

Abstract
Context
Identifying families at high risk for the Lynch syndrome (ie, hereditary non-polyposis colorectal cancer) is critical for both genetic counseling and cancer prevention. Current clinical guidelines are effective but limited by applicability and cost.
Objective
To develop and validate a genetic counseling and risk prediction tool that estimates the probability of carrying a deleterious mutation in mismatch repair genes MLH1, MSH2, or MSH6 and the probability of developing colorectal or endometrial cancer.
Design, Setting, and Patients
External validation of the MMRpro model was conducted on 279 individuals from 226 clinic-based families in the United States, Canada, and Australia (referred between 1993–2005) by comparing model predictions with results of highly sensitive germline mutation detection techniques. MMRpro models the autosomal dominant inheritance of mismatch repair mutations, with parameters based on meta-analyses of the penetrance and prevalence of mutations and of the predictive values of tumor characteristics. The model’s prediction is tailored to each individual’s detailed family history information on colorectal and endometrial cancer and to tumor characteristics including microsatellite instability.
Main Outcome Measure
Ability of MMRpro to correctly predict mutation carrier status, as measured by operating characteristics, calibration, and overall accuracy.
Results
In the independent validation, MMRpro provided a concordance index of 0.83 (95% confidence interval, 0.78–0.88) and a ratio of observed to predicted cases of 0.94 (95% confidence interval, 0.84–1.05). This results in higher accuracy than existing alternatives and current clinical guidelines.
Conclusions
MMRpro is a broadly applicable, accurate prediction model that can contribute to current screening and genetic counseling practices in a high-risk population. It is more sensitive and more specific than existing clinical guidelines for identifying individuals who may benefit from MMR germline testing. It is applicable to individuals for whom tumor samples are not available and to individuals in whom germline testing finds no mutation.
The lynch syndrome (ie, hereditary nonpolyposis colorectal cancer [HNPCC]) is the most common familial colorectal cancer (CRC).1,2 It can be caused by germline deleterious mutations of DNA mismatch repair (MMR) genes, including MLH1,3,4 MSH2,5 MSH6,6 and several others.7 Screening for individuals likely to carry a deleterious mutation of these genes has traditionally relied on examination of family history, as per the Amsterdam Criteria,8–10 and has recently moved toward multistep algorithms combining family history with molecular tumor characteristics such as microsatellite instability (MSI),11 as per the Bethesda Guidelines.2,12
The latter were developed to help recognize those individuals who would potentially benefit from a more detailed molecular diagnostic workup, including MSI and subsequent germline testing. This approach is useful, but not without important limitations. First, MSI tests can only be performed on affected patients for whom a tumor block is available. This limits the preventive usefulness of the approach. Second, the algorithm calls for MSI testing on a large population with colorectal cancer, which increases the cost per mutation carrier detected. Third, for individuals tested with commercial germline testing techniques that have imperfect sensitivity, this approach cannot offer guidance for decision making when no mutation is found.
Extensive knowledge is now available about the Lynch syndrome: the mode of inheritance of the genes is autosomal dominant,13 prevalence and penetrance have been studied, and their clinical/molecular manifestation in tumors is well characterized. The translation of such knowledge into clinically useful algorithms can benefit greatly from a systematic, quantitative, and objective approach. Application of such an approach to a specific family should use as much clinical and biological information about the pedigree as possible, to provide an individualized assessment of risks. Insurance companies may demand an objective assessment of mutation probability when considering reimbursement for genetic testing, and they often accept a statistical estimate of mutation probability as a justification.
A step in this direction is the Leiden model,14 which estimates the combined probability of carrying a mutation of an MMR gene using a logistic regression based on 3 variables: fulfillment of the Amsterdam Criteria, mean age of CRC diagnoses, and presence of any endometrial cancers in the family. This model is useful but does not use all available biological knowledge and does not provide risk predictions.
The translational goals described above are more fully achieved by formulating an explicit genetic model,15 as was done successfully for the BRCA genes.16–19 To this end, we developed MMRpro, a model that estimates the probability that an individual carries a deleterious mutation in an MMR gene. This probability is evaluated on the basis of detailed family history of colorectal and endometrial cancer, including information for the individual being counseled and each of his or her first- and second-degree relatives, as described in the BOX. In addition, MMRpro provides estimates of future cancer risks for unaffected individuals, including known mutation carriers, untested individuals, and individuals in whom no mutation is found. MMRpro is appropriate for prediction in both population-based and clinic-based families. It is useful to clinical geneticists in parallel with clinical criteria, especially when current guidelines are inadequate to address the particularity of a given family and when the possibility of a heritable defect cannot be directly addressed in the laboratory.
Box. Family History as Inputs to the MMRpro Model and Resulting Output
Input (for the counselee and each first- or second-degree relative)*
Exact relation to the counselee
Colorectal cancer status (affected or unaffected)
Age at diagnosis (in years) of colorectal cancer if affected
Endometrial cancer status (affected or unaffected)
Age at diagnosis (in years) of endometrial cancer if affected
Current age or age at last follow-up (in years) if unaffected
Result of microsatellite instability testing (instability present or not present) or immunohistochemical staining (loss of expression or present) if tumor available
Result of previous germline testing of MLH1, MSH2, or MSH6 (positive or not found)
Output (for the counselee)†
Probability, by gene, that the counselee carries a deleterious mutation of MLH1, MSH2, and MSH6
Probability, in yearly intervals, that the counselee, if asymptomatic, will develop colorectal or endometrial cancers in the future
*Any input can be left unspecified if not available, with the exception of relation to the counselee.
†Prediction can be made for any member in the family by designating that member as the counselee.
METHODS
This study included the development and validation of MMRpro. Development consisted of estimating genetic parameters from published studies and specifying a mutation-prediction algorithm as well as a cancer-risk prediction algorithm. External validation consisted of comparing model predictions with germline testing results in a sample of families.
Parameter Estimation
MMRpro relies on estimates of mutation prevalence and penetrance of MMR genes and on sensitivity and specificity of MSI and germline testing. We obtained these via comprehensive literature reviews summarized here and described in detail at http://astor.som.jhmi.edu/~sining/documents/MMRpro_Supplement.pdf.
Penetrance refers to the age-specific risks of developing colorectal and endometrial tumors among mutation carriers, by gene and by sex. We restricted our meta-analysis to 5 studies20–24 that used design and analysis plans giving unbiased risk estimates. Among these, 4 are population-based; the fifth used an adjustment for ascertainment bias. We estimated prevalence of MMR mutations indirectly, from penetrance estimates obtained from the literature as described above; CRC incidence from the Surveillance, Epidemiology, and End Results registry; and carrier prevalence among cases reported in the literature.
Testing for MSI is not perfectly sensitive in predicting MMR mutations because of uncertainty in the classification of the instability and its lower prevalence in tumors with certain mutations, such as mutations on MSH6. It is also not perfectly specific because of MLH1 promoter hypermethylation. Mutation analysis is also not always sensitive because of the complex spectrum of MMR mutations. To accurately account for the predictive ability of these tests in predicting a true germline mutation, these sensitivities and specificities were derived based on a comprehensive published meta-analysis25 of 16 studies.26–41 The sensitivity and specificity of immunohistochemical testing was set to equal that of MSI testing because its predictive value of loss of protein expression is similar.42–45
To represent a specific population (different mutation prevalence/penetrance) or assay (different sensitivity/specificity), it is straightforward for users to replace the default values described above with preferred ones.
Model Specification
MMRpro translates estimates of mutation prevalence and penetrance of MMR genes into mutation predictions based on a general mendelian modeling approach, described in detail elsewhere.46 In summary, the model involves the application of the Bayes rule15,16,47–49 and mendelian laws as follows: Prgenotype|history = (Prgenotype × Prhistory|genotype)/Prhistory, where Pr denotes probability, genotype denotes counselee’s genotype (deleterious mutations of MLH1, MSH2, or MSH6 by gene), history denotes family history (refers to the information presented in the Box), and Pr(A|B) denotes the probability of A given that B is observed.
In the numerator, the probability of the counselee’s genotype is derived from the mutations’ prevalences, while the probability of family history given the counselee’s genotype is evaluated as a weighted average of the probabilities of family history conditional on all possible relatives’ genotypes, where the weights are the mendelian autosomal dominant probabilities of each genotype configuration. The probability of the family history under each genotype configuration can be broken down into the product of each relative’s probability of phenotype given his or her own genotype. Each term is calculated based on the penetrance for affected relatives or the probability of censoring for unaffected relatives. When a family member undergoes MSI, immunohistochemical, or germline testing, the corresponding probabilities of test results given the genotype come from the sensitivity and specificity of that test in detecting true genotype. To evaluate the denominator, we sum the probabilities at the numerator over all possible genotypes for the counselee. Finally, risks of developing colorectal and endometrial cancers for unaffected individuals are estimated based on averages of the carriers’ and noncarriers’ risks, weighted by carrier probability.46
Validation
Patients
Our validation sample included 279 germline-tested individuals from 226 families in 3 clinic-based groups. All study patients provided written informed consent for this study, and the study was approved by the institutional review board at each participating institution.
The first group included 81 individuals from 59 families from the Johns Hopkins Colorectal Cancer Risk Assessment Clinic and Hereditary Colorectal Cancer Registry, Baltimore, Md. These individuals were either self-referred or referred to the clinic by health care workers between 1993–1996 because of a personal and/or family history of colorectal cancer. Patients and families were entered in the registry if at least 2 family members had colon or rectal cancer or if an individual developed colorectal or uterine cancer before age 50 years. Most patients came from Maryland. All individuals were screened extensively for MLH1 and MSH2 mutations using techniques including direct sequencing, protein truncation, conversion, and, in some individuals, more than 1 of the above.50–52 Testing for MSI was performed on a subset of cases.
The second group of patients was recruited since 1997 through 4 centers (the Mayo Clinic and the University of Southern California Consortium in the United States, Cancer Care Ontario in Canada, and the University of Queensland in Australia) belonging to the National Cancer Institute Colon Cancer Family Registry (Colon CFR). The purpose was studying the clinical usefulness of conversion analysis.53 Patients were included if they had a prior diagnosis of CRC, had an available EpsteinBarr virus–transformed cell line, and met any of 3 criteria: (1) were members of a family meeting the Amsterdam Criteria I; (2) had at least 2 first-or second-degree relatives with CRC, or 1 relative with endometrial cancer and at least 1 other with CRC; and (3) were otherwise diagnosed with CRC before age 50 years. In addition to meeting these criteria, all patients had prior evidence of a defect in MMR due to having either a tumor classified as MSI-high or loss of expression of an MMR protein demonstrated by immunohistochemistry. Individuals from the Colon Cancer Family Registry were uniformly tested by both DNA sequencing and conversion. Southern blotting was performed to detect large genomic deletions. Microsatellite instability testing and immunohistochemical analysis was performed on all cases.53
The third group consisted of 144 individuals from the Memorial Sloan-Kettering Cancer Center. These individuals were referred from within the center and by outside physicians to the center’s Clinical Genetics Service between November 1994 and December 2005 for the possible presence of an MMR mutation. Criteria for referral varied, but generally included either early age at onset of colon cancer or multiple cases of colon cancer in multiple generations. Among all patients who underwent MMR gene testing, a randomly selected subset of 144 was used in the validation. This group included approximately equal numbers of patients referred for clinical testing and enrolled into 3 clinical protocols using different ascertainment criteria. Testing for MSI was performed when tumors were available. MLH1 and MSH2 mutational analysis was carried out on all index CRC patients from whom a blood sample could be obtained. MSH6 mutation analysis was performed on all patients enrolled in one of the clinical protocols. MMR mutations were tested using a combination of denaturing high-performance liquid chromatography and semiquantitative fluorescent multiplex–polymerase chain reaction analysis.11,54–56
Family history and demographic information from all 3 groups are summarized by source in TABLE 1. The families include those at high risk (eg, Amsterdam Criteria), moderate risk (eg, HNPCC-like or Bethesda Guidelines), and others who do not fulfill any of these criteria. Overall, this mix reflects the population presenting for clinical and genetic counseling and for whom mutation prediction models are most useful. All individuals across the 3 clinic groups were searched extensively for germline mutations with highly sensitive techniques that are capable of detecting large-scale deletions, exon duplications, and monoallelic mutations.
Table 1.
Demographic Information for the Independent Validation Sample, by Source
| No. | ||||
|---|---|---|---|---|
| Characteristic | JHU | Colon CFR | MSKCC | Total |
| Ascertainment | ≥2 CRCs or age <50 y | Highly selected | Physician referred | |
| Genotyping method | ≥1 of direct sequencing, PTT, conversion | Both direct sequencing and conversion | DHPLC or SQF-PCR | |
| Total germline-tested individuals | 81 | 54 | 144 | 279 |
| Total families | 59 | 54 | 113 | 226 |
| Total individuals in all families (counseled + first- and second-degree relatives) | 1197 | 1548 | 3249 | 5994 |
| Tested male individuals | 39 | 33 | 62 | 134 |
| Tested individuals with CRC | 53 | 53 | 70 | 176 |
| Tested individuals with EC | 1 | 5 | 14 | 20 |
| Mean CRC cases per family (first- and second-degree only) | 3.7 | 4.9 | 2.7 | 3.4 |
| Mean EC cases per family (first- and second-degree only) | 0.34 | 0.61 | 0.60 | 0.54 |
| Mean age at diagnoses of CRC in all affected members, y | 50.9 | 43.6 | 51.4 | 48.6 |
| Mean age at diagnoses of EC in all affected members, y | 59.5 | 42.7 | 50.9 | 50.4 |
| Families fulfilling Amsterdam Criteria II | 32 | 48 | 65 | 145 |
| Individuals fulfilling revised Bethesda Guidelines | 52 | 53 | 61 | 166 |
| Mutations found
MLH1 |
14 |
27 |
10 |
51 |
| MSH2 | 10 | 18 | 35 | 63 |
| MSH6 | 0 | 0 | 7 | 7 |
| MSI tests performed | 15 | 46 | 59 | 120 |
| MSI test results, mutations/tests
MSI-H |
15/15 |
39/46 |
23/29 |
77/90 |
| MSI-L | −/0 | −/0 | 1/2 | 1/2 |
| MSS | −/0 | −/0 | 1/21 | 1/21 |
| Indeterminate | −/0 | −/0 | 2/7 | 2/7 |
Abbreviations: Colon CFR, Colon Cancer Family Registry; CRC, colorectal cancer; DHPLC, denaturing high-performance liquid chromatography; EC, endometrial cancer; JHU, Johns Hopkins University; MSKCC, Memorial Sloan-Kettering Cancer Center; MSI, microsatellite instability; MSI-H, microsatellite instability–high; MSI-L, microsatellite instability–low; MSS, microsatellite stability; PTT, protein truncation testing; SQF-PCR, semiquantitative fluorescent multiplex–polymerase chain reaction.
Data Analysis
We computed MMR-pro and Leiden mutation predictions on all germline-tested individuals in the validation set. MMRpro predicts risks for mutations on MLH1, MSH2, and MSH6. For individuals screened only for MLH1 and MSH2 mutations, we used the predictions on these 2 genes for comparability with the mutation analysis performed. We calculated MMRpro probabilities both with and without MSI testing. In both cases we used the full set of individuals and only incorporated MSI information when available. We evaluated the models’ refinement, calibration, and overall performance.57,58 Refinement is the ability to discriminate between mutation-positive and -negative individuals and is measured by the receiver operating characteristic (ROC) curve, which is summarized by the area under the curve (AUC), ie, the concordance index. Calibration is the correspondence between the number of mutation-positive individuals predicted and the number found and is quantified by the ratio of observed to expected positive results. Overall performance is quantified by mean squared error of prediction, a measure of distance between the predicted probabilities and the observed mutation carrier status. Confidence intervals are evaluated using the bootstrap method59 with 95% coverage.
We also evaluated sensitivity and specificity of Amsterdam Criteria II9 and revised Bethesda Guidelines12 (hereafter, “II” and “revised” are implied), examining whether each patient’s family history fulfilled the criteria. For evaluating Bethesda Guidelines followed by MSI testing, we defined positive individuals as those fulfilling the Bethesda Guidelines and showing MSI. Since we did not find individuals who fulfilled the Bethesda Guidelines but were not tested for MSI to be a biased subsample, we imputed their MSI status according to the proportions observed among Bethesda Guidelines–positive individuals whose MSI status is available.
RESULTS
MMRpro Software
Software for performing MMRpro calculations is open source and available free of charge via either the mendelian risk prediction package BayesMendel46 at http://astor.som.jhmi.edu/BayesMendel/ or the genetic counseling package CancerGene60 at http://www4.utsouthwestern.edu/breasthealth/cagene/.
Clinical Application
To demonstrate the application of the MMRpro model in a counseling setting, we consider 5 variations of the pedigree in FIGURE 1. The scenarios are hypothetical but realistic. In each, we give the MMR mutation probability for the counselee (arrow), as estimated by MMRpro, before and after MSI testing. As a reference, the mutation probability of a random individual for whom no information is available is 0.002. Meanwhile, the mutation probability for the counselee alone, with no information on relatives, is 0.05 prior to MSI testing and 0.34 after a positive result. This level at 0.34 may be sufficient to justify germline testing or increased colonoscopic screening, even though the counselee alone would not meet any clinical criteria.
Figure 1.

Comparisons of Carrier Probability Estimation Approaches on 5 Pedigree Variations
Numbers below each family member indicate current age, or age at death, for unaffected individuals and age at diagnosis for affected individual. The counselee is identified by the arrow. MS indicates microsatellite; MSI, microsatellite instability; MSS, microsatellite stable.
*Information in these columns refers to MSI testing for the counselee.
In scenario 1, the counselee and her father were both diagnosed with CRC in their 50s. This family history does not fulfill the Bethesda Guidelines, but yields a significant mutation probability of 0.15. Should the counselee have MSI testing, her possible post-MSI mutation probabilities would differ substantially (0.65 if MSI and 0.04 if microsatellite stable), indicating that MSI testing is highly informative for her. MMRpro results can justify insurance coverage for her MSI test. Another important use of the MMR-pro estimate is to interpret an inconclusive test result quantitatively. If the counselee has an MSI-positive tumor and no mutation was found by germline testing using sequencing, her postsequencing probability of an undetected germline mutation would be as high as 0.36. Counselees may be less likely to undertake screening when they are informed that no mutation was found. In such a case, immunohistochemical analysis for MSH6 expression or conversion analysis, for example, would be additional considerations justified by this model. Also in this example, the counselee’s 30-year-old son has a 0.18 mutation probability. This leads to a 15% chance of developing CRC by age 70 years, making him a likely candidate for intensive screening despite his mother’s inconclusive test result.
In scenario 1, the counselee’s paternal grandmother died at age 30 years without a cancer diagnosis. Had she lived longer, knowledge of whether she was affected or not would have a stronger effect on the counselee’s mutation probability. For example, in scenario 2, she lived cancer-free until age 71 years, providing evidence against autosomal dominant transmission. This results in a reduction in mutation probability from 0.15 to 0.10 in the counselee.
In scenario 3, the paternal grandmother is affected with CRC at 71 years, and the diagnoses of the father and the paternal aunt are switched. Now the counselee has 2 relatives diagnosed with CRC, fulfilling the Bethesda Guidelines. However, the father is cancer-free until age 79 years, reducing the chance that the counselee’s cancer is due to inherited mutation, and her mutation probability is one third of that in scenario 1.
Scenario 4 presents an additional case of endometrial cancer on the maternal side. Although endometrial cancer is highly predictive of a mutation, the maternal lineage of this tumor is independent of the mendelian dominant transmission pattern in the paternal lineage. So, while this additional diagnosis changes the counselee’s Bethesda Guidelines status from scenario 1, her mutation probability does not increase significantly.
In scenario 5, endometrial cancer occurs in the counselee’s sister. This is stronger evidence of a transmitted mutation, so the pretest probability increases to 0.82. For counselees at such high risk, the use of a prediction model can help them proceed directly to immunohistochemical analysis or germline testing. Even if the tumor does not show MSI, the posttest probability is still high. This suggests that the family may still be genetically susceptible.
External Validation
MMRpro predicted the presence of approximately 129 mutations. This shows a close correspondence (calibration) with the observed 121 mutations (ratio of observed to expected results, 0.94), as shown in TABLE 2. The slight overprediction by MMRpro may be an indication of its ability to predict germline mutations that are not detected by the mutation analysis performed here, such as mutations in PMS2. On the other hand, the Leiden model predicts a substantially lower number of cases than are observed (3 cases are observed for every 2 that are predicted). A possible explanation is that it was developed using results from conventional mutation analysis techniques, which may have missed a large fraction of MMR mutations.50,53
Table 2.
Summary of Validation Results
| Concordance Index 5% CI)* | O/E Ratio (95% CI)† | Mean Squared Error (95% CI) | ||
|---|---|---|---|---|
| MMRpro + MSI testing‡ | 0.83 (0.78 to 0.87) | 0.94 (0.84 to 1.05) | 0.18 (0.15 to 0.22) | |
| MMRpro | 0.79 (0.74 to 0.84) | 0.97 (0.86 to 1.08) | 0.19 (0.16 to 0.23) | |
| Leiden | 0.77 (0.71 to 0.83) | 1.54 (1.35 to 1.80) | 0.24 (0.20 to 0.28) | |
| Difference between MMRpro + MSI and Leiden | 0.06 (−0.02 to 0.14) | Not applicable§ | 0.06 (0.005 to 0.11) | |
| Sensitivity | Specificity | |||
| Revised Bethesda Guidelines + MSI|| | 0.72 | 0.69 | ||
| Revised Bethesda Guidelines | 0.77 | 0.54 | ||
| Amsterdam Criteria|| | 0.75 | 0.62 | ||
Abbreviations: CI, confidence interval; MSI, microsatellite instability.
Concordance index is equal to the area under the receiver operating characteristic curve.
Ratio between the observed number of carriers and the total number of predicted carriers.
Represents MMRpro prediction after taking into account the MSI test results, when available.
Computing the difference in this case is not an appropriate comparison, as each of the ratios should be compared directly with the reference value of 1.
Refers to applying the revised Bethesda Guidelines, testing the individuals who fulfill the guidelines for MSI, and referring those individuals testing positive for MSI to germline testing.
MMRpro provides better discriminatory ability than both the Leiden model and the Bethesda Guidelines. FIGURE 2 shows ROC curves for the MMRpro and Leiden models with and without MSI, as well as sensitivity and specificity of the Bethesda Guidelines with and without MSI testing. The corresponding AUCs are presented (as concordance indexes) in Table 2. The difference between the AUC of MMRpro with MSI testing and that of the Leiden model is 0.06 (95% confidence interval, −0.02 to 0.14), an important difference in the AUC scale. MMRpro with MSI testing discriminates better than the Leiden model in 93% of the bootstrap replicates. When calibration and discrimination are combined into a single evaluation using the mean squared error of prediction, the MMR-pro model shows significantly improved performance compared with the Leiden model (Table 2).
Figure 2.
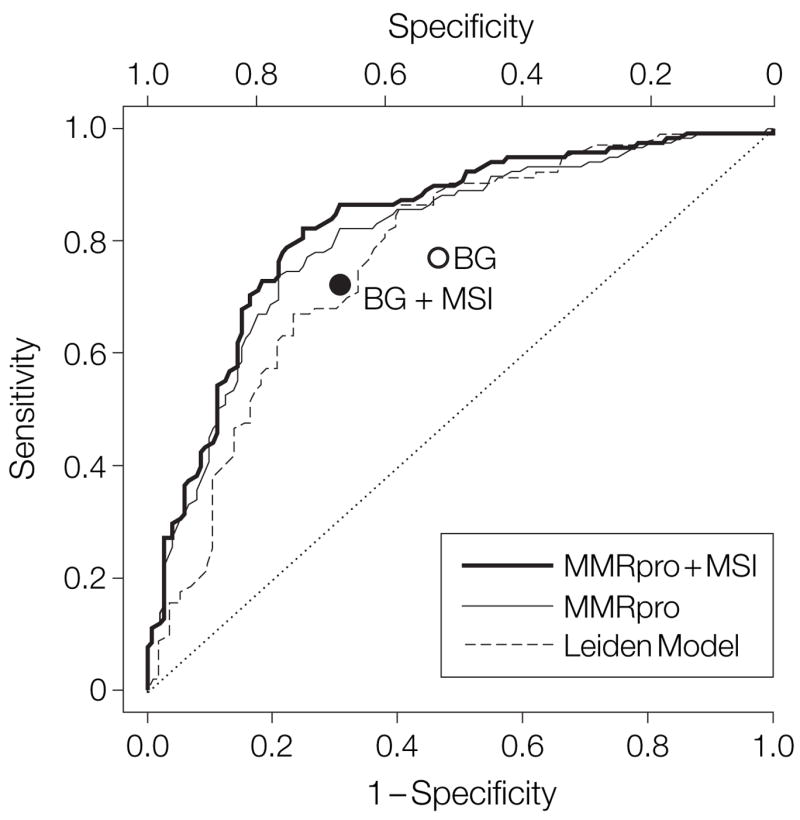
Receiver Operating Characteristic Curves for the MMRpro and Leiden Models on the Validation Data Set
Also shown are the estimated true-positive (y-axis) and false-positive (x-axis) fractions associated with the Bethesda Guidelines (BG). MSI indicates microsatellite instability.
The point corresponding to the Bethesda Guidelines without MSI testing lies below the ROC curve for MMR-pro without MSI testing (Figure 2), indicating that MMRpro performs better than the Bethesda Guidelines in selecting individuals who may carry a germline mutation. Similar results apply when comparing the point for the Bethesda Guidelines with MSI testing to the curve for MMRpro with MSI testing. Using a cutoff of 0.35 on the MMRpro probability, one can achieve the same specificity as the Bethesda Guidelines with MSI testing and higher sensitivity, while 0.62 will provide the same sensitivity and higher specificity. Cutoffs within this range provide both higher sensitivity and higher specificity. While the specific cutoffs may depend on the mix of families and the proportion of MSI-tested individuals in our sample, the positioning of the Bethesda Guidelines points below the corresponding MMRpro curves is likely to be robust.
The comparison of the ROC curves between MMRpro and MMRpro with MSI testing suggests that MMRpro correctly takes advantage of MSI test results, as predictions improve almost uniformly. However, the improvement is moderate. This may be due to the fact that not all cases have been tested for MSI. It may also indicate that in situations in which family history is very informative, MSI testing provides only limited additional information. In 99 of 120 MSI-tested individuals in our sample, MSI results changed probability by less than 0.10; in 88 individuals, by less than 0.05; and in 51 individuals, by less than 0.01. In such cases, MSI testing may not be the best course of action to determine the presence of a mutation. When used to assess the usefulness of MSI testing, MMRpro can lead to significant cost savings.
Finally, we considered how often MMRpro leads to a different classification compared with the Leiden model, Bethesda Guidelines, and Amsterdam Criteria. The results are shown in TABLE 3. MMRpro leads to reclassification of a significant fraction of individuals. In all 3 comparisons, correctly reclassified individuals outnumber those reclassified incorrectly. In the table, both MMRpro and Leiden predictions are dichotomized at 0.5. This threshold is chosen for illustrative purposes. In practice, thresholds should be chosen based on individual circumstances. However, MMRpro will always reclassify correctly more often than incorrectly, irrespective of the threshold chosen, based on ROC results.
Table 3.
Comparison of MMRpro-Based Probability With Leiden-Based Classification, Bethesda Guidelines, and Amsterdam Criteria*
| Carriers |
Noncarriers |
|||||
|---|---|---|---|---|---|---|
| MMRpro Pr ≥0.5 | MMRpro Pr <0.5 | MMRpro Pr ≥0.5 | MMRpro Pr <0.5 | Reclassification Rate, % | ||
| Leiden | ||||||
| ≥0.5 | 54 | 3† | 9 | 17‡ | ] | 33 |
| <0.5 | 39‡ | 22 | 23† | 80 | ||
| Bethesda Guidelines | ||||||
| + | 83 | 10† | 28 | 37‡ | ] | 24 |
| − | 1‡ | 18 | 4† | 63 | ||
| Amsterdam Criteria | ||||||
| + | 76 | 5† | 26 | 11‡ | ] | 15 |
| − | 17‡ | 23 | 6† | 89 | ||
Abbreviation: MMRpro Pr, MMRpro-based probability.
MMRpro and Leiden predictions dichotomized at 0.5 for illustrative purposes.
Patients classified differently using MMRpro, compared with the model in the corresponding row.
Patients reclassified correctly using MMRpro.
COMMENT
This article introduces MMRpro, a model for prediction of genetic susceptibility in the Lynch syndrome, which makes efficient use of family history and tumor information and provides individualized evaluations. Because model-based prediction algorithms are increasingly used in genetic counseling and prevention activities, MMRpro is a timely tool for identifying and counseling families at risk for the Lynch syndrome and can improve current genetic counseling and early detection practice. In an independent validation, MMRpro demonstrated a better ability to predict mutation carriers than both the Leiden model and Bethesda Guidelines–based screening.
Current Bethesda Guidelines–based screening practice aims at identifying individuals likely to harbor tumors with MSI. An important limitation is that these criteria are not applicable when a tumor block is unavailable or to unaffected individuals concerned by family history and considering genetic testing and secondary prevention. Among individuals with tumors, the Bethesda Guidelines are sensitive but not highly specific and rely on MSI testing to improve specificity. However, even when tumor samples are available, MSI testing may not be the optimal course of action for all families. The high-resolution quantitative assessment obtained by setting a high threshold on MMRpro offers the option of performing germline testing directly, without MSI testing, in selected families.46 This strategy can both increase specificity and decrease costs. In our validation sample, MMRpro provides equal or better sensitivity and specificity with less MSI testing. We also estimated that a large fraction of families in the validation sample can reach an informed decision on whether to undertake germline testing without testing for MSI. For others, such as small families, families with older ages at diagnosis, and some families not meeting the Bethesda Guidelines, MSI testing is highly informative.
Some clinics also use immunohistochemical staining, because the loss of a protein product is highly predictive of the presence of mutations.61 In our model we can account for either immunohistochemical or MSI results. Given the technical complexity of MSI analysis, which involves microdissection and DNA extraction, pathology departments may first perform immunohistochemical analysis and reserve MSI analysis for cases with a strong clinical suggestion. In that scenario, MMR-pro may also be useful, in that it would allow for setting a threshold for performing such additional analyses.
When germline mutation tests with relatively low sensitivity are used, MMRpro posttest probabilities are useful for individuals in whom, despite strong evidence of predisposition, no mutation is found. Before more sensitive techniques become available or new genetic factors are identified, the cancer risk predictions for such individuals provided by MMRpro help to guide subsequent clinical management. This feature is also valuable for counselees who do not wish to be genotyped but would still like to consider preventative measures.
Along with other screening approaches,62,63 risk prediction based on family history is routinely used to identify individuals for CRC screening. Due to the imperfect sensitivity of sequencing, it remains likely that high-risk in dividuals who are untested or who receive “no mutation found” results will undergo routine screening.When asymptomatic individuals and their family members age without developing CRC, their chance of carrying deleterious mutations decreases. MMRpro can be used to help adaptively update their screening choices.
While useful, our model has limitations, some of which will be addressed in future updates as new studies provide the necessary information. Currently, MMRpro considers only endometrial cancer, the most common type of extracolonic tumor associated with the Lynch syndrome. The spectrum is wider, but at present, data on penetrance for these cancers are insufficient for modeling purposes. Colorectal adenomas and polyps and their histological features may also be predictive of MMR mutations, but their predictive value is difficult to quantify. The predictive value of MSI may vary with age because of the age-related increase in hypermethylation of the MLH1 promoter region.64,65 The MSI status of extracolonic cancers is likely to have different predictive abilities, depending on the site as well. Lifestyle risk factors also have yet to enter the model, as doprophylactic surgeries that reduce risks significantly.66 In model-based risk counseling, variability in the estimates should be recognized. Uncertainty remains on the risk conferred by MMR mutation (as illustrated by the supplementary figure at http://astor.som.jhmi.edu/~sining/documents/MMRpro_Supplement.pdf), calling attention to the importance of more extensive investigations of penetrance.
Finally, in decision making for germline testing, a mathematical model can be informative for the reasons we have described. However, decision making regarding germline testing should reflect a broader range of factors, including the effectiveness and cost of genotyping; the available means and efficacy of measures for early detection and risk reduction; and possible psychological, social, and ethical implications. This should be done in concert with a health care professional experienced in cancer genetics67–69 who can also advise on the choice of cutoffs appropriate for individual circumstances. The informed consent process will ensure that patients consider all of these issues prior to testing.70
Supplementary Material
Acknowledgments
We thank John Hopper, PhD, and Mark Jenkins, PhD, Centre for Molecular, Environmental, Genetic and Analytic Epidemiology, University of Melbourne, Victoria, Australia, for providing unpublished details about their article23 and Amanda Blackford, MSc, Sidney Kimmel Comprehensive Cancer Center, for assistance with data analysis. Drs Hopper and Jenkins received no compensation for their contributions; Ms Blackford’s work is supported by Johns Hopkins SPORE in Gastrointestinal Cancer grant P50CA62924.
Funding/Support: This research was supported by Johns Hopkins SPORE in Gastrointestinal Cancer grant P50CA62924; National Cancer Institute (NCI) grant R01CA105090; the Cancer Research Foundation of America; the John G. Rangos, Sr, Charitable Foundation; the Clayton Fund; the Niehaus-Southworth-Weissenbach Research Fund; and the Evan Frankel Fellowship. The work of the Colon Cancer Family Registry (Colon CFR) is supported by the NCI, National Institutes of Health, under Request for Applications No. CA-96-001 and through cooperative agreements with member of the Colon CFR and principal investigators.
Footnotes
Author Contributions: Dr Chen had full access to all of the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Study concept and design: Chen, Wang, S. Lee, Watson, Gruber, Euhus, Kinzler, Parmigiani.
Acquisition of data: Nafa, J. Lee, Romans, Watson, Gruber, Jass, Gallinger, Lindor, Casey, Ellis, Giardiello.
Analysis and interpretation of data: Chen, Wang, S. Lee, Watson, Gruber, Ellis, Giardiello, Parmigiani.
Drafting of the manuscript: Chen, Romans, Gruber, Giardiello, Parmigiani.
Critical revision of the manuscript for important intellectual content: Chen, Wang, S. Lee, Nafa, J. Lee, Watson, Gruber, Euhus, Kinzler, Jass, Gallinger, Lindor, Casey, Ellis, Giardiello, Parmigiani.
Statistical analysis: Chen, S. Lee, Parmigiani.
Obtained funding: Kinzler, Jass, Gallinger, Lindor, Parmigiani.
Administrative, technical, or material support: Nafa, J. Lee, Romans, Gruber, Euhus, Giardiello.
Study supervision: Chen, Gruber, Giardiello, Parmigiani.
Financial Disclosures: None reported.
Publisher's Disclaimer: Disclaimer: The content of this article does not necessarily reflect the views or policies of the NCI or any of the collaborating centers in the Colon CFR, nor does mention of trade names, commercial products or organizations imply endorsement by the US Government or the Colon CFR.
References
- 1.Lynch HT, de la Chapelle A. Genetic susceptibility to non-polyposis colorectal cancer. J Med Genet. 1999;36:801–818. [PMC free article] [PubMed] [Google Scholar]
- 2.Rodriguez-Bigas MA, Boland CR, Hamilton SR, et al. A National Cancer Institute workshop on hereditary nonpolyposis colorectal cancer syndrome: meeting highlights and Bethesda guidelines. J Natl Cancer Inst. 1997;89:1758–1762. doi: 10.1093/jnci/89.23.1758. [DOI] [PubMed] [Google Scholar]
- 3.Papadopoulos N, Nicolaides NC, Wei YF, et al. Mutation of a mutL homolog in hereditary colon cancer. Science. 1994;263:1625–1629. doi: 10.1126/science.8128251. [DOI] [PubMed] [Google Scholar]
- 4.Bronner CE, Baker SM, Morrison PT, et al. Mutation in the DNA mismatch repair gene homologue hMLH1 is associated with hereditary non-polyposis colon cancer. Nature. 1994;368:258–261. doi: 10.1038/368258a0. [DOI] [PubMed] [Google Scholar]
- 5.Leach FS, Nicolaides NC, Papadopoulos N, et al. Mutations of a mutS homolog in hereditary nonpolyposis colorectal cancer. Cell. 1993;75:1215–1225. doi: 10.1016/0092-8674(93)90330-s. [DOI] [PubMed] [Google Scholar]
- 6.Miyaki M, Konishi M, Tanaka K, et al. Germline mutation of MSH6 as the cause of hereditary nonpolyposis colorectal cancer. Nat Genet. 1997;17:271–272. doi: 10.1038/ng1197-271. [DOI] [PubMed] [Google Scholar]
- 7.Nicolaides NC, Papadopoulos N, Liu B, et al. Mutations of two PMS homologues in hereditary non-polyposis colon cancer. Nature. 1994;371:75–80. doi: 10.1038/371075a0. [DOI] [PubMed] [Google Scholar]
- 8.Vasen HFA, Mecklin JP, Khan PM, Lynch HT. The International Collaborative Group on Hereditary Non-Polyposis Colorectal Cancer (ICG-HNPCC) Dis Colon Rectum. 1991;34:424–425. doi: 10.1007/BF02053699. [DOI] [PubMed] [Google Scholar]
- 9.Vasen HF, Watson P, Mecklin JP, Lynch H. New clinical criteria for hereditary nonpolyposis colorectal cancer (HNPCC, Lynch syndrome) proposed by the International Collaborative group on HNPCC. Gastroenterology. 1999;116:1453–1456. doi: 10.1016/s0016-5085(99)70510-x. [DOI] [PubMed] [Google Scholar]
- 10.Syngal S, Fox E, Eng C, Kolodner R, Garber J. Sensitivity and specificity of clinical criteria for hereditary non-polyposis colorectal cancer associated mutations in MSH2 and MLH1. J Med Genet. 2000;37:641–645. doi: 10.1136/jmg.37.9.641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Boland CR, Thibodeau SN, Hamilton SR. A National Cancer Institute Workshop on Microsatellite Instability for cancer detection and familial predisposition: development of international criteria for the determination of microsatellite instability in colorectal cancer. Cancer Res. 1998;58:5248–5257. [PubMed] [Google Scholar]
- 12.Umar A, Boland C, Terdiman J, et al. Revised Bethesda guidelines for hereditary nonpolyposis colorectal cancer (Lynch syndrome) and microsatellite instability. J Natl Cancer Inst. 2004;96:261–268. doi: 10.1093/jnci/djh034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Watson P, Lynch H. Extracolonic cancer in hereditary nonpolyposis colorectal cancer. Cancer. 1993;71:677–685. doi: 10.1002/1097-0142(19930201)71:3<677::aid-cncr2820710305>3.0.co;2-#. [DOI] [PubMed] [Google Scholar]
- 14.Wijnen JT, Vasen HFA, Khan PM, et al. Clinical findings with implications for genetic testing in families with clustering of colorectal cancer. N Engl J Med. 1998;339:511–518. doi: 10.1056/NEJM199808203390804. [DOI] [PubMed] [Google Scholar]
- 15.Murphy EA, Mutalik GS. The application of Bayesian methods in genetic counseling. Hum Hered. 1969;19:126–151. [Google Scholar]
- 16.Berry DA, Parmigiani G, Sanchez J, Schildkraut J, Winer E. Probability of carrying a mutation of breast-ovarian cancer gene BRCA1 based on family history. J Natl Cancer Inst. 1997;89:227–238. doi: 10.1093/jnci/89.3.227. [DOI] [PubMed] [Google Scholar]
- 17.Parmigiani G, Berry DA, Aguilar O. Determining carrier probabilities for breast cancer susceptibility genes BRCA1 and BRCA2. Am J Hum Genet. 1998;62:145–158. doi: 10.1086/301670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Antoniou AC, Gayther SA, Stratton JF, Ponder BA, Easton DF. Risk models for familial ovarian and breast cancer. Genet Epidemiol. 2000;18:173–190. doi: 10.1002/(SICI)1098-2272(200002)18:2<173::AID-GEPI6>3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]
- 19.Nanda R, Schumm LP, Cummings S, et al. Genetic testing in an ethnically diverse cohort of high-risk women: a comparative analysis of BRCA1 and BRCA2 mutations in American families of European and African ancestry. JAMA. 2005;294:1925–1933. doi: 10.1001/jama.294.15.1925. [DOI] [PubMed] [Google Scholar]
- 20.Dunlop MG, Farrington SM, Carothers AD, et al. Cancer risk associated with germline DNA mismatch repair gene mutations. Hum Mol Genet. 1997;6:105–110. doi: 10.1093/hmg/6.1.105. [DOI] [PubMed] [Google Scholar]
- 21.Quehenberger F, Vasen HFA, van Houwelingen HC. Risk of colorectal and endometrial cancer for carriers of mutations of the hMLH1 and hMSH2 gene: correction for ascertainment. J Med Genet. 2005;42:491–496. doi: 10.1136/jmg.2004.024299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hampel H, Stephens JA, Pukkala E, et al. Cancer risk in hereditary nonpolyposis colorectal cancer syndrome: later age of onset. Gastroenterology. 2005;129:415–421. doi: 10.1016/j.gastro.2005.05.011. [DOI] [PubMed] [Google Scholar]
- 23.Jenkins MA, Baglietto L, Dowty JG, et al. Cancer risks for mismatch repair gene mutation carriers: a population-based early onset case-family study. Clin Gastroenterol Hepatol. 2006;4:489–498. doi: 10.1016/j.cgh.2006.01.002. [DOI] [PubMed] [Google Scholar]
- 24.Buttin BM, Powell M, Mutch D, et al. Penetrance and expressivity of MSH6 germline mutations in seven kindreds not ascertained by family history. Am J Hum Genet. 2004;74:1262–1269. doi: 10.1086/421332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chen S, Watson P, Parmigiani G. Accuracy of MSI testing in predicting germline mutations of MSH2 and MLH1: a case study in Bayesian meta-analysis of diagnostic tests without a gold standard. Biostatistics. 2005;6:450–464. doi: 10.1093/biostatistics/kxi021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bapat BV, Madlensky L, Temple LK, et al. Family history characteristics, tumor microsatellite instability and germline MSH2 and MLH1 mutations in hereditary colorectal cancer. Hum Genet. 1999;104:167–176. doi: 10.1007/s004390050931. [DOI] [PubMed] [Google Scholar]
- 27.Calistri D, Presciuttini S, Buonsanti G, et al. Microsatellite instability in colorectal cancer patients with suspected genetic predisposition. Int J Cancer. 2000;89:87–91. doi: 10.1002/(sici)1097-0215(20000120)89:1<87::aid-ijc14>3.0.co;2-9. [DOI] [PubMed] [Google Scholar]
- 28.Debniak T, Kurzawski G, Gorski B, Kladny J, Domagala W, Lubinski J. Value of pedigree/clinical data, immunohistochemistry and microsatellite instability analyses in reducing the cost of determining hMLH1 and hMSH2 gene mutations in patients with colorectal cancer. Eur J Cancer. 2000;36:49–54. doi: 10.1016/s0959-8049(99)00208-7. [DOI] [PubMed] [Google Scholar]
- 29.Dieumegard B, Grandjouan S, Sabourin JC, et al. Extensive molecular screening for hereditary non-polyposis colorectal cancer. Br J Cancer. 2000;82:871–880. doi: 10.1054/bjoc.1999.1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lamberti C, Kruse R, Ruelfs C, et al. Microsatellite instability—a useful diagnostic tool to select patients at high risk for hereditary non-polyposis colorectal cancer: a study in different groups of patients with colorectal cancer. Gut. 1999;44:839–843. doi: 10.1136/gut.44.6.839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Liu T, Wahlberg S, Burek E, Lindblom P, Rubio C, Lindblom A. Microsatellite instability as a predictor of a mutation in a DNA mismatch repair gene in familial colorectal cancer. Genes Chromosomes Cancer. 2000;27:17–25. doi: 10.1002/(sici)1098-2264(200001)27:1<17::aid-gcc3>3.0.co;2-y. [DOI] [PubMed] [Google Scholar]
- 32.Scartozzi M, Bianchi F, Rosati S, et al. Mutations of hMLH1 and hMSH2 in patients with suspected hereditary nonpolyposis colorectal cancer: correlation with microsatellite instability and abnormalities of mismatch repair protein expression. J Clin Oncol. 2002;20:1203–1208. doi: 10.1200/JCO.2002.20.5.1203. [DOI] [PubMed] [Google Scholar]
- 33.Cederquist K, Golovleva I, Emanuelsson M, Stenling R, Gronberg H. A population based cohort study of patients with multiple colon and endometrial cancer: correlation of microsatellite instability (MSI) status, age at diagnosis and cancer risk. Int J Cancer. 2001;91:486–491. doi: 10.1002/1097-0215(200002)9999:9999<::aid-ijc1093>3.0.co;2-p. [DOI] [PubMed] [Google Scholar]
- 34.Ponz de Leon M, Benatti P, Di Gregorio C, et al. Genetic testing among high-risk individuals in families with hereditary nonpolyposis colorectal cancer. Br J Cancer. 2004;90:882–887. doi: 10.1038/sj.bjc.6601529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Terdiman JP, Gum JR, Jr, Conrad PG, et al. Efficient detection of hereditary non-polyposis colorectal cancer gene carriers by screening for tumor microsatellite instability before germline genetic testing. Gastroenterology. 2001;120:21–30. doi: 10.1053/gast.2001.20874. [DOI] [PubMed] [Google Scholar]
- 36.Wang Y, Friedl W, Lamberti C, et al. HNPCC: frequent occurrence of large genomic deletions in MSH2 and MLH1. Int J Cancer. 2003;103:636–641. doi: 10.1002/ijc.10869. [DOI] [PubMed] [Google Scholar]
- 37.Percesepe A, Borghi F, Menigatti M, et al. Molecular screening for hereditary non-polyposis colorectal cancer: a prospective, population-based study. J Clin Oncol. 2001;19:3944–3950. doi: 10.1200/JCO.2001.19.19.3944. [DOI] [PubMed] [Google Scholar]
- 38.Salahshor S, Kressner U, Fischer H, et al. Microsatellite instability in sporadic colorectal cancer is not an independent prognostic factor. Br J Cancer. 1999;81:190–193. doi: 10.1038/sj.bjc.6690676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dietmaier W, Wallinger S, Bocker T, Kullmann F, Fishel R, Ruschoff J. Diagnostic microsatellite instability: definition and correlation with mismatch repair protein expression. Cancer Res. 1997;57:4749–4756. [PubMed] [Google Scholar]
- 40.Salovaara R, Loukola A, Kristo P, et al. Population-based molecular detection of hereditary nonpolyposis colorectal cancer. J Clin Oncol. 2000;18:2193–2200. doi: 10.1200/JCO.2000.18.11.2193. [DOI] [PubMed] [Google Scholar]
- 41.Loukola A, Salovaara R, Kristo P, et al. Microsatellite instability in adenomas as a marker for hereditary nonpolyposis colorectal cancer. Am J Pathol. 1999;155:1849–1853. doi: 10.1016/S0002-9440(10)65503-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Marcus VA, Madlensky L, Gryfe R, et al. Immunohistochemistry for hMLH1 and hMSH2: a practical test for DNA mismatch repair-deficient tumors. Am J Surg Pathol. 1999;23:1248–1255. doi: 10.1097/00000478-199910000-00010. [DOI] [PubMed] [Google Scholar]
- 43.Lindor NM, Burgart LJ, Leontovich O, et al. Immunohistochemistry versus microsatellite instability testing in phenotyping colorectal tumors. J Clin Oncol. 2002;20:1043–1048. doi: 10.1200/JCO.2002.20.4.1043. [DOI] [PubMed] [Google Scholar]
- 44.Engel C, Forberg J, Holinski-Feder E, et al. Novel strategy for optimal sequential application of clinical criteria, immunohistochemistry and microsatellite analysis in the diagnosis of hereditary nonpolyposis colorectal cancer. Int J Cancer. 2006;118:115–122. doi: 10.1002/ijc.21313. [DOI] [PubMed] [Google Scholar]
- 45.Caldés T, Godino J, Sanchez A, et al. Immunohistochemistry and microsatellite instability testing for selecting MLH1, MSH2 and MSH6 mutation carriers in hereditary non-polyposis colorectal cancer. Oncol Rep. 2004;12:621–629. [PubMed] [Google Scholar]
- 46.Chen S, Wang W, Broman K, Parmigiani G. Bayes-Mendel: an R environment for Mendelian risk prediction. [Accessibility verified August 30, 2006];Stat Appl Genet Mol Biol. 2004 3 doi: 10.2202/1544-6115.1063. [published online September 17, 2004] Article21. http://www.bepress.com/jhubiostat/paper39/ [DOI] [PMC free article] [PubMed]
- 47.Elston RC, Stewart J. A general model for the genetic analysis of pedigree data. Hum Hered. 1971;21:523–542. doi: 10.1159/000152448. [DOI] [PubMed] [Google Scholar]
- 48.Szolovits P, Pauker S. Pedigree analysis for genetic counseling. In: Lun KC, Degoulet P, Piemme TE, Rienhoff O, editors. MEDINFO-92 Proceedings of the Seventh Conference on Medical Informatics. New York, NY: Elsevier; 1992. pp. 679–683. [Google Scholar]
- 49.Offit K, Brown K. Quantitating familial cancer risk: a resource for clinical oncologists. J Clin Oncol. 1994;12:1724–1736. doi: 10.1200/JCO.1994.12.8.1724. [DOI] [PubMed] [Google Scholar]
- 50.Yan H, Papadopoulos N, Marra G, et al. Conversion of diploidy to haploidy. Nature. 2000;403:723–724. doi: 10.1038/35001659. [DOI] [PubMed] [Google Scholar]
- 51.Liu B, Parsons R, Papadopoulos N, et al. Analysis of mismatch repair genes in hereditary non-polyposis colorectal cancer patients. Nat Med. 1996;2:169–174. doi: 10.1038/nm0296-169. [DOI] [PubMed] [Google Scholar]
- 52.Papadopoulos N, Leach FS, Kinzler KW, Vogelstein B. Monoallelic mutation analysis (MAMA) for identifying germline mutations. Nat Genet. 1995;11:99–102. doi: 10.1038/ng0995-99. [DOI] [PubMed] [Google Scholar]
- 53.Casey G, Lindor NM, Papadopoulos N, et al. Conversion analysis for mutation detection in MLH1 and MSH2 in patients with colorectal cancer. JAMA. 2005;293:799–809. doi: 10.1001/jama.293.7.799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Charbonnier F, Raux G, Wang Q, et al. Detection of exon deletions and duplications of the mismatch repair genes in hereditary nonpolyposis colorectal cancer families using multiplex polymerase chain reaction of short fluorescent fragments. Cancer Res. 2000;60:2760–2763. [PubMed] [Google Scholar]
- 55.Fredricks DN, Relman DA. Paraffin removal from tissue sections for digestion and PCR analysis. Biotechniques. 1999;26:198–200. doi: 10.2144/99262bm04. [DOI] [PubMed] [Google Scholar]
- 56.Peterlongo P, Nafa K, Lerman GS, et al. MSH6 germline mutations are rare in colorectal cancer families. Int J Cancer. 2003;107:571–579. doi: 10.1002/ijc.11415. [DOI] [PubMed] [Google Scholar]
- 57.DeGroot MH, Fienberg SE. The comparison and evaluation of forecasters. Statistician. 1983;32:12–22. [Google Scholar]
- 58.Harrell FE, Jr, Califf RM, Pryor DB, Lee KL, Rosati RA. Evaluating the yield of medical tests. JAMA. 1982;247:2543–2546. [PubMed] [Google Scholar]
- 59.Efron B. The bootstrap and modern statistics. J Am Stat Assoc. 2000;95:1293–1296. [Google Scholar]
- 60.Euhus DM. Understanding mathematical models for breast cancer risk assessment and counseling. Breast J. 2001;7:224–232. doi: 10.1046/j.1524-4741.2001.20012.x. [DOI] [PubMed] [Google Scholar]
- 61.Shia J, Klimstra DS, Nafa K, et al. Value of immunohistochemical detection of DNA mismatch repair proteins in predicting germline mutation in hereditary colorectal neoplasms. Am J Surg Pathol. 2005;29:96–104. doi: 10.1097/01.pas.0000146009.85309.3b. [DOI] [PubMed] [Google Scholar]
- 62.Frazier AL, Colditz GA, Fuchs CS, Kuntz KM. Cost-effectiveness of screening for colorectal cancer in the general population. JAMA. 2000;284:1954–1961. doi: 10.1001/jama.284.15.1954. [DOI] [PubMed] [Google Scholar]
- 63.Ransohoff DF, Sandler RS. Clinical practice: screening for colorectal cancer. N Engl J Med. 2002;346:40–44. doi: 10.1056/NEJMcp010886. [DOI] [PubMed] [Google Scholar]
- 64.Nakagawa H, Nuovo GJ, Zervos EE, et al. Age-related hypermethylation of the 5′ region of MLH1 in normal colonic mucosa is associated with microsatellite-unstable colorectal cancer development. Cancer Res. 2001;61:6991–6995. [PubMed] [Google Scholar]
- 65.Kakar S, Burgart LJ, Thibodeau SN, et al. Frequency of loss of hMLH1 expression in colorectal carcinoma increases with advancing age. Cancer. 2003;97:1421–1427. doi: 10.1002/cncr.11206. [DOI] [PubMed] [Google Scholar]
- 66.Schmeler KM, Lynch HT, Chen LM, et al. Prophylactic surgery to reduce the risk of gynecologic cancers in the Lynch syndrome. N Engl J Med. 2006;354:261–269. doi: 10.1056/NEJMoa052627. [DOI] [PubMed] [Google Scholar]
- 67.National Action Plan on Breast Cancer; American Society of Clinical Oncology. [Accessibility verified August 23, 2006];Hereditary susceptibility to breast and ovarian cancer: an outline of the basic fundamental knowledge needed by all health care professionals. 1997 February; http://www.4woman.gov/napbc/catalog.wci/napbc/hsedcurr.htm.
- 68.Li FP. Identification and management of inherited cancer susceptibility. Environ Health Perspect. 1995;103(suppl 8):297–300. doi: 10.1289/ehp.95103s8297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.American Society of Clinical Oncology. Policy statement update: genetic testing for cancer susceptibility recommendations pertaining to clinical aspects of genetic testing for cancer susceptibility. J Clin Oncol. 2003;21:2397–2406. doi: 10.1200/JCO.2003.03.189. [DOI] [PubMed] [Google Scholar]
- 70.Trepanier A, Ahrens M, McKinnon W, et al. Genetic cancer risk assessment and counseling: recommendations of the national society of genetic counselors. J Genet Couns. 2004;13:83–114. doi: 10.1023/B:JOGC.0000018821.48330.77. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.