

Abstract
The ability of photo-activated rhodopsin to achieve the enzymatically active metarhodopsin II conformation is exquisitely sensitive to bilayer hydrophobic thickness. The sensitivity of rhodopsin to the lipid matrix has been explained by the hydrophobic matching theory which predicts that lipid bilayers adjust elastically to the hydrophobic length of transmembrane helices. Here, we examined if bilayer thickness adjusts to the length of the protein or if the protein alters its conformation to adapt to the bilayer. Purified bovine rhodopsin was reconstituted into a series of monounsaturated phosphatidylcholines with 14 to 20 carbons per hydrocarbon chain. Changes of hydrocarbon chain length were measured by 2H-NMR and protein helical content quantified by synchrotron radiation circular dichroism and conventional circular dichroism. Experiments were conducted on dark-adapted rhodopsin, the photointermediates metarhodopsin I/II/III, and opsin. Changes of bilayer thickness upon rhodopsin incorporation and photoactivation were mostly absent. In contrast, the helical content of rhodopsin increased with membrane hydrophobic thickness. Helical content did not change measurably upon photoactivation. The increase of bilayer thickness and helicity of rhodopsin are accompanied by higher Metarhodopsin II/Metarhodopsin I ratios, faster rates of Metarhodopsin II formation, an increase of tryptophan fluorescence, and higher temperatures of rhodopsin denaturation. The data suggest a surprising adaptability of this G protein-coupled membrane receptor to properties of the lipid matrix.
Keywords: rhodopsin, solid state NMR, membrane protein, membrane, hydrophobic matching, fluorescence, DSC, Circular Dichroism
1. Introduction
Rhodopsin, the mammalian dim-light photoreceptor, is the best characterized G protein-coupled receptor (GPCR) and the only one for which several high resolution structures are available in both the dark adapted 1 and photoactivated 2 states. Absorption of light leads to the formation of an active metarhodopsin II state (MII) in equilibrium with an inactive metarhodopsin I state (MI). Membrane composition strongly modulates the early steps of the visual process in rod cells by mechanisms that are still a matter of debate. Membrane hydrophobic thickness 3, 4, acyl chain unsaturation 5, and lipid headgroups 6, 7 are key factors in MII formation and Gt activation.
Exposure of hydrophobic sidechains of proteins to water is energetically unfavorable. Since membranes are known to deform elastically, it was proposed that membrane hydrophobic thickness adjusts to the length of hydrophobic stretches on transmembrane helices of proteins, also referred to as hydrophobic matching 8. Indeed, there is evidence from work on single helix, membrane-spanning peptide molecules which supports the notion that bilayers may adjust at least partially to transmembrane helices (see 9 and references therein). Another possibility is that membrane-incorporated peptides and proteins adjust to the hydrophobic thickness of lipid bilayers, see e.g. Killian at al. 9 and Jensen and Mouritsen 10. Recently it was suggested that transmembrane helix I of lactose permease may flex as well as tilt to satisfy the hydrophobic mismatch with the lipid matrix 11.
Here we examined whether dramatic changes of bilayer thickness result in matching of the lipid matrix to the hydrophobic length of the 7-helix bundle of rhodopsin or if rhodopsin adjusts to the bilayer. Experiments were conducted on bovine rhodopsin reconstituted into a series of mixed-chain lipids with saturated hydrocarbon chains of chain length from C14 to C20 in position sn-1 and matching monounsaturated chains with the same number of carbon atoms in position sn-2. Lipid matrix properties were investigated by 2H NMR order parameter measurements on the perdeuterated sn-1 chain lipids. Secondary structure of rhodopsin was investigated by circular dichroism (CD) measurements. Key experiments were repeated with synchrotron radiation CD (SRCD) to take advantage of the higher sensitivity of SRCD and the reduced perturbation of spectra by light scattering at low wavelength.
The response of dark adapted rhodopsin to changes of bilayer thickness was monitored by measurement of intrinsic tryptophan fluorescence and by measurement of the temperature of thermal denaturation. Photoactivation of rhodopsin was monitored by UV-Visible spectroscopy and by flash photolysis experiments to explore the rates of MII formation. The data show that rhodopsin structure and function are exquisitely sensitive to changes of membrane hydrophobic thickness. The helical content of rhodopsin increased incrementally with bilayer thickness, while the bilayers adjusted their thickness to the protein very little or not at all.
2. Experimental Methods
Preparation of reconstituted membranes
Sample preparation was carried out in complete darkness. The phospholipids 1-perdeutero-myristoyl-2-myristoleoyl-sn-glycero-3-phosphocholine (14:0d27-14:1-PC), 1-perdeutero-palmitoyl-2-palmitoleoyl-sn-glycero-3-phosphocholine (16:0d31-16:1-PC), 1-perdeutero-stearoyl-2-oleoyl-sn-glycero-3-phosphocholine (18:0d35-18:1-PC) and 1-perdeutero-arachidoyl-2-eicosanoyl-sn-glycero-3-phosphocholine (20:0d39-20:1-PC) were synthesized by Avanti Polar Lipids Inc. (Alabaster, AL).
Rhodopsin was purified from bovine retinas using procedures that were developed by the Litman laboratory 12. Rhodopsin fractions in 3 wt% octylglucoside (OG) which gave a UV/Visible absorption intensity ratio at 280 nm/500 nm of 1.8 were used. To eliminate the possibility that minor differences in rhodopsin purity may influence results, experiments were conducted using one batch of rhodopsin for reconstitution of the entire series of lipids. For rhodopsin reconstitution a glass round bottom flask was coated with phospholipids by slow rotation and removal of solvent in a stream of pure nitrogen gas. The rhodopsin-OG solution was added to lipid-OG mixed micelles so that the OG/lipid molar ratio was 10/1 and the rhodopsin/phospholipid molar ratio was 1/250. The sample was vortexed to complete solubilization of the lipid and then equilibrated for 12 hours under argon. Subsequently, this rhodopsin/lipid solution was added dropwise at a rate of approximately 400 μL/min to deoxygenated PIPES buffer (10 mM Pipes, 100 mM NaCl, 50 μm DTPA, pH = 7.0) under rapid stirring, resulting in formation of unilamellar proteoliposomes. Typically, the final OG concentration was 7.2 mM which is well below the critical micelle concentration. For all experiments but NMR, the proteoliposome dispersion (2.5-3 mL) was then dialyzed against 1 L of PIPES buffer (Slide-A-Lyzer membrane, 10 kD cut- off (Pierce, Rockford, IL)). The buffer was exchanged 3 times over 16 hours. Final rhodopsin concentration in the samples was measured by light absorption at 500 nm assuming a molar extinction coefficient ε500= 40,600 M-1cm-1 13. The concentration of residual OG in the lipid bilayers was lower than 0.4 mol% of the lipid concentration as determined by high resolution 1H NMR.
For NMR experiments on dark adapted rhodopsin, tubular rhodopsin-containing bilayers were formed by extrusion of the proteoliposomes after rapid dilution through porous anodic aluminum oxide (AAO) filters (Anopore, Whatman) with a nominal pore diameter of 0.2 μm and a thickness of 60 μm. Extrusion was performed at ambient temperature which is well above the main phase transition temperature of 14:0d27-14:1-PC, 16:0d31-16:1-PC and 18:0d35-18:1-PC, and at 30 °C in a heated glove box for the 20:0d39-20:1-PC membranes. For each experiment, one AAO filter (diameter 25 mm) and one polycarbonate filter (nominal pore size 0.8 μm) were stacked and flushed with several milliliters of PIPES buffer before extruding the dispersion of proteoliposomes. The 3 mL of proteoliposome suspension (0.3 - 0.7 mg lipids/mL) were then extruded ten times through the stack of filters at a rate of 0.01 mL/s resulting in entrapment of multilamellar structures inside AAO pores. All but a single tubular bilayer covering the inner AAO pore surface, as well as residual OG in membranes, were removed by flushing the filters with 5-10 mL of PIPES buffer at a rate of 0.2 mL/s as reported earlier 14, 15.
For NMR experiments with photoactivation, the dialyzed proteoliposomes were pelleted by centrifugation at 500,000xg and 10°C for 12 hours in a TLX-Optima centrifuge (Beckmann-Coulter Inc, Fullerton, CA, USA) and the pellet transferred to a 50 μL NMR tube. Rhodopsin was bleached by illuminating samples
NMR experiments
Solid state 2H NMR experiments were carried out on a Bruker AV800 spectrometer equipped with a double resonance Bruker probe (Bruker Biospin, Karlsruhe, Germany) operating at a 2H NMR resonance frequency of 122.8 MHz. A home-made flat coil was used for experiments on oriented bilayers, and a 4 mm solenoid coil for experiments on proteoliposomes with photoactivation. Data were acquired at a temperature of 27°C with a quadrupolar echo pulse sequence, d1-90°x-τ-90°y-τ-acq, a relaxation delay time, d1=250 ms (d1 was reduced to 100 ms in the photoactivation experiments), a 4.25-μs or 3-μs 90° pulse (flat coil or solenoid coil, respectively), a delay time, τ=50 μs, and a 200 kHz spectral width. Typically, 100,000 transients were acquired.
Order parameter profiles, mosaic spread of bilayer orientations, and resonance line width were determined by fitting the spectra with a program written in Mathcad (PTC, Needham, MA). The program reported a smoothed order parameter profile of lipid hydrocarbon chains, the orientational distribution function of bilayer normals (assumed to be Gaussian), and the resonance line width of all resolved quadrupolar splittings.
CD experiments.
Far-UV CD spectra of rhodopsin were recorded at 21°C with a JASCO J-810 spectrometer or by SRCD at beamline U11 of the National Synchrotron Light Source, Brookhaven National Laboratory. Rhodopsin concentration was about 5 μM unless stated otherwise, and the rhodopsin/lipid molar ratio was 1/250 except for a control experiment conducted at a molar ratio of 1/1,000. The exact rhodopsin concentrations were measured by light absorption at 500 nm. Cells with an optical pathlength of 0.01cm were used to minimize light scattering. Control experiments with cuvettes of 0.05 cm pathlength were conducted as well. Spectra on the JASCO J-810 were recorded from 260-180 nm with a data pitch of 0.2 nm, a bandwidth of 1 nm and an integration time of 4 s. The SRCD spectra were acquired with a Wadsworth monochromator at wavelength increments of 1 nm and a bandwidth of 1 nm. Four data points per wavelength were collected at a time constant of 1 s per data point.
Experiments with photobleaching of rhodopsin were conducted by recording CD spectra before and after illumination of samples with the light of a high powered green laser pointer for 30 s which results in complete bleaching. Measurements were conducted at a constant wavelength of 222 nm as a function of time.
CD intensities are expressed as the mean residue molar ellipticity [θ], calculated from the equation below, where θobs is the measured ellipticity in millidegrees, l is the optical path length, c is the final molar concentration of rhodopsin, and n is the number of amino acid residues.
The percentage of helicity was calculated from the residue ellipticity according to the method of Chen 16, using the following empirical equation to correct for helix length dependence,
where [θ]222 is the observed mean residue molar ellipticity at 222 nm, is the theoretical mean residue molar ellipticity for a helix of infinite length (-39,500 at 222 nm), η is an effective number of amino acid residues per helical segment of rhodopsin, and k is a wavelength-dependent constant (2.57 for 222 nm).
Fluorescence experiments.
The tryptophan fluorescence of rhodopsin was measured with a PTI QuantaMaster™ spectrofluorimeter (Birmingham, NJ). Briefly, rhodopsin-containing proteoliposomes (rhodopsin concentration 0.5-2.0 μM) were equilibrated at 25°C for 5 minutes in dark. The excitation and emission slits were set at 1 nm and 8 nm, respectively, to minimize bleaching of rhodopsin during measurement. The sample was excited at 295 nm and the emission from 305 to 400 nm was recorded. The sample was then fully bleached and, the emission spectrum of bleached rhodopsin was acquired at the same instrumental settings.
DSC experiments.
Differential scanning calorimetry (DSC) measurements were conducted with a Nano-scan II calorimeter (Calorimetry Sciences Co., Provo, UT) equipped with capillary cells which minimize the sedimentation of proteoliposomes during heating or cooling processes. Membrane suspensions were degassed for 5 minutes prior to loading into the sample cell in complete darkness. The reference cell was loaded with buffer only. The cell was sealed under a stream of argon and pressurized to 2.8 atm. At least two heating scans and two cooling scans were collected at a scan rate of 1.0 °C/min from 25°C to 85°C for each sample.
Measurement of MII/MI ratio after photoactivation
The equilibrium constant for MI ↔ MII, Keq, was determined from rapidly acquired spectra of the MI-MII equilibrium as previously described 17. Briefly, vesicles were diluted to a rhodopsin concentration of 0.3 mg/mL in pH 7.0 PBS buffer and equilibrated at 37°C in a thermally regulated sample holder. A set of four absorption spectra were collected sequentially in an Agilent 8453 diode array spectrophotometer. These included the spectra acquired 1) after the sample was equilibrated in the dark at 37°C; 2) 3 s after the sample was 15-20% bleached by a 520-nm flash; 3) 10 min after addition of 30 mM hydroxylamine to convert bleached rhodopsin to opsin and retinal oxime; and 4) after complete bleach of the sample. Individual MI and MII spectra were deconvolved from spectra of their equilibrium mixture; [MI] and [MII] were determined using extinction coefficients at their absorbance maximum of 44,000 cm-1 and 38,000 cm-1, respectively.
Rate of MII formation.
The rate of MII formation was determined from flash photolysis measurements at 380 nm acquired with an LKS60 spectrophotometer (Applied Photophysics, Leatherhead, UK). The actinic flash was provided by a Nd-YAG laser (Surelite™ I, Continuum, Santa Clara, CA) frequency doubled to 532 nm. Changes in photomultipier tube output were acquired by a 600 MHz Infiniium digital oscilloscope (Agilent Technologies, Santa Clara, CA), at 2 μs per point. A microcuvette containing 130 μL of proteoliposomes with 0.2-0.3 mg/mL rhodopsin was incubated in the sample holder at 37°C for 5 minutes. The 0% transmission level (baseline signal) and 100% transmission level were determined prior to the activation of rhodopsin by a laser flash. The signal was differentially amplified and recorded by the digital oscilloscope in the oversampling mode, which collects 32,000 data points/trace. The data were reduced to 2,000 points using boxcar averaging. The vendor software Laser (Applied Photophysics, Leatherhead, UK) allocates 10% of the points as pretrigger data points, which were used to convert the voltage signal into absorbance. The transient absorbance was calculated as: ΔA = log VA/VA’, where VA is the voltage of the pre-trigger data and VA’ is the voltage of the transient signal. Samples were measured in triplicate and the transient absorption traces were averaged.
3. Results and Discussion
Influence of rhodopsin on bilayer thickness.
According to the hydrophobic matching hypothesis, exposure of hydrophobic residues of protein or lipid to water is energetically unfavorable. Because acyl chains of fluid lipid phases are considered disordered and flexible, it has been hypothesized that they may stretch to match the hydrophobic length of the protein or compress and tilt if they are too short. Such a change in effective acyl chain length is easily detected by 2H NMR on perdeuterated, saturated hydrocarbon chains as a change of quadrupolar splittings.
The structural changes of lipids are largest near the protein, and perturbations decay with increasing distance from the protein/lipid interface. 2H NMR order parameters are measured on the relatively long timescale of ten microseconds. Since only one set of quadrupolar splittings per deuterated methylene segment has been observed in this and other studies (see e.g. 3, 14), lipids are in rapid exchange between the rhodopsin annulus and the bulk phase. Consequently, quadrupolar splittings are the geometric mean over lipid order near the protein and in the bulk. Because the quadrupolar splittings are measured with a resolution of a fraction of one percent, even small changes of lipid order near the protein are detectable.
Bilayer hydrophobic thickness depends on the number of methylene segments per hydrocarbon chain and on lipid order. The 2H NMR order parameters of methylene segments were converted into a time averaged hydrophobic thickness of bilayers as originally proposed by Schindler and Seelig 18. More accurate procedures for prediction of bilayer thickness from order parameter measurements have been introduced 19. Nevertheless, the conversion is likely to remain somewhat model dependent, in particular for membranes of rather different hydrocarbon chain length, and in the presence of membrane proteins. Thus we analyzed the data using the original approach 18. When differences of bilayer thickness are calculated as in this study, all methods essentially yield the same result. Even when assuming that the entire change of lipid order stems only from the contribution of the first lipid layer surrounding rhodopsin, corresponding to about 10 % of all lipids at a protein/lipid ratio of 1/250, the methods employed would still be able to detect thickness changes in the lipid annulus of less than 1 Å.
We incorporated dark-adapted rhodopsin into phosphatidylcholine membranes with sn-1 saturated and sn-2 monounsaturated acyl chains of 14 to 20 methylene segments in length (rhodopsin/lipid molar ratio 1/250). 2H NMR spectra were aquired on single lipid bilayers supported by porous anodic aluminum oxide as reported earlier 14 (Fig 1A-E). The order parameter measurements show that hydrophobic thickness between 14:0d27-14:1-PC and 20:0-20:1d39-PC bilayers differs by 12 Å (Table 1). The increase stems primarily from the increase of carbon atoms per hydrocarbon chain. However, there is also a small contribution, less than 10%, from a reduction of lateral area per lipid, seen as an incremental increase of average chain order with increasing chain length (Fig. 2). The latter is probably caused by an increase of attractive van der Waals interaction between hydrocarbon chains.
Figure 1.
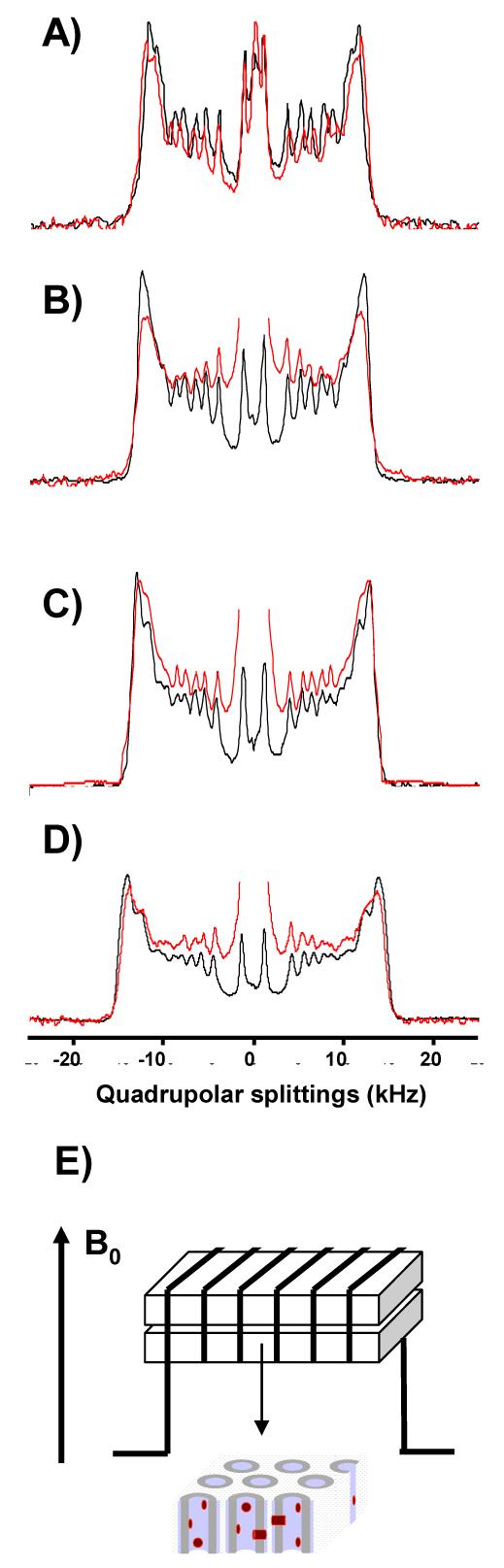
2H NMR spectra of sn-1 chain perdeuterated 14:0d27-14:1-PC (A), 16:0d31-16:1-PC (B), 18:0d35-18:1-PC (C), 20:0d39-20:1-PC (D) with (red trace) and without rhodopsin (black trace) at 27°C. The rhodopsin/lipid ratio is 1/250. Rhodopsin was reconstituted by rapid dilution from octylglucoside micellar solution. The proteoliposomes were extruded through porous aluminum oxide filters to form single, tubular lipid bilayers that line the pores (E). Filters were oriented in the magnetic field such that the axis of pores is oriented parallel to B0.
Table 1.
Variation of membrane and rhodopsin properties as a function of lipid chain length. The average order parameter (ΔSave), estimated hydrophobic thickness (d), rhodopsin molar ellipticity at λ= 222 nm [θ]222, increase in helical content relative to helical content in 14:0d27-14:1-PC (& relative to18:0d35-18:1-PC) , the ratio MII/MI after photoactivation at 37°C, Keq, apparent time constant of MII formation at 37°C, Tapp, and temperature of the midpoint of rhodopsin thermal denaturation, Td, as a function of lipid chain length.
| 14:0-14:1-PC | 16:0-16:1-PC | 18:0-18:1-PC | 20:0-20:1-PC | ||
|---|---|---|---|---|---|
| ΔSave | Rho- | 0.142±0.002 | 0.161±0.002 | 0.174±0.002 | 0.188±0.002 |
| Rho+ | 0.148±0.002 | 0.161±0.002 | 0.174±0.002 | 0.188±0.002 | |
| d* (Å) | Rho- | 21.20±0.07 | 25.20±0.08 | 29.20±0.09 | 33.20±0.10 |
| Rho+ | 21.40±0.07 | 25.20±0.08 | 29.20±0.09 | 33.20±0.10 | |
|
[θ]222 (deg.cm2. dmol-1 x10-4) |
CD | -1.66 ± 0.09 | -1.78 ± 0.05 | -1.94 ± 0.11 | NA |
| -1.8 ± 0.05# | -1.96 ± 0.05# | ||||
| SRCD | -1.71 ± 0.12 | ND | -2.04 ± 0.15 | ND | |
| Keq =MII/MI | 0.95 (± 0.04) | 1.71 (± 0.02) | 1.88 (± 0.02) | 2.18 (± 0.08) | |
| τapp MII (ms) | 2.33 (± 0.28) | 1.61 (± 0.17) | 0.38 (± 0.03) | 0.18 (± 0.02) | |
| Td(°C) | 67.6±0.1 | 68.9±0.1 | 71.1±0.1 | 72.6±0.1 | |
The hydrophobic thickness d, defined as the distance between acyl chain carbonyls of the two apposing monolayers was calculated as in (17).
Temperature was 27°C (all other CD, SRCD experiments were conducted at 21°C).
Figure 2.
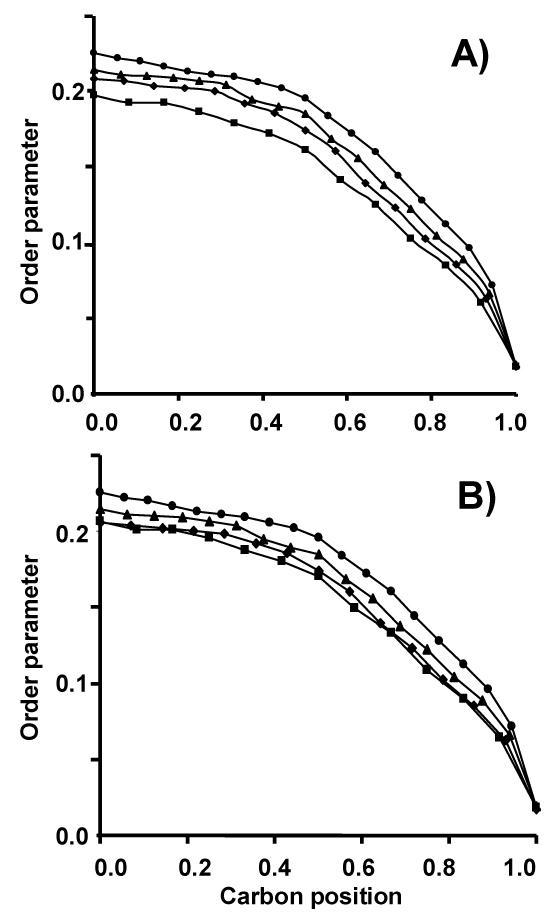
Order parameter profiles of perdeuterated sn-1 chains without (A) and in the presence of rhodopsin, lipid/rhodopsin=250/1, mol/mol (B) recorded at 27°C. Chain order increases with increasing chain length. ■ 14:0d27-14:1-PC, ◆ 16:0d31-16:1-PC; ▲18:0d35-18:1-PC, • 20:0d39-20:1-PC.
For lipids with 16, 18 and 20 methylene segments, changes of lipid order in the presence of dark adapted rhodopsin are smaller than the detection limit, translating into a membrane thickness variation of less than 0.5 Å near the protein (Fig 1, 2, Table 1).
The only bilayer for which the average order parameter changed measurably upon addition of protein was 14:0d27-14:1-PC (see Fig. 1A). Still, even for the bilayer with lowest hydrophobic thickness the increase of lipid order was very small, corresponding to an average bilayer stretch of only 0.2 Å, equivalent to a stretch of 2 Å or less in the first layer of lipids surrounding the protein.
Hydrophobic thickness after photoactivation
The pressure dependence of the MI-MII equilibrium as well as surface plasmon resonance studies suggest that MII has a larger volume, and possibly longer transmembrane helices 6, 20. If so, one would expect an additional increase of lipid C-D bond order parameters after photoactivation at pH 6 where MII formation is favored over MI. To test this hypothesis, order parameters were measured before and after photoactivation at 1-minute intervals. Experiments were conducted on 14:0d27-14:1-PC multilamellar proteoliposomes prepared by dialysis and ultracentrifugation to achieve higher sample densities. Samples were bleached by exposure to the light of a high-powered laser pointer (532 nm) for one minute at ambient temperature. According to UV-Vis absorption spectra, typical levels of bleaching are 98%. No changes of lipid order parameters were detected upon bleaching. This observation rules out any significant adjustment of membrane hydrophobic thickness to formation of MII. Sensitivity was such that changes in the lipid annulus of rhodopsin equal to or larger than 0.5 Å were detectable. Also, under conditions where the MI-MII equilibrium is greatly shifted toward MI, no measurable change of bilayer thickness occurred. At longer times after bleaching, when rhodopsin had converted predominantly to Meta III or opsin, no changes were observed as well.
Rhodopsin structural changes.
CD spectra of dark adapted rhodopsin were recorded in all four lipid matrices. To reduce light scattering, experiments were conducted at a very short optical path length. Measurements for every lipid were repeated several times to confirm observations, and 14:0d27-14:1-PC and 18:0d35-18:1-PC samples were investigated by SRCD which permitted recording spectra to a wavelength of 190 nm without exceeding the limits of high tension voltage for a linear response of the photomultiplier tube (Fig. 3). Spectra recorded with the JASCO J-810 spectrometer are in agreement with SRCD spectra in the wavelength range from 260 — 200 nm. Also, CD and SRCD spectra recorded at lower rhodopsin content (rhodopsin/lipid=1/1,000, mol/mol) are identical over this wavelength range after adjustment for concentration differences and the longer optical pathlength, suggesting that light scattering has negligible influence on results in the 260-200 nm range.
Figure 3.
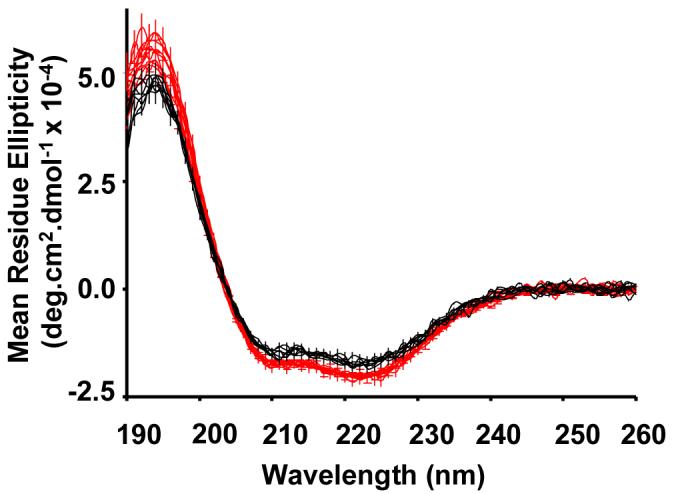
SRCD spectra of rhodopsin reconstituted into 14:0d27-14:1-PC (black traces) and 18:0d35-18:1-PC membranes (red traces) at 21°C. The spectra of three samples of both lipids, each recorded four times, are superimposed. Vertical bars reflect the experimental uncertainty of [θ] derived from precision of concentration measurements. In order to reduce scattering, experiments were conducted with a slide setup of 0.01 cm pathlength at a rhodopsin concentration of 5 μM.
At 21°C, taking the helical content of 14:0d27-14:1-PC of 48.9 %, determined by SRCD as base level, helicity increased by 3.2 ±1.5 % (CD) for 16:0d31-16:1-PC and by 7.7 ±3% (CD) or 8.0 ±3.5% (SRCD) for 18:0d35-18:1-PC. The increase of helicity from 18:0d35-18:1-PC to 20:0d39-20:1-PC is 4.6 ±1.5% (CD). The latter experiments were conducted at 27°C to be well above the main phase transition of 20:0d39-20:1-PC bilayers. The increase in helicity with increasing bilayer thickness suggests that rhodopsin helical content increases incrementally with increasing hydrophobic thickness of bilayers.
Helical content was calculated assuming an effective number of amino acids per helix of η=25. If it is assumed that the increase of helical content of 8% (SRCD) from 14:0d27-14:1-PC to 18:0d35-18:1-PC occurs entirely in the transmembrane helices of rhodopsin, each helix is elongated by one turn or ∼6 Å, roughly equivalent to the difference in membrane hydrophobic thickness between those bilayers.
Our spectra predict similar helical content as SRCD spectra of rhodopsin in DHPC/DMPC bicelles that were reported recently 21, but show some differences in the 215 - 190 nm spectral range that require further evaluation. In our spectra the minimum at 208 nm is not as deep and intensity at 195 nm is somewhat higher. The latter suggests that differences are not caused by light scattering that would have lowered intensity more at the lower wavelength. The lower intensity at 208 nm compared to 222 nm may indicate that the effective number of amino acids per helical segment, η, is in the range of twenty or fewer amino acids per helix as in myoglobin 16 rather than the 25 amino acids per transmembrane helix assumed on the basis of rhodopsin models.
Increasing bilayer thickness also stabilized dark adapted rhodopsin to temperature-induced denaturation as measured by DSC (Table 1). The increase is incremental, about 2°C per 3 Å increase of bilayer thickness. Furthermore, the tryptophan fluorescence of dark adapted rhodopsin is sensitive to differences between 14:0d27-14:1-PC and 18:0d35-18:1-PC bilayers. Excitation of rhodopsin in the dark state at 295 nm produces a weak fluorescence signal with an emission maximum at 326 nm that stems from the five tryptophan residues. Fluorescence intensity stems mostly from tryptophans 35 (helix I) and 175 (helix IV) located on the N-terminal side of the protein in the lipid-water interface. The other tryptophans are efficiently quenched by retinal 22. The fluorescence intensity was lower in 14:0d27-14:1 PC bilayers compared to those formed from 18:0d35-18:1 PC by 20% (Fig. 4).
Figure 4.
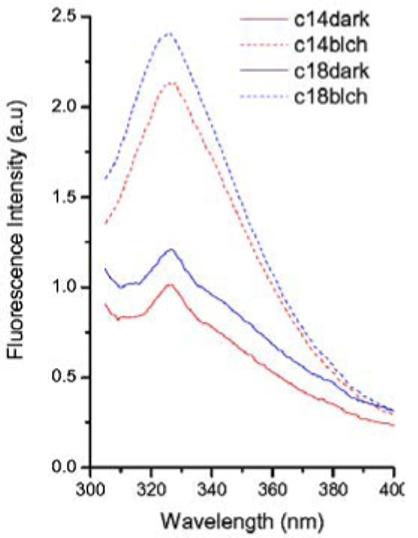
Fluorescence emission spectra of dark and bleached rhodopsin in 14:0d27-14:1-PC and 18:0d35-18:1-PC vesicles. The spectra were obtained at 25°C in PIPES buffer, pH 7.0. The solid lines are emission spectra of dark rhodopsin in 14:0d27-14:1-PC (red) and 18:0d35-18:1-PC (blue). The dashed lines are emission spectra of dark rhodopsin in 14:0d27-14:1-PC (red) and 18:0d35-18:1-PC (blue).
The lower intensity may result from collisional quenching by water suggesting that tryptophans are more accessible to solvent in the thinner bilayers. Alternatively, the structural change of rhodopsin itself may be responsible for the altered fluorescence yield.
Taken together, the increase of helicity observed by CD, the higher temperature of thermal denaturation, and the higher intensity of fluorescence spectra with increasing bilayer thickness suggest that the hydrophobic mismatch between a lipid bilayer and the GPCR rhodopsin is compensated for by an increase in the length of transmembrane helices of rhodopsin rather than a substantial modulation of bilayer thickness.
Influence on rhodopsin function
How do these bilayer-induced changes in helix length impact rhodopsin function? We measured the equilibrium concentration of MII by UV-Visible spectroscopy for the series of lipids and calculated Keq=MII/MI (Fig. 5). As reported previously 3, an increase of bilayers thickness raised the amount of MII (Table 1). The Keq rises incrementally with increasing bilayer thickness.
Figure 5.
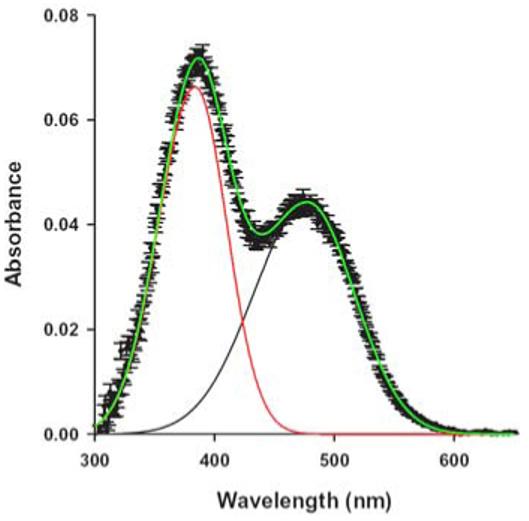
Deconvolved UV-VIS difference spectra of rhodopsin in 16:0d31-16:1-PC bilayers showing MI and MII photointermediates. The equilibrium concentrations of MI and MII were determined following a brief, actinic flash. Red trace: MII, black trace: MI, Green trace: total fit, MI + MII. Spectra of dark-adapted rhodopsin were subtracted from spectra acquired ∼2 seconds after a brief flash with green light. Spectra were acquired in 0.3 s with a diode array instrument. Absorbance due to unblached rhodopsin in this difference spectrum was subtracted by using a set of 3 difference spectra containing only rhodopsin and retinal oxime (absorbance maximum at 365 nm) to analytically determine the bandshape of the unbleached rhodopsin.
Experiments on the MI↔MII equilibrium were complemented by measurements of rates of MII formation. Results of flash-induced, time-resolved absorbance increases are shown in Fig. 6. The post-flash increase in absorbance at 380 nm is almost entirely due to the MII conformational state. The transient absorbance increase of MII formation, ΔA, was analyzed in terms of a sum of three exponentials, ΔA=Σai(1-exp(-kit)), shown as solid curves. For simplicity, the kinetics of MII formation was characterized in terms an apparent time constant, τapp, which is the time when the best-fit curve had risen to (1-1/e) of the plateau value, where the plateau value is a1+a2+a3(Table 1). The time constant of MII formation after photoactivation by flash photolysis declined progressively with increasing bilayer thickness. The change in bilayer thickness appears to have a differential influence on the time constants of subtransitions, a subject that requires a separate investigation. In summary, the MII/MI equilibrium and the rate of MII formation clearly demonstrate that rhodopsin photointermediates sense differences in membrane hydrophobic thickness as well.
Figure 6.
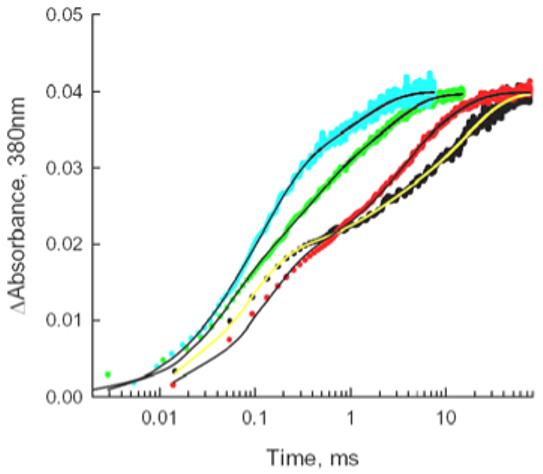
Flash-induced absorbance increases at 380 nm showing the effects of membrane thickness on the time course of MII formation. Traces were scaled to the same amplitude to facilitate comparison of the variation in kinetics. Blue points: 20:0d39-20:1-PC, green points: 18:0d35-18:1-PC, red points: 16:0d31-16:1-PC, black points: 14:0d27-14:1-PC. Smooth curves through the data points are best-fit curves of the sum of three exponential terms.
Experiments were conducted at the rhodopsin/lipid molar ratio of 1/250 to minimize any influence on results from rhodopsin oligomerization. In earlier experiments, conducted as a function of rhodopsin concentration, a dependence of the MI↔MII equilibrium as well as of lipid dynamics on rhodopsin concentration at higher rhodopsin content were observed 14, 23. Brown and coworkers reported recently that association of rhodopsin is promoted by a reduction of membrane thickness as well as by an increase in protein/lipid molar ratio 3. Molecular simulations conducted as a function of bilayer thickness at the rhodopsin/lipid molar ratio of 1/100 also showed some formation of rhodopsin oligomers, in particular for lipids with shorter hydrocarbon chains 24. But in bilayers of 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine rhodopsin, fluorescence resonance energy transfer between rhodopsin molecules at protein/lipid molar ratio of 1/200 and lower had low efficiency, suggesting that rhodopsin molecules were mostly monomeric 3. While we may not exclude that some oligomerization has occured in our experiments, the sensitivity of rhodopsin structure and function to changes of membrane hydrophobic thickness indicate that rhodopsin molecules remain well exposed to the lipid matrix. Any rhodopsin-rhodopsin association must be low scale and temporary to be compatible with experimental observations. Indeed, freeze-fracture electron micrographs of rhodopsin in similar membranes of 14:0-14:0-PC 4, 25 and 18:0-22:6-PC (Kachar, Polozov & Gawrisch, unpublished results) investigated at a rhodopsin/lipid ratio of 1/100 did not show any large aggregates of protein. However, formation of dimers or trimers may not be excluded, and these could explain recent experimental and theoretical observations on rhodopsin oligomerization as a function of membrane thickness 3, 24.
4. Conclusion
The results presented here underline the importance of hydrophobic matching between proteins and membranes and promote a novel view of hydrophobic matching. Rather then observing the anticipated adjustment of membrane hydrophobic thickness to match the hydrophobic length of rhodopsin’s transmembrane helices, we observed an increase of helical content with increasing bilayer thickness. This suggests, then, that transmembrane helices of rhodopsin have an underappreciated plasticity for matching bilayer thickness. Furthermore, there is no evidence that helical content or bilayer thickness undergo an adjustment when photoactivated rhodopsin undergoes the conformation changes required for G-protein binding.
It is well known that the stability of helical turns at the ends of helices is generally lower since they are not as well stabilized by intrahelical hydrogen bonding 26. Therefore, it is plausible that the protein responds to changes of bilayer thickness by increasing or decreasing the length of transmembrane helices. Those bilayer-induced structural changes persist after photoactivation. The altered helical content of rhodopsin could be directly responsible for changes in the amount of MII, and of rates of MII formation.
Supplementary Material
Acknowledgement
This work was supported by the Intramural Research Program of NIAAA, NIH. We thank Kirk Hines for extraction and purification of rhodopsin. The SRCD experiments were performed at beamline U11 at the National Synchrotron Light Source which is supported by the Office of Biological and Environmental Research, U.S. Department of Energy. The NSLS is supported by the Office of Basic Energy Science of the USDOE. We thank John G. Trunk, NSLS, for assistance and support.
Supporting Information Available: Figure showing the thermal denaturation of rhodopsin in 14:0d27-14:1-PC bilayers recorded by DSC. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Li J, Edwards PC, Burghammer M, Villa C, Schertler GFX. J. Mol. Biol. 2004;343:1409–1438. doi: 10.1016/j.jmb.2004.08.090. [DOI] [PubMed] [Google Scholar]; Okada T, Sugihara M, Bondar AN, Elstner M, Entel P, Buss V. J. Mol. Biol. 2004;342:571–583. doi: 10.1016/j.jmb.2004.07.044. [DOI] [PubMed] [Google Scholar]; Palczewski K, Kumasaka T, Hori T, Behnke CA, Motoshima H, Fox BA, Le Trong I, Teller DC, Okada T, Stenkamp RE, Yamamoto M, Miyano M. Science. 2000;289:739–745. doi: 10.1126/science.289.5480.739. [DOI] [PubMed] [Google Scholar]
- 2.Ruprecht JJ, Mielke T, Vogel R, Villa C, Schertler GFX. EMBO J. 2004;23:3609–3620. doi: 10.1038/sj.emboj.7600374. [DOI] [PMC free article] [PubMed] [Google Scholar]; Salom D, Lodowski DT, Stenkamp RE, Le Trong I, Golczak M, Jastrzebska B, Harris T, Ballesteros JA, Palczewski K. Proc. Natl. Acad. Sci. USA. 2006;103:16123–16128. doi: 10.1073/pnas.0608022103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Botelho AV, Huber T, Sakmar TP, Brown MF. Biophys. J. 2006;91:4464–4477. doi: 10.1529/biophysj.106.082776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Baldwin PA, Hubbell WL. Biochemistry. 1985;24:2633–2639. doi: 10.1021/bi00332a007. [DOI] [PubMed] [Google Scholar]
- 5.Mitchell DC, Niu SL, Litman BJ. J. Biol. Chem. 2001;276:42801–42806. doi: 10.1074/jbc.M105772200. [DOI] [PubMed] [Google Scholar]; Niu SL, Mitchell DC, Lim SY, Wen ZM, Kim HY, Salem N, Litman BJ. J. Biol. Chem. 2004;279:31098–31104. doi: 10.1074/jbc.M404376200. [DOI] [PubMed] [Google Scholar]; Niu SL, Mitchell DC, Litman BJ. J. Biol. Chem. 2001;276:42807–42811. doi: 10.1074/jbc.M105778200. [DOI] [PubMed] [Google Scholar]
- 6.Brown MF. Chem. Phys. Lip. 1994;73:159–180. doi: 10.1016/0009-3084(94)90180-5. [DOI] [PubMed] [Google Scholar]
- 7.Wang Y, Botelho AV, Martinez GV, Brown MF. J. Am. Chem. Soc. 2002;124:7690–7701. doi: 10.1021/ja0200488. [DOI] [PubMed] [Google Scholar]
- 8.Mouritsen OG, Bloom M. Biophys. J. 1984;46:141–153. doi: 10.1016/S0006-3495(84)84007-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Killian JA, Nyholm TKM. Curr. Op. Struct. Biol. 2006;16:473–479. doi: 10.1016/j.sbi.2006.06.007. [DOI] [PubMed] [Google Scholar]
- 10.Jensen MO, Mouritsen OG. Biochim. Biophys. Acta. 2004;1666:205–226. doi: 10.1016/j.bbamem.2004.06.009. [DOI] [PubMed] [Google Scholar]
- 11.Yeagle PL, Bennett M, Lemaitre V, Watts A. Biochim. Biophys. Acta. 2007;1768:530–537. doi: 10.1016/j.bbamem.2006.11.018. [DOI] [PubMed] [Google Scholar]
- 12.Litman BJ. Method Enzymol. 1982;81:150–153. doi: 10.1016/s0076-6879(82)81025-2. [DOI] [PubMed] [Google Scholar]
- 13.Wald G, Brown PK. J. Gen. Physiol. 1953;37:189–200. doi: 10.1085/jgp.37.2.189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Soubias O, Polozov IV, Teague WE, Yeliseev AA, Gawrisch K. Biochemistry. 2006;45:15583–90. doi: 10.1021/bi061416d. [DOI] [PubMed] [Google Scholar]
- 15.Gaede HC, Luckett KM, Polozov IV, Gawrisch K. Langmuir. 2004;20:7711–7719. doi: 10.1021/la0493114. [DOI] [PubMed] [Google Scholar]
- 16.Chen YH, Yang JT, Chau KH. Biochemistry. 1974;13:3350–3359. doi: 10.1021/bi00713a027. [DOI] [PubMed] [Google Scholar]
- 17.Straume M, Mitchell DC, Miller JL, Litman BJ. Biochemistry. 1990;29:9135–9142. doi: 10.1021/bi00491a006. [DOI] [PubMed] [Google Scholar]
- 18.Schindler H, Seelig J. Biochemistry. 1975;14:2283–2287. doi: 10.1021/bi00682a001. [DOI] [PubMed] [Google Scholar]
- 19.Nagle JF. Biophys. J. 1993;64:1476–1481. doi: 10.1016/S0006-3495(93)81514-5. [DOI] [PMC free article] [PubMed] [Google Scholar]; Petrache HI, Dodd SW, Brown MF. Biophys. J. 2000;79:3172–3192. doi: 10.1016/S0006-3495(00)76551-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Salamon Z, Wang Y, Soulages JL, Brown MF, Tollin G. Biophys. J. 1996;71:283–294. doi: 10.1016/S0006-3495(96)79224-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.McKibbin C, Farmer NA, Jeans C, Reeves PJ, Khorana HG, Wallace BA, Edwards PC, Villa C, Booth PJ. J. Mol. Biol. 2007;374:1319–1332. doi: 10.1016/j.jmb.2007.10.018. [DOI] [PubMed] [Google Scholar]
- 22.Farrens DL, Khorana HG. J. Biol. Chem. 1995;270:5073–5076. doi: 10.1074/jbc.270.10.5073. [DOI] [PubMed] [Google Scholar]
- 23.Niu SL, Mitchell DC. Biophys. J. 2005;89:1833–1840. doi: 10.1529/biophysj.105.061812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Periole X, Huber T, Marrink SJ, Sakmar TP. J. Am. Chem. Soc. 2007;129:10126–10132. doi: 10.1021/ja0706246. [DOI] [PubMed] [Google Scholar]
- 25.Chen YS, Hubbell WL. Exp. Eye Res. 1973;17:517–532. doi: 10.1016/0014-4835(73)90082-1. [DOI] [PubMed] [Google Scholar]
- 26.Aurora R, Srinivasan R, Rose GD. Science. 1994;264:1126–1130. doi: 10.1126/science.8178170. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.