

Abstract
Apigenin, a plant flavone, potentially activates wild-type p53 and induces apoptosis in cancer cells. We conducted detailed studies to understand its mechanism of action. Exposure of human prostate cancer 22Rv1 cells, harboring wild-type p53, to growth-suppressive concentrations (10–80 μM) of apigenin resulted in the stabilization of p53 by phosphorylation on critical serine sites, p14ARF-mediated downregulation of MDM2 protein, inhibition of NF-κB/p65 transcriptional activity, and induction of p21/WAF-1 in a dose- and time-dependent manner. Apigenin at these doses resulted in ROS generation, which was accompanied by rapid glutathione depletion, disruption of mitochondrial membrane potential, cytosolic release of cytochrome c, and apoptosis. Interestingly, we observed accumulation of a p53 fraction to the mitochondria, which was rapid and occurred between 1 and 3 h after apigenin treatment. All these effects were significantly blocked by pretreatment of cells with the antioxidant N-acetylcysteine, p53 inhibitor pifithrin-α, and enzyme catalase. Apigenin-mediated p53 activation and apoptosis were further attenuated by p53 antisense oligonucleotide treatment. Exposure of cells to apigenin led to a decrease in the levels of Bcl-XL and Bcl-2 and increase in Bax, triggering caspase activation. Treatment with the caspase inhibitors Z-VAD-FMK and DEVD-CHO partially rescued these cells from apigenin-induced apoptosis. In vivo, apigenin administration demonstrated p53-mediated induction of apoptosis in 22Rv1 tumors. These results indicate that apigenin-induced apoptosis in 22Rv1 cells is initiated by a ROS-dependent disruption of the mitochondrial membrane potential through transcriptional-dependent and -independent p53 pathways.
Keywords: Reactive oxygen species, Apoptosis, p53, Prostate cancer, Apigenin, Free radicals
Cancer prevention is profoundly dependent on the p53 tumor suppressor protein, as it functions in regulating cellular growth and protecting cells from malignant transformation [1,2]. Several studies have demonstrated the anticancer activities of p53, and in fact, p53 is the most frequently altered protein in human cancers [2,3]. Approximately 50% of all human malignancies harbor mutations or deletions in the TP53 gene that disable the tumor suppressor function of the encoded protein [4,5]. Most other human cancers express wild-type p53 protein but encode alternate defects in the p53 signaling pathway that play critical roles in tumorigenesis [4–6]. The role that the p53 gene plays in preventing the onset of cancer involves the elimination of damaged or infected cells by causing cell cycle arrest and/or apoptosis. The suppression of cellular proliferation by p53 occurs through two different mechanisms: apoptosis and cell cycle arrest. In abnormally proliferating cells, induction of p53 leads to programmed cell death or apoptosis, whereas in normal fibroblasts p53 induces G1 arrest in response to genotoxic agents, presumably to allow the cells to perform critical repair functions before progressing through the cell cycle [7]. Apoptosis mediated by p53 in response to DNA damage is predominantly attributable to the transcriptional activation of genes that regulate the cell cycle and apoptosis, such as the BH3-only proteins Noxa and Puma, p21/WAF-1, MDM2, gadd45, Bax, p53AIP1, and PERP [8–10], and is additionally attributable to transcriptional repression, involving inhibition of the expression of a number of genes, including c-fos, DNA polymerase α, microtubule-associated protein A, IGF-IR, NF-κB/p65/RelA, Bcl-2, and survivin [11,12]. Wild-type p53 protein activity is associated with apoptosis induced by DNA-damaging agents and growth hormone depletion [13]. In addition to its roles in such transcriptional regulation, recent evidence has suggested the existence of a transcription-independent pathway of p53-mediated apoptosis [4]. A fraction of the p53 molecules that accumulate in damaged cells translocate to mitochondria, and this translocation is sufficient for p53 to induce permeabilization of the outer mitochondrial membrane through formation of complexes with the protective proteins Bcl-XL and Bcl-2, resulting in the release of cytochrome c into the cytosol [15,16]. Cytochrome c then binds to apoptosis protease-activating factor-1 (Apaf-1) and activates caspase-3 through caspase-9, resulting in execution of apoptosis [17]. Consequently, regulation of the expression of the p53 gene may be of critical importance to the induction of apoptosis in premalignant and malignant cells, and agents that can influence this process may prove to be valuable in the prevention and/or therapeutic management of cancer.
In recent years plant flavonoids have gained considerable attention as anticancer agents, particularly apigenin (4′,5,7-trihydroxyflavone), which is abundantly present in common fruits and vegetables and which has shown considerable promise for development as a chemo-preventive agent [18,19]. Recent studies from our group have shown that apigenin is capable of selectively inhibiting cell growth and inducing apoptosis in cancer cells without affecting normal cells [20]. In addition, the anticancer properties of apigenin in animals have been demonstrated by inhibition of tumor initiation induced by various carcinogens [21,22]. Apigenin has been shown to suppress angiogenesis in melanoma and carcinoma of the breast, skin, and colon [23–26]. Apigenin is a potent inhibitor of several protein tyrosine kinases, including epidermal growth factor receptor and Src tyrosine kinase [27,28]. Apigenin has also been shown to modulate the expression of phosphatidylinositol 3-kinase, protein kinase B/Akt, mitogen-activated protein kinase, ERK1/2, casein kinase-2, and other upstream kinases involved in the development and progression of cancer [29–31]. Recently, our laboratory has demonstrated that apigenin sensitizes tumor cells to TNF-α-induced apoptosis through inhibition of NF-κB [32]. We also observed that apigenin induces apoptosis in solid tumors through upregulation of IGFBP-3 [33]. More recently, we have demonstrated that apigenin-induced suppression of tumor proliferation correlates with downregulation of cyclin D1 expression and cdk4-mediated Rb phosphorylation, induction of p21/WAF-1, and p53 stabilization [34]. However, the mechanisms by which apigenin influences p53-mediated apoptosis are not clearly understood. We undertook studies of human prostate cancer cell lines as well as studies of prostate cancer xenografts in athymic nude mice to investigate the role of apigenin in p53-mediated apoptosis, examining its influence in both p53-dependent and p53-independent activation of apoptotic mechanisms that culminate in cell death.
Materials and methods
Reagents and antibodies
Apigenin (>95% purity) was obtained from A.G. Scientific (San Diego, CA, USA), N-acetyl- L-cysteine (NAC) was from Sigma–Aldrich (St. Louis, MO, USA), and pifithrin-α, Z-VAD-FMK, and DEVD-CHO were from Alexis (San Diego, CA, USA). Fetal bovine serum was obtained from Life Technologies (Gaithersburg, MD, USA). Antibodies recognizing full-length poly(ADP)ribose polymerase and its cleaved product, p53 (No. 9282); phospho-p53, Ser6 (No. 9285); phospho-p53, Ser9 (No. 9288); phospho-p53, Ser15 (No. 9284); phospho-p53, Ser20 (No. 9287); phospho-p53, Ser37 (No. 9289); phospho-p53, Ser46 (No. 2521); phospho-p53, Ser392 (No. 9281); p14ARF (No. 2407); caspase-3 (No. 9662); caspase-8, 1C12 (No. 9746); and Bcl-XL (No. 2762) were purchased from Cell Signaling Technology (Fremont, CA, USA). MDM2, D-12 (No. sc-5304); Apaf-1, H324 (No. sc-8339); Bax, 6A7 (No. sc-23959); Bcl2, C-2 (No. sc-7382); cytochrome c, 7H8 (No. sc-13560); caspase-9, p10(F-7) (No. sc-17784); and actin (1–19) (No. sc-1616) were procured from Santa Cruz Biotechnology (Santa Cruz, CA, USA), whereas monoclonal antibodies for WAF-1/p21 were obtained from Lab Vision Corp. (Fremont, CA, USA).
Cell culture and treatment
Human prostate cancer 22Rv1 cells were purchased from the American Type Culture Collection (Manassas, VA, USA). Additionally, we used other cell lines which were isogenic prostate cells PC-3 (p53−/−) and PC-3 (p53+/+); the only difference was in their p53 status. Cells were cultured in RPMI 1640 medium with 10% heat-inactivated fetal bovine serum (FBS),100 μg/ml penicillin–streptomycin (Invitrogen, Carlsbad, CA, USA), and maintained in an incubator with a humidified atmosphere of 95% air and 5% CO2 at 37°C. The cells were treated with varying concentrations of apigenin dissolved in DMSO, which was provided to the control group within permissible concentrations.
Cell growth inhibition by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) assay
The effects of apigenin on cell viability/proliferation were determined using the MTT assay, as described previously [31]. Briefly,1 × 104 cells/well were plated in 96-well culture plates. After overnight incubation, the cells were treated with varying concentrations of apigenin (0, 10, 20, 40, or 80 μM) for 12, 24, and 48 h. The cells were treated with 50 μl of 5 mg/ml MTT and the resulting formazan crystals were dissolved in dimethyl sulfoxide (200 μl). Absorbance was recorded at 540 nm with a reference at 650 nm serving as the blank. The effect of apigenin on cell viability was assessed as percentage cell viability compared to vehicle-treated control cells, which were arbitrarily assigned 100% viability.
Preparation of total cell lysates and mitochondrial lysates
After treatment of the cells, the medium was aspirated and the cells were washed twice with cold phosphate-buffered saline (PBS; 10 mM, pH 7.4). Ice-cold lysis buffer (50 mM Tris–HCl, 150 mM NaCl, 1 mM EGTA, 1 mM EDTA, 20 mM NaF, 100 mM Na3VO4, 0.5% NP-40, 1% Triton X-100, 1 mM PMSF, 10 mg/ml aprotinin, 10 mg/ml leupeptin, pH 7.4) was added to the plates, which were then placed over ice for 30 min. The cells were scraped, and the lysate was collected in a microfuge tube and passed through a 21-gauge needle to break up the cell aggregates. The lysate was cleared by centrifugation at 14,000 g for 15 min at 4°C and the supernatant (total cell lysate) was either used immediately or stored at −80°C. Mitochondria were isolated using an ApoAlert cell fractionation kit following the manufacturer’s protocol (BD Clontech, USA). Cells were trypsinized, collected, and centrifuged at 600 g for 5 min at 4 °C. After being washed, the cell pellet was resuspended in 0.8 ml ice-cold fractionation buffer mix and incubated on ice for 10 min followed by homogenization in an ice-cold Dounce homogenizer. The homogenate was centrifuged at 700 g for 10 min. The supernatant was again centrifuged at 10,000 g for 25 min at 4°C. Supernatant (cytosol) was collected and the pellet (mitochondria) was resuspended in 50 μl of cell fractionation buffer. Purity was checked by COX-IV immunoblotting. The protein concentration was determined by the DC Bio-Rad assay using the manufacturer’s protocol (Bio-Rad Laboratories).
Catalase treatment
22Rv1 cells were pretreated with 400 units/ml concentration of catalase, which was followed by the addition of 1 mM H2O2 or 40 μM apigenin for 3 h. The cells were washed with PBS and then total cell lysate and cytosolic and mitochondrial fractions were prepared according to previously described methods.
Enzyme-linked immunoabsorbent assay (ELISA) for detection of apoptosis
22Rv1 cells were treated with varying concentrations of apigenin for 12, 24, and 48 h, then apoptotic cells were evaluated by using the Cell Death Detection ELISAplus kit (Roche Molecular Biochemicals, Indianapolis, IN, USA). The assay is based on the enrichment of mono-and oligonucleosomes in the cytoplasm of the apoptotic cells due to degradation of DNA by endogenous endonuclease (that cleaves double-stranded DNA at the internucleosomal linker regions). Cell lysates and the assay procedures were performed according to the manufacturer’s protocol. Absorbance at 405 nm was measured as the indicator of apoptotic cells. The reference wavelength was 490 nm. The enrichment factor (total amount of apoptosis) was calculated by dividing the absorbance of the sample (A405 nm) by the absorbance of the controls without treatment (A490 nm).
Immunoblot analysis
22Rv1 cells were cultured on 100-mm dishes and grown to approximately 60% confluence. The cells were then treated with apigenin and other agents and harvested at various time intervals. Tumor lysates were prepared and subjected to immunoblot analysis. Fifty micrograms of protein from total cell lysates or tumor lysates was resolved by 10% SDS–PAGE and transferred to nitrocellulose membrane. After being blocked with blocking buffer (PBS containing 0.1% Tween 20 and 10% FBS) for 2 h, the membrane was incubated with primary antibody for 2 h at room temperature. The membrane was then incubated with HRP-conjugated secondary antibodies for another 2 h at room temperature. The protein was detected by ECL substrate reagents (Amersham Biosciences, Arlington Heights, IL, USA).
Assay for mitochondrial membrane potential
Mitochondrial membrane potential was quantitatively determined by flow cytometry using the lipophilic cationic probe JC-1 dye (5,5′,6,6′-tetrachloro-1,1′,3,3′-tetraethylbenzimidazolcarbocyanine iodide) detection kit, following the manufacturer’s instructions. In brief, 60% confluent 22Rv1 cells were treated with apigenin (0, 20, and 40 μM) for 12 h, harvested, and washed with PBS buffer and 1 × 106 cells were incubated in 1 ml PBS containing 10 mg JC-1 dye for 15 min at 37°C in the dark. Stained cells were washed, resuspended in 500 ml PBS, and used for immediate FACS analysis.
Measurement of reactive oxygen species (ROS)
22Rv1 cells were maintained 75–80% in RPMI 1640 medium with 10% FBS and were treated or not with NAC for 3 h. The treatment medium was removed and the monolayer was exposed to PBS containing 10 μM 2′7′-dichlorofluorescein diacetate (DCF; Molecular Probes, Eugene, OR, USA), a dye that fluoresces when ROS are generated. The cells were incubated with the dye for 20 min, after which fluorescence intensity was determined with an adherent cell laser cytometer using 488 nm for excitation and 560 nm for detection of fluorescence emission. The values, expressed in percentage arbitrary fluorescence units, were compared across treatment groups.
Transfections and p53 luciferase reporter assay
22Rv1 cells (2 × 105 per well) were plated in 35-mm dishes and cotransfected the next day using Lipofectamine transfection reagent (Invitrogen) according to the protocol of the manufacturer done with luciferase p53-responsive reporter plasmid. Twenty-four hours later, cells were treated with various concentrations of apigenin. The preparation of cell lysates and luciferase activity measurements were made with the Luciferase Assay Kit (Promega, Madison, WI, USA) according to the instructions of the manufacturer. The luciferase activities were normalized to β-galactosidase activity (Promega). Data shown are from one of at least two independent experiments with similar results.
Analysis of DNA fragmentation by agarose gel electrophoresis
The 22Rv1 cells were grown to about 70% confluence and treated with apigenin (1, 10, 20, 40, and 80 μM concentration) for 48 h. After these treatments, the cells were washed twice with phosphate-buffered saline (10 mM Tris, pH 7.5, 150 mM NaCl, 5 mM MgCl2, and 0.5% Triton X-100), left on ice for 15 min, and pelleted by centrifugation (14,000 g) at 4°C. The pellet was incubated with DNA lysis buffer (10 mM Tris, pH 7.5, 400 mM NaCl, 1 mM EDTA, and1% Triton X-100) for 30 min on ice and then centrifuged at 14,000 g in 4°C. The supernatant obtained was incubated overnight with RNase (0.2 mg/ml) at room temperature and then with proteinase K (0.1 mg/ml) for 2 h at 37°C. DNA was extracted with phenol: chloroform (1:1) and precipitated with 95% ethanol for 2 h at 80°C. The DNA precipitate was centrifuged at 14,000 g at 4°C for 15 min and the pellet was air dried and dissolved in 20 μl of Tris–EDTA buffer (10 mM Tris–HCl, pH 8.0, and 1 mM EDTA). The total amount of DNA was resolved over 1.5% agarose gel, containing 0.3 μg/ml ethidium bromide in TBE buffer (pH 8.3, 89 mM Tris, 89 mM boric acid, and 2 mM EDTA) (Bio Whittaker, Walkersville, MD, USA). The bands were visualized under UV trans-illuminator (Model TM-36; UVP, San Gabriel, CA, USA) followed by Polaroid photography (MP-4 Photographic System, Fotodyne, Hartland, WI, USA).
ELISA for NF-κB/p65/RelA activity
ELISA was performed for NF-κB/p65/RelA activity. The commercially available Trans-AM NF-κB assay kit was obtained from Active Motif North America (Carlsbad, CA, USA) for assay of NF-κB/p65 activity according to the vendor’s protocol. Briefly, the assay uses an oligonucleotide containing the NF-κB consensus site (5′-GGGACTTTCC-3′) that binds to the cell extract and can detect NF-κB, which can recognize an epitope on p65 activated and bound to its target DNA. This assay is specific for NF-κB activation and is highly sensitive.
Estimation of cellular glutathione (GSH)
Cellular GSH was estimated according to the vendor’s protocol. Cell extracts were prepared by sonication and deproteination. Total GSH was detected by measuring the product of glutathionylated 5,5′-dithio-bis(2-nitrobenzoic acid) by UV spectrophotometer at 405 nm. The cellular GSH contents were calculated using the standard curve generated in parallel experiments.
Experimental design for tumor xenograft studies
22Rv1 tumors were grown subcutaneously in athymic nude mice. Approximately 1 million 22Rv1 cells suspended in 0.05 ml of medium and mixed with 0.05 ml of Matrigel were subcutaneously injected into the left and right flank of each mouse to initiate tumor growth. The first group received only 0.2 ml of vehicle material by gavage daily and served as a control group. The second and third groups of animals received 20 and 50 μg/mouse/day doses of apigenin in vehicle, respectively, for 10 weeks. These doses are comparable to the daily consumption of flavonoid in humans as reported in previously published studies [33,34]. Apigenin feeding was started 2 weeks before cell inoculation and was continued for 10 weeks. Animals were monitored daily, and their body weights were recorded weekly throughout the studies. Once the tumors started growing, their sizes were measured twice weekly in two dimensions with calipers. At the termination of the experiment, tumors were excised and weighed to record wet tumor weight. A portion of the tumors from control and treated animals was used for preparation of tumor lysate used in further experiments.
Statistical analysis
Changes in tumor volume and body weight during the course of the experiments were visualized by scatter plot. Differences in tumor volume (mm3) and body weight at the termination of the experiment among three treatment groups were examined using analysis of variance (ANOVA) followed by Tukey’s multiple comparison procedure. The statistical significance of differences between control and treatment groups was determined by simple ANOVA followed by multiple comparison tests. All tests were two-tailed and p values less than 0.05 were considered to be statistically significant.
Results
Apigenin induces inhibition of cell growth, decrease in NF-κB/p65 activity, and apoptosis in 22Rv1 cells
To study the effect of apigenin on prostate cancer, human prostate cancer 22Rv1 cells were exposed to 10–80 μM concentration of apigenin for 12, 24, and 48 h and their viability was determined posttreatment by MTT assay. We observed that doses higher than 10 μM apigenin were effective in inhibiting cell growth in that 30% inhibition was observed at 24 h and 50% at 48 h exposure with 20 μM apigenin (Fig. 1A). Exposure of 22Rv1 cells to 20 μM apigenin caused a decrease in NF-κB/p65 transcriptional activity by 24% at 12 h, which was further decreased to 41% at 24 h (data not shown). To determine if the loss of cell viability and NF-κB/p65 transcriptional activity induced by apigenin resulted in apoptosis, we measured apoptosis in 22Rv1 cells by ELISA and DNA fragmentation assays. Apigenin-induced apoptosis was observed as early as 12 h posttreatment, which was significant for 40 and 80 μM apigenin. We observed significant apoptosis in 22Rv1 cells after 24 and 48 h of treatment with 20, 40, and 80 μM apigenin (Fig. 1B). The exposure of 22Rv1 cells for 48 h to apigenin further resulted in DNA laddering in a dose-dependent manner (Fig. 1C). These results indicate that apigenin inhibits cell growth, which could be a result of decreased NF-κB activity inducing apoptosis in 22Rv1 cells as a function of dose and time.
Fig. 1.
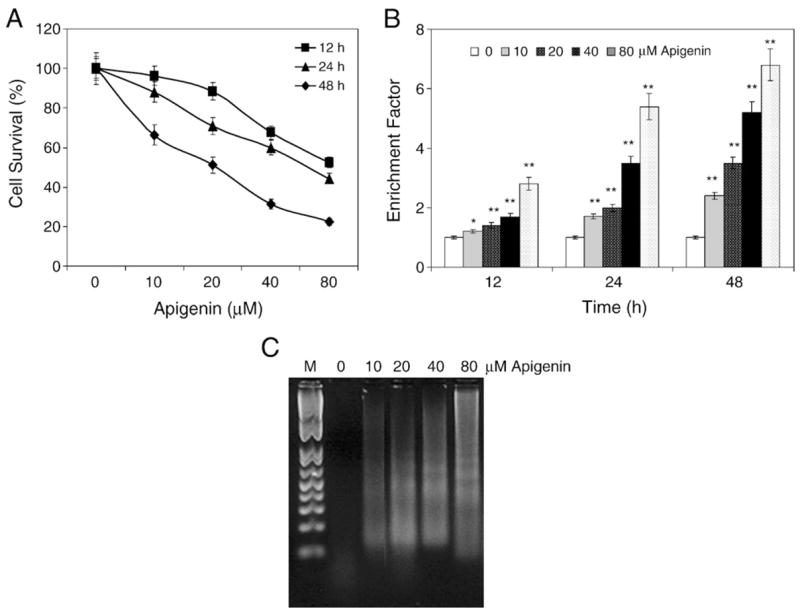
Apigenin treatment causes cell growth inhibition and apoptosis in 22Rv1 cells. (A) Percentage of cells surviving after treatment with apigenin at different concentrations for 12, 24, and 48 h. Representative data, means±SE, n=8, repeated twice with similar results. (B) Apoptosis in the lysates from 22Rv1 cells treated for 12, 24, and 48 h with various concentrations of apigenin. Values are represented as enrichment factor described under Materials and methods. Data, means± SE, n=3. Significantly different from control: *p<0.05 and **p<0.001. (C) Apoptosis as demonstrated by DNA fragmentation assay of 22Rv1 cells treated for 48 h with the indicated concentrations of apigenin. The experiment was performed twice with similar results.
Apigenin increases the expression and transcriptional activation of p53 in 22Rv1 cells
To investigate whether apigenin has an effect on p53 protein expression, we determined the p53 levels in 22Rv1 cells treated with 10–80 μM doses of apigenin for 12, 24, and 48 h. Immunoblot analysis showed that apigenin exposure increased the protein expression of p53. The increased p53 protein expression correlated with an increase in the levels of its transcriptional target p21/WAF-1 (Fig. 2A). The relative densities of these proteins exhibit an increase in the levels of p53 and p21/WAF-1 with 24 h of exposure to 20 μM apigenin (Fig. 2B). To examine if increased p53 expression resulted in concurrent upregulation of its transcriptional activity, 22Rv1 cells were transfected with the PG-13 luciferase plasmid, which contains p53 consensus binding sites. For normalizing transfection efficiency, 22Rv1 cells were co-infected with a β-galactosidase plasmid. After 24 h transfection, the cells were treated with 10–80 μM apigenin and luciferase activity was determined 24 h postexposure. We observed a linear increase in the transcriptional activity of p53 as a function of apigenin dose (Fig. 2C).
Fig. 2.
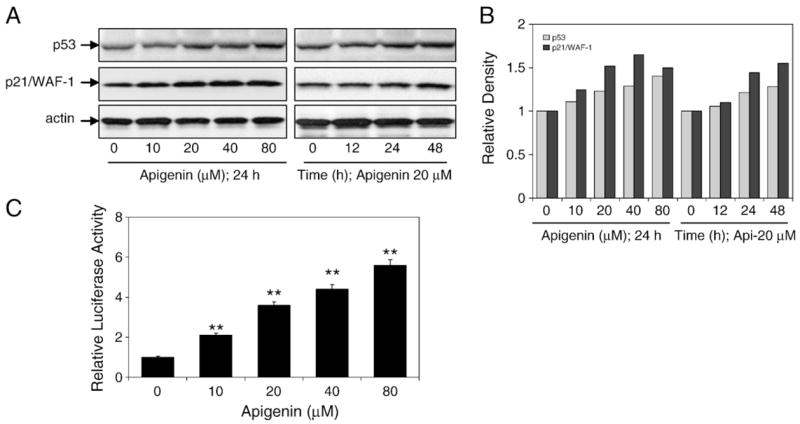
Effects of apigenin treatment on p53 expression and transcriptional activity in 22Rv1 cells. (A) Immunoblots for p53 and p21/WAF-1 using lysates from 22Rv1 cells treated with various concentrations of apigenin for the indicated time periods. The blot was stripped and reprobed with antiactin antibody to ensure equal protein loading. (B) Densitometric analysis for p53 and p21/WAF-1. (C) Transcriptional activation of p53 using p53-dependent PG-13 promoter. At 24 h posttransfection with the PG-13 luciferase plasmid, 22Rv1 cells were treated with the indicated concentrations of apigenin. Data are means±SE, n=3, repeated twice with similar results. Significantly different from control: **p<0.001. Luciferase assay was performed as described under Materials and methods.
Apigenin induces stabilization of p53 in 22Rv1 cells
The stabilization and activation of the p53 protein as a transcriptional activator occur through a series of phosphorylation and acetylation events at critical serine residues [15–17]. To determine the mechanism by which apigenin increases the p53 levels, we examined the phosphorylation status of p53 at various serine residues that have been documented to increase the half-life of this protein. As shown in Fig. 3A, 24 h of apigenin exposure at 20 and 40 μM doses resulted in phosphorylation of p53 at serines 6, 15, 20, 37, and 392, whereas no changes were observed in serines 9 and 46. Adriamycin, which induces p53 and phosphorylates it at various serine residues, was used as a positive control. We further analyzed the protein levels of MDM2–p14ARF, because stabilization of p53 can occur via this pathway. MDM2 is a ubiquitin ligase that binds and targets p53 to ubiquitin-dependent proteolysis. The interaction of MDM2 with p53 is antagonized by the tumor suppressor p14ARF, which averts the MDM2-mediated ubiquitination and degradation of p53. To determine the role of MDM2–p14ARF in apigenin-induced p53 stabilization, we measured the levels of these proteins after apigenin exposure. As shown in Fig. 3B, apigenin exposure resulted in an increase in p14ARF and a concomitant decrease in MDM2 levels as a function of dose and time. These results suggest that the apigenin-induced p53 stabilization through serine phosphorylation is via p14ARF-mediated down-regulation of MDM2 protein.
Fig. 3.
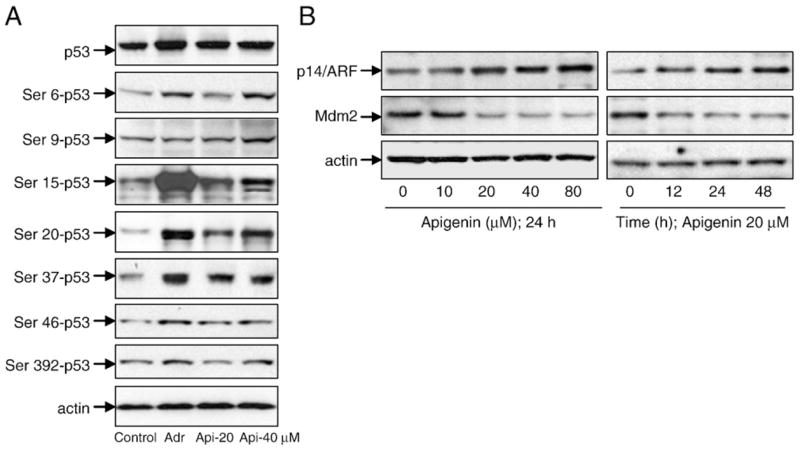
Apigenin treatment causes stabilization of p53 in 22Rv1 cells. (A) Immunoblots for p53 at various phosphorylation sites using lysates from 22Rv1 cells treated with various doses of apigenin for 24 h. (B) Immunoblots for p14ARF and MDM2 using lysates from 22Rv1 cells treated with various doses of apigenin for the indicated time periods. The blot was stripped and reprobed with anti-actin antibody to ensure equal protein loading.
p53 is a potential target for apigenin action in 22Rv1 cells
To determine if p53 protein is specifically involved in apigenin-induced apoptosis, 22Rv1 cells were treated with 20 μM p53-specific antisense oligonucleotide for 8 h before the addition of 20 μM apigenin for 16 h. As expected, treatment of 22Rv1 cells with p53-specific antisense oligonucleotide resulted in a decrease in the protein levels of p53 as well as its transcriptional regulator p21/WAF-1. In contrast, treatment of 22Rv1 cells with 20 μM apigenin for 16 h markedly increased the levels of these proteins. Further, treatment of cells with a combination of p53-specific antisense oligonucleotide and apigenin for 16 h attenuated the effects of apigenin-induced p53 and p21/WAF-1 (Fig. 4A). Under this condition apigenin-induced apoptosis was attenuated by the p53-specific antisense oligonucleotide and significantly decreased apoptosis in these cells (Fig. 4C).
Fig. 4.
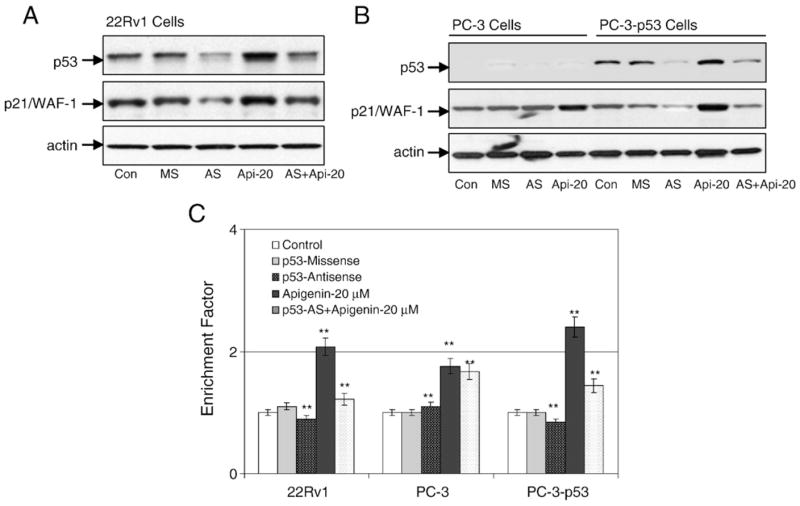
Inhibition of p53 causes a decrease in apigenin-mediated apoptosis. (A) Immunoblots for p53 and p21/WAF-1 using lysates from 22Rv1 cells pretreated with 10 μM p53-specific antisense and mismatch oligonucleotides for 8 h, followed by 20 μM apigenin treatment for 16 h and combination of p53-specific antisense oligonucleotide and apigenin for 16 h. (B) Immunoblots for p53 and p21/WAF-1 using lysates from PC-3 (p53−/−) and PC-3 (p53+/+) cells treated with 10 μM p53-specific antisense and mismatch oligonucleotides for 8 h, followed by 20 μM apigenin treatment for 16 h and combination of p53-specific antisense oligonucleotide and apigenin for 16 h. The blots were stripped and reprobed with anti-actin antibody to ensure equal protein loading. (C) Apoptosis in the lysates from 22Rv1, PC-3 (p53−/−), and PC-3 (p53+/+) cells with the indicated treatments. Values are represented as enrichment factor described under Materials and methods. Data are means±SE, n=3. Significantly different from control: **p<0.001.
To further evaluate the requirement for p53 in apigenin-mediated apoptosis, we used isogenic prostate cancer PC-3 cells. PC-3 (p53−/−) and PC-3 (p53+/+) differ only in their p53 status. Introduction of wild-type p53 into PC-3 cells renders these cells more susceptible to apigenin exposure and causes a significant increase in apoptosis compared to the parental cells (Fig. 4C). Indeed, apigenin-induced increase in p53 protein was effectively inhibited by treatment with p53-specific antisense oligonucleotide, which was attenuated by addition of 20 μM apigenin (Fig. 4B). These results indicate that p53 protein plays a critical role in apigenin-induced apoptotic cell death.
Apigenin treatment induces reactive oxygen species generation in 22Rv1 cells
Next we evaluated the hypothesis that apigenin-induced apoptosis is initiated by ROS, which have been implicated in apoptosis induction by various stimuli, including ionizing radiation, hyperoxia, and therapeutic/chemopreventive agents [13–16]. Intracellular ROS levels in 22Rv1 cells after apigenin exposure was assessed using fluorometric analysis. We used the fluorescence probe 2′,7′-chlorofluorescein diacetate, which converts to highly fluorescent DCF in the presence of intracellular ROS. A time-course study using 40 μM apigenin showed an increase in ROS generation as early as 1 h after apigenin treatment, which peaked at approximately 3–4 h, followed by a decreasing trend (data not shown). Therefore, we next assessed the levels of ROS after 3 h of apigenin treatment. As shown in Fig. 5A, exposure of 22Rv1 cells to 10–80 μM apigenin for 3 h resulted in a significant increase in ROS generation. Apigenin-treated 22Rv1 cells exhibited a dose-dependent increase in DCF fluorescence compared to vehicle-treated control. This increase in intracellular ROS by apigenin positively correlated with apoptosis (Fig. 5B).
Fig. 5.
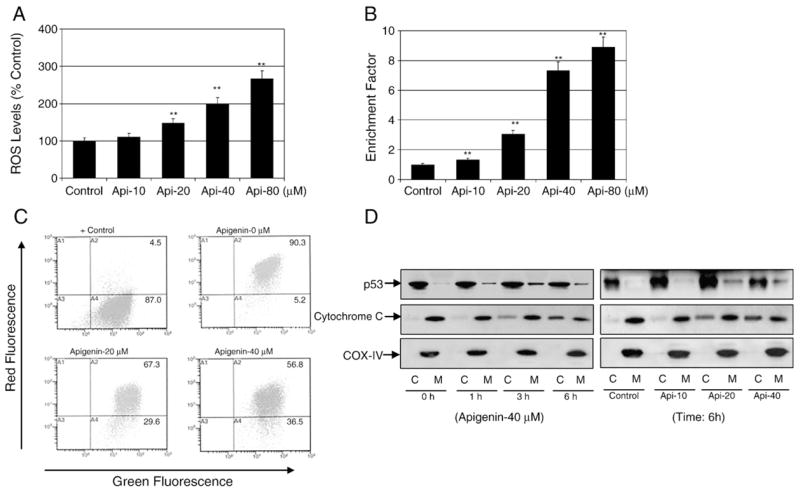
Apigenin treatment causes ROS generation, disruption of mitochondrial membrane potential, translocation of p53 to the mitochondria, and cytochrome c release into the cytosol in 22Rv1 cells. (A) Percentage of cells with high DCF fluorescence in cell culture after 3 h treatment with the indicated concentrations of apigenin. Representative data, means±SE, n=3, from a single experiment, which was repeated twice with similar results. Significantly different from control: **p<0.001. (B) Apoptosis in the lysates from 22Rv1 cells treated with various concentrations of apigenin for 16 h. Values are represented as enrichment factor described under Materials and methods. Data are means±SE, n=3. Significantly different from control: **p<0.001. (C) Flow histograms depicting JC-1 dye probe after 6 h treatment with various indicated concentrations of apigenin. Values in the right-hand lower section indicate the mitochondrial membrane depolarization after apigenin treatment, repeated twice with similar results. (D) Immunoblots for p53 and cytochrome c in the cytosolic and mitochondrial fractions obtained after treatment for various times with the indicated doses of apigenin. The blot was stripped and reprobed with anti-COX-IV antibody to ensure equal mitochondrial protein loading.
Apigenin treatment causes mitochondrial membrane depolarization in 22Rv1 cells
As apigenin exposure to 22Rv1 cells resulted in increased intracellular ROS generation, we sought to determine whether apigenin-induced apoptosis was associated with a disruption of the mitochondrial membrane potential. The effect of apigenin exposure on mitochondrial membrane depolarization was determined by flow cytometry after staining with the mitochondrial potential-sensitive dye JC-1. As shown in Fig. 5C, treatment of 22Rv1 cells with apigenin for 8 h resulted in a significant increase in the number of green fluorescence-positive cells. For instance, green fluorescence-positive cells treated with 20 and 40 μM apigenin increased to 29.6 and 36.5%, compared to 5.2% noted with vehicle-treated control.
Apigenin treatment causes translocation of the p53 fraction to the mitochondria in 22Rv1 cells
Next we investigated the possibility that apigenin-initiated apoptosis may utilize p53 translocation to the mitochondria as previously demonstrated during stress-induced p53-dependent apoptosis after DNA or hypoxic damage [15–17]. As shown in Fig. 5D, exposure of 22Rv1 cells to 40 μM concentration of apigenin resulted in a time-course increase in translocation of the p53 fraction to the mitochondria, with highest accumulation observed 3 h posttreatment. Simultaneously, apigenin-induced release of cytochrome c in the cytosol was observed at 3 h with increasing trend up to 6 h. Dose-dependent studies with 10–40 μM apigenin exposure for 6 h exhibited similar increases in p53 in the mitochondrial fraction along with the release of cytochrome c into the cytosol (Fig. 5D).
NAC pretreatment protects against apigenin-induced ROS generation and apoptosis in 22Rv1 cells
To test whether augmented ROS levels play a role in mediating the death signal of p53, 22Rv1 cells were pretreated with a well-known antioxidant, NAC, at 5 mM concentration or 25 μM pifithrin-α, a well-known p53 inhibitor, for 3 h. Both NAC and pifithrin-α treatment exhibited protective effects against apigenin-induced and p53-mediated apoptosis as well as attenuating the intracellular ROS generation in 22Rv1 cells, albeit at different levels (Figs. 6A and B).
Fig. 6.
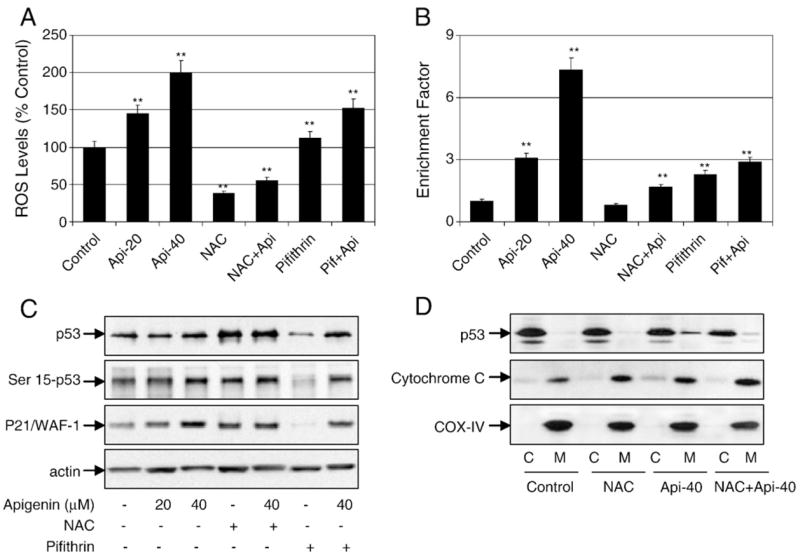
Apigenin-induced ROS generation and apoptosis are inhibited by antioxidants and p53 inhibition in 22Rv1 cells. (A) Percentage of cells with high DCF fluorescence in cell culture after 3 h treatment with the indicated concentrations of apigenin, 5 mM NAC, or 25 μM pifithrin-α and their combination. Representative data, means±SE, n=3, from a single experiment, which was repeated twice with similar results. Significantly different from control: **p<0.001. (B) Apoptosis in the lysates from 22Rv1 cells treated with various concentrations of apigenin, 5 mM NAC, or 25 μM pifithrin-α and their combination for 16 h. Values are represented as enrichment factor described under Materials and methods. Data are means±SE, n=3. Significantly different from control: **p<0.001. (C) Immunoblots for p53, Ser15-p53, and p21/WAF-1 after the indicated treatments with apigenin, NAC, and pifithrin-α for 16 h. The blots were stripped and reprobed with anti-actin antibody to ensure equal protein loading. (D) Immunoblots for p53 and cytochrome c in the cytosolic and mitochondrial fractions from 22Rv1 cells treated with apigenin, NAC, and their combination. The blot was stripped and reprobed with anti-COX-IV antibody to ensure equal mitochondrial protein loading.
To determine the relationship between ROS and the apoptotic cascade of p53 after apigenin exposure, we used NAC and pifithrin-α to further test whether they could influence p53 levels and apoptosis in 22Rv1 cells. As shown in Fig. 6C, treatment of cells with apigenin before NAC exposure caused an increase in p53-Ser15 phosphorylation and induction of p21/WAF-1, suggesting that ROS and p53 regulate the apoptotic process of 22Rv1 cells after apigenin treatment. Further, NAC pretreatment inhibited the translocation of p53 to the mitochondria and the release of cytochrome c into the cytosol of 22Rv1 cells after exposure to apigenin (Fig. 6D).
Apigenin treatment causes glutathione depletion in 22Rv1 cells
Because apigenin induced cytotoxicity by causing excessive ROS accumulation, we next investigated the mechanisms by which apigenin caused an increase in ROS generation. Based on the important role of GSH as a major cellular antioxidant, we postulated that the active ROS generation in 22Rv1 cells would render them highly dependent on GSH to maintain redox balance and that a depletion of GSH by apigenin would result in excessive ROS generation, triggering p53 stabilization, which may drive the damaged cells to apoptosis [35]. To test this possibility we first examined the effects of apigenin on GSH content. Exposure of 22Rv1 cells to 40 μM apigenin led to a depletion of cellular GSH by 67% in 1 h, which was further increased to 81% in 3 h (data not shown).
Catalase causes attenuated ROS generation and apoptosis induction by apigenin in 22Rv1 cells
Next we treated 22Rv1 cells with 1 mM concentration of hydrogen peroxide (H2O2), which is considered the most stable ROS and an important mediator of apoptosis. As expected, exposure of 22Rv1 cells to H2O2 up to 3 h caused a significant increase in ROS generation and triggered apoptosis (Fig. 7A). Furthermore it was observed that H2O2 caused a transient rise in p53 protein expression as well as sustained elevation of p21/WAF-1 in 22Rv1 cells. Interestingly, exogenous addition of catalase effectively abolished H2O2-induced ROS increase and cell death, which correlated with the reductions in the protein levels of p53 and p21/WAF-1 (Fig. 7B). To further affirm the role of apigenin and its comparison with H2O2-mediated ROS generation and cell death, 22Rv1 cells were treated with 1 mM concentration of H2O2 and 40 μM apigenin. Responses similar to those to H2O2 were observed with apigenin treatment. In addition, enzyme catalase, when added to apigenin-treated cells, caused a significant decrease in ROS induction and apoptosis along with a decrease in the protein expression of p53 and p21/WAF-1, similar to that previously observed with pretreatment of cells with NAC before apigenin exposure (Fig. 7C). Additionally, catalase treatment inhibited the translocation of p53 to the mitochondria and the release of cytochrome c into the cytosol of 22Rv1 cells after exposure to apigenin (Fig. 7D).
Fig. 7.
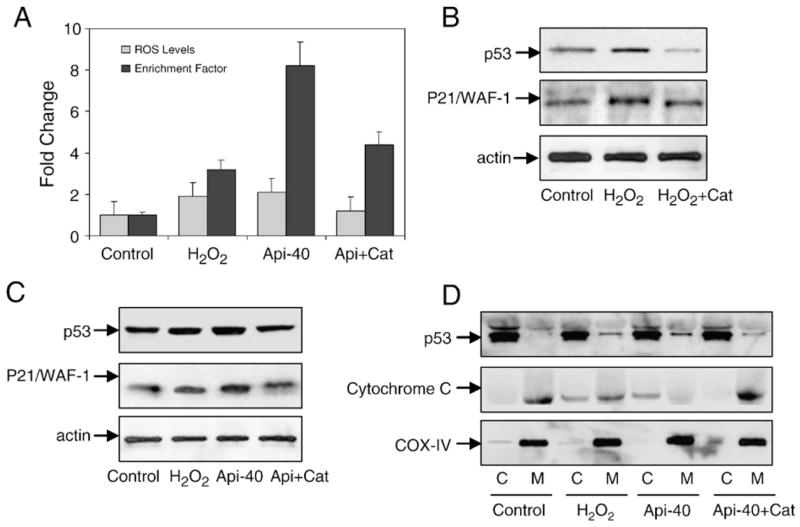
Apigenin-induced ROS generation, p53 translocation, and cytochrome c release into the cytosol are reversed by treatment with catalase in 22Rv1 cells. (A) Fold change in ROS levels and enrichment factor after 3 h treatment of cells with 1 mM H2O2 or 40 μM apigenin and pretreatment with 400 units/ml recombinant catalase followed by apigenin. Values are represented as means±SE, n=3, from a single experiment, which was repeated twice with similar results. (B) Immunoblots for p53 and p21/WAF-1 after treatment with H2O2 and its combination with catalase. (C) Immunoblots for p53 and p21/WAF-1 after treatment with H2O2, apigenin, and apigenin combined with catalase. The blot was stripped and reprobed with anti-actin antibody to ensure equal protein loading. (D) Immunoblots for p53 and cytochrome c in the cytosolic and mitochondrial fractions from 22Rv1 cells treated with H2O2, apigenin, and apigenin combined with catalase. The blot was stripped and reprobed with anti-COX-IV antibody to ensure equal mitochondrial protein loading.
Apigenin increases the ratio of Bax/Bcl-2 and cytochrome c release in 22Rv1 cells
The ratio of proapoptotic Bax to antiapoptotic Bcl-2 is critical for apoptosis; therefore we next measured the levels of these proteins after apigenin treatment. As shown in Fig. 8A, exposure of 22Rv1 cells to apigenin resulted in a marked increase in Bax protein along with a simultaneous decrease in Bcl-2 levels. Densitometric analysis indicated that the shift in Bax/Bcl-2 ratio in favor of apoptosis was a function of dose and time (Fig. 8B). Apigenin treatment also resulted in a decrease in Bcl-XL protein and an increase in cytochrome c and Apaf-1 levels in the cytosol (Figs. 8A and B).
Fig. 8.
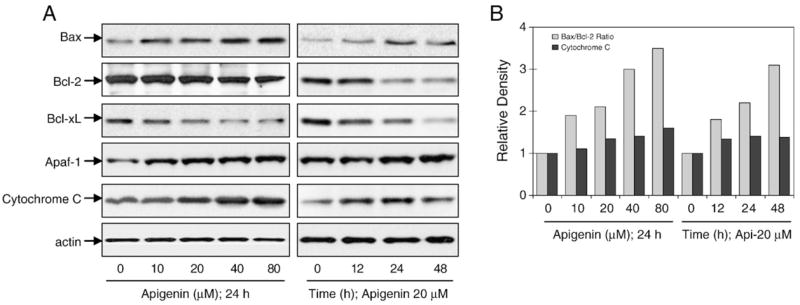
Apigenin treatment caused a modulation in Bcl-2 family member proteins, Apaf-1, and cytochrome c in 22Rv1 cells. (A) Immunoblots for Bax, Bcl-2, Bcl-XL, Apaf-1, and cytochrome c using lysates from 22Rv1 cells treated with various concentrations of apigenin for the indicated time periods. The blots were stripped and reprobed with anti-actin antibody to ensure equal protein loading. (B) Densitometric analysis for proteins.
Apigenin induces caspase activation and PARP cleavage in 22Rv1 cells
We demonstrated earlier that apigenin-induced apoptosis in 22Rv1 cells is associated with a shift in the Bax/Bcl-2 ratio and disruption of the mitochondrial membrane potential, leading to the release of cytochrome c and other apoptogenic molecules from the mitochondria to the cytosol (Figs. 8A and B). Once in the cytosol, cytochrome c binds to Apaf-1 and recruits and activates caspase-9 in the apoptosome. Active caspase-9 cleaves and activates executioner caspases, including caspase-3. We therefore determined caspase activation after apigenin treatment. Exposure of 22Rv1 cells for 24 h to apigenin led to increased levels of active caspase-9 and -3 and caused cleavage of PARP as a function of dose and time. In addition, apigenin exposure also increased the levels of active caspase-8 in 22Rv1 cells, suggesting the involvement of both extrinsic and intrinsic pathways of apoptosis (Figs. 9A and B).
Fig. 9.
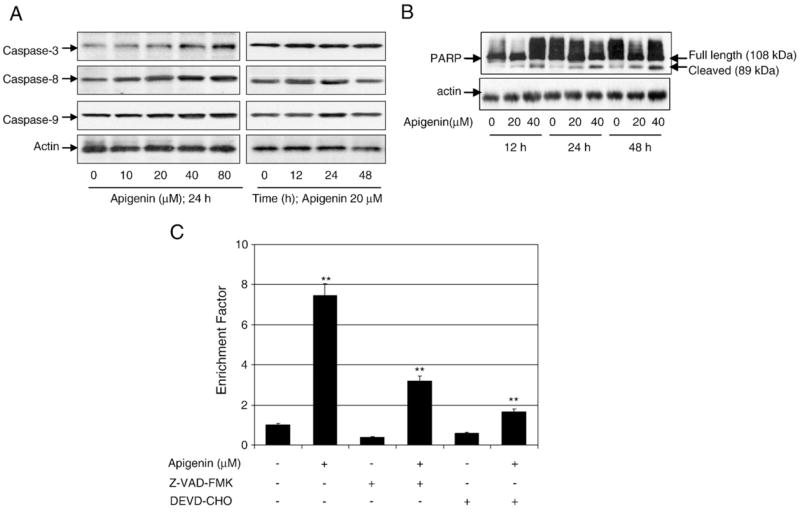
Apigenin treatment causes caspase activation, PARP cleavage, and apoptosis, which was partially protected against by caspase inhibitors in 22Rv1 cells. (A) Immunoblots for caspase-3, -8, and -9 using lysates from 22Rv1 cells treated with various concentrations of apigenin for the indicated time periods. (B) Cleavage of poly(ADP)ribose polymerase in 22Rv1 cells after treatment with various concentrations of apigenin for the indicated time periods. Full-length (108 kDa) and cleaved products (89 kDa) are depicted. (C) Apoptosis as demonstrated by ELISA in 22Rv1 cells after treatment of cells with 40 μM apigenin as well as pretreatment with the general caspase inhibitor Z-VAD-FMK and the caspase-3-specific inhibitor DEVD-CHO, followed by apigenin. Values are means±SE, n=3, from a single experiment, which was repeated twice with similar results. Significantly different from control: **p<0.001.
Next we evaluated whether blocking caspases could inhibit apigenin-mediated p53-dependent apoptosis in these cells. Exposure of cells to the general caspase inhibitor Z-VAD-FMK before apigenin treatment partially blocked apoptosis. Further, use of the caspase-3 inhibitor DEVD-CHO partially rescued the cells from apigenin-induced apoptosis, at higher levels than Z-VAD-FMK (Fig. 9C). None of these inhibitors, however, affected the p53 serine phosphorylation in 22Rv1 cells (data not shown).
Apigenin intake inhibits growth of 22Rv1 xenografts in athymic nude mice
Apigenin has been shown to be effective in cell culture and to cause p53-dependent apoptosis in 22Rv1 cells; therefore we further extended our study to determine whether these events occur in vivo using a xenograft mouse model. We designed a protocol that simulates a prevention regimen, wherein apigenin was administered at 20 and 50 μg/mouse/day 2 weeks before cell inoculation and was continued for 8 weeks. In this experimental protocol, prior intake of apigenin delayed the establishment of tumor xenograft at both doses of apigenin. As shown in Figs. 10A and B, tumor volume was inhibited by 44 and 59% (p<0.002 and 0.0001) and the wet weight of tumor was decreased by 41 and 53% (p<0.05), respectively, at the termination of the experiment. Further, apigenin administration to these mice did not seem to induce any adverse effects as judged by monitoring body weight gain, dietary intake, and prostate weight (data not shown).
Fig. 10.
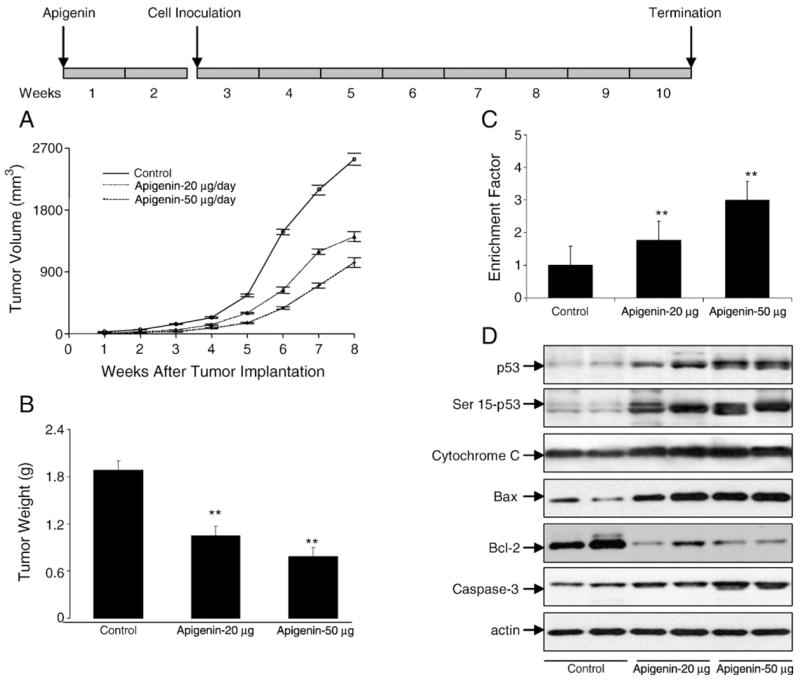
Apigenin administration inhibits 22Rv1 tumor growth in athymic nude mice via modulation of p53 signaling pathways. Approximately 1 million cells were injected into both flanks of each mouse to initiate ectopic prostate tumor growth, and apigenin was provided to the animals 2 weeks before cell inoculation. Mice were fed ad libitum with Teklad 8760 autoclaved high-protein diet. Apigenin was provided with 0.5% methyl cellulose and 0.025% Tween 20 as vehicle to these animals perorally on a daily basis. Group I, control, received 0.2 ml vehicle only, Group II received 20 μg apigenin per mouse in 0.2 ml vehicle, and Group III received 50 μg apigenin per mouse in 0.2 ml vehicle daily for 8 weeks. Once the tumor xenografts started growing, their sizes were measured twice weekly in two dimensions throughout the study. (A) Tumor volume (mm3) in control and treated groups. (B) Wet weight of tumors is represented as the mean of 8–10 tumors from each group. (C) Apoptosis as demonstrated by ELISA for 22Rv1 tumors after apigenin intake at the indicated doses. Values are means±SE, n=6–8, repeated twice with similar results. (D) Immunoblots for p53, Ser15-p53, cytochrome c, Bax, Bcl-2, and caspase-3 in tumor lysates after apigenin intake at the indicated doses. The blots were stripped and reprobed with anti-actin antibody to ensure equal protein loading. Significantly different from control: **p<0.001.
Apigenin intake causes apoptosis in 22Rv1 tumors through a p53-dependent pathway
Earlier results in cell culture demonstrated that apigenin induces apoptosis in 22Rv1 cells; therefore we evaluated the effects of apigenin intake on the induction of apoptosis in tumor xenografts. As shown in Fig. 10C, peroral administration of apigenin at doses of 20 and 50 μg/mouse/day resulted in a marked induction of apoptosis in 22Rv1 tumor xenografts as shown by ELISA. Compared to vehicle-treated control,1.7- and 2.9-fold increases (p<0.0001) in the induction of apoptosis were observed in 22Rv1 tumors at 20 and 50 μg/day apigenin treatment. Further, consistent with the findings in cell culture, apigenin administration to nude mice resulted in a dose-dependent increase in the expression of wild-type p53 protein, p53-Ser15 phosphorylation, cytochrome c, and cleaved caspase 3, compared to mice receiving vehicle treatment. An increase in the protein levels of Bax and a simultaneous decrease in Bcl-2 levels were observed after apigenin administration, indicating that inhibition of growth of 22Rv1 tumor xenografts is due to induction of apoptosis, which is mediated via p53 pathways (Fig. 10D).
Discussion
In this study, we demonstrated that apigenin is able to induce apoptosis in 22Rv1 cells and in in vivo tumors. This induction of apoptosis is accompanied by increases in ROS production, p53 phosphorylation, and p21/WAF-1 induction; alterations in MDM2/p14ARF; a decrease in NF-κB/p65 transcriptional activity leading to loss of mitochondrial membrane potential; and caspase activation. Although our study and prior studies have shown that apigenin-induced p53-dependent apoptosis occurs in prostate cancer and other malignancies, the pathways whereby p53 leads to execution of the apoptosis program are not well characterized.
There is accumulating evidence that apigenin can suppress the growth of malignant prostate cells as well as tumor xenografts in vivo by causing cell cycle arrest and apoptosis [31–34,36]. In this study we have demonstrated that the p53-associated pathway is required for apigenin-mediated apoptosis, as evidenced by the p53 antisense oligonucleotide experiment. Apigenin-induced p53 response in prostate cancer cells is primarily achieved through DNA damage via initiation and generation of ROS. We observed that exposure of 22Rv1 cells to growth-suppressive concentrations of apigenin resulted in ROS generation as indicated by an increase in oxidation of DCF. This increase in ROS production leads to activation and increase in the cellular levels of p53. This increase in p53 expression was accompanied by phosphorylation of p53 at serines 6, 15, 20, 37, and 392, but not at serines 9 and 46. Phosphorylation of p53 at serines 15 and 20 has been shown to stimulate p53-dependent transcriptional activation [37] and also results in reduced interaction of p53 with its negative regulator MDM2 both in vivo and in vitro [24,38]. Apigenin treatment also resulted in increased expression of p14ARF, a tumor suppressor that negatively regulates MDM2 [39]. Our results indicate that apigenin stabilizes p53 via both phosphorylation of p53 and modulation of the p14ARF–MDM2 pathway. Interestingly, we observed a decrease in the levels of MDM2 protein in response to apigenin treatment. We speculate that this decrease may be directly mediated by apigenin at the transcriptional level [40] or may be a result of an apigenin-mediated increase in p14ARF levels [41].
The importance of p53 in apigenin-mediated apoptosis was further demonstrated by the introduction of wild-type p53 in PC-3 cells, which have a p53-null status. Compared to parental PC-3 cells, the p53-PC-3 cells were more sensitive to apigenin-mediated apoptosis, similar to 22Rv1 cells, which harbor wild-type p53. Additionally, apigenin-induced stabilization of p53 caused an increase in its transcriptional activity, thereby resulting in an upregulation of its downstream target p21/WAF-1. p21/WAF-1 can inhibit cdk’s 2, 4, and 6, causing cell cycle arrest [42]. Earlier studies from our laboratory have demonstrated that apigenin-mediated upregulation of p21/WAF-1 is critical in inducing both cell cycle arrest and apoptosis [31,34]. The mechanisms by which p21 promotes apoptosis are not currently understood, but may be related to its ability to interact with and possibly regulate components of the DNA repair machinery [43].
Several recent reports suggest a role for ROS in apoptosis induced by drugs, including certain chemopreventive agents, through the engagement of downstream proteins involved in execution of apoptosis [44–46]. Intracellular generation of H2O2, the most stable ROS, has been considered an important mediator of apoptosis, and even exogenous addition of H2O2 is a potent activator of the apoptotic machinery [47]. In our studies, sublethal concentrations of H2O2 caused an increase in p53 protein expression along with sustained elevation of p21/WAF-1, which correlated with increased apoptosis observed in these cells. These effects were alleviated by pretreatment of cells with 400 units/ml concentration of catalase. There is evidence that mitochondrial catalase provides protective effects against exogenously added H2O2, which suggests that some of the added H2O2 diffuses into the mitochondria and perhaps causes damage to the mitochondrial membrane [47,48]. This suggests that mitochondrially produced H2O2 diffuses into the cytosol where it may exert cytotoxic effects. It seems that catalase produced in any cellular compartment might act as a sink for H2O2 and further addition of catalase may promote H2O2 movement down to its concentration gradient. Further reports suggest that catalase added to the culture medium might protect cells against oxidant-induced injury, whereas this may reflect some uptake of catalase into the cells by endocytosis and/or removal of diffusible H2O2 from the cells [48]. Under physiological conditions, H2O2 may play an important role in signal transduction pathways and activation of p53 [49]. Our studies indicate that intracellular H2O2 generation induced by exogenous H2O2 or apigenin was suppressed by the addition of catalase, which in turn inhibited the release of cytochrome c into the cytosol as well as translocation of p53 to the mitochondria.
It is well established that p53 is a transcriptional regulator and that p53-mediated apoptosis in response to DNA damage is predominantly attributable both to the transcriptional activation of genes that encode apoptotic effectors, such as Bax, and to transcriptional repression of genes for antiapoptotic proteins such as Bcl-2 [8–10]. In this context, upregulation of Bcl-2 by NF-κB might be critical in compromising the apoptotic abilities of p53. We have shown that apigenin inhibits the transcriptional activity of NF-κB/p65, downregulates the levels of Bcl-2 and Bcl-XL, and concomitantly upregulates the levels of Bax protein, shifting the Bax/Bcl-2 ratio in favor of apoptosis. In addition to such translational regulation, recent findings have demonstrated the existence of a transcription-independent pathway of p53-mediated apoptosis, wherein a fraction of the p53 molecule accumulates in the damaged cells and translocates to mitochondria in quantities sufficient to induce permeabilization of the outer mitochondrial membrane, resulting in the release of cytochrome c [15,16]. Our studies demonstrate that apigenin causes the translocation of p53 to the mitochondria as early as 1 h, peaking at 3 h post-apigenin treatment. This increased translocation of p53 to the mitochondria leads to cytochrome c release observed as early as 3 h and increasing up to 24 h post-apigenin treatment. Because apigenin-induced p53 activation and translocation are dependent on ROS generation, which was evident as early as 1 h posttreatment, whereas the translocation of p53 and cytochrome c release were not observed until 3–6 h after apigenin exposure, we conclude that ROS act as upstream signaling molecules to initiate p53-mediated cell death responses. This hypothesis is further supported by our findings that pretreatment of 22Rv1 cells with NAC not only prevents ROS generation but also confers almost complete protection against apigenin-induced p53-translocation to the mitochondria, cytochrome c release, and cell death. Moreover, inhibition of p53 by the use of pifithrin-α protected tumor cells from apigenin-induced apoptosis, further demonstrating the involvement of p53 in cell death. ROS generation in apoptosis induction by some agents has been shown to occur downstream of cytochrome c release; however, apigenin-induced release of cytochrome c is significantly inhibited by pretreatment with NAC. These results indicate that ROS acts as a trigger for apigenin-induced apoptosis upstream of both p53 and cytochrome c.
Many apoptotic cascades in eukaryotic cells utilize mitochondria as a nodal point at which diverse death stimuli translate from initiation to execution of apoptosis [50,51]. Mitochondria play an essential role in death signal transduction through the permeability transition pore opening and collapse of the mitochondrial membrane potential, resulting in the rapid release of caspase activators such as cytochrome c into the cytoplasm. Cytochrome c then binds to Apaf-1 and activates caspase-3 through caspase-9, culminating in cell death [50,51]. We demonstrated that the release of cytochrome c into the cytosol increased within 3 h of apigenin treatment, a time frame parallel to that for initiation of apoptosis. Furthermore, the increase in cytochrome c paralleled Apaf-1 levels observed between 3 and 24 h of apigenin exposure, which led to the activation of caspase-3 and cleavage of poly(ADP)ribose polymerase. In addition, we also observed an increase in caspase-8, albeit at higher doses of apigenin treatment. The general caspase inhibitor, Z-VAD-FMK, led to the protection of 22Rv1 cells against apoptosis, indicating that apigenin-mediated apoptosis is caspase dependent. Our data suggest that the caspase-8 and the caspase-9 activation pathways contributed to the activation of the executioner caspase-3. In our studies, the protective effect of the caspase-3 inhibitor, DEVD-CHO, against apigenin-induced apoptosis was greater than that of Z-VAD-FMK, indicating the involvement of additional caspases in execution of cell death by apigenin. Further work is required to examine the detailed mechanism(s) of apigenin with respect to the death receptor cascades as well as the endoplasmic reticulum-stress activated caspases.
In our cell culture studies, growth inhibition and apoptosis induction by apigenin were observed at 10–40 μM concentrations, which provided mechanistic insights; however, demonstration that these effects are also operative in vivo is required to establish a potential for clinical development. Our in vivo studies using 20 and 50 μg/day apigenin administration to mice with prostate cancer xenografts confirmed that apigenin administration significantly inhibited tumor growth, without any apparent signs of toxicity. Consistent with the findings in cell culture, apigenin intake resulted in p53 activation and its phosphorylation at serine 15, cytochrome c release, increase in the levels of cleaved caspase-3, and altered Bax and Bcl-2 protein levels, favoring apoptosis, compared to vehicle-treated animals. Based on these observations it is apparent that apigenin most likely exerts its cancer preventive/therapeutic effects directly through the p53 signaling pathway, utilizing mitochondria as a universal key effector of cell death. Understanding the modes of action of apigenin should provide useful information for its possible application in cancer prevention and perhaps in therapy for prostate cancer.
Acknowledgments
This research was supported by the Athymic Animal Core Facility of the Comprehensive Cancer Center of Case Western Reserve University and University Hospitals Case Medical Center (P30 CA43703). This work was also supported by grants from the U.S. Public Health Services, RO1 CA108512, RO1 AT002709, RO3 CA094248, and RO3 CA099049, and received partial support of funds from the Cancer Research and Prevention Foundation to S.G. The authors are thankful to Dr. Pingfu Fu for performing statistical evaluations for this study.
Abbreviations
- Apaf-1
apoptosis protease-activating factor-1
- ROS
reactive oxygen species
- ERK
extracellular signal-regulated kinase
- NF-κB
nuclear factor-κB
- IGFBP
insulin-like growth factor binding protein
- PARP
poly(ADP)ribose polymerase
- MDM
murine double minute
- GSH
glutathione
- DCF
dichlorofluorescein
- NAC
N-acetylcysteine
Footnotes
This article appeared in a journal published by Elsevier. The attached copy is furnished to the author for internal non-commercial research and education use, including for instruction at the authors institution and sharing with colleagues. Other uses, including reproduction and distribution, or selling or licensing copies, or posting to personal, institutional or third party websites are prohibited. In most cases authors are permitted to post their version of the article (e.g. in Word or Tex form) to their personal website or institutional repository. Authors requiring further information regarding Elsevier’s archiving and manuscript policies are encouraged to visit:
References
- 1.Selivanova G. p53: fighting cancer. Curr Cancer Drug Targets. 2004;4:385–402. doi: 10.2174/1568009043332934. [DOI] [PubMed] [Google Scholar]
- 2.Oren M. Decision making by p53: life, death and cancer. Cell Death Differ. 2003;10:431–442. doi: 10.1038/sj.cdd.4401183. [DOI] [PubMed] [Google Scholar]
- 3.Bode AM, Dong Z. Post-translational modification of p53 in tumorigenesis. Nat Rev, Cancer. 2004;4:793–805. doi: 10.1038/nrc1455. [DOI] [PubMed] [Google Scholar]
- 4.Russo A, Bazan V, Iacopetta B, Kerr D, Soussi T, Gebbia N TP53–CRC Collaborative Study Group. The TP53 Colorectal Cancer International Collaborative Study on the prognostic and predictive significance of p53 mutation: influence of tumor site, type of mutation, and adjuvant treatment. J Clin Oncol. 2005;23:7518–7528. doi: 10.1200/JCO.2005.00.471. [DOI] [PubMed] [Google Scholar]
- 5.Roncuzzi L, Brognara I, Baiocchi D, Amadori D, Gasperi-Campani A. Loss of heterozygosity at 17p13.3–ter, distal to TP53, correlates with negative hormonal phenotype in sporadic breast cancer. Oncol Rep. 2005;14:471–474. [PubMed] [Google Scholar]
- 6.Downing SR, Russell PJ, Jackson P. Alterations of p53 are common in early stage prostate cancer. Can J Urol. 2003;10:1924–1933. [PubMed] [Google Scholar]
- 7.Jackson JG, Pereira-Smith OM. p53 is preferentially recruited to the promoters of growth arrest genes p21 and GADD45 during replicative senescence of normal human fibroblasts. Cancer Res. 2006;66:8356–8360. doi: 10.1158/0008-5472.CAN-06-1752. [DOI] [PubMed] [Google Scholar]
- 8.Bohm M, Wolff I, Scholzen TE, Robinson SJ, Healy E, Luger TA, Schwarz T, Schwarz A. Alpha-melanocyte-stimulating hormone protects from ultraviolet radiation-induced apoptosis and DNA damage. J Biol Chem. 2005;280:5795–5802. doi: 10.1074/jbc.M406334200. [DOI] [PubMed] [Google Scholar]
- 9.Baus F, Gire V, Fisher D, Piette J, Dulic V. Permanent cell cycle exit in G2 phase after DNA damage in normal human fibroblasts. EMBO J. 2003;22:3992–4002. doi: 10.1093/emboj/cdg387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Flatt PM, Price JO, Shaw A, Pietenpol JA. Differential cell cycle checkpoint response in normal human keratinocytes and fibroblasts. Cell Growth Differ. 1998;9:535–543. [PubMed] [Google Scholar]
- 11.Shen Y, Shenk T. Relief of p53-mediated transcriptional repression by the adenovirus E1B 19-kDa protein or the cellular Bcl-2 protein. Proc Natl Acad Sci U S A. 1994;91:8940–8944. doi: 10.1073/pnas.91.19.8940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hoffman WH, Biade S, Zilfou JT, Chen J, Murphy M. Transcriptional repression of the anti-apoptotic survivin gene by wild type p53. J Biol Chem. 2002;277:3247–3257. doi: 10.1074/jbc.M106643200. [DOI] [PubMed] [Google Scholar]
- 13.Caelles C, Helmberg A, Karin M. p53-dependent apoptosis in the absence of transcriptional activation of p53-target genes. Nature. 1994;370:220–223. doi: 10.1038/370220a0. [DOI] [PubMed] [Google Scholar]
- 14.Haupt Y, Rowan S, Shaulian E, Vousden KH, Oren M. Induction of apoptosis in HeLa cells by trans-activation-deficient p53. Genes Dev. 1995;9:2170–2183. doi: 10.1101/gad.9.17.2170. [DOI] [PubMed] [Google Scholar]
- 15.Marchenko ND, Zaika A, Moll UM. Death signal-induced localization of p53 protein to mitochondria: a potential role in apoptotic signaling. J Biol Chem. 2000;275:16202–16212. doi: 10.1074/jbc.275.21.16202. [DOI] [PubMed] [Google Scholar]
- 16.Arima Y, Nitta M, Kuninaka S, Zhang D, Fujiwara T, Taya Y, Nakao M, Saya H. Transcriptional blockade induces p53-dependent apoptosis associated with translocation of p53 to mitochondria. J Biol Chem. 2005;280:19166–19176. doi: 10.1074/jbc.M410691200. [DOI] [PubMed] [Google Scholar]
- 17.Schuler M, Bossy-Wetzel E, Goldstein JC, Fitzgerald P, Green DR. p53 induces apoptosis by caspase activation through mitochondrial cytochrome c release. J Biol Chem. 2000;275:7337–7342. doi: 10.1074/jbc.275.10.7337. [DOI] [PubMed] [Google Scholar]
- 18.Ren W, Qiao Z, Wang H, Zhu L, Zhang L. Flavonoids: promising anticancer agents. Med Res Rev. 2003;23:519–534. doi: 10.1002/med.10033. [DOI] [PubMed] [Google Scholar]
- 19.Patel D, Shukla S, Gupta S. Apigenin and cancer chemoprevention: progress, potential and promise (review) Int J Oncol. 2007;30:233–245. [PubMed] [Google Scholar]
- 20.Gupta S, Afaq F, Mukhtar H. Selective growth-inhibitory, cell-cycle deregulatory and apoptotic response of apigenin in normal versus human prostate carcinoma cells. Biochem Biophys Res Commun. 2001;287:914–920. doi: 10.1006/bbrc.2001.5672. [DOI] [PubMed] [Google Scholar]
- 21.Wei H, Tye L, Bresnick E, Birt DF. Inhibitory effect of apigenin, a plant flavonoid, on epidermal ornithine decarboxylase and skin tumor promotion in mice. Cancer Res. 1990;50:499–502. [PubMed] [Google Scholar]
- 22.Tatsuta A, Iishi H, Baba M, Yano H, Murata K, Mukai M, Akedo H. Suppression by apigenin of peritoneal metastasis of intestinal adenocarcinomas induced by azoxymethane in Wistar rats. Clin Exp Metastasis. 2000;18:657–662. doi: 10.1023/a:1013133803806. [DOI] [PubMed] [Google Scholar]
- 23.Liu LZ, Fang J, Zhou Q, Hu X, Shi X, Jiang BH. Apigenin inhibits expression of vascular endothelial growth factor and angiogenesis in human lung cancer cells: implication of chemoprevention of lung cancer. Mol Pharmacol. 2005;68:635–643. doi: 10.1124/mol.105.011254. [DOI] [PubMed] [Google Scholar]
- 24.Fang J, Xia C, Cao Z, Zheng JZ, Reed E, Jiang BH. Apigenin inhibits VEGF and HIF-1 expression via PI3K/AKT/p70S6K1 and HDM2/p53 pathways. FASEB J. 2005;19:342–353. doi: 10.1096/fj.04-2175com. [DOI] [PubMed] [Google Scholar]
- 25.Trochon V, Blot E, Cymbalista F, Engelmann C, Tang RP, Thomaidis A, Vasse M, Soria J, Lu H, Soria C. Apigenin inhibits endothelial-cell proliferation in G(2)/M phase whereas it stimulates smooth-muscle cells by inhibiting P21 and P27 expression. Int J Cancer. 2000;85:691–696. doi: 10.1002/(sici)1097-0215(20000301)85:5<691::aid-ijc15>3.0.co;2-q. [DOI] [PubMed] [Google Scholar]
- 26.Schindler R, Mentlein R. Flavonoids and vitamin E reduce the release of the angiogenic peptide vascular endothelial growth factor from human tumor cells. J Nutr. 2006;136:1477–1482. doi: 10.1093/jn/136.6.1477. [DOI] [PubMed] [Google Scholar]
- 27.Huang YT, Kuo ML, Liu JY, Huang SY, Lin JK. Inhibitionsof protein kinase C and proto-oncogene expressions in NIH 3T3 cells by apigenin. Eur J Cancer. 1996;32:146–151. doi: 10.1016/0959-8049(95)00540-4. [DOI] [PubMed] [Google Scholar]
- 28.Agullo G, Gamet-Payrastre L, Manenti S, Viala C, Remesy C, Chap H, Payrastre B. Relationship between flavonoid structure and inhibition of phosphatidylinositol 3-kinase: a comparison with tyrosine kinase and protein kinase C inhibition. Biochem Pharmacol. 1997;53:1649–1657. doi: 10.1016/s0006-2952(97)82453-7. [DOI] [PubMed] [Google Scholar]
- 29.Yin F, Giuliano AE, Law RE, Van Herle AJ. Apigenin inhibits growth and induces G2/M arrest by modulating cyclin-CDK regulators and ERK MAP kinase activation in breast carcinoma cells. Anticancer Res. 2001;21:413–420. [PubMed] [Google Scholar]
- 30.Llorens F, Miro FA, Casanas A, Roher N, Garcia L, Plana M, Gomez N, Itarte E. Unbalanced activation of ERK1/2 and MEK1/2 in apigenin-induced HeLa cell death. Exp Cell Res. 2004;299:15–26. doi: 10.1016/j.yexcr.2004.05.006. [DOI] [PubMed] [Google Scholar]
- 31.Shukla S, Gupta S. Apigenin-induced cell cycle arrest is mediated by modulation of MAPK, PI3K-Akt, and loss of cyclin D1 associated retinoblastoma depho-sphorylation in human prostate cancer cells. Cell Cycle. 2007;6:1102–1114. doi: 10.4161/cc.6.9.4146. [DOI] [PubMed] [Google Scholar]
- 32.Shukla S, Gupta S. Suppression of constitutive and tumor necrosis factor alpha-induced nuclear factor (NF)-kappaB activation and induction of apoptosis by apigenin in human prostate carcinoma PC-3 cells: correlation with down-regulation of NF-kappaB-responsive genes. Clin Cancer Res. 2004;10:3169–3178. doi: 10.1158/1078-0432.ccr-03-0586. [DOI] [PubMed] [Google Scholar]
- 33.Shukla S, Mishra A, Fu P, MacLennan GT, Resnick MI, Gupta S. Up-regulation of insulin-like growth factor binding protein-3 by apigenin leads to growth inhibition and apoptosis of 22Rv1 xenograft in athymic nude mice. FASEB J. 2005;19:2042–2044. doi: 10.1096/fj.05-3740fje. [DOI] [PubMed] [Google Scholar]
- 34.Shukla S, Gupta S. Molecular targets for apigenin-induced cell cycle arrest and apoptosis in prostate cancer cell xenograft. Mol Cancer Ther. 2006;5:843–852. doi: 10.1158/1535-7163.MCT-05-0370. [DOI] [PubMed] [Google Scholar]
- 35.Galati G, Chan T, Wu B, O’Brien PJ. Glutathione-dependent generation of reactive oxygen species by the peroxidase-catalyzed redox cycling of flavonoids. Chem Res Toxicol. 1999;12:521–525. doi: 10.1021/tx980271b. [DOI] [PubMed] [Google Scholar]
- 36.Zheng PW, Chiang LC, Lin CC. Apigenin induced apoptosis through p53-dependent pathway in human cervical carcinoma cells. Life Sci. 2005;76:1367–1379. doi: 10.1016/j.lfs.2004.08.023. [DOI] [PubMed] [Google Scholar]
- 37.Dumaz N, Meek DW. Serine15 phosphorylation stimulates p53 transactivation but does not directly influence interaction with HDM2. EMBO J. 1999;18:7002–7010. doi: 10.1093/emboj/18.24.7002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shieh SY, Ikeda M, Taya Y, Prives C. DNA damage-induced phosphorylation of p53 alleviates inhibition by MDM2. Cell. 1997;91:325–334. doi: 10.1016/s0092-8674(00)80416-x. [DOI] [PubMed] [Google Scholar]
- 39.Weber JD, Taylor LJ, Roussel MF, Sherr CJ, Bar-Sagi D. Nucleolar Arf sequesters Mdm2 and activates p53. Nat Cell Biol. 1999;1:20–26. doi: 10.1038/8991. [DOI] [PubMed] [Google Scholar]
- 40.Levites Y, Amit T, Youdim MB, Mandel S. Involvementof protein kinase C activation and cell survival/cell cycle genes in green tea polyphenol ())-epigallocatechin 3-gallate neuroprotective action. J Biol Chem. 2002;277:30574–30580. doi: 10.1074/jbc.M202832200. [DOI] [PubMed] [Google Scholar]
- 41.Jackson MW, Lindstrom MS, Berberich SJ. MdmX binding to ARF affects Mdm2 protein stability and p53 transactivation. J Biol Chem. 2001;276:25336–25341. doi: 10.1074/jbc.M010685200. [DOI] [PubMed] [Google Scholar]
- 42.Bates S, Vousden KH. Mechanisms of p53-mediated apoptosis. Cell Mol Life Sci. 1999;55:28–37. doi: 10.1007/s000180050267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gartel AL, Tyner AL. The role of the cyclin-dependent kinase inhibitor p21 in apoptosis. Mol Cancer Ther. 2002;1:639–649. [PubMed] [Google Scholar]
- 44.Hirpara JL, Clément MV, Pervaiz S. Intracellular acidification triggered by mitochondrial-derived hydrogen peroxide is an effector mechanism for drug-induced apoptosis in tumor cells. J Biol Chem. 2001;276:514–521. doi: 10.1074/jbc.M004687200. [DOI] [PubMed] [Google Scholar]
- 45.Trachootham D, Zhou Y, Zhang H, Demizu Y, Chen Z, Pelicano H, Chiao PJ, Achanta G, Arlinghaus RB, Liu J, Huang P. Selective killing of oncogenically transformed cells through a ROS-mediated mechanism by beta-phenylethyl isothiocyanate. Cancer Cell. 2006;10:241–252. doi: 10.1016/j.ccr.2006.08.009. [DOI] [PubMed] [Google Scholar]
- 46.Singh SV, Srivastava SK, Choi S, Lew KL, Antosiewicz J, Xiao D, Zeng Y, Watkins SC, Johnson CS, Trump DL, Lee YJ, Xiao H, Herman-Antosiewicz A. Sulforaphane-induced cell death in human prostate cancer cells is initiated by reactive oxygen species. J Biol Chem. 2005;280:19911–19924. doi: 10.1074/jbc.M412443200. [DOI] [PubMed] [Google Scholar]
- 47.Dini L. Apoptosis induction in DU-145 human prostate carcinoma cells. Tissue Cell. 2005;37:379–384. doi: 10.1016/j.tice.2005.06.006. [DOI] [PubMed] [Google Scholar]
- 48.Bai J, Rodriguez AM, Melendez JA, Cederbaum AI. Overexpression of catalase in cytosolic or mitochondrial compartment protects HepG2 cells against oxidative injury. J Biol Chem. 1999;274:26217–26224. doi: 10.1074/jbc.274.37.26217. [DOI] [PubMed] [Google Scholar]
- 49.Chen QM, Liu J, Merrett JB. Apoptosis or senescence-like growth arrest: influence of cell-cycle position, p53, p21 and bax in H2O2 response of normal human fibroblasts. Biochem J. 2000;347:543–551. doi: 10.1042/0264-6021:3470543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Cheng WC, Berman SB, Ivanovska I, Jonas EA, Lee SJ, Chen Y, Kaczmarek LK, Pineda F, Hardwick JM. Mitochondrial factors with dual roles in death and survival. Oncogene. 2006;25:4697–4705. doi: 10.1038/sj.onc.1209596. [DOI] [PubMed] [Google Scholar]
- 51.Armstrong JS. The role of the mitochondrial permeability transition in cell death. Mitochondrion. 2006;6:225–234. doi: 10.1016/j.mito.2006.07.006. [DOI] [PubMed] [Google Scholar]