

Abstract
Acidification of the phagosome is considered to be a major mechanism used by macrophages against bacteria, including Mycobacterium tuberculosis (Mtb). Mtb blocks phagosome acidification1, but interferon-γ (IFN-γ) restores acidification and confers antimycobacterial activity2, 3. Nonetheless, it remains unclear whether acid kills Mtb, whether the intrabacterial pH of any pathogen falls when it is in the phagosome and whether acid resistance is required for mycobacterial virulence. In vitro at pH 4.5, Mtb survived in a simple buffer and maintained intrabacterial pH. Therefore Mtb resists phagolysosomal concentrations of acid. Mtb also maintained its intrabacterial pH and survived when phagocytosed by IFN-γ –activated macrophages. We used transposon mutagenesis to identify genes responsible for Mtb’s acid resistance. A strain disrupted in Rv3671c, a previously uncharacterized gene encoding a membrane-associated protein, was sensitive to acid and failed to maintain intrabacterial pH in acid in vitro and in activated macrophages. Growth of the mutant was also severely attenuated in mice. Thus, Mtb is able to resist acid, owing in large part to Rv3671c, and this resistance is essential for virulence. Disruption of Mtb’s acid resistance and intrabacterial pH maintenance systems is an attractive target for chemotherapy.
When Metchnikoff discovered phagocytosis, he speculated that macrophages kill most ingested microbes by acidifying them and that Mtb uses its waxy cell wall to resist this acidification4. Subsequent studies established that after macrophages ingest particles or microbes, phagosomes with an initial pH of ~6.2 fuse with lysosomes, and their pH falls to ~4.55, 6. Mtb blocks phagolysosomal fusion7–9; however, activation of macrophages by the T cell–derived cytokine IFN-γ overcomes the block via induction of the GTPase Lrg-47, and thus the mycobacterium-containing phagosomes acidify1–3, 10. Additionally, IFN-γ activation enhances the antimicrobial capacity of macrophages11 and is essential for control of mycobacterial infection in mice and people12–15. Thus, acidification of the phagosome may represent a major antimycobacterial mechanism. However, IFN-γ induces hundreds of genes in macrophages16, among them other pathways with antimycobacterial activity, such as inducible nitric oxide synthase17, 18. Because Mtb is killed to an extent, but not eradicated, in acidic phagosomes, it is unclear whether Mtb should be regarded as acid sensitive or acid resistant1, 19, 20. In addition, mutants of Mtb and Mycobacterium bovis BCG that fail to prevent phagosome acidification are not necessarily compromised for survival in macrophages, suggesting that the bacterium can resist acid21–23. To resolve this issue, we asked whether phagolysosomal concentrations of acid kill Mtb in pure culture. Then we addressed two questions pertinent to a wide range of intracellular microbial pathogens—does phagosomal acidification affect intrabacterial pH, and are acid-sensitive mutants attenuated in vivo?
We first monitored survival of Mtb in standard 7H9 growth medium, which routinely contains Tween 80 as a dispersal agent so that colony-forming units (CFU) can be enumerated. When suspended in this medium at pH 6.6, Mtb grows logarithmically (Supplementary Fig. 1 online). However, when we acidified the medium to pH 4.5 (7H9-Tw-4.5 medium), a culture containing 5 × 106 CFU ml−1 Mtb was almost sterilized by day 15 (Fig. 1a). Mtb is highly susceptible to killing by free fatty acids24–26, and it is possible that acid hydrolyzes Tween 80, releasing oleic acid. Alternatively, Tween 80’s ability to strip the bacterium’s glycoprotein capsule27 might render Mtb sensitive to acid. To avoid Tween-associated artifacts, we examined survival of Mtb at pH 4.5 in 7H9 medium containing nonhydrolyzable Tyloxapol as the dispersing agent (7H9-Ty-4.5 medium). Killing in 7H9-Ty-4.5 medium was reduced, but not eliminated (Fig. 1b). Finally, in phosphate-citrate buffer at pH 4.5 containing Tyloxapol (pcit-Ty-4.5 buffer), 100% of Mtb survived (Fig. 1b). Thus, at least two components of the standard growth medium become toxic to Mtb at pH 4.5: one associated with Tween and another associated with albumin (data not shown), and both may release free fatty acids at low pH.
Figure 1.
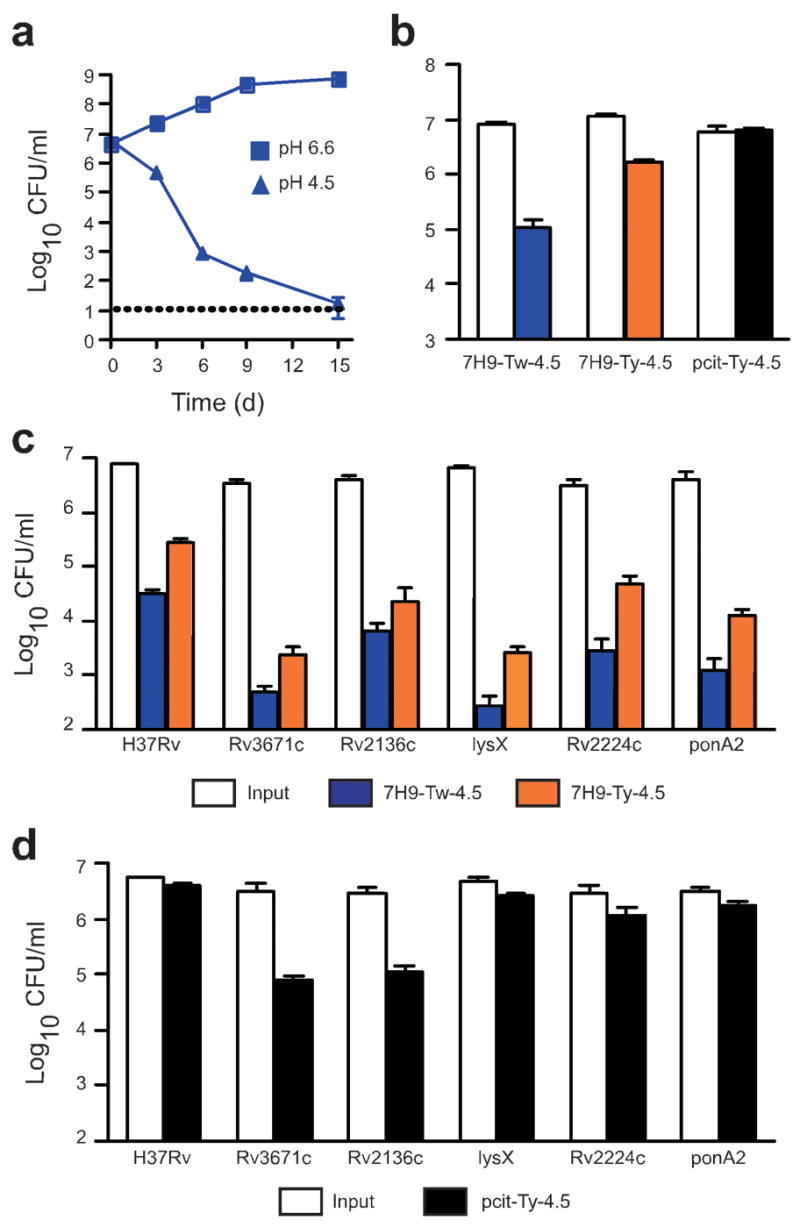
Survival of wild-type Mtb and transposon mutants at pH 4.5. (a) Quantification of CFU of wild-type Mtb (H37Rv) at pH 4.5 or pH 6.6 in 7H9-Tw medium. Horizontal dotted line indicates limit of detection. (b) Impact of medium on survival of Mtb. Mtb were plated after 6 d incubation in 7H9-Tw-4.5, 7H9-Ty-4.5 or pcit-Ty-4.5 medium, and CFU were quantified.(c) Wild-type Mtb and five acid-sensitive mutants were plated after 6 d incubation in 7H9-Tw-4.5 or 7H9-Ty-4.5 medium, and CFU were quantified. LysX is predicted to encode a lysyl-tRNA synthetase, Rv2224c a secreted protease and ponA2 a transglycosylase/transpeptidase (d) Wild-type Mtb and five acid-sensitive mutants were plated after 6 d incubation in pcit-Ty-4.5 medium, and CFU were quantified. The pH of all test media remained at pH 4.5 throughout the experiment. Bacterial input was 0.5–1 × 107 CFU ml−1. Data are means ± s.d. of triplicate cultures and represent two or three independent experiments. In some panels, error bars are too small to be seen.
Aside from the artifactual effects arising from the use of 7H9-Tw-4.5, we found that wild-type Mtb was highly resistant to acid at pH 4.5, in that the bacteria were not killed in pcit-Ty-4.5 buffer. Therefore, we sought to identify the genes responsible, and at the same time we sought to learn which genes conferred relative resistance to the combination of acid and 7H9-Tw medium, because many experiments with Mtb are carried out in this medium. We screened 10,100 Mtb transposon mutants28 individually for impaired ability to recover from a 6-d exposure to 7H9-Tw-4.5 medium and identified 34 mutants containing transposons in 21 genes. Mutants with disruptions of the same gene carried independent transposon insertions at distinct sites (Supplementary Table 1 online). Fifteen (71%) of the affected genes were annotated as involved in cell wall functions, such as peptidoglycan and lipoarabinomanan biosynthesis29 (for example, pbpA, ponA2, glnA2, Rv2136c and ppm1). Mutants in only 5 of the 21 genes remained hypersensitive to pH 4.5 when we replaced Tween with Tyloxapol (Fig. 1c and data not shown), and only two remained hypersensitive when we replaced 7H9 with pcit (Fig. 1d). None of the mutants showed a growth defect in 7H9-Tw medium at pH 6.6 (Supplementary Fig. 1 and data not shown).
The two mutants that were acid sensitive in all three test media contained transposon insertions in Rv2136c and Rv3671c. Rv2136c encodes the Mtb homolog of Escherichia coli BacA, an undecaprenol phosphatase30 involved in peptidoglycan assembly and resistance to bacitracin; the gene family contributes to in vivo survival in several bacterial species, including Mycobacterium smegmatis31. Rv3671c is predicted to be a serine protease with conserved aspartate, histidine and serine active site residues (Supplementary Fig. 2 online) and four transmembrane domains29, 32.
Next we sought to determine the intrabacterial pH (pHIB) of wild-type and mutant Mtb strains in near-neutral conditions, in acidified medium and when the bacilli resided in macrophages that were either nonactivated or IFN-γ –activated. We transformed each strain with a plasmid encoding a pH-sensitive ratiometric GFP (pH-GFP)33, which allowed for noninvasive measurements of pHIB on live cells, and monitored pHIB at intervals after transfer to pcit-Ty buffer. Basal pHIB, calculated from measured ratio values, was 7.68 ± 0.02 in wild-type Mtb, 7.65 ± 0.08 in the Rv3671c mutant and 7.55 ± 0.06 in the Rv2136c mutant (mean ± s.d., n = 3 experiments; Fig. 2a). After 8 h in pcit-Ty-7.4 buffer, the pHIB fell slightly in all strains tested (Fig. 2a). In contrast, after an 8-h incubation in pcit-Ty-4.5 buffer, the pHIB of wild-type Mtb dropped only to 7.20 ± 0.11, whereas the pHIB of the Rv3671c and Rv2136c mutants fell to 6.44 ± 0.11 and 6.37 ± 0.15, respectively (Fig. 2a). Intrabacterial acidification preceded (Fig. 2b and Supplementary Fig. 3 online) and was associated with (Fig. 1d) a marked decline in viability. Therefore, the rapid influx of protons (<15 min) was the probable cause of death.
Figure 2.
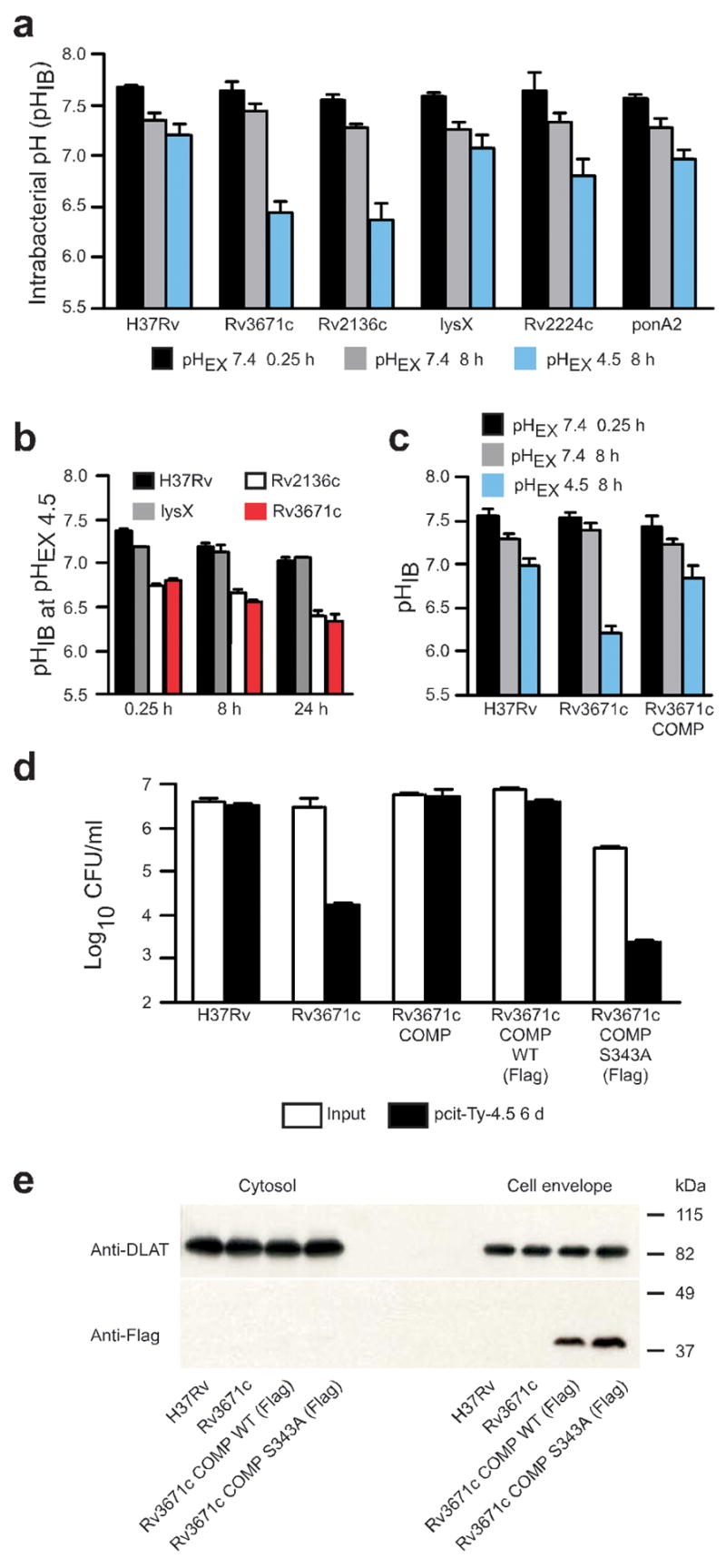
pHIB measurements of acid-sensitive mutants and complementation of the Rv3671c mutant. (a) pHIB determined at two extracellular pHs (pHEX) in pcit-Ty buffer: pHEX of 7.4 and 4.5 after incubation for the indicated times. (b) pHIB measured after incubation for the indicated times in pcit-Ty buffer at an extracellular pH of 4.5. (c) pHIB at 8 h of wild-type Mtb, Rv3671c mutant and complemented mutant (Rv3671c COMP) in pcit-Ty-4.5. (d) Quantification of CFU at 6 d of culturing wild-type Mtb, Rv3671c mutant, complemented mutant and mutant complemented with Flag-tagged wild-type Rv3671c (WT) or S343A Rv3671c in pcit-Ty-4.5. [AU: Ok as edited?] (e) Immunoblot of cytosol and cell envelope fractions probed with a Flag-specific antibody (Anti-Flag). An immunoblot for dihydrolipoamide S-acyltransferase (Anti-DLAT) serves as a loading control. Data are means ± s.d. of three experiments, each in triplicate in a and c; data are means ± s.d. of triplicate cultures and represent two or three independent experiments in b, d and e.
Because Rv2136c is part of a putative operon, and because the mutant phenotypes did not revert when we provided the wild-type allele in trans (data not shown), we focused further studies on the Rv3671c mutant. Transformation of the Rv3671c mutant with an integrative plasmid encoding a wild-type Rv3671c allele restored pHIB homeostasis (Fig. 2c) and survival at pH 4.5 (Fig. 2d). Mutation of the Rv3671c active site serine (Ser343) to alanine abolished complementation of the Rv3671c mutant (Fig. 2d). Rv3671c protein was detected in equivalent amounts in the cell envelope fraction prepared from bacterial lysates of the complemented strains, both those with and those without the predicted active site serine (Fig. 2e). These data indicate that Rv3671c indeed encodes a membrane-associated protein whose function requires Ser343.
We next performed what to our knowledge are the first measurements of the pHIB of bacteria residing in macrophage phagosomes. Wild-type Mtb maintained its pHIB in both nonactivated and IFN-γ –activated macrophages at pH 6.76–7.50 (Fig. 3a–d). In contrast, the Rv3671c mutant failed to control its pHIB in IFN-γ –activated macrophages, but was able to do so in nonactivated macrophages (Fig. 3a–d). Most mutant bacteria had a pHIB of 5.5 or lower in phagosomes of IFN-γ –activated macrophages (Fig. 3d). Complementation of the Rv3671c mutant restored the defect in intraphagosomal pHIB maintenance (Fig. 3a–d). Likewise, the Rv3671c mutant was killed by IFN-γ –activated but not nonactivated macrophages (Fig. 3e, f). Kinetic analysis showed that the proportion of Rv3671c mutant bacteria that were acidified increased from 4 to 72 h in IFN-γ –activated macrophages, whereas both wild-type Mtb and the complemented mutant maintained pHIB even at 72 h after infection (Supplementary Fig. 4 online). We did not observe any defect in the pHIB of the strains in nonactivated macrophages at 72 h after infection (Supplementary Fig. 4). The ability of the Rv3671c mutant to maintain pHIB in nonactivated macrophages indicates that it remains capable of restricting phagosome acidification. As observed for the Rv3671c mutant in vitro, in IFN-γ –activated macrophages, intrabacterial acidification preceded a decline in viability of the bacteria and is probably the primary cause of death of the mutant. Accordingly, treatment of IFN-γ –activated macrophages with the alkalinizing weak base NH4Cl (ref. 34), which prevents acidification of phagosomes, protected pHIB of the mutant (Fig. 3g) and restored its survival (Fig. 3h).
Figure 3.

The Rv3671c mutant fails to maintain pHIB and is killed within activated macrophages. (a, b) Transmitted images with overlays of bacteria in green (left), fluorescent bacteria (center) and pseudocolored images of the 410:470 excitation ratio (right) of nonactivated (a) and IFN-γ –activated (b) macrophages 24 h after infection with wild-type Mtb, the Rv3671c mutant and the complemented mutant, each expressing pH-GFP. Scale bar, 10 μm. (c, d) Number of wild-type Mtb, Rv3671c mutant and complemented mutant bacterial groups plotted against their pHIB in nonactivated (c) and IFN-γ activated (d) macrophages at 24 h after infection. There were 105–130 bacterial groups examined, and data represent three independent experiments. (e, f) Quantification of CFU of wild-type Mtb, the Rv3671c mutant and the complemented mutant recovered from nonactivated (e) and IFN-γ –activated (f) macrophages at the indicated time points. Data are means ± s.d. of two independent experiments, each in triplicate, and are representative of four independent experiments. *P < 0.005 between wild-type and Rv3671c mutant bacteria, as determined by Student’s two tailed t-test. (g) Number of Rv3671c mutant bacterial groups plotted against their pHIB at 24 h after infection in IFN-γ –activated macrophages left untreated or treated with 10 mM NH4Cl. There were 185 bacterial groups examined, and data represent two independent experiments. (h) Quantification of CFU of wild-type Mtb, the Rv3671c mutant and the complemented mutant recovered from IFN-γ –activated macrophages treated with 10 mM NH4Cl. Data are means ± s.d. of two independent experiments, each in triplicate.
The Rv3671c mutant was severely impaired for growth in mice infected by inhalation (Fig. 4a). At 21 d after infection, titers of the Rv3671c mutant in lungs and spleens were more than 1.0 log lower than for wild-type Mtb (Fig. 4a), despite a 0.6-log higher inoculum. In C57BL/6 mice, a robust adaptive immune response is initiated within about 21 d after inhalation of low numbers of Mtb, at which point IFN-γ is produced, bacterial growth is restricted and Mtb persists at a near-constant titer for about a year35. In contrast, when we infected mice with the Rv3671c mutant, the mutant was killed progressively in the lung and spleen after day 21, such that at least 3.6-log and 3.0-log fewer mutant bacteria were recovered from lungs and spleens, respectively, at day 98 as compared to wild-type bacteria (Fig. 4a). This indicates that Rv3671c is required not only for exponential growth of Mtb but also for its persistence in the face of an activated immune system. The Rv3671c mutant induced markedly less pulmonary pathology than wild-type bacteria (Fig. 4b, c). Complementation restored full virulence indicating that attenuation of the Rv3671c mutant in mice was solely due to the disruption of Rv3671c (Fig. 4a, b).
Figure 4.
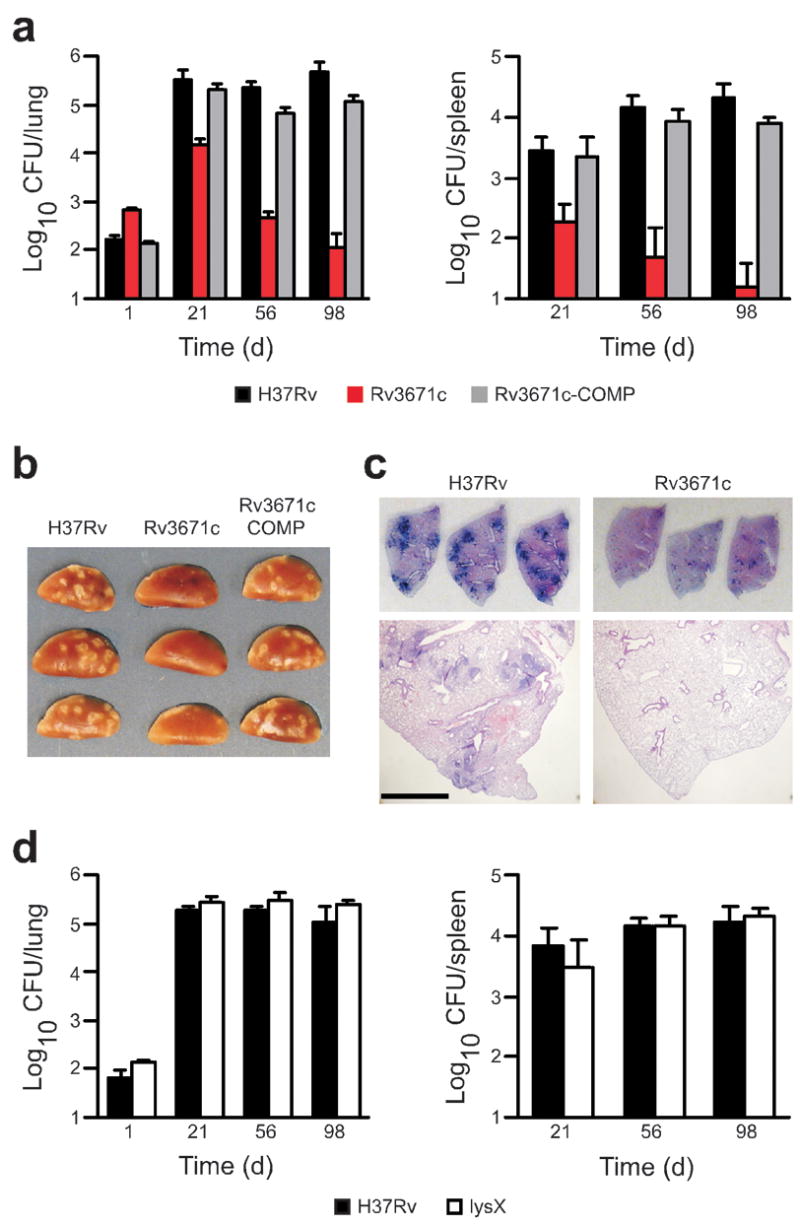
Rv3671c is required for Mtb growth and persistence in vivo. (a) Lung (left) and spleen (right) bacterial loads from mice infected with wild-type Mtb, the Rv3671c mutant or the complemented mutant at the indicated time points. At days 56 and 98, the CFU from two and three out of five Rv3671c mutant infected mice, respectively, were below the limit of detection in the spleen. Data are means ± s.d. from five mice per time point per group and represent three independent experiments; the limit of detection was 10 CFU per lung. (b) Gross pathology of lungs infected with wild-type Mtb, the Rv3671c mutant and the complemented mutant at day 56. The length of each lung lobe is 1.2 cm (c) Lung sections stained with H&E from mice infected with wild-type Mtb and Rv3671c mutant bacteria at day 56. Top images show unmagnified sections. The length of each lung lobe is 1.2 cm. Scale bar, 1.0 mm for bottom images. (d) Lung (left) and spleen (right) bacterial loads from mice infected with wild-type Mtb and the lysX mutant at the indicated time points. Data are means ± s.d. from four mice per time point per group and represent two independent experiments.
Rv3671c is a membrane protein but does not contain a PDZ protein-protein interaction domain, and as such is distinct from bacterial periplasmic HtrA (high temperature requirement A) proteins, of which Mtb is predicted to have at least three29, 36. It may protect Mtb from acid by modifying the bacterial cell envelope, regulating protein or lipid quality control and/or serving in signaling pathways that help the bacterium resist extracellular stresses. Expression of Rv3671c mRNA was constitutive and not induced after exposure of Mtb to low pH in vitro or in nonactivated and IFN-γ –activated macrophages (data not shown and ref. 37). Treatment of the Rv3671c mutant with the protonophore carbonyl cyanide m-chlorophenylhydrazone or the ionophore monensin further reduced the mutant’s pHIB and further attenuated its survival in pcit-Ty-4.5 medium (Supplementary Fig. 5 online), indicating that Rv3671c-independent mechanisms of pHIB homeostasis exist. Despite its susceptibility to extracellular protons, the Rv3671c mutant’s cell wall was not grossly altered, as the mutant bacterium resembled the wild-type Mtb in its colony morphology, formation of cords and cell structure as visualized by transmission and scanning electron microscopy at both neutral and acidic pHs (Supplementary Fig. 6 online). However, the Rv3671c mutant was hypersensitive to the cell wall–damaging detergent SDS and to the lipophilic antibiotics erythromycin and rifampin, suggesting that it has some defect in cell wall function (Supplementary Figs. 7 and 8 online). A mutant containing a transposon in lysX, which is annotated to encode a lysyl-tRNA synthetase, was also hypersensitive to erythromycin and rifampin (Supplementary Fig. 7) and as susceptible as the Rv3671c mutant to acid in 7H9-Tw-4.5 and 7H9-Ty-4.5 media (Fig. 1c). However, the mutant was capable of maintaining pHIB and was as viable as wild-type Mtb in pcit-Ty-4.5 medium (Figs. 2a and 1d). The lysX mutant was also fully virulent in mouse lungs and spleens (Fig. 4d). Thus, a defect in pHIB maintenance was associated with severe attenuation in vivo.
Mtb survives within macrophages by preventing fusion of phagosomes with lysosomes, but it also persists within acidic phagolysosomes in activated macrophages. We used ratiometric fluorescence measurements on live bacteria to show that Mtb is acid resistant and is able to control its pHIB in acid in vitro and within both nonactivated and activated macrophages. In the absence of Rv3671c, Mtb was unable to maintain pHIB and survive within activated macrophages. As acid promotes the activity of numerous host defenses, such as lysosomal hydrolases and reactive oxygen and nitrogen intermediates, the marked attenuation of the Rv3671c mutant in vivo is probably due to the synergistic interaction of phagosomal acid with other macrophage products. For instance, nitric oxide, whose accumulating auto-oxidation product nitrite (NO2−) can (re)generate the radicals NO and NO2 at low pH38, is likely to become more potent within the acidified bacterial cytosol of the Rv3671c mutant. The vulnerability of the mutant to the host environment recommends the Rv3671c protein as a drug target, notwithstanding that the mutant’s normal growth in vitro under standard conditions would exclude the protein’s candidacy by conventional criteria39. Identification of molecular pathways used by intracellular pathogens for pHIB homeostasis in acidic compartments of host cells is likely to reveal new targets for chemotherapy.
METHODS
Strains and media
We grew Mtb in Middlebrook 7H9 medium containing 0.2% glycerol, 0.5% BSA, 0.2% dextrose, 0.085% NaCl and 0.05% Tween-80 or 0.02% Tyloxapol, or on Middlebrook 7H10 or 7H11 agar containing 10% oleic acid–albumin-dextrose-catalase. The Rv3671c mutant showed reduced growth on 7H11 agar and was cultured on 7H10 agar. We acidified 7H9-Tw and 7H9-Ty to pH 4.5 with 2 N HCl and prepared phosphate-citrate (pcit) buffers from 200 mM sodium phosphate and 100 mM citric acid.
Screen
We grew the φMycoMarT7 transposon mutants28 in 96-well plates to stationary phase in 7H9-Tw-6.6 medium. We diluted aliquots (5 μl) 40-fold in 7H9-Tw-4.5 medium to ~5 × 106 CFU ml−1. After 6 d, we subcultured 10-μl aliquots in 200 μl 7H9-Tw-6.6 medium. We measured optical densities at an absorbance of 580 nm (OD580) 2–3 weeks later, when wild-type Mtb reached OD580 ~0.8–1.0, and rescreened mutants with OD580 < 0.1 five times. We analyzed chromosomal DNA from colony-purified mutants by Southern blotting with the kanamycin gene as a probe to confirm single transposon insertions, and then we sequenced the insertion sites.
Measurement of acid sensitivity
We washed early–log-phase cultures with 7H9-Tw-4.5, 7H9-Ty-4.5 or pcit-Ty-4.5 medium and centrifuged at 120g. for 10 min. We adjusted single-cell suspensions to ~5 × 106 CFU ml−1 in 7H9-Tw-4.5, 7H9-Ty-4.5 or pcit-Ty-4.5 medium and incubated them at 37 °C. We determined CFU by plating serial dilutions of the suspensions on 7H10 or 7H11 agar plates.
Intrabacterial pH measurements
We cloned pH-GFP33 downstream of the mycobacterial promoter Psmyc (ref. 40) and transformed Mtb strains with it. We adjusted early–log-phase cultures to an OD580 of 0.5 in 7H9-Tw medium, centrifuged at 3000g and resuspended pellets in an equal volume of pcit-Ty-7.4 or pcit-Ty-4.5 medium. We performed all pHIB measurements after incubation of bacteria at an OD580 of 0.5 in the respective buffer. We incubated triplicate 1-ml aliquots at 37 °C and concentrated them fivefold by centrifugation to increase the GFP signal. We analyzed 100-μl aliquots in a Molecular Devices M5 plate reader, exciting at absorbances of 395 nm and 475 nm and recording emission at an absorbance of 510 nm. We derived the pHIB by interpolating the 395:475 absorbance ratios on a standard curve (Supplementary Fig. 9 online). For pHIB measurements of intraphagosomal bacteria, we plated 1.5 × 105 bone marrow–derived mouse macrophages (BMDMs) in glass-bottom No. 1.5 thickness poly-D-lysine–coated 35-mm culture dishes (MatTek). We infected BMDMs with Mtb at an multiplicity of infection of two for 2 h, after which we washed twice with PBS. For microscopy, we placed BMDMs in DMEM without phenol red (GibcoBRL) supplemented with 1% FBS, 0.58 g l−1 L-glutamine, 1 mM sodium pyruvate and 10 mM HEPES. We used a Leica DMIRB inverted fluorescence microscope fitted with a 63× 1.4 numerical aperture objective and Chroma Technology pH-sensitive GFP filter set (exciters D410/30X and D470/20X, beamsplitter 500DCXR, emitter 535/50M). We performed image acquisition and analysis with a Photometrics CoolSnap HQ digital camera and MetaMorph v. 6.2r6 image analysis software (Universal Imaging). We acquired and analyzed all images within an experiment under identical conditions. For display in histograms, we determined average bacterial group ratio intensities (Fig. 3c, d, g, and Supplementary Fig. 4). A bacterial group was defined as at least one bacterium, but may consist of 2–5 bacilli. Pseudocolor images show pixel-by-pixel ratio intensities (Fig. 3a, b). All images at a given wavelength are shown at the same intensity settings or pseudocolor scale. We derived the pHIB by interpolating the 410:470 absorbance ratios on a standard curve (Supplementary Fig. 9).
Immunoblotting
We prepared lysates from mid–log-phase cultures in PBS containing protease inhibitor cocktail (Roche) by bead beating. We centrifuged lysates at 18,000g for 2 h at 4 °C to pellet cell walls. We centrifuged supernatants at 100,000g for 1 h at 4 °C to pellet cell membranes. We washed and resuspended pellets in SDS sample buffer. For SDS-PAGE, we loaded 10 μg of cytosolic fractions and an equal volume of cell envelope fractions based on total volume of the fractions. We used Flag-specific (Sigma) and dihydrolipoamide S-acyltransferase–specific rabbit antisera.
Complementation and mutagenesis
We cloned wild-type Rv3671c on an integrative vector conferring streptomycin resistance. We transformed the Rv3671c mutant by electroporation and selected transformants with 20 μg ml−1 streptomycin. We generated Flag-tagged and S343A mutant Rv3671c by PCR.
Mycobacterium tuberculosis survival in macrophages
We differentiated BMDMs in DMEM (GibcoBRL) containing 20% L-cell medium, 10% FBS, 0.58 g l−1 L-glutamine, 1 mM sodium pyruvate and 10 mM HEPES, providing a nearly pure macrophage population, as assessed by morphology and cell surface staining of the macrophage markers CD14, F4/80, Fc-γ receptor II/III and major histocompatibility complex class II, the latter after IFN-γ activation. We seeded BMDMs with or without 10 ng ml−1 murine IFN-γ (R&D Systems). Sixteen hours later, we infected macrophages at a multiplicity of infection of 0.1 and then washed with PBS 4 h later. We replaced medium every 48 h. We lysed BMDMs with 0.5% Triton X-100 and enumerated bacteria by plating serial dilutions of the lysate on 7H10 or 7H11 agar plates. Where indicated, we added 10 mM NH4Cl with IFN-γ before infection.
Mouse infections
We infected C57BL/6 mice (Jackson Laboratories) using an Inhalation Exposure System (Glas-Col) with early–log-phase Mtb to deliver ~100–200 bacilli per mouse or more where stated. We plated serial dilutions of organ homogenates from 4 or 5 mice per data point on 7H10 or 7H11 agar plates to quantify CFU. We fixed the upper left lung lobes in 10% buffered formalin, embedded them in paraffin and stained them with H&E. Procedures involving mice were reviewed and approved by the Institutional Animal Care and Use Committee of Weill Cornell Medical College. Statistical analyses. Statistical significance of the difference between experimental groups was determined by the two-tailed Student's t-test using PRISM Software. P values less than 0.05 were considered significant.
Supplementary Material
Acknowledgments
We thank G. Miesenböck (Oxford University) for providing ratiometric pH-GFP; F. Maxfield, J. Roberts and T. Odaira for guidance and support; and L. Cohen-Gould and T. Labissiere at the Electron Microscopy and Histology Core Facilities at Weill Cornell Medical College and Hospital for Special Surgery for assistance with electron microscopy. This work is supported by the US National Institutes of Health (grant PO1 AI056293 to C.F.N.) and the I.T. Hirschl Trust (S.E.). The Department of Microbiology and Immunology acknowledges the support of the William Randolph Hearst Foundation.
References
- 1.MacMicking JD, Taylor GA, McKinney JD. Immune control of tuberculosis by IFN-γ –inducible LRG-47. Science. 2003;302:654–659. doi: 10.1126/science.1088063. [DOI] [PubMed] [Google Scholar]
- 2.Schaible UE, Sturgill-Koszycki S, Schlesinger PH, Russell DG. Cytokine activation leads to acidification and increases maturation of Mycobacterium avium–containing phagosomes in murine macrophages. J Immunol. 1998;160:1290–1296. [PubMed] [Google Scholar]
- 3.Via LE, et al. Effects of cytokines on mycobacterial phagosome maturation. J Cell Sci. 1998;111:897–905. doi: 10.1242/jcs.111.7.897. [DOI] [PubMed] [Google Scholar]
- 4.Metchnikoff E. Immunity to Infective Disease. Cambridge University Press; Cambridge, London, New York: 1905. [Google Scholar]
- 5.Huynh KK, Grinstein S. Regulation of vacuolar pH and its modulation by some microbial species. Microbiol Mol Biol Rev. 2007;71:452–462. doi: 10.1128/MMBR.00003-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ohkuma S, Poole B. Fluorescence probe measurement of the intralysosomal pH in living cells and the perturbation of pH by various agents. Proc Natl Acad Sci USA. 1978;75:3327–3331. doi: 10.1073/pnas.75.7.3327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Armstrong JA, Hart PD. Response of cultured macrophages to Mycobacterium tuberculosis, with observations on fusion of lysosomes with phagosomes. J Exp Med. 1971;134:713–740. doi: 10.1084/jem.134.3.713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sturgill-Koszycki S, et al. Lack of acidification in Mycobacterium phagosomes produced by exclusion of the vesicular proton-ATPase. Science. 1994;263:678–681. doi: 10.1126/science.8303277. [DOI] [PubMed] [Google Scholar]
- 9.Clemens DL, Horwitz MA. Characterization of the Mycobacterium tuberculosis phagosome and evidence that phagosomal maturation is inhibited. J Exp Med. 1995;181:257–270. doi: 10.1084/jem.181.1.257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sibley LD, Franzblau SG, Krahenbuhl JL. Intracellular fate of Mycobacterium leprae in normal and activated mouse macrophages. Infect Immun. 1987;55:680–685. doi: 10.1128/iai.55.3.680-685.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nathan CF, Murray HW, Wiebe ME, Rubin BY. Identification of interferon-γ as the lymphokine that activates human macrophage oxidative metabolism and antimicrobial activity. J Exp Med. 1983;158:670–689. doi: 10.1084/jem.158.3.670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nathan CF, et al. Local and systemic effects of intradermal recombinant interferon-γ in patients with lepromatous leprosy. N Engl J Med. 1986;315:6–15. doi: 10.1056/NEJM198607033150102. [DOI] [PubMed] [Google Scholar]
- 13.Cooper AM, et al. Disseminated tuberculosis in interferon-γ gene-disrupted mice. J Exp Med. 1993;178:2243–2247. doi: 10.1084/jem.178.6.2243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Flynn JL, et al. An essential role for interferon-γ in resistance to Mycobacterium tuberculosis infection. J Exp Med. 1993;178:2249–2254. doi: 10.1084/jem.178.6.2249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dorman SE, et al. Clinical features of dominant and recessive interferon-γ receptor 1 deficiencies. Lancet. 2004;364:2113–2121. doi: 10.1016/S0140-6736(04)17552-1. [DOI] [PubMed] [Google Scholar]
- 16.Ehrt S, et al. Reprogramming of the macrophage transcriptome in response to interferon-γ and Mycobacterium tuberculosis: signaling roles of nitric oxide synthase-2 and phagocyte oxidase. J Exp Med. 2001;194:1123–1140. doi: 10.1084/jem.194.8.1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Xie QW, et al. Cloning and characterization of inducible nitric oxide synthase from mouse macrophages. Science. 1992;256:225–228. doi: 10.1126/science.1373522. [DOI] [PubMed] [Google Scholar]
- 18.MacMicking JD, et al. Identification of nitric oxide synthase as a protective locus against tuberculosis. Proc Natl Acad Sci USA. 1997;94:5243–5248. doi: 10.1073/pnas.94.10.5243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Armstrong JA, Hart PD. Phagosome-lysosome interactions in cultured macrophages infected with virulent tubercle bacilli. Reversal of the usual nonfusion pattern and observations on bacterial survival. J Exp Med. 1975;142:1–16. doi: 10.1084/jem.142.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gomes MS, et al. Survival of Mycobacterium avium and Mycobacterium tuberculosis in acidified vacuoles of murine macrophages. Infect Immun. 1999;67:3199–3206. doi: 10.1128/iai.67.7.3199-3206.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.MacGurn JA, Cox JS. A genetic screen for Mycobacterium tuberculosis mutants defective for phagosome maturation arrest identifies components of the ESX-1 secretion system. Infect Immun. 2007;75:2668–2678. doi: 10.1128/IAI.01872-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pethe K, et al. Isolation of Mycobacterium tuberculosis mutants defective in the arrest of phagosome maturation. Proc Natl Acad Sci USA. 2004;101:13642–13647. doi: 10.1073/pnas.0401657101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Stewart GR, Patel J, Robertson BD, Rae A, Young DB. Mycobacterial mutants with defective control of phagosomal acidification. PLoS Pathog. 2005;1:269–278. doi: 10.1371/journal.ppat.0010033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dubos R. The effect of lipids and serum albumin on bacterial growth. J Exp Med. 1947;85:9–22. doi: 10.1084/jem.85.1.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kanai K, Kondo E. Antibacterial and cytotoxic aspects of long-chain fatty acids as cell surface events: selected topics. Jpn J Med Sci Biol. 1979;32:135–174. doi: 10.7883/yoken1952.32.135. [DOI] [PubMed] [Google Scholar]
- 26.Vandal OH, Gelb MH, Ehrt S, Nathan CF. Cytosolic phospholipase A2 enzymes are not required by mouse bone marrow–derived macrophages for the control of Mycobacterium tuberculosis in vitro. Infect Immun. 2006;74:1751–1756. doi: 10.1128/IAI.74.3.1751-1756.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ortalo-Magne A, et al. Identification of the surface-exposed lipids on the cell envelopes of Mycobacterium tuberculosis and other mycobacterial species. J Bacteriol. 1996;178:456–461. doi: 10.1128/jb.178.2.456-461.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Darwin KH, Ehrt S, Gutierrez-Ramos JC, Weich N, Nathan CF. The proteasome of Mycobacterium tuberculosis is required for resistance to nitric oxide. Science. 2003;302:1963–1966. doi: 10.1126/science.1091176. [DOI] [PubMed] [Google Scholar]
- 29.Cole ST, et al. Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence. Nature. 1998;393:537–544. doi: 10.1038/31159. [DOI] [PubMed] [Google Scholar]
- 30.El Ghachi M, Bouhss A, Blanot D, Mengin-Lecreulx D. The bacA gene of Escherichia coli encodes an undecaprenyl pyrophosphate phosphatase activity. J Biol Chem. 2004;279:30106–30113. doi: 10.1074/jbc.M401701200. [DOI] [PubMed] [Google Scholar]
- 31.Rose L, Kaufmann SH, Daugelat S. Involvement of Mycobacterium smegmatis undecaprenyl phosphokinase in biofilm and smegma formation. Microbes Infect. 2004;6:965–971. doi: 10.1016/j.micinf.2004.05.011. [DOI] [PubMed] [Google Scholar]
- 32.Rawlings ND, Morton FR, Barrett AJ. MEROPS: the peptidase database. Nucleic Acids Res. 2006;34:D270–D272. doi: 10.1093/nar/gkj089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Miesenbock G, De Angelis DA, Rothman JE. Visualizing secretion and synaptic transmission with pH-sensitive green fluorescent proteins. Nature. 1998;394:192–195. doi: 10.1038/28190. [DOI] [PubMed] [Google Scholar]
- 34.Hart PD, Young MR. Ammonium chloride, an inhibitor of phagosome-lysosome fusion in macrophages, concurrently induces phagosome-endosome fusion, and opens a novel pathway: studies of a pathogenic mycobacterium and a nonpathogenic yeast. J Exp Med. 1991;174:881–889. doi: 10.1084/jem.174.4.881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.North RJ, Jung YJ. Immunity to tuberculosis. Annu Rev Immunol. 2004;22:599–623. doi: 10.1146/annurev.immunol.22.012703.104635. [DOI] [PubMed] [Google Scholar]
- 36.Mohamedmohaideen NN, et al. Structure and function of the virulence-associated high-temperature requirement A of Mycobacterium tuberculosis. Biochemistry. 2008;47:6092–6102. doi: 10.1021/bi701929m. [DOI] [PubMed] [Google Scholar]
- 37.Rohde KH, Abramovitch RB, Russell DG. Mycobacterium tuberculosis invasion of macrophages: linking bacterial gene expression to environmental cues. Cell Host Microbe. 2007;2:352–364. doi: 10.1016/j.chom.2007.09.006. [DOI] [PubMed] [Google Scholar]
- 38.Stuehr DJ, Nathan CF. Nitric oxide. A macrophage product responsible for cytostasis and respiratory inhibition in tumor target cells. J Exp Med. 1989;169:1543–1555. doi: 10.1084/jem.169.5.1543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nathan C. Antibiotics at the crossroads. Nature. 2004;431:899–902. doi: 10.1038/431899a. [DOI] [PubMed] [Google Scholar]
- 40.Ehrt S, et al. Controlling gene expression in mycobacteria with anhydrotetracycline and Tet repressor. Nucleic Acids Res. 2005;33:e21. doi: 10.1093/nar/gni013. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.