased collagen content in periodontal tissue [4]. The MMP protein level in gingival crevicular fluid in these patients was higher than that found in individuals without periodontal disease [5]. Moreover, it has been shown that polymorphism in the promoter region of the MMP-1 gene, which leads to increased MMP-1 production, is associated with a severe chronic periodontitis phenotype in nonsmokers [6]. Among several MMPs expressed in periodontal tissue, MMP-1 (fibroblast-type collagenase) and MMP-8 (neutrophil-type collagenase) are considered to play an essential role in periodontal tissue destruction [2, 3]. MMP expression is largely regulated at the transcriptional level, and LPS, derived from oral Gram-negative bacteria, is a potent stimulator for MMP expression [7, 8]. LPS up-regulates the expression of MMPs and proinflammatory cytokines including TNF-α, IL-1β, and IL-6 by mononuclear cells to promote inflammation. Given the critical role of LPS in stimulation of MMP expression for inflammation and tissue destruction, researchers have long sought to discover agents that are potent in blocking LPS-stimulated, proinflammatory gene expression.
Statins, a class of drugs that lowers cholesterol by inhibiting 3-hydroxyl-3-methylglutaryl (HMG)-CoA reductase, have been widely prescribed to hyperlipidemic patients who have risk for cardiovascular disease [9]. Clinical trials have demonstrated that statin therapy significantly reduces cardiovascular events [9]. In addition to its cholesterol-lowering effect, statins have pleiotropic properties such as improvement of endothelial function, stabilization of atherosclerotic plaques, controlling thrombosis, and the reduction of oxidative stress [10]. As inflammation plays a central role in cardiovascular disease, the effect of statins on inflammation has been investigated [11]. The documented anti-inflammatory effects of statins include inhibition of reactive oxygen species formation, decreased expression of adhesion molecules, and proinflammatory cytokines such as TNF-α, IL-1β, IL-6, CD40 ligand, growth factors and chemokines, decreased T cell activation, and NO synthesis [12, 13].
Although it is known that statins have anti-inflammatory properties, their effect on LPS-stimulated, inflammatory gene expression has not been well-established, and conflicting observations were reported [14,15,16,17,18]. Although many publications have shown that statins blunt the effect of LPS on proinflammatory gene expression, several studies demonstrated that statins augment LPS-triggered, inflammatory responses. Therefore, more studies are necessary to define the action of statins on LPS-stimulated inflammation. In the present study, we demonstrated that simvastatin effectively inhibited LPS-stimulated MMP expression by human U937 mononuclear cells. We also showed that simvastatin inhibits LPS-stimulated MMP expression by inhibiting the protein isoprenylation-mediated ERK pathway.
MATERIALS AND METHODS
Cell culture
U937 mononuclear cells [19] were purchased from American Type Culture Collection (Manassas, VA, USA). The cells were cultured in a 5% CO2atmosphere in RPMI-1640 medium (Gibco, Invitrogen Corp., Carlsbad, CA, USA) containing 10% FCS, 1% MEM nonessential amino acid solution, and 0.6 g/100 ml HEPES. The medium was changed every 2–3 days.
ELISA
MMPs in conditioned medium were quantified using sandwich ELISA kits according to the protocol provided by the manufacturer (R&D Systems, Minneapolis, MN, USA).
Real-time PCR
Total RNA was isolated from cells using the RNeasy Plus Mini kit (Qiagen, Santa Clarita, CA, USA). During the process of RNA purification, genomic DNA was removed effectively in a single centrifugation step. RNA concentration was determined using a Quant-iT™ RNA assay kit from Invitrogen Corp. RNA purity was determined by the ratio of absorbance readings of samples at 260 and 280 nm. First-strand cDNA (cDNA) was synthesized with the iScript™ cDNA synthesis kit (Bio-Rad, Hercules, CA, USA) using 20 μl reaction mixture containing 0.25 μg total RNA, 4 μl 5× iScript reaction mixture, and 1 μl iScript RT. The complete reaction was cycled for 5 min at 25°C, 30 min at 42°C, and 5 min at 85°C using a PTC-200 DNA engine (MJ Research, Waltham, MA, USA). The RT reaction mixture was then diluted 1:10 with nuclease-free water and used for PCR amplification of MMP cDNA in the presence of the primers. The Beacon designer software (Premier Biosoft International, Palo Alto, CA, USA) was used for primer designing. Primers for MMP-1 (5′ primer: CTGGGAAGCCATCACTTACCTTGC; 3′ primer: GTTTCTAGAGTCGCTGGGAAGCTG) were synthesized (Integrated DNA Technologies, Inc., Coralville, IA, USA), and real-time PCR was performed in duplicate using 25 μl reaction mixture containing 1.0 μl RT mixture, 0.2 μM both primers, and 12.5 μl iQ™ SYBR Green Supermix (Bio-Rad). Real-time PCR was run in the iCycler™ real-time detection system (Bio-Rad) with a two-step method. The hot-start enzyme was activated (95°C for 3 min), and cDNA was then amplified for 40 cycles consisting of denaturation at 95°C for 10 s and annealing/extension at 53°C for 45 s. A melt-curve assay was then performed (55°C for 1 min; then temperature was increased by 0.5°C every 10 s) to detect the formation of primer-derived trimers and dimmers. GAPDH (5′ primer: GAATTTGGCTACAGCAACAGGGTG; 3′ primer: TCTCTTCCTCTTGTGCTCTTGCTG) served as a control. Data were analyzed with the iCycler iQ™ software. The average starting quantity (SQ) of fluorescence units was used for analysis. Quantification was calculated using the SQ of MMP-1 cDNA relative to that of GAPDH cDNA in the same sample.
Treatment of cells with the inhibitor of the ERK pathway, geranylgeranyl pyrophosphate (GGPP), farnesyl pyrophosphate (FPP), farnesyl transferase inhibitor (FTI), or geranylgeranyl transferase inhibitor (GGTI)
In the studies to determine the effects of the specific inhibitor of the ERK signaling pathways on LPS-stimulated MMP-1 secretion, U937 cells were treated with 100 ng/ml LPS (Sigma-Aldrich, St. Louis, MO, USA) in the absence or presence of 10 μM PD98059 (Calbiochem/EMD Biosciences, Inc., San Diego, CA, USA) for 24 h. After the treatment, MMP-1 in the conditioned medium was quantified using ELISA. In the studies to determine the effect of GGPP and FPP on simvastatin-inhibited MMP-1 secretion, U937 cells were treated with different concentrations (0, 5, 10, 50 μM) of GGPP or FPP (Sigma-Aldrich) in the presence of 100 ng/ml LPS and 10 μM simvastatin for 24 h, and MMP-1 secreted in culture medium was quantified. In the studies to determine the effect of GGTI and FTI on LPS-stimulated MMP-1 expression, U937 cells were treated with different concentrations (0, 5, 15, 30 μM) of GGTI or FTI (Calbiochem/EMD Biosciences, Inc.) in the presence of 100 ng/ml LPS for 24 h, and MMP-1 secreted in culture medium was assayed afterwards.
Extraction of membrane and nuclear proteins
Membrane and cytosol fractions of U937 cells were extracted using a kit from BioVision Research Products (Mountain View, CA, USA). Nuclear protein was extracted using the NE-PER™ nuclear and cytoplasmic extraction kit (Pierce, Rockford, IL, USA). The protein concentration in the membrane, cytosol, and nuclear fractions was determined using a protein assay kit (Bio-Rad).
Immunoblotting of MAPK and G-proteins
Each sample (50 μg) was electrophoresed in a 10% polyacrylamide gel. After transferring proteins to a polyvinylidene difluoride membrane, total and phosphorylated MAPK were immunoblotted with antitotal and antiphosphorylated MAPK primary antibodies (Cell Signaling Technology, Danvers, MA, USA), respectively, and HRP-conjugated secondary antibody. MAPK was detected by incubating the membrane with a chemiluminescence reagent (NEN Life Science Products) for 1 min and exposing it to X-ray film for 1–10 min. The X-ray films were scanned using an Epson scanner (Perfection 1200U), and the density of bands on the images was quantified using NIH Image Version 1.63. The results were presented as the ratios of phosphorylated MAPK versus total MAPK. Cellular and membrane-associated G-proteins were detected using anti-Ras, Rac1, or RhoA antibody (BD Biosciences, Palo Alto, CA, USA).
Ras, Rac1, and RhoA activation assays
U937 cells were first treated with simvastatin, GGPP, or FPP for 6 h. After that, they were treated with LPS for 15 min. Cells were washed twice with ice-cold PBS and lysed in Mg2+ lysis/wash buffer (MLB; 25 mM HEPES, 150 mM sodium chloride, 1% Nonidet P-40, 10 mM magnesium chloride, 1 mM EDTA, 2% glycerol, 0.3 mg/ml PMSF, 1.0 mM sodium vanadate, and protease inhibitors; pH 7.5). Cell lysates were cleared by centrifugation and incubated at 4°C for 45 min with Raf-1 Ras-binding domain (RBD)-agarose (for Ras pulldown), p21-activated kinase 1-binding domain (PBD)-agarose (for Rac1 pulldown), or rhotekin-binding domain-agarose beads (for RhoA pulldown; Millipore Corp., Temecula, CA, USA). The beads were washed four times with MLB buffer. Pull-down and total cellular levels of Ras, Rac1, and RhoA proteins were detected by immunoblotting, using the corresponding antibodies.
EMSA
Nuclear proteins (10 μg) were used for EMSA to determine AP-1 or NF-κB DNA-binding activity. DNA–protein-binding reactions were performed at room temperature for 20 min in a buffer containing 10 mM Trizma base (pH 7.9), 50 mM NaCl, 5 mM MgCl2, 1 mM EDTA, 1 mM DTT, 1 μg polydeoxyinosinic-deoxycytidylic, 5% (v/v) glycerol, and ∼0.3 pmole AP-1 or NF-κB oligonucleotide (Promega, Madison, WI, USA) labeled with digoxigenin (DIG)-didesoxy-UTP using terminal deoxynucleotidyl transferase (Roche Molecular Biochemicals, Indianapolis, IN, USA). Protein–DNA complexes were resolved from protein-free DNA in 5% polyacrylamide gels at room temperature in 50 mM Tris, pH 8.3, 0.38 M glycine, and 2 mM EDTA and electroblotted onto positively charged nylon membranes. The chemiluminescence detection of DIG-labeled probes was conducted by following the instruction provided by Roche Molecular Biochemicals.
Transcription factor activity assays
Nuclear protein (2 μg) of each sample was applied to the assay for AP-1 and NF-κB activity using the TransAM kits produced by Active Motif (Carlsbad, CA, USA), according to the protocol provided by the manufacturer. These kits contain a 96-stripwell plate, to which the consensus-binding site oligonucleotides were immobilized. Activated nuclear extract is added to each well, and the transcription factor AP-1 or NF-κB binds specifically to bound oligonucleotides. TransAM assays are up to 100 times more sensitive than EMSA and detect transcription factors with specific antibodies to AP-1 or NF-κB.
Statistical analysis
Data were presented as mean ± sd. Student’s t-tests were performed to determine the statistical significance of cytokine expression among different experimental groups. A value of P < 0.05 was considered significant.
RESULTS
Simvastatin suppresses LPS-stimulated MMP-1 expression by U937 mononuclear cells
Our initial study examined the effect of simvastatin on LPS-stimulated MMP-1 expression by U937 cells. Results showed that LPS markedly stimulated MMP-1 secretion, and 10 μM simvastatin inhibited LPS-stimulated MMP-1 secretion by 80% (Fig. 1A). Real-time PCR further demonstrated a similar inhibition of LPS-stimulated MMP-1 mRNA expression by simvastatin (Fig. 1, B and C), suggesting that the inhibition on MMP-1 secretion by simvastatin was a result of the suppression of MMP-1 mRNA expression. Figure 1D showed that simvastatin inhibited LPS-stimulated MMP-1 secretion in a concentration-dependent manner. In addition to MMP-1, simvastatin significantly inhibited LPS-stimulated MMP-8 and MMP-9 mRNA expression (Fig. 1E).
Fig. 1.

Simvastatin inhibits LPS-stimulated MMP expression by U937 mononuclear cells. (A and B) U937 cells were treated with 10 μM simvastatin (Simv) in the presence or absence of 100 ng/ml LPS for 24 h, and MMP-1 secreted into medium and MMP-1 mRNA level were then quantified by ELISA (A) and real-time PCR (B), respectively. (C) Real-time PCR graphs for MMP-1 and GAPDH. Curve A, LPS; Curve B, LPS + simvastatin; Curve C, control; Curve D, control + simvastatin. CF, curve fit; RFU, relative fluorescence unit. (D) The concentration-dependent effect of simvastatin on MMP-1 secretion. U937 cells were treated with different concentrations of simvastatin in the presence or absence of 100 ng/ml LPS for 24 h, and MMP-1 secreted into medium was then quantified by ELISA. (E) The effect of simvastatin on LPS-stimulated MMP-8 and MMP-9 secretion by U937 mononuclear cells. The conditioned medium from the experiment to quantify MMP-1 was used in ELISA to quantify secreted MMP-8 and MMP-9. The data (mean±sd) presented are representative of three independent experiments with similar results.
Simvastatin suppressed LPS-stimulated MMP-1 expression by inhibiting protein isoprenylation
Statins inhibit HMG-CoA reductase and as a result, block the production of mevalonate [10]. Thus, we determined whether the inhibition of LPS-stimulated MMP-1 expression by statin is a result of the reduction of mevalonate production. Results showed that although the addition of mevalonate (1–1000 μM) to cells had no effect on the baseline MMP-1 secretion and LPS-stimulated MMP-1 secretion, it lessened the inhibitory effect of simvastatin on LPS-stimulated MMP-1 secretion in a concentration-dependent manner (Fig. 2A). Furthermore, we determined whether the inhibition of LPS-stimulated MMP-1 expression by simvastatin is related to the reduction of isoprenoid intermediates, such as FPP and GGPP, of the mevalonate pathway as a result of the blockade of mevalonate production by simvastatin. Results showed that the addition of GGPP counteracted the inhibitory action of simvastatin on LPS-stimulated MMP-1 secretion in a concentration-dependent manner (Fig. 2B). The addition of FPP also increased MMP-1 secretion, but the increase did not reach the statistical significance (Fig. 2B). Given the role of GGPP in protein isoprenylation, these results suggest that simvastatin inhibited LPS-stimulated MMP-1 expression by blocking protein isoprenylation. To further verify the role of protein isoprenylation in LPS-stimulated MMP-1 expression, we treated U937 cells with LPS in the presence of GGTI, which inhibited protein isoprenylation by blocking GGPP transfer. Results showed that GGTI significantly suppressed LPS-stimulated MMP-1 secretion (Fig. 2C).
Fig. 2.

The effect of mevalonate, FPP, GGPP, FTI, and GGTI on MMP-1 expression. (A) U937 mononuclear cells were treated with or without different concentrations of mevalonate (Mev; 1–1000 μM) in the presence or absence of 10 μM simvastatin and 100 ng/ml LPS for 24 h, and MMP-1 secreted into culture medium was quantified by ELISA. (B) U937 mononuclear cells were treated with or without FPP (5–50 μM) or GGPP (5–50 μM) in the presence or absence of 10 μM simvastatin and 100 ng/ml LPS for 24 h, and MMP-1 secreted into culture medium was quantified by ELISA. Ctr, Control. (C) U937 mononuclear cells were treated with or without FTI (5–30 μM) or GGTI (5–30 μM) in the presence or absence of 100 ng/ml LPS for 24 h, and MMP-1 secreted into culture medium was quantified by ELISA. The data presented are representative of two independent experiments with similar results.
Simvastatin suppressed LPS-induced MMP-1 expression by blocking LPS-stimulated Ras and Rac isoprenylation
Small G-proteins such as Ras, Rac, and Rho are isoprenylated by isoprenoid intermediates such as FPP and GGPP [20]. Isoprenylated G-proteins are translocated to membranes, where they are activated for signaling transduction and subsequent gene expression. Thus, we determined if the membrane association of these G-proteins as a result of isoprenylation was regulated by simvastatin. Results showed that LPS stimulated and simvastatin inhibited membrane association of Ras and Rac but not Rho (Fig. 3). FPP abolished the inhibitory effect of simvstatin on Ras, whereas GGPP and FPP lessened the effect of simvastatin on Rac (Fig. 3). In contrast, LPS and simvastatin had no effect on Rho membrane translocation. To assess Ras activation, we further performed pull-down assays with Raf-1 RBD-agarose beads. Similarly, we evaluated Rac1 and RhoA activation with PBD-agarose and rhotekin-binding domain-agarose beads, respectively. Results showed that LPS increased active Ras and Rac1 and decreased RhoA, and simvastatin inhibited LPS-stimulated Ras and Rac1 and increased RhoA (Fig. 3B). The addition of GGPP or FPP reversed the inhibitory effect of simvastatin (Fig. 3B). These results indicate that simvastatin inhibits LPS-induced MMP-1 expression by blocking LPS-stimulated Ras and Rac isoprenylation. To further determine if Ras and Rac are essential in LPS-stimulated MMP-1 expression, U937 cells were pretreated with 100 μM Ras inhibitor farnesylthiosalicylic acid (FTS) or 200 μM Rac inhibitor NSC23766 prior to the exposure to LPS. Both inhibitors at the concentrations applied to the cells did not inhibit cell viability as demonstrated by DNA assay (data not shown) but significantly inhibited baseline and LPS-stimulated MMP-1 secretion (Fig. 4).
Fig. 3.

The effect of LPS, simvastatin, and isoprenoid intermediates (GGPP and FPP) on Ras, Rac1, and RhoA activation. (A) U937 cells were treated with 10 μM simvastatin, simvastatin plus 30 μM GGPP, or simvastatin plus 30 μM FPP in the presence of 100 ng/ml LPS for 24 h. After the treatment, membrane protein was extracted and used for immunoblotting of Ras, Rac1, and RhoA. TLR4 was immunoblotted and served as a control. The blots presented are representative of two independent experiments with similar results. (B) U937 cells were treated with 10 μM simvastatin, simvastatin plus 30 μM GGPP, or simvastatin plus 30 μM FPP in the presence of 100 ng/ml LPS for 24 h, and the cell lysate was used in the pull-down assay to determine Ras, Rac1, and RhoA activities as described in Materials and Methods. Total Ras, Rac1, and RhoA in cell lysate were also immunoblotted.
Fig. 4.

Inhibition of LPS-stimulated MMP-1 expression by Ras inhibitor FTS (A) and Rac inhibitor NSC23766 (NSC; B). U937 mononuclear cells were treated with 100 μM FTS for 24 h in the absence or presence of 100 ng/ml LPS or pretreated with 200 μM NSC23766 for 1 h, followed by incubation with 100 ng/ml LPS for 24 h. After the treatment, MMP-1 in culture medium was quantified by ELISA. The data presented are reprsentative of two independent experiments with similar results.
Simvastatin inhibited LPS-stimulated AP-1 activity
Activation of transcription factors AP-1 and NF-κB is a downstream event following activation of MAPK and NF-κB signaling pathways induced by LPS [21]. To determine if AP-1 or NF-κB is suppressed by simvastatin, EMSA was performed. Results showed that simvastatin reduced LPS-stimulated AP-1 DNA-binding activity (Fig. 5A). Furthermore, the addition of GGPP neutralized the effect of simvastatin (Fig. 5A), indicating a role of protein isoprenylation in the inhibition of LPS-stimulated AP-1 activity by simvastatin. In contrast, although LPS increased NF-κB DNA-binding activity markedly, simvastatin did not inhibit it (Fig. 5B), suggesting that the NF-κB pathway is not involved in the inhibition of LPS-stimulated MMP-1 expression by simvastatin [22].
Fig. 5.

The effect of simvastatin and GGPP on LPS-stimulated AP-1 (A) or NF-κB (B) DNA-binding activity. U937 cells were treated with 10 μM simvastatin, 30 μM GGPP, or simvastastin plus GGPP in the presence or absence of 100 ng/ml LPS for 4 h. After the treatment, nuclear protein was extracted and used for EMSA to determine AP-1 or NF-κB DNA-binding activities as described in Materials and Methods. The amount of shifted AP-1 (A) or NF-κB (B) was quantified by densitometric scanning of the shifted bands. The EMSA presented is representative of two independent experiments with similar results.
Simvastatin inhibited LPS-stimulated MMP-1 expression by inhibiting ERK pathway activation
As the NF-κB pathway has been excluded by the above study, we focused on MAPK pathways, including the ERK, JNK, or p38 MAPK cascade. Figure 6 showed that LPS stimulated ERK phosphorylation at 10, 30, and 60 min, but simvastatin plus LPS led to a significantly reduced ERK phosphorylation. To demonstrate the role of the ERK pathway in MMP-1 expression, U937 cells were treated with LPS in the presence of PD98059, a specific inhibitor of the ERK pathway. Results showed that 10 μM PD98059 inhibited LPS-stimulated MMP-1 secretion by 80% (Fig. 7A), indicating that the ERK pathway is essential for LPS-stimulated MMP-1 expression. As simvastatin inhibits LPS-stimulated ERK activation (Fig. 6), it is likely that simvastatin inhibits LPS-stimulated MMP-1 expression by blocking the ERK activation. We also performed EMSA to determine the role of ERK pathway in AP-1 activation. Results showed that PD98059 significantly inhibited LPS-stimulated AP-1 DNA-binding activity (Fig. 7B). In addition to ERK, the effect of simvastatin on JNK and p38 MAPK pathways was investigated. Results showed that although LPS stimulated p38 MAPK activation at 10 and 30 min, simvastatin did not inhibit LPS-stimulated p38 MAPK activation (Fig. 8). Similarly, although LPS increased JNK phosphorylation at 60 min, the addition of simvastatin had no effect on LPS-stimulated JNK activation (Fig. 8).
Fig. 6.

The effect of simvastatin on LPS-stimulated ERK phosphorylation. U937 cells were treated with 10 μM simvastatin, with or without 100 ng/ml LPS for 10, 30, and 60 min. After the treatment, cellular protein was extracted and subjected to immunobloting to detect phosphorylated ERK (P-ERK) and total ERK (T-ERK). The amount of P-ERK and T-ERK was quantified by densitometric scanning of the blot, which is representative of three independent experiments with similar results. N, no treatment; S, simvastatin; L, LPS.
Fig. 7.

The inhibitory effect of PD98059 on LPS-stimulated MMP-1 expression and AP-1 DNA-binding activity. U937 cells were treated with 100 ng/ml LPS in the presence or absence of 10 μM PD98059 for 24 h. After the treatment, MMP-1 in culture medium was quantified by ELISA (A). Nuclear proteins were extracted from cells for EMSA to determine AP-1 DNA-binding activity (B) as described in Materials and Methods. The data presented are representative of three independent experiments with similar results.
Fig. 8.

The effect of simvastatin, LPS, or simvastatin plus LPS on p38 MAPK and JNK phosphorylation. U937 cells were treated with 10 μM simvastatin. with or without 100 ng/ml LPS for 10, 30, and 60 min. After the treatment, cellular protein was extracted and subjected to immunobloting to detect total p38, total JNK, phosphorylated p38. and phosphorylated JNK. The blot presented is representative of two independent experiments with similar results. C, control; S, simvastatin; L, LPS.
Involvement of protein isoprenylation in the regulation of LPS-stimulated ERK activation by simvastatin
Our above studies have shown that simvastatin suppresses MMP-1 expression by inhibiting protein isoprenylation and ERK activation. To demonstrate the relationship between protein isoprenylation and ERK activation, U937 mononuclear cells were treated with LPS in the presence of GGTI, simvastatin, or simvastatin plus GGPP, and the ERK phosphorylation was then examined. Results showed that LPS-stimulated ERK activation was inhibited by GGTI (Fig. 9), indicating that the stimulation of ERK activation by LPS requires protein isoprenylation. Furthermore, simvastatin inhibited LPS-stimulated ERK phosphorylation, but the addition of GGPP partially neutralized the inhibitory effect of simvastatin (Fig. 9). These results indicate that protein isoprenylation is essential for ERK activation stimulated by LPS and suppressed by simvastatin.
Fig. 9.
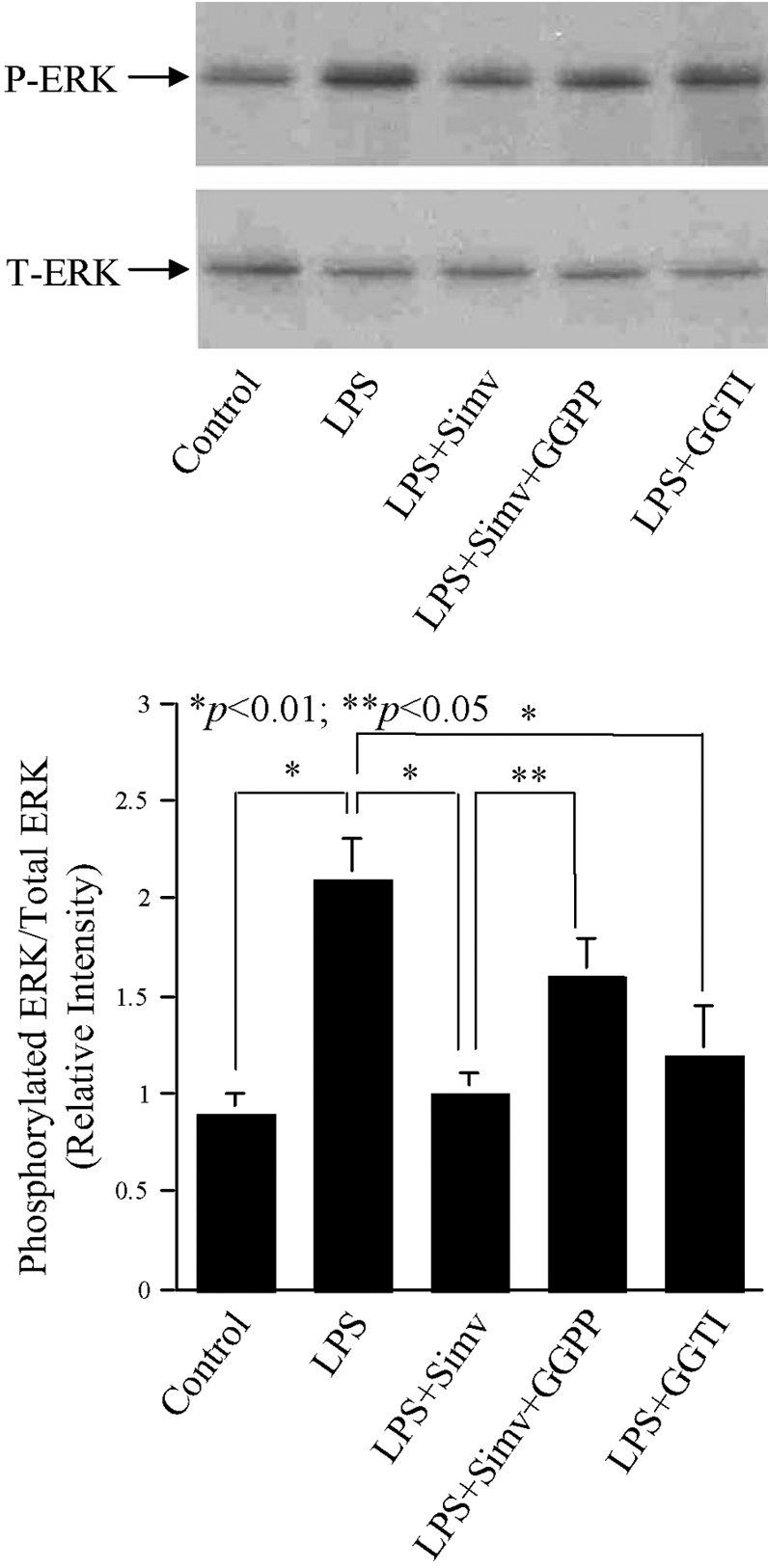
The effect of simvastatin, GGPP, and GGTI on LPS-stimulated ERK phosphorylation. U937 cells were treated with 10 μM simvastatin, 30 μM GGTI, or simvastatin plus GGPP in the presence of 100 ng/ml LPS for 30 min. After the treatment, cellular protein was extracted and used for immunobloting to detect total ERK and phosphorylated ERK. The amount of T-ERK and P-ERK was quantified by densitometric scanning. The blot presented is representative of two independent experiments with similar results.
Inhibition of LPS-stimulated MMP-1 expression in THP-1 mononuclear cells by simvastatin
To determine if simvastatin inhibits LPS-stimulated MMP-1 expression in other mononuclear cells, we examined the effect of simvastatin on THP-1 monocytic cells. Results (Fig. 10) showed that simvastatin inhibited LPS-stimulated MMP-1 secretion in a concentration-dependent manner, and it requires higher concentrations to inhibit LPS-stimulated MMP-1 secretion as compared with U937 cells (Fig. 1D). To further determine if simvastatin also inhibits LPS-stimulated AP-1 activity, AP-1 activity in cells treated with simvastain, LPS, or LPS plus simvastatin was examined. Results (Fig. 11) showed that simvastatin inhibits LPS-stimulated AP-1 (c-Jun and c-Fos) activity but had no effect on LPS-stimulated NF-κB (p65 and p50) activity, which is similar to what we observed in U937 cells (Fig. 5B).
Fig. 10.

The concentration-dependent effect of simvastatin on LPS-stimulated MMP-1 secretion from THP-1 cells, which were treated with an increasing concentration of simvastatin (0–100 μM) in the absence or presence of 100 ng/ml LPS for 24 h, and MMP-1 released into culture medium was quantified using ELISA. The data presented are representative of two independent experiments with similar results.
Fig. 11.

The effect of simvastatin on AP-1 and NF-κB activities in THP-1 cells. THP-1 cells were treated with 100 μM simvastatin, 100 ng/ml LPS, or both for 4 h. After the treatment, nuclear protein was extracted and used for transcription factors AP-1 (c-Jun and c-Fos) and NF-κB (p65 and p50) activity assay as described in Materials and Methods. The data presented are representative of two independent experiments with similar results.
DISCUSSION
Although it has been well documented that statins have anti-inflammatory effects [11,12,13], the reports about the action of statins on LPS-stimulated proinflammatory gene expression are conflicting. Kopp and coworkers [23] showed that simvastatin suppressed the inflammatory response of human monocytes to LPS and blunted tissue factor expression. We also reported recently [16] that simvastatin inhibited LPS-stimulated expression of IL-1β, TNF-α, and IL-6 in U937 mononuclear cells. Furthermore, Methe and coworkers [24] have shown that statins decreased the expression of TLR4, a receptor for LPS, and downstream signaling in human CD14+ monocytes. In contrast, Monick and coworkers [18] demonstrated that treatment of murine RAW 264.7 macrophages with lovastatin increased LPS-induced TNF-α production. Aderem and coworkers [17] also showed that simvastatin augments LPS-induced proinflammatory responses in the same cell line by differential regulation of the c-Fos and c-Jun transcription factors. The reason for these conflicting findings remains unknown, although it is possible that human monocytes/macrophages may be different from murine RAW 264.7 macrophages in response to the combined treatment with statins and LPS. It is likely that statins antagonize LPS in human monocytes/macrophages but act in concert with LPS in murine RAW 264.7 macrophages on gene expression. Further studies are necessary to investigate the underlying mechanism. In the present study, we further documented the antagonizing effect of statin on LPS-stimulated gene expression by demonstrating that simvastatin effectively inhibited LPS-stimulated MMP-1 expression in human U937 mononuclear cells through targeting protein isoprenylation-mediated ERK activation.
It is known that NF-κB and MAPK cascades are the major signaling pathways that are activated by LPS and mediate the LPS-stimulated proinflammatory gene expression [21]. In our study, it appears that the ERK pathway, one of the major pathways for MAPK activation, is targeted specifically by simvastatin for the inhibition on LPS-stimulated MMP-1 expression. Interestingly, our results showed that simvastatin or LPS by itself stimulated ERK1/2 activation (Fig. 6). However, when simvastatin was applied in combination with LPS, the stimulatory effect of LPS on ERK1/2 activation was abolished completely (Fig. 6), suggesting that simvastatin antagonizes LPS in ERK1/2 activation, although it stimulates ERK1/2 by itself. Matsumura and coworkers [25] also showed that statins activated peroxisome proliferator-activated receptor-γ by activating ERK1/2 and p38 MAPK in murine macrophages. Furthermore, the antagonizing effect of statins on LPS-activated MAPK pathways seems to be specific for ERK1/2, as although simvastatin also stimulated p38 MAPK phosphorylation by itself, it did not inhibit LPS-stimulated p38 MAPK (Fig. 8).
It is known that LPS activates the NF-κB signaling pathway that leads to the translocation of the NF-κB transcription factor from the cytoplasma to the nucleus and resultant gene expression [21]. Indeed, our EMSA (Fig. 5B) showed that LPS induced nuclear NF-κB DNA-binding activity. However, simvastatin had no inhibitory effect on LPS-stimulated NF-κB DNA-binding activity, indicating that the NF-κB signaling pathway was not involved in the inhibition of LPS-stimulated MMP-1 expression by simvastatin. In contrast to NF-κB, simvastatin inhibited LPS-stimulated AP-1 DNA-binding activity (Fig. 5A). More importantly, addition of GGPP reversed the inhibitory effect of simvastatin, suggesting that protein isoprenylation is involved in the AP-1 inhibition by simvastatin. As AP-1 activation is a downstream event following the activation of the ERK1/2 pathway, it is likely that simvastatin suppressed AP-1 transcriptional activity by inhibiting LPS-induced ERK1/2 activation.
It has been well established that small GTP-binding proteins (G-proteins) including Ras, Rho, Rac, and their downstream MAPKs (ERK, JNK, p38 MAPK) play a crucial role in the regulation of gene expression and cellular events such as proliferation, apoptosis, and migration [26]. All of these small G-proteins require post-translational modification by isoprenylation for membrane association. In this study, we provided several lines of evidence to demonstrate that the inhibition of LPS-stimulated MMP-1 expression by simvastatin is a result of blocking protein isoprenylation. First, we showed that the addition of GGPP, an isoprenoid intermediate for protein isoprenylation, to U937 cells led to partial reversal of the inhibitory effect of simvastatin on LPS-stimulated MMP-1 secretion (Fig. 2B). Second, we demonstrated that addition of GGTI, an inhibitor for protein isoprenylation, inhibited LPS-stimulated MMP-1 secretion (Fig. 2C). Third, we showed that addition of GGPP diminished the inhibitory effect of simvastatin on ERK1/2 activation (Fig. 9) that mediates MMP-1 expression (Fig. 7). Finally, we demonstrated that the inhibitory effect of simvastatin on LPS-stimulated Ras and Rac membrane association and activation was reduced by GGPP (Fig. 3, A and B). Taken together, these results demonstrate that simvastatin inhibits MMP-1 expression by targeting Ras and Rac isoprenylation and ERK1/2 activation.
Ras is an important molecule in the ERK signaling pathway that is essential for MMP-1 expression [27]. Therefore, it is expected that inhibition of Ras would lead to inhibition of MMP-1 expression as shown in Figure 4. In addition to Ras, our results showed that Rac activation is inhibited by simvastatin (Fig. 3), and Rac inhibition results in a suppression of MMP-1 expression (Fig. 4). It is known that Rac as a member of the Rho family activates the JNK pathway and to a much lesser extent, the p38 MAPK pathway [26]. Additionally, it cooperates with Raf-1 to activate the ERK pathway [26]. It was reported that human neutrophils use a Rac/Cdc42-dependent ERK pathway to direct intracellular granule mobilization toward ingested microbial pathogens [28]. Furthermore, it was demonstrated that expression of an interfering Rac mutant decreases the activation of ERK by activated Ras [26]. Thus, Rac is likely to be involved in the activation of not only the JNK and p38 MAPK cascades but also the ERK pathway. In our study, although it was found that Rac isoprenylation was inhibited by simvastatin (Fig. 3), we did not observe the inhibitory effect of simvastatin on p38 and JNK (Fig. 8). It is possible that although p38 and JNK activations are negatively controlled by simvastatin through Rac, they may also be controlled by another factor that neutralizes the inhibition by simvastatin. More studies are required to determine the underlying mechanism. In contrast to p38 and JNK, our data clearly demonstrated that simvastatin inhibited LPS-stimulated ERK phosphorylation. Given that Ras and Rac isoprenylation is involved in ERK activation, it is likely that simvastatin inhibits LPS-stimulated MMP-1 expression by blocking not only Ras but also Rac isoprenylation. In conclusion, this study demonstrates that simvastatin suppresses LPS-induced MMP-1 expression in U937 mononuclear cells by targeting protein isoprenylation-mediated ERK activation as illustrated and summarized in Figure 12.
Fig. 12.

The mechanism potentially involved in the inhibition of LPS-stimulated MMP-1 expression by simvastatin.
Acknowledgments
This work was supported by National Institutes of Health Grant DE16353 and a Merit Review Grant from the Department of Veterans Affairs (to Y. H.).
References
- Mealey B. Diabetes and periodontal diseases. J Periodontol. 1999;70:935–949. doi: 10.1902/jop.1999.70.8.935. [DOI] [PubMed] [Google Scholar]
- Aiba T, Akeno N, Kawane T, Okamoto H, Horiuchi N. Matrix metalloproteinase-1 and -8 and TIMP-1 mRNA levels in normal and diseased human gingivae. Eur J Oral Sci. 1996;104:562–569. doi: 10.1111/j.1600-0722.1996.tb00142.x. [DOI] [PubMed] [Google Scholar]
- Romanelli R, Mancini S, Laschinger C, Overall C M, Sodek J, McCulloch C A. Activation of neutrophil collagenase in periodontitis. Infect Immun. 1999;67:2319–2326. doi: 10.1128/iai.67.5.2319-2326.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ejeil A L, Igondjo-Tchen S, Ghomrasseni S, Pettat B, Godeau G, Gogly B. Expression of matrix metalloproteinases (MMPs) and tissue inhibitors of metalloproteinases (TIMPs) in healthy and diseased human gingival. J Periodontol. 2003;74:188–195. doi: 10.1902/jop.2003.74.2.188. [DOI] [PubMed] [Google Scholar]
- Tuter G, Kurtis B, Serdar M. Effects of phase I periodontal treatment on gingival crevicular fluid levels of matrix metalloproteinase-1 and tissue inhibitor of metalloproteinase-1. J Periodontol. 2002;73:487–493. doi: 10.1902/jop.2002.73.5.487. [DOI] [PubMed] [Google Scholar]
- De Souza A P, Trevilatto P C, Scarel-Caminaga R M, Brito R B, Line S R. MMP-1 promoter polymorphism: association with chronic periodontitis severity in a Brazilian population. J Clin Periodontol. 2003;30:154–158. doi: 10.1034/j.1600-051x.2003.300202.x. [DOI] [PubMed] [Google Scholar]
- Shankavaram U T, DeWitt D L, Wahl L M. Lipopolysaccharide induction of monocytes matrix metalloproteinases is regulated by the tyrosine phosphorylation of cytosolic phospholipase A2. J Leukoc Biol. 1998;64:221–227. doi: 10.1002/jlb.64.2.221. [DOI] [PubMed] [Google Scholar]
- Pierce R A, Sandefur S, Doyle G A, Welgus H G. Monocytic cell type-specific transcriptional induction of collagenase. J Clin Invest. 1996;97:1890–1899. doi: 10.1172/JCI118620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heart Protection Study Collaborative Group MRC/BHF Heart Protection Study of cholesterol lowering with simvastatin in 20,536 high-risk individuals: a randomized placebo-controlled trial. Lancet. 2002;360:7–22. doi: 10.1016/S0140-6736(11)61125-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takemoto M, Liao J K. Pleiotropic effects of 3-hydroxy-3-methylglutaryl coenzyme A reductase inhibitors. Arterioscler Thromb Vasc Biol. 2001;21:1712–1719. doi: 10.1161/hq1101.098486. [DOI] [PubMed] [Google Scholar]
- Endres M. Statins: potential new indications in inflammatory conditions. Atherosclerosis. 2006;7:31–35. doi: 10.1016/j.atherosclerosissup.2006.01.005. [DOI] [PubMed] [Google Scholar]
- Liao J K. Statins: potent vascular anti-inflammatory agents. Int J Clin Pract Suppl. 2004;143:41–48. doi: 10.1111/j.1368-504x.2004.00375.x. [DOI] [PubMed] [Google Scholar]
- Forrester J S, Libby P. The inflammation hypothesis and its potential relevance to statin therapy. Am J Cardiol. 2007;99:732–738. doi: 10.1016/j.amjcard.2006.09.125. [DOI] [PubMed] [Google Scholar]
- Guijarro C, Kim Y, Schoonover C M, Massy Z A, O'Donnell M P, Kasiske B L, Keane W F, Keane W F, Kashtan C E. Lovastatin inhibits lipopolysaccharide-induced NF-κB activation in human mesangial cells. Nephrol Dial Transplant. 1996;11:990–996. [PubMed] [Google Scholar]
- Hilgendorff A, Muth H, Parviz B, Staubitz A, Haberbosch W, Tillmanns H, Hölschermann H. Statins differ in their ability to block NF-κB activation in human blood monocytes. Int J Clin Pharmacol Ther. 2003;41:397–401. doi: 10.5414/cpp41397. [DOI] [PubMed] [Google Scholar]
- Nareika A, Maldonado A, He L, Game B A, Slate E H, Sanders J J, London S D, Lopes-Virella M F, Huang Y. High glucose-boosted inflammatory responses to lipopolysaccharide are suppressed by statin. J Periodontal Res. 2007;42:31–38. doi: 10.1111/j.1600-0765.2006.00911.x. [DOI] [PubMed] [Google Scholar]
- Matsumoto M, Einhaus D, Gold E S, Aderem A. Simvastatin augments lipopolysaccharide-induced proinflammatory responses in macrophages by differential regulation of the c-Fos and c-Jun transcription factor. J Immunol. 2004;172:7377–7384. doi: 10.4049/jimmunol.172.12.7377. [DOI] [PubMed] [Google Scholar]
- Monick M M, Powers L S, Butler N S, Hunninghake G W. Inhibition of Rho family GTPases results in increased TNF-α production after lipopolysaccharide exposure. J Immunol. 2003;171:2625–2630. doi: 10.4049/jimmunol.171.5.2625. [DOI] [PubMed] [Google Scholar]
- Sundstrom C, Nilsson K. Establishment and characterization of a human histiocytic lymphoma cell line (U937) Int J Cancer. 1976;17:565–577. doi: 10.1002/ijc.2910170504. [DOI] [PubMed] [Google Scholar]
- Shirai H, Autieri M, Eguchi S. Small GTP-binding proteins and mitogen-activated protein kinases as promising therapeutic targets of vascular remodeling. Curr Opin Nephrol Hypertens. 2007;16:111–115. doi: 10.1097/MNH.0b013e3280148e4f. [DOI] [PubMed] [Google Scholar]
- Guha M, Mackman N. LPS induction of gene expression in human monocytes. Cell Signal. 2001;13:85–94. doi: 10.1016/s0898-6568(00)00149-2. [DOI] [PubMed] [Google Scholar]
- Vincenti M P, Brinckerhoff C E. Transcriptional regulation of collagenase (MMP-1, MMP-13) genes in arthritis: integration of complex signaling pathways for the recruitment of gene-specific transcription factors. Arthritis Res. 2002;4:157–164. doi: 10.1186/ar401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steiner S, Speidl W S, Pleiner J, Seidinger D, Zorn G, Kaun C, Wojta J, Huber K, Minar E, Wolzt M, Kopp C W. Simvastatin blunts endotoxin-induced tissue factor in vivo. Circulation. 2005;111:1841–1846. doi: 10.1161/01.CIR.0000158665.27783.0C. [DOI] [PubMed] [Google Scholar]
- Methe H, Kim J, Kofler S, Nabauer M, Weis M. Statins decrease Toll-like receptor 4 expression and downstream signaling in human CD14+ monocytes. Arterioscler Thromb Vasc Biol. 2005;25:1439–1445. doi: 10.1161/01.ATV.0000168410.44722.86. [DOI] [PubMed] [Google Scholar]
- Yano M, Matsumura T, Senokuchi T, Ishii N, Murata Y, Taketa K, Motoshima H, Taguchi T, Sonoda K, Kukidome D, Takuwa Y, Kawada T, Brownlee M, Nishikawa T, Araki E. Statins activate peroxisome proliferator-activated receptor γ through extracellular signal-regulated kinase 1/2 and p38 mitogen-activated protein kinase-dependent cyclooxygenase-2 expression in macrophages. Circ Res. 2007;100:1442–1451. doi: 10.1161/01.RES.0000268411.49545.9c. [DOI] [PubMed] [Google Scholar]
- Frost J A, Steen H, Shapiro P, Lewis T, Ahn N, Shaw P, Cobb M H. Cross-cascade activation of ERKs and ternary complex factor by Rho family proteins. EMBO J. 1997;16:6426–6438. doi: 10.1093/emboj/16.21.6426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komatsu M, Ruoslahti E. R-Ras is a global regulator of vascular regeneration that suppresses intimal hyperplasia and tumor angiogenesis. Nat Med. 2005;11:1346–1350. doi: 10.1038/nm1324. [DOI] [PubMed] [Google Scholar]
- Zhong B, Jiang K, Gilvary D L, Epling-Burnette P K, Ritchey C, Liu J, Jackson R J, Hong-Geller E, Wei S. Human neutrophils utilize a Rac/Cdc42-dependent MAPK pathway to direct intracellular granule mobilization toward ingested microbial pathogens. Blood. 2003;101:3240–3248. doi: 10.1182/blood-2001-12-0180. [DOI] [PubMed] [Google Scholar]