

Abstract
The most widely used modern mass spectrometers face severe performance limitations with molecules larger than a few kDa. For far larger biomolecules, a common practice has been to break these up chemically or enzymatically into fragments that are sufficiently small for the instrumentation available. With its many sophisticated recent enhancements, this “bottom-up” approach has proved highly valuable, such as for the rapid, routine identification and quantitation of DNA-predicted proteins in complex mixtures. Characterization of smaller molecules, however, has always measured the mass of the molecule and then that of its fragments. This “top-down” approach has been made possible for direct analysis of large biomolecules by the uniquely high (>105) mass resolving power and accuracy (~1 ppm) of the Fourier-transform mass spectrometer. For complex mixtures, isolation of a single component’s molecular ions for MS/MS not only gives biomolecule identifications of far higher reliability, but directly characterizes sequence errors and posttranslational modifications. Protein sizes amenable for current MS/MS instrumentation are increased by a “middle-down” approach in which limited proteolysis forms large (e.g., 10 kDa) polypeptides that are then subjected to the top-down approach, or by “prefolding dissociation”. The latter, which extends characterization to proteins >200 kDa, was made possible by greater understanding of how molecular ion tertiary structure evolves in the gas phase.
Introduction
Today, biomolecular research benefits greatly from the unique capabilities of modern mass spectrometry (MS). For protein and DNA/RNA identification and characterization by MS, two complementary strategies have evolved during the past 15 years: the “bottom-up” [1–6] and “top-down” [1, 5, 7–15] approaches. In the most commonly used bottom-up approach, the mixture of large biomolecules of interest is first digested in solution, and then their combined small (generally <3 kDa) products are analyzed by MS and MS/MS to yield molecular weight (Mr) and fragment mass values, respectively. For the top-down approach, on the other hand, a mixture of proteins is directly introduced into the mass spectrometer for determination of their Mr values. Molecular ions of an individual component can then be mass-isolated and dissociated (MS/MS) in the mass spectrometer, yielding product masses related directly to its Mr value. A component’s multiple isotopic peaks of far higher (even 50 kDa or greater) mass values require correspondingly high resolving power and accuracy, for which electrospray ionization (ESI) [16] and the relatively expensive Fourier-transform (FT) MS instrument [17, 18] have been generally used (the recently-introduced “Orbitrap” shows promising capabilities [19, 20]). Special software is also required, e.g., THRASH converts m/z values from multiply-charged ion to mass values and averages isotopic peak intensity values for higher signal/noise (S/N) [21], and ProSight uses these values to match those of DNA-predicted proteins and possible posttranslational modifications [22].
Ion dissociation for MS/MS (or MSn) generally has employed “threshold activation” methods with sequential activation events (e.g., energetic collisions, IR photon absorption) that energize the ion above its dissociation threshold and cleave the weakest bonds first. A newer method, “electron capture dissociation” (ECD) [23–27] suddenly adds a larger amount of energy (~5 eV) that cleaves the protein backbone bonds almost stochastically, thus providing far higher sequence coverage. Although ECD is mainly applicable to FTMS instruments, its recently developed off-shoot “electron-transfer dissociation” (ETD) by the Hunt research group [19, 20, 28, 29] makes a similar capability available on linear and 3D ion trap instruments used for the bottom-up approach. Citations of these original descriptions [23, 28] show that their adoption for both bottom-up and top-down proteomics has been particularly impressive (Figure 1).
Figure 1.

Cumulative citations of the first publication on ECD [23], ETD [28], and “top-down” [7], and the cumulative number of publications with “top-down” in the title. Ref. 23 is one of the most highly cited JACS papers of the last decade, while ref. 28 shows an even faster initial growth in number of citations. The values for 2007 cumulative use of “ECD” and “ETD” in titles are 243 and 56, respectively. Data are from SciFinder, American Chemical Society.
Another advantage of ECD/ETD is that only backbone bonds are cleaved, preserving side-chain modifications such as glycosylation and phosphorylation. However, even tertiary non-covalent bonds can be preserved, so that these must be cleaved separately using “activated ion” (AI) ECD for protein ions >~20 kDa to obtain fragmentation information. For ions larger than ~50 kDa, dissociation in the mass spectrometer has been ineffective using any gas-phase dissociation method, with only bottom-up applicable. The recently described “prefolding dissociation” (PFD) extends top-down capabilities (but not ECD) to proteins larger than 200 kDa [11].
Origins of top-down mass spectrometry
When J. J. Thomson described his first mass spectrometry instrument almost a century ago [30], he predicted that the measured masses of a molecule and its fragments would provide uniquely valuable structural information, e.g., the masses 12, 16, and 28 for carbon monoxide. The basic idea of top-down MS was also established more than a half century ago for electron ionization mass spectra, and this application has been extensively developed [31]. In pioneering research, Biemann showed that such MS data from peptides could provide unique sequence information [32]. For protein characterization, these basic principles were discussed [33] and applied in a series of papers [34–36] and the useful description “protein fragment ladder” was proposed [36]. However, the later “top-down” terminology [7] clearly differentiates this from the far more widely used “bottom-up” MS proteomics method, a major reason for the immediate popularity of such a designation (Figure 1). This is only new nomenclature, not new science, with its use in titles of scientific articles now surpassing the citation of the original reference.
Top-Down Methodology
Ionizing a molecule and dissociating its molecular ions in the mass spectrometer can provide mass data indicating the molecular weight and elemental composition of the molecule and of its pieces. Structural interpretation is greatly simplified for linear (noncyclic) molecules, as cleavage of only one bond is necessary to produce two pieces of the molecule. Rearranged product ions that complicate mass spectral interpretation [31] are usually minimal for MS/MS of linear proteins [37–39] and DNA/RNA [5, 12–14], and the extent of secondary fragmentation can be reduced by lowering the dissociation power or removing the products from further activation [40]. The primary product ions of linear proteins are complementary fragment ion pairs, each fragment containing one terminus of the molecule; thus if two product ion masses differ by the mass of an amino acid, there is a strong possibility that this residue is located that distance in mass from a terminus, although this could also arise by a coincidence in mass values of products containing different termini. In contrast, proteolysis in bottom-up produces peptides with no mass information concerning their location in the protein. For top-down, differences between the experimental Mr value and that of a DNA-predicted protein gives an immediate indication of the molecular heterogeneity and sequence errors for that protein assignment. Such mass differences can also indicate specific post-translational [37] or post-transcriptional [5] modifications (PTM, e.g. 79.9663 Da for phosphorylation, 14.0156 Da for methylation) [38, 41, 42]. Further, any complementary pair of fragment ions, those whose mass value total corresponds to that of the parent molecular ion, narrows the location(s) of modification, which can be furthered with MS3, etc. Such 100% sequence coverage is rare in the bottom-up approach.
Biomolecular samples typically are complex mixtures of compounds, and their purification becomes more difficult with increasing molecular size using conventional methods. The powerful capabilities of FTMS for separation by molecular weight can “purify” individual molecular ion species, such as the 1 Da resolution in the ESI mass spectrum of chondroitinases I and II, MW 111,713 and 112,508 [43]. MS/MS of such resolved molecular ions of a mixture was first demonstrated for small peptides [44, 45] and larger peptides [46], and then proteins [34–36] and DNA/RNA [12, 13]. As an example of the potential of this method, ESI of a copolymer mixture gave >130 molecular ion species with assigned elemental compositions [47]. The MS/MS spectra from the separate isolation of five of these components, each <1% relative abundance, clearly showed that the manufacturer’s structural assignment as block copolymers was inaccurate. ESI of a mixture of proteins isolated from Arabidopsis thaliana gave accurate Mr values for 14 separated molecular species [48], and MS/MS and MS3 on the MS separated molecular ions of components of ~5% abundance gave identifications and sequence corrections.
Top-down instrumentation
The extraction of extensive sequence information from the high-mass MS data relies on the separation and accurate mass assignment of all individual ion signals in the mass spectrum. Most top-down MS studies reported to date use FTMS instrumentation [17, 18, 49] because of its uniquely high resolving power and mass accuracy. More than 575 FT-ICR mass spectrometers have been installed worldwide, and this number continues to increase with the increasing demand for sensitive and accurate mass spectrometry measurements in biology and related disciplines. A front-end input capability for ion storage and mass separation, such as the linear trap quadrupole (LTQ), provides valuable additional control permitting software selection of specific MS and MS/MS experiments on a ~1 s timescale for fractions eluted from chromatographic separation [19, 37, 42]. Of further value, the LTQ can control the total number of charges of ions loaded into the FTMS cell to maximize the mass accuracy using external calibration.
Of additional high promise is the recently-introduced “Orbitrap” mass spectrometer [19, 20, 28, 29] with mass accuracy and resolving power only exceeded by FTMS. The Orbitrap does not need the expensive super-conducting magnet and liquid helium supply required for FTMS, important factors in its widespread current adoption. Proteins exceeding its resolving power can be subjected first to limited proteolysis/degradation, with the resulting large polypeptides then subjected individually to top-down MS and MS/MS [19, 20], the “middle-down” approach. The Orbitrap is also proving valuable for the bottom-up approach, for example providing peptide Mr values whose higher accuracy restricts the possible protein assignments.
Special software for reduction of high-resolution data and correlation of fragment masses with DNA-predicted sequences has also been critical to the efficient application of the top-down methodology. For large molecular ions, heavy isotopes of carbon and other elements produce multiple isotopic peaks with ~1 Da spacings. With [13C]/[12C] = 1.1%, the ions 12C100, 12C9913C1, 12C98, 13C2, and 12C97C13C3 will give isotopic peaks of relative abundances 100:110:60:22, with the most abundant peak one Da above the monoisotopic peak. For carbonic anhydrase, 29 kDa, the most abundant isotopic peak is 17 Da higher than the monoisotopic peak, and its abundance is vanishingly small [50]. For this multitude of peaks in such an “isotopic cluster”, the THRASH [21] program converts the m/z values of these spacings to m values and compares the relative abundance values of the isotopic cluster peaks to those predicted in order to assign the cluster’s monoisotopic mass value. THRASH also combines the abundances of the isotopic peaks, thus maximizing signal/noise for detection of that ion species. The ProSight algorithm [22] for proteins combines an extended THRASH version with software to match molecular and fragment mass values with those derived from DNA predicted amino acid sequences. The user can also suggest possible PTMs with which the program modifies mass values to find additional matches.
Methods for dissociation of multiply-charged biomolecular ions
MS/MS was originally accomplished by allowing metastable ions to dissociate in field-free regions of the mass spectrometer [44, 51, 52]. Later, it was found more efficient [53] to activate stable ions to above the dissociation threshold using methods such as collisionally activated dissociation (CAD) [40, 54] (also termed collision induced dissociation) [10, 55], infrared multiphoton dissociation (IRMPD) [56], or blackbody infrared dissociation (BIRD) [57]. For these “threshold” or “slow heating” [58] methods, the incremental activation by collisions or photons raises the internal energy slowly (μs, ms) to above the dissociation threshold for various bonds of the ion. Generally the first to dissociate are the noncovalent bonds of the tertiary structure (which produces no lower-mass fragment ions), followed by the weakest covalent bonds. Thus product ions from these cleavages are strongly favored in the CAD, IRMPD, or BIRD spectra of proteins, and dominate their metastable ion spectra.
A far larger proportion of interresidue bonds is cleaved using the related techniques of ECD [23–27, 38, 59–62] and ETD [19, 20, 28, 29]. In ECD, a low energy (≪1 eV) electron is captured by a multiply protonated protein, (M + nH)n+, to form a reduced (M + nH)(n−1)+• radical ion; for ETD the electron is supplied by an aromatic radical anion. The resulting charge recombination energy is so large (~5 eV) that it can cause backbone dissociation of the newly formed radical ion before this energy is randomized over the large ion (“nonergodic” dissociation). Backbone cleavage is dominant in ECD, so specific that there is negligible dissociation of side-chain and other covalent bonds of the protein ion, and even of its noncovalent bonds, as discussed further below. This ~5 eV energy gain also makes the <1 eV differences in interresidue bond dissociation energies have far less influence on product abundances. Thus ECD and ETD backbone cleavage is much less affected by the identity of the amino acids (except for proline, vide infra) framing the cleavage site, and in this way it provides more extensive sequence coverage and PTM location specificity [25, 59].
In contrast to the threshold methods such as CAD and IRMPD that produce amide bond cleavages (b, y product ions), with ECD and ETD the reduced molecular ions dissociate into complementary pairs of c and z• (~90%) or a• and y (~10%) fragment ions (Scheme 1) [25]. Proline has two N-Cα bonds, so that this ECD cleavage of only one results in no c and z• product ions. As another unique feature of the ECD mechanism advantageous for protein sequencing, fragment ions containing either the N- or C-terminus can be distinguished. The N-terminal fragment ions actually contain an extra hydrogen atom from the H+ neutralized by electron capture; the mass sum of the “complementary” c and z• pair is actually 1 Da greater than the Mr value. Thus if such a complementary c/z• pair is indicated by their mass sum, the N-terminal c fragment is identified directly by its +1 Da mass discrepancy. This N- or C-terminal identification can also be made in the same way if a single b or y ion from a threshold dissociation corresponds to a single c or z• ion by a mass difference corresponding to the –NH- difference in their formation mechanisms (Scheme 1). Such “golden complementary pairs” are valuable for the complete de novo sequencing of smaller proteins with ECD [39].
Scheme 1.

“Internal” fragment ions, those that contain neither of the protein termini, complicate spectral interpretation. These ions arise from two or more successive cleavages within the same precursor ion, which is much less probable for ECD/ETD than for CAD/IRMPD/BIRD. The latter methods energize all vibrational modes of the ion, so that a primary product ion may still be sufficiently energized for simultaneous or secondary dissociation, and the product ion could also undergo further excitation (although this is minimized with SORI-CAD [40]). Nonergodic ECD occurs before energy randomization, so that the primary excitation energy not expended in cleavage provides little relative increase to the internal energy of the ECD primary products when spread over their many vibrational modes. Further, electron capture is dependent on the square of the ion charge, reducing the probability of secondary ECD of lower charge fragment ions versus that of the protein ion. As a general strategy, more extensive and reliable sequence coverage is obtained from two separate MS/MS spectra, one with conditions that maximize the formation of different terminal fragment ions and the other with conditions that minimize the formation of internal fragment ions.
ECD of course requires positive, not negative ions, a limitation for its applicability to the analysis of acidic proteins or nucleic acids. Their net charge in solution is negative, resulting in significantly lower ion yields using ESI of the opposite polarity [63]. However, ECD of doubly-protonated nucleic acids [64] or CAD of multiply-charged protein anions [7] can provide useful structural information. An alternative to ECD is electron detachment dissociation (EDD) [61], which has been applied to negatively charged ions of polypeptides [65] and nucleic acids [66, 67]. In EDD, higher energy (>20 eV) electrons impact on multiply deprotonated proteins or nucleic acids, (M - nH)n−, to form (M - nH)(n−1)-• radical ions [65]. The dominant fragmentation channel in EDD of polypeptide anions is cleavage of the Cα-C=O backbone bond, yielding complementary a•/x ion pairs [65]. EDD of polypeptides can provide extensive sequence coverage and can be used for the analysis of PTMs [61]. Nucleic acid fragmentation in EDD is only beginning to be explored, and involves backbone cleavage with only minimal nucleobase loss [66].
ECD Retains Unstable PTM Side-Chains
MS/MS methods have been marginal for locating glycosylation, phosphorylation, etc. The covalent bonds connecting such side chains can be sufficiently weak that the common threshold dissociation methods such as CAD and IRMPD cleave them as well as the backbone bonds, destroying the PTM information. ECD, instead, preserves side-chain bonds while effecting near-exclusive cleavage of covalent protein backbone bonds (vide supra) [68–70]. The enzyme γ-glutamylcarboxylase, as studied by the Christopher Walsh group [69], modifies zymogens of serine proteases in the blood coagulation cascade. Mechanistic investigations were complicated by the fact that these zymogens contain up to 12 γ-carboxyglutamic acids within a protein section of 45 residues, and that both CAD and IRMPD produced complete loss of side-chain CO2. However, ECD retained the side chain while producing cleavages that directly localized this labile modification.
Activated Ion ECD
Adding electrons to cold protein ions larger than 20 kDa only reduces their charge state sequentially. Attempted ECD of the 32+ ions of a 49 kDa protein gave only (M + 32 H)25–31+ ions [71]. As described above, ECD can cleave protein backbone bonds without appreciably affecting the far weaker noncovalent bonds typical of compact tertiary ion structures. If the resulting fragment ion pairs are still joined by noncovalent bonds, ECD will have reduced their charge without noticeably changing the molecular ion mass [27, 62, 72]. In general, protein ions of increasing size have more complex tertiary structures, so that “activated ion” ECD is necessary for these. In AI-ECD, the protein ion’s internal energy is raised by energetic collisions with background gas or irradiation with IR photons to cause sufficient cleavage of the intramolecular noncovalent bonds of the ion’s tertiary structure; this can be done before, during, or after ECD [71]. Systematic ECD studies on cytochrome c [62] and ubiquitin [27] validated this approach; thermal unfolding of the gaseous 7+ ions of ubiquitin increases the extent of sequence information from 17% at 25 ºC to 77% at 155 ºC [27]. AI-ECD is required for proteins larger than ~20 kDa [38, 71], and can even delineate the transmembrane domains of integral membrane proteins [73]. For larger proteins, different ion activation conditions cleave different tertiary noncovalent bonds, yielding different ECD product ions. Combining 25 ECD spectra using a variety of ion activation conditions gave product ions from the cleavage of 250 of the 258 interresidue bonds of bovine carbonic anhydrase [74]. EDD of nucleic acids also appears to cleave covalent bonds without affecting the intramolecular noncovalent bonding [66], and AI-EDD of a 15-mer DNA using an infrared laser for ion activation increased the extent of sequence information from 0% to 47% [67].
However, even with techniques in top-down MS that effectively destroy the gas phase tertiary structure of smaller ions, the relative extent of sequence information generally decreases with increasing mass of the biomolecular ions [10] (Figure 2). AI-ECD of larger (>250 residues) multiply protonated protein ions gives extensive sequence information from the terminal protein regions, but coverage in the central region is typically poor [38, 71]. As the cross section of an ion for electron capture is proportional to the square of the charge, additional electron irradiation to maximize molecular ion dissociation causes the larger, more highly charged fragment ions to become favored for secondary dissociation. This provides improved sequence coverage at the ends of the protein at the expense of coverage in its center. For proteins larger than ~50 kDa (Figure 2), special proteolysis can be employed that yields far larger polypeptides, followed by their individual top-down characterization [75–77], the “middle-down” approach. “Prefolding dissociation” (vide infra) can be used [11].
Figure 2.

Extent of sequence information obtained in protein ECD (open squares) and AI-ECD (filled squares) experiments versus the number of the protein’s residues (data from reference [71]); lines are meant to guide the eye.
The “plasma” ECD method [78] attempts to maximize the extent of AI unfolding for ECD while minimizing the extent of secondary electron capture. From opposing entrances, electrons and ions enter the pressurized FTMS cell, both through high (~+10 eV) cation trapping potentials. The ions are accelerated to the grounded cell center, with collisions denaturing the ion conformation. After the cell center the ions are slowed and then reversed and accelerated by the other trapping potential. This potential accelerates electrons approaching the cell, but on entrance the electrons are retarded electrostatically and by collisions. Now an electron that is slowing down can be traveling in the same direction and in the same path as an accelerating ion. When one overtakes the other, the velocity difference of this electron/ion pair will be minimum, maximizing the cross-section for ECD. ECD will yield product ions, however, of different velocities, reducing the probability that they will capture another electron; most electrons have similar velocity gradient pathways [78]. A single such spectrum of carbonic anhydrase (259 residues) gave 512 m/z values, corresponding to 183 interresidue cleavages. The missed 75 cleavages are in the center of the protein, indicating that the larger product ions representing these missing cleavages have undergone secondary dissociation. Plasma ECD using lower electron currents produced more of these large product ions, but found fewer total cleavages. Note, however, that MS/MS with this version of plasma ECD requires mass selection exterior to the FTMS ion cell. A technique in which the precursor ions are mass selected within the FTMS cell and then accelerated along its magnetic axis to effect plasma ECD with incoming electrons has not yet been developed.
Charge Density Effects
Consistent with the H+ reduction mechanism (Scheme 1), increasing the number of positive charges on a protein molecular ion increases, on average, the number of ECD cleavages. For example, increasing the ubiquitin ion’s charge from 5+ to 13+ increases sequence coverage from ECD under standard conditions at 25 ºC from 0% to 72% [27]. The charges per residue ratio of electrosprayed protein ions is somewhat dependent on composition, e.g., increasing with an increasing proportion of basic residues and decreasing for integral residue proteins [73]. In general, the maximum charge state of peptide and protein ions electrosprayed from denaturing solutions increases almost linearly with the number of residues [79], with a slope of ~0.15 charges per residue (Figure 3). However, the width of the charge state distribution also increases with increasing number of residues [11, 55] to an extent that the average charge actually decreases with increasing number of residues. In other words, larger protein ions from ESI carry, on average, fewer charges per residue than smaller protein ions. In general, the larger the protein ion, the more complex can be its tertiary structure; a greater proportion of its conformers have more complex folded regions that accommodate fewer charges. Williams and co-workers [80] have found electrospray conditions that increase protein cation charge densities. Further, if sample amount is not limiting, lower charge states can be ejected from the cell during ESI to raise the proportion of higher charge states.
Figure 3.
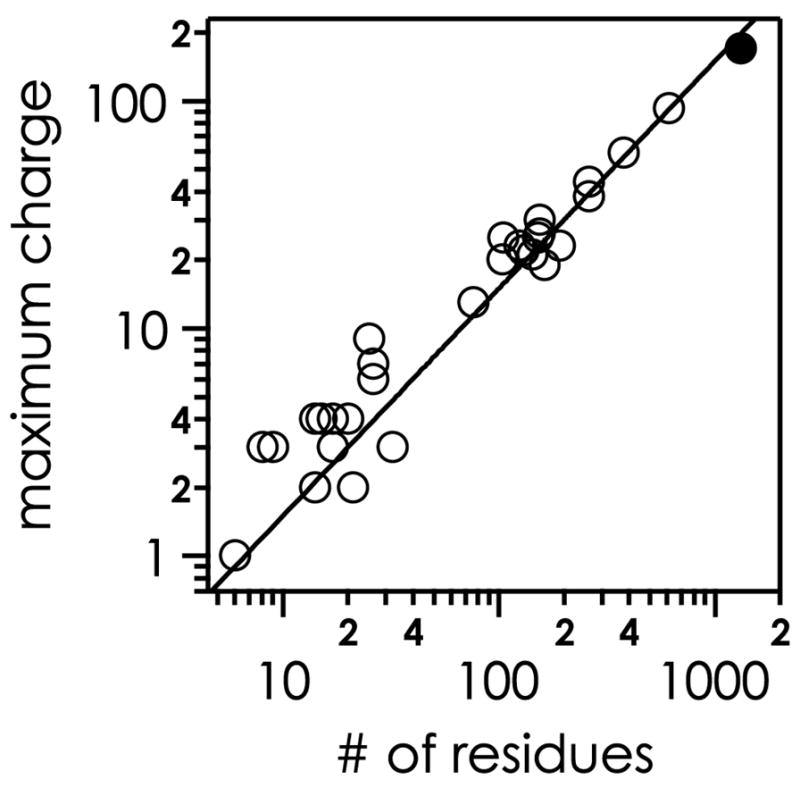
Maximum charge state of peptide and protein ions electrosprayed from denaturing solutions (~50% methanol or acetonitrile) versus the number of protein residues; open circles from reference [79], filled circles from reference [11].
Unfolding of a region during or after desolvation after electrospray can also produce different charge densities within the protein ion, the phenomenon of “asymmetric charge partitioning” [81]. An unfolded region with newly-exposed basic residues can become more highly charged by attracting protons from more compact regions. Evidence from native electron capture dissociation (NECD) [82–84] shows that desolvation during ESI causes stepwise unfolding of the protein in the gas phase followed by intramolecular proton transfer to the newly unfolded region. Each unfolding leaves the more compact protein region more depleted of charge, which renders its subsequent unfolding even more difficult because of reduced Coulombic repulsion. For example, the unfolding enthalpies of the 6+ and 9+ ions of the 76-residue protein ubiquitin are 30 and 6 kJ/mol, respectively, consistent with the loss of ECD sequence information with decreasing the ubiquitin ion’s charge from 13+ to 5+ [27].
Gaseous Protein Ion Conformations
A basic structure of these protein ions subjected to MS/MS is now indicated by a variety of experiments, in particular by H/D exchange [85–88], ion mobility cross sections [89–92], ECD spectra, and infrared photodissociation (IRPD) spectra [88, 93, 94]. For gaseous ubiquitin ions, the number of D atoms exchanged to near-completion by D2O actually decreases from ~70 for 7+ ions to ~48 for 12+ ions; the latter also exhibiting a conformer of ~24 D exchange [95]. The 13+ ions, which contain a proton at each basic site, exchange only ~15 D [87, 88]. Initial ion mobility studies of the Clemmer group [89, 92] showed an ion cross section value of 13+ ubiquitin ions consistent with that of an α-helix. The 10+ - 13+ ion values of 1700–2100 Ǻ2 are classified as “elongated”, the 8+ and 9+ ion values of 1100–1500 Ǻ2 as “partially folded”, and the 5+ - 7+ ions of ~1000 Ǻ2 as “compact”. These descriptions generally agree with conformers proposed from ECD experiments [27, 88, 94]. The 13+ conformer is indicated to be an α-helix in which the protonated side chains are solvated back into the helix to charge and stabilize its lengthwise H-bonding network; this also is consistent with D-substitution almost exclusively at the 13 protonated sites [87, 88]. ECD data indicate that this stiffened 13+ helix bends for 12+ - 10+ conformers at four sites of H+ loss. For 9+, 8+ conformers, the helix C-terminal region folds over onto itself to form side-by-side H-bonded helical regions with dipole-dipole stabilization, while further proton loss allows an additional N-terminal fold-over to form a 3-helix bundle (7+, 6+ conformers). Consistent with this general picture, IRPD spectra [94] indicate unusually strong hydrogen-bonding of nearly all of the gaseous protein N-H and O-H groups, with surprising spectral similarities for different proteins and for different charge states and temperatures.
Prefolding Dissociation
Activation of larger (1314, 1714, and 2153 residues) protein ions by IRMPD in the trapped ion cell of a FT-ICR instrument produced no backbone cleavages, even under conditions (27 W CO2-laser power, 80 ms irradiation time) far more strenuous than those required for the complete dissociation of smaller proteins [11]. However, NECD of cytochrome c dimer ions showed that removal of the aqueous solvent in ESI first causes denaturing of one monomer, with breakage of hydrophobic bonds (i.e., those formed from “fear” of water) in a first step [82–84]. Moreover, unfolding of the solution structure in the gas phase was found to occur in essentially the reversed order of the unfolding in solution, meaning that the solution structure is thermodynamically unstable in the gas phase [72]. Several experiments indicate that this unfolding is followed by the rapid formation of stable gas phase structures; possibly the loss of hydrophobic bonding exposes hydrophilic sites that immediately can participate in forming new electrostatic bonds [11, 95]. For ubiquitin 7+ ions stored in the FTMS cell, fast (<0.2 s) denaturing with an IR laser pulse showed a variety of refolding pathways occurring in 1 s to >10 min [27].
Fortunately, such longer folding times allow special techniques to interfere and compete with this process before the ion becomes too stable, thus allowing “prefolding dissociation” (PFD) that extends the upper limit of top-down MS to proteins larger than 200 kDa [11]. The unfolding of the solution conformers’ hydrophobic bonding starts in the capillary that introduces electrosprayed ions into the mass spectrometer vacuum system; thus heating this capillary is a first technique to delay any fast folding. Just after the capillary and before the skimmer is a high pressure region in which the newly desolvated ions undergo many collisions (nozzle-skimmer dissociation [34]), so that increased accelerating voltage here also delays refolding by increasing the proportion of energetic collisions. In the lower pressure post-skimmer region (only used for PFD), far higher energy collisions can cause backbone CAD before refolding prevents dissociation. Additives such as ammonium tartrate in the ESI solution gave a surprising additional 50% in backbone cleavages, possibly by replacing water molecules adducted to hydrophilic protein sites and thus delaying the fast folding.
This successive activation approach immediately following desolvation takes advantage of the phase transition of the ions from solution to gas phase, by which the order of local protein stability is altered [72, 83, 84]. The change in local stability is the driving force for conformational rearrangements, apparently with transient structures that are locally unfolded. Various activation conditions during the phase transition apparently produce differences in this unfolding-refolding process, as different activations yield fragmentation in different local regions. For a 1314-residue protein for which 100 backbone cleavages were the most produced in a single PFD spectrum, PFD techniques varied in 21 spectra produced 287 backbone cleavages. These data revealed a posttranslational modification that removed the N-terminal Met, correcting the molecular weight prediction from 143,635 to 143,504, in agreement with the measured value of 143,500 ± 23 [11]. However, these extended activation conditions produced no dissociation in the ~800-residue central region of the protein. Apparently, increasing the gaseous protein ion’s internal energy causes preferential unfolding of the terminal regions away from the center while leaving the rest relatively intact, a “ball of spaghetti” structure [11].
A disadvantage of PFD [11, 95] is that its backbone cleavages are effected by collisional activation in the ESI source region, which generally favors threshold-energy dissociation channels [55]. Although PFD is thus applicable to the analysis of stable PTMs (e.g., methylation, oxidation), it is less useful for the characterization of labile PTMs (e.g., phosphorylation, glycosylation). For a 200 kDa protein with ~7% glycosylation, >99.9% of the latter was removed by PFD [11]; however, the resulting PFD spectrum made possible SH versus S-S bond assignments to all of its 27 Cys residues. A way out of the dilemma that extensive backbone fragmentation and sequence coverage requires ion internal energies far too high to retain the labile PTMs would be to effect backbone cleavage by a separate dissociation method that is not based on slow ion activation. Reactions with electrons (ECD/ETD) could dissociate the denatured ends without PTM loss. Further, ECD could also cleave the protein internally, with the strenuous CAD then forcing out and dissociating these new chain ends.
Conclusions
Although the amazingly high resolving power and MS/MS capabilities of FTMS have made possible exciting new applications for characterizing complex petroleum samples [96], the great majority of the ~575 FTMS instruments now available are in proteomics laboratories. Most of these instruments are producing uniquely useful research and protein characterization data, yet relatively few are used in extensive top-down applications, especially those with automated introduction of separated (e.g., LC, CE) fractions of complex protein mixtures, online data reduction to select molecular ions for CAD or ECD, direct PTM characterization, etc. [42, 97]. Thus the impressive Figure 1 growth curve for the top-down methodology does not appear to be limited by the equipment costs involved, which are still attractive versus investments for modern molecular characterization techniques such as NMR, imaging, and synchrotron radiation. With the even more impressive current research in top-down fundamentals and applications from many highly capable laboratories, top-down should continue to become an increasingly important approach for biomolecule identification and characterization.
Acknowledgments
We thank Tadhg Begley, Barry Carpenter, Giuseppe Infusini, Neil Kelleher, Xianglei Kong, and Harold Scheraga for helpful discussions and cooperation, and the General Medical Institute of the National Institutes of Health (grant GM 16609) for generous financial support.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Chait BT. Mass Spectrometry: Bottom-up or Top-down? Science. 2006;314:65–66. doi: 10.1126/science.1133987. [DOI] [PubMed] [Google Scholar]
- 2.Aebersold R, Mann M. Mass Spectrometry-based Proteomics. Nature. 2003;422:198–207. doi: 10.1038/nature01511. [DOI] [PubMed] [Google Scholar]
- 3.Henzel WJ, Watanabe C, Stults JT. Protein Identification: the Origins of Peptide Mass Fingerprinting. J Am Soc Mass Spectrom. 2003;14:931–942. doi: 10.1016/S1044-0305(03)00214-9. [DOI] [PubMed] [Google Scholar]
- 4.McDonald WH, Yates JR., 3rd Shotgun Proteomics: Integrating Technologies to Answer Biological Questions. Curr Opin Mol Ther. 2003;5:302–309. [PubMed] [Google Scholar]
- 5.Meng Z, Limbach PA. Mass Spectrometry of RNA: Linking the Genome to the Proteome. Brief Funct Genomic Proteomic. 2006;5:87–95. doi: 10.1093/bfgp/ell012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hossain M, Limbach PA. Mass Spectrometry-based Detection of Transfer RNAs by Their Signature Endonuclease Digestion Products. RNA. 2007;13:295–303. doi: 10.1261/rna.272507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kelleher NL, Lin HY, Valaskovic GA, Aaserud DJ, Fridriksson EK, McLafferty FW. Top-down versus Bottom-up Protein Characterization by Tandem High-resolution Mass Spectrometry. J Am Chem Soc. 1999;121:806–812. [Google Scholar]
- 8.McLafferty FW, Fridriksson EK, Horn DM, Lewis MA, Zubarev RA. Biomolecule Mass Spectrometry. Science. 1999;284:1289–1290. doi: 10.1126/science.284.5418.1289. [DOI] [PubMed] [Google Scholar]
- 9.Reid GE, McLuckey SA. ‘Top-down’ Protein Characterization via Tandem Mass Spectrometry. J Mass Spectrom. 2002;37:663–675. doi: 10.1002/jms.346. [DOI] [PubMed] [Google Scholar]
- 10.Bogdanov B, Smith RD. Proteomics by FT-ICR Mass Spectrometry: Top-down and Bottom-up. Mass Spectrom Rev. 2005;24:168–200. doi: 10.1002/mas.20015. [DOI] [PubMed] [Google Scholar]
- 11.Han X, Jin M, Breuker K, McLafferty FW. Extending Top-down Mass Spectrometry to Proteins with Masses Greater than 200 Kilodaltons. Science. 2006;314:109–112. doi: 10.1126/science.1128868. [DOI] [PubMed] [Google Scholar]
- 12.Little DP, Aaserud DJ, Valaskovic GA, McLafferty FW. Sequence Information from 42–108mer DNAs (Complete for a 50mer) by Tandem Mass Spectrometry. J Am Chem Soc. 1996;118:9352–9359. [Google Scholar]
- 13.McLafferty FW, Aaserud DJ, Guan Z, Little DP, Kelleher NL. Double Stranded DNA Sequencing by Tandem Mass Spectrometry. Int J Mass Spectrom Ion Processes. 1997;165/166:457–466. [Google Scholar]
- 14.Hofstadler SA, Sannes-Lowery KA, Hannis JC. Analysis of Nucleic Aids by FTICR MS. Mass Spectrom Rev. 2005;24:265–285. doi: 10.1002/mas.20016. [DOI] [PubMed] [Google Scholar]
- 15.McLafferty FW, Breuker K, Jin M, Han X, Infusini G, Jiang H, Kong X, Begley TP. Top-down MS, a Powerful Complement to the High Capabilities of Proteolysis Proteomics. FEBS J. 2007;274:6256–6268. doi: 10.1111/j.1742-4658.2007.06147.x. [DOI] [PubMed] [Google Scholar]
- 16.Fenn JB, Mann M, Meng CK, Wong SF, Whitehouse CM. Electrospray Ionization for Mass Spectrometry of Large Biomolecules. Science. 1989;246:64–71. doi: 10.1126/science.2675315. [DOI] [PubMed] [Google Scholar]
- 17.Comisarov MB, Marshall AG. Fourier Transform Ion Cyclotron Resonance Spectroscopy. Chem Phys Lett. 1974;25:282–283. [Google Scholar]
- 18.Marshall AG, Hendrickson CL, Jackson GS. Fourier Transform Ion Cyclotron Mass Spectrometry: A Primer. Mass Spectrom Rev. 1998;17:1–35. doi: 10.1002/(SICI)1098-2787(1998)17:1<1::AID-MAS1>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- 19.McAlister GC, Phanstiel D, Good DM, Berggren WT, Coon JJ. Implementation of Electron-transfer Dissociation on a Hybrid Linear Ion Trap-Orbitrap Mass Spectrometer. Anal Chem. 2007;79:35253534. doi: 10.1021/ac070020k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Phanstiel D, Brumbaugh J, Berggren WT, Conard K, Feng X, Levenstein ME, McAlister GC, Thomson JA, Coon JJ. Mass Spectrometry Identifies and Quantifies 74 Unique Histone H4 Isoforms in Differentiating Human Embryonic Stem Cells. Proc Natl Acad Sci USA. 2008;105:4093–4098. doi: 10.1073/pnas.0710515105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Horn DM, Zubarev RA, McLafferty FW. Automated Reduction and Interpretation of High Resolution Electrospray Mass Spectra of Large Molecules. J Am Soc Mass Spectrom. 2000;11:320–332. doi: 10.1016/s1044-0305(99)00157-9. [DOI] [PubMed] [Google Scholar]
- 22.Zamdborg L, LeDuc RD, Glowacz KJ, Kim YB, Viswanathan V, Spaulding IT, Early BP, Bluhm EJ, Babai S, Kelleher NL. ProSight PTM 2.0: Improved Protein Identification and Characterization for Top Down Mass Spectrometry. Nucleic Acids Res. 2007;35:701–706. doi: 10.1093/nar/gkm371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zubarev RA, Kelleher NL, McLafferty FW. Electron Capture Dissociation of Multiply Charged Protein Cations. A Nonergodic Process. J Am Chem Soc. 1998;120:3265–3266. [Google Scholar]
- 24.Kruger NA, Zubarev RA, Horn DM, McLafferty FW. Electron Capture Dissociation of Multiply Charged Peptide Cations. Int J Mass Spectrom. 1999;185/186/187:787–793. [Google Scholar]
- 25.Zubarev RA, Horn DM, Fridriksson EK, Kelleher NL, Kruger NA, Lewis MA, Carpenter BK, McLafferty FW. Electron Capture Dissociation for Structural Characterization of Multiply Charged Protein Cations. Anal Chem. 2000;72:563–573. doi: 10.1021/ac990811p. [DOI] [PubMed] [Google Scholar]
- 26.McLafferty FW, Horn DM, Breuker K, Ge Y, Lewis MA, Cerda B, Zubarev RA, Carpenter BK. Electron Capture Dissociation of Gaseous Multiply Charged Ions by Fourier Transform Ion Cyclotron resonance. J Am Soc Mass Spectrom. 2001;12:245–249. doi: 10.1016/S1044-0305(00)00223-3. [DOI] [PubMed] [Google Scholar]
- 27.Breuker K, Oh HB, Horn DM, Cerda BA, McLafferty FW. Detailed Unfolding and Folding of Gaseous Ubiquitin Ions Characterized by Electron Capture Dissociation. J Am Chem Soc. 2002;124:6407–6420. doi: 10.1021/ja012267j. [DOI] [PubMed] [Google Scholar]
- 28.Syka JEP, Coon JJ, Schroeder MJ, Shabanowitz J, Hunt DF. Peptide and Protein Sequence Analysis by Electron Transfer Dissociation Mass Spectrometry. Proc Natl Acad Sci USA. 2004;101:9528–9533. doi: 10.1073/pnas.0402700101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Coon JJ, Ueberheide B, Syka JEP, Dryhurst DD, Ausio J, Shabanowitz J, Hunt DF. Protein Identification Using Sequential Ion/ion Reactions and Tandem Mass Spectrometry. Proc Natl Acad Sci USA. 2005;102:9463–9468. doi: 10.1073/pnas.0503189102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Thomson JJ. Rays of Positive Electricity and their Application to Chemical Analysis. Longmans, Green; London: 1913. [Google Scholar]
- 31.McLafferty FW, Turecek F. Interpretation of Mass Spectra. 4. University Science Books; Mill Valley, CA: 1993. [Google Scholar]
- 32.Biemann K. Mass Spectrometry Organic Chemical Applications. McGraw-Hill; New York: 1962. pp. 260–296. [Google Scholar]
- 33.Henry KD, Williams ER, Wang B-H, McLafferty FW, Shabanowitz J, Hunt DF. Fourier-Transform Mass Spectrometry of Large Molecules by Electrospray Ionization. Proc Natl Acad Sciences USA. 1989;86:9075–9078. doi: 10.1073/pnas.86.23.9075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Loo JH, Edmonds CG, Smith RD. Primary Sequence Information from Intact Proteins by Electrospray Ionization Tandem Mass Spectrometry. Science. 1990;248:201–204. doi: 10.1126/science.2326633. [DOI] [PubMed] [Google Scholar]
- 35.Loo JA, Quinn JP, Ryu SI, Henry KD, Senko MW, McLafferty FW. High-Resolution Tandem Mass Spectrometry of Large Biomolecules. Proc Natl Acad Sci USA. 1992;89:286–289. doi: 10.1073/pnas.89.1.286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chait BT, Wang R, Beavis RC, Kent SBH. Protein Ladder Sequencing. Science. 1993;262:89–92. doi: 10.1126/science.8211132. [DOI] [PubMed] [Google Scholar]
- 37.Meng F, Forbes AJ, Miller LM, Kelleher NL. Detections and Localization of Protein Modifications by High Resolution Tandem Mass Spectrometry. Mass Spectrom Rev. 2005;24:126–134. doi: 10.1002/mas.20009. [DOI] [PubMed] [Google Scholar]
- 38.Ge Y, Lawhorn BG, ElNaggar M, Strauss E, Park JH, Begley TP, McLafferty FW. Top Down Characterization of Larger Proteins (45 kDa) by Electron Capture Dissociation Mass Spectrometry. J Am Chem Soc. 2002;124:672–678. doi: 10.1021/ja011335z. [DOI] [PubMed] [Google Scholar]
- 39.Horn DM, Zubarev RA, McLafferty FW. Automated de Novo Sequencing of Proteins by Tandem High-Resolution Mass Spectrometry. Proc Natl Acad Sci USA. 2000;97:10313–10317. doi: 10.1073/pnas.97.19.10313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gauthier JW, Trautman TR, Jacobson DB. Sustained Off-resonance Irradiation for Collision-activated Dissociation Involving Fourier Transform Mass Spectrometry. Collision-activated Dissociation Technique that Emulates Infrared Multiphoton Dissociation. Anal Chim Acta. 1991;246:211–225. [Google Scholar]
- 41.Garcia BA, Joshi S, Thomas CE, Chitta RK, Diaz RL, Busby SA, Andrews PC, Ogorzalek-Loo RR, Shabanowitz J, Kelleher NL, Mizzen CA, Allis CD, Hunt DF. Comprehensive Phosphoprotein Analysis of Linker Histone H1 from Tetrahymena thermophila. Mol Cell Proteomics. 2006;5:1593–1609. doi: 10.1074/mcp.M600086-MCP200. [DOI] [PubMed] [Google Scholar]
- 42.Jiang L, Smith JN, Anderson SL, Ma P, Mizzen CA, Kelleher NL. Global Assessment of Combinatorial Post-translational Modification of Core Histones in Yeast Using Contemporary Mass Spectrometry. LYS4 Trimethylation Correlates with Degree of Acetylation on the Same H3 Tail. J Biol Chem. 2007;21:27923–27934. doi: 10.1074/jbc.M704194200. [DOI] [PubMed] [Google Scholar]
- 43.Kelleher NL, Senko MW, Siegel MW, McLafferty FW. Unit Resolution Mass Spectra of 112 kDa Molecules with 3 Da Accuracy. J Am Soc Mass Spectrom. 1997;8:380–383. [Google Scholar]
- 44.McLafferty FW, Venkataraghavan R, Irving P. Determination of Amino Acid Sequences in Peptide Mixtures by Mass Spectrometry. Biochem Biophys Res Commun. 1970;39:274–278. doi: 10.1016/0006-291x(70)90789-8. [DOI] [PubMed] [Google Scholar]
- 45.Wipf H-K, Irving P, McCamish M, Venkataraghavan R, McLafferty FW. Mass Spectrometric Studies of Peptides. V. Determination of Amino Acid Sequences in Peptide Mixtures by Mass Spectrometry. J Am Chem Soc. 1973;95:3369–3375. doi: 10.1021/ja00791a048. [DOI] [PubMed] [Google Scholar]
- 46.Williams ER, Furlong JJP, McLafferty FW. Efficiency of Collisionally Activated Dissociation and 193 nm Photodissociation of Peptide Ions in Fourier-Transform Mass Spectrometry. J Am Soc Mass Spectrom. 1990;1:288–294. doi: 10.1016/1044-0305(90)85003-5. [DOI] [PubMed] [Google Scholar]
- 47.Cerda BA, Horn DM, Breuker K, McLafferty FW. Sequencing of Specific Copolymer Oligomers by Electron-Capture-Dissociation Mass Spectrometry. J Am Chem Soc. 2002;124:9287–9291. doi: 10.1021/ja0123756. [DOI] [PubMed] [Google Scholar]
- 48.Zabrouskov V, Giacomelli L, van Wijk KJ, McLafferty FW. A New Approach for Plant Proteomics. Characterization of Chloroplast Proteins of Arabidopsis thaliana by Top-Down Mass Spectrometry. Mol Cell Proteomics. 2003;2:1253–1260. doi: 10.1074/mcp.M300069-MCP200. [DOI] [PubMed] [Google Scholar]
- 49.Marshall AG, Hendrickson CL, Shi SDH. Scaling MS Plateaus with High-resolution FT-ICRMS. Anal Chem. 2002;74:252A–259A. doi: 10.1021/ac022010j. [DOI] [PubMed] [Google Scholar]
- 50.McLafferty FW. High-Resolution Tandem FT Mass Spectrometry Above 10 kDa. Acc Chem Res. 1994;27:379–386. [Google Scholar]
- 51.Beynon JH. Mass Spectrometry and Its Applications to Organic Chemistry. Elsevier; Amsterdam: 1960. pp. 251–262. [Google Scholar]
- 52.Shannon TW, McLafferty FW. Identification of Gaseous Organic Ions by the Use of “Metastable Peaks”. J Am Chem Soc. 1966;88:5021–5022. [Google Scholar]
- 53.Haddon WF, McLafferty FW. Metastable Ion Characteristics. VII. Collision Induced Metastables. J Am Chem Soc. 1968;90:4745–4746. [Google Scholar]
- 54.Senko MW, Speir JP, McLafferty FW. Collisional Activation of Large Multiply Charged Ions Using Fourier Transform Mass Spectrometry. Anal Chem. 1994;66:2801–2808. doi: 10.1021/ac00090a003. [DOI] [PubMed] [Google Scholar]
- 55.Gabelica V, de Pauw ED. Internal Energy and Fragmentation of Ions Produced in Electrospray Sources. Mass Spectrom Rev. 2005;24:566–587. doi: 10.1002/mas.20027. [DOI] [PubMed] [Google Scholar]
- 56.Little DP, Speir JP, Senko MW, O’Connor PB, McLafferty FW. Infrared Multiphoton Dissociation of Large Multiply-Charged Ions for Biomolecule Sequencing. Anal Chem. 1994;66:2809–2815. doi: 10.1021/ac00090a004. [DOI] [PubMed] [Google Scholar]
- 57.Price WD, Schnier PD, Williams ER. Tandem Mass Spectrometry of Large Biomolecule Ions by Blackbody Infrared Radiative Dissociation. Anal Chem. 1996;68:859–866. doi: 10.1021/ac951038a. [DOI] [PubMed] [Google Scholar]
- 58.McLuckey SA, Goeringer DE. Slow Heating Methods in Tandem Mass Spectrometry. J Mass Spectrom. 1997;32:461–474. [Google Scholar]
- 59.Kruger NA, Zubarev RA, Carpenter BK, Kelleher NL, Horn DM, McLafferty FW. Electron Capture versus Energetic Dissociation of Protein Ions. Int J Mass Spectrom. 1999;182/183:1–5. [Google Scholar]
- 60.Cooper HJ, Håkansson K, Marshall AG. The Role of Electron Capture Dissociation in Biomolecular Analysis. Mass Spectrom Rev. 2005;24:201–222. doi: 10.1002/mas.20014. [DOI] [PubMed] [Google Scholar]
- 61.Zubarev RA. Reactions of Polypeptide Ions with Electrons in the Gas Phase. Mass Spectrom Rev. 2003;22:57–77. doi: 10.1002/mas.10042. [DOI] [PubMed] [Google Scholar]
- 62.Horn DM, Breuker K, Frank AJ, McLafferty FW. Kinetic Intermediates in the Folding of Gaseous Protein Ions Characterized by Electron Capture Dissociation Mass Spectrometry. J Am Chem Soc. 2001;123:9792–9799. doi: 10.1021/ja003143u. [DOI] [PubMed] [Google Scholar]
- 63.Pan P, Gunawardena HP, Xia Y, McLuckey SA. Nanoelectrospray Ionization of Protein Mixtures: Solution pH and Protein pI. Anal Chem. 2004;76:1165–1174. doi: 10.1021/ac035209k. [DOI] [PubMed] [Google Scholar]
- 64.Håkansson K, Hudgins RR, Marshall AG, O’Hair RAJ. Electron Capture Dissociation and Infrared Multiphoton Dissociation of Oligodeoxynucleotide Dications. J Am Soc Mass Spectrom. 2003;14:23–41. doi: 10.1016/S1044-0305(02)00708-0. [DOI] [PubMed] [Google Scholar]
- 65.Kjeldsen F, Silivra OA, Ivonin IA, Haselmann KF, Gorshkov M, Zubarev RA. Cα-C Backbone Fragmentation Dominates in Electron Detachment Dissociation of Gas Phase Polypeptide Polyanions. Chem Eur J. 2005;11:1803–1812. doi: 10.1002/chem.200400806. [DOI] [PubMed] [Google Scholar]
- 66.Yang J, Mo J, Adamson JT, Håkansson K. Characterization of Oligodeoxynucleotides by Electron Detachment Dissociation Fourier Transform Ion Cyclotron Resonance Mass Spectrometry. Anal Chem. 2005;77:1876–1882. doi: 10.1021/ac048415g. [DOI] [PubMed] [Google Scholar]
- 67.Mo J, Håkansson K. Characterization of Nucleic Acid Higher Order Structure by High Resolution Tandem Mass Spectrometry. Anal Bioanal Chem. 2006;386:675–681. doi: 10.1007/s00216-006-0614-z. [DOI] [PubMed] [Google Scholar]
- 68.Mirgorodskaya E, Roepstorff P, Zubarev RA. Localization of O-glycosylation Sites in Peptides by Electron Capture Dissociation in a Fourier Transform Mass Spectrometer. Anal Chem. 1999;71:4431–4436. doi: 10.1021/ac990578v. [DOI] [PubMed] [Google Scholar]
- 69.Kelleher NL, Zubarev RA, Bush K, Furie B, Furie BC, McLafferty FW, Walsh CT. Localization of Labile Posttranslational Modifications by Electron Capture Dissociation: The Case of γ-carboxyglutamic Acid. Anal Chem. 1999;71:4250–4253. doi: 10.1021/ac990684x. [DOI] [PubMed] [Google Scholar]
- 70.Shi SDH, Hemling ME, Carr SA, Horn DM, Lindh IF, McLafferty FW. Phosphopeptide/phosphoprotein Mapping by Electron Capture Dissociation Mass Spectrometry. Anal Chem. 2001;73:19–22. doi: 10.1021/ac000703z. [DOI] [PubMed] [Google Scholar]
- 71.Horn DM, Ge Y, McLafferty FW. Activated Ion Electron Capture Dissociation for Mass Spectral Sequencing of Larger (42 kDa) Proteins. Anal Chem. 2000;72:4778–4784. doi: 10.1021/ac000494i. [DOI] [PubMed] [Google Scholar]
- 72.Breuker K. Protein Structure and Folding in the Gas Phase: Ubiquitin and Cytochrome c. In: Laskin J, Lifshitz C, editors. Principles of Mass Spectrometry Applied to Biomolecules. John Wiley & Sons, Inc.; Hoboken, NJ: 2006. p. 177. [Google Scholar]
- 73.Zabrouskov V, Whitelegge J. Increased Coverage in the Transmembrane Domain with Activated-Ion Electron Capture Dissociation for Top-Down Fourier-Transform Mass Spectrometry of Integral Membrane Proteins. J Proteome Res. 2007;6:2205–2210. doi: 10.1021/pr0607031. [DOI] [PubMed] [Google Scholar]
- 74.Sze SK, Ge Y, Oh HB, McLafferty FW. Top Down Mass Spectrometry of a 29 kDa Protein for Characterization of Any Posttranslational Modification to within One Residue. Proc Natl Acad Sci USA. 2002;99:1774–1779. doi: 10.1073/pnas.251691898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Demirev PA, Ramirez J, Fenselau C. Tandem Mass Spectrometry of Intact Proteins for Characterization of Biomarkers from Bacillus cereus T Spores. Anal Chem. 2001;73:5725–5731. doi: 10.1021/ac010672n. [DOI] [PubMed] [Google Scholar]
- 76.Forbes AJ, Mazur M, Patel HM, Walsh CT, Kelleher NL. Toward Efficient Analysis of >70 kDa Proteins with 100% Sequence Coverage. Proteomics. 2001;1:927–933. doi: 10.1002/1615-9861(200108)1:8<927::AID-PROT927>3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]
- 77.Garcia BA, Pesavento JJ, Mizzen CA, Kelleher NL. Pervasive Combinatorial Modification of Histone H3 in Human Cells. Nat Methods. 2007;4:487–489. doi: 10.1038/nmeth1052. [DOI] [PubMed] [Google Scholar]
- 78.Sze SK, Ge Y, Oh HB, McLafferty FW. Plasma Electron Capture Dissociation for the Characterization of Large Proteins by Top Down Mass Spectrometry. Anal Chem. 2003;75:1599–1603. doi: 10.1021/ac020446t. [DOI] [PubMed] [Google Scholar]
- 79.Schnier PD, Gross DS, Williams ER. On the Maximum Charge State and Proton Transfer Reactivity of Peptide and Protein Ions Formed by Electrospray Ionization. J Am Soc Mass Spectrom. 1995;6:1086–1097. doi: 10.1016/1044-0305(95)00532-3. [DOI] [PubMed] [Google Scholar]
- 80.Iavarone AT, Williams ER. Mechanism of Charging and Supercharging Molecules in Electrospray Ionization. J Am Chem Soc. 2003;125:2319–2327. doi: 10.1021/ja021202t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Jurchen JC, Williams ER. Origin of Asymmetric Charge Partitioning in the Dissociation of Gas-phase Protein Homodimers. J Am Chem Soc. 2003;125:2817–2826. doi: 10.1021/ja0211508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Breuker K, McLafferty FW. Native Electron Capture Dissociation for the Structural Characterization of Noncovalent Interactions in Native Cytochrome c. Angew Chem Int. 2003;42:4900–4904. doi: 10.1002/anie.200351705. [DOI] [PubMed] [Google Scholar]
- 83.Breuker K, McLafferty FW. The Thermal Unfolding of Native Cytochrome c in the Transition from Solution to Gas Phase Probed by Native Electron Capture Dissociation. Angew Chem Int Ed. 2005;44:4911–4914. doi: 10.1002/anie.200500668. [DOI] [PubMed] [Google Scholar]
- 84.Breuker K. Segmental Charge Distributions of Cytochrome c on Transfer into the Gas Phase. Int J Mass Spectrom. 2006;253:249–255. [Google Scholar]
- 85.Cassady CJ, Carr SR. Elucidation of Isomeric Structures for Ubiquitin [M + 12H]12+ Ions Produced by Electrospray Ionization Mass Spectrometry. J Mass Spectrom. 1996;31:247–254. doi: 10.1002/(SICI)1096-9888(199603)31:3<247::AID-JMS285>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- 86.McLafferty FW, Guan Z, Haupts U, Wood TD, Kelleher NL. Gaseous Conformational Structures of Cytochrome c. J Am Chem Soc. 1998;120:4732–4740. [Google Scholar]
- 87.Freitas MA, Hendrickson CL, Emmett MR, Marshall AG. Gas-phase bovine ubiquitin cation conformations resolved by gas-phase hydrogen/deuterium exchange rate and extent. Int J Mass Spectrom. 1999;185/186/187:565–575. [Google Scholar]
- 88.Oh HB, Breuker K, Sze SK, Ge Y, Carpenter BK, McLafferty FW. Secondary and Tertiary Structures of Gaseous Protein Ions Characterized by Electron Capture Dissociation Mass Spectrometry and Photofragment Spectroscopy. Proc Natl Acad Sci USA. 2002;99:15863–15868. doi: 10.1073/pnas.212643599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Hoaglund-Hyzer CS, Counterman AE, Clemmer DE. Anhydrous Protein Ions. Chem Rev. 1999;99:3037–3079. doi: 10.1021/cr980139g. [DOI] [PubMed] [Google Scholar]
- 90.Kohtani M, Jarrold MF. Water Molecule Adsorption on Short Alanine Peptides: How Short Is the Shortest Gas-Phase Alanine-Based Helix? J Am Chem Soc. 2004;126:8454–8458. doi: 10.1021/ja049298+. [DOI] [PubMed] [Google Scholar]
- 91.Robinson EW, Williams ER. Multidimensional Separations of Ubiquitin Conformers in the Gas Phase: Relating Ion Cross Sections to H/D Exchange Measurements. J Am Soc Mass Spectrom. 2005;16:1427–1437. doi: 10.1016/j.jasms.2005.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Koeniger SL, Clemmer DE. Resolution and Structural Transitions of Elongated States of Ubiquitin. J Am Soc Mass Spectrom. 2007;18:322–331. doi: 10.1016/j.jasms.2006.09.025. [DOI] [PubMed] [Google Scholar]
- 93.Oh HB, Lin C, Hwang HY, Zhai H, Breuker K, Zabrouskov V, Carpenter BK, McLafferty FW. Infrared Photodissociation Spectroscopy of Electrosprayed Ions in a Fourier-Transform Mass Spectrometer. J Am Chem Soc. 2005;127:4076–4083. doi: 10.1021/ja040136n. [DOI] [PubMed] [Google Scholar]
- 94.Lin C, Infusini G, Oh HB, Hwang HY, Breuker K, Kong X, Carpenter BK, McLafferty FW. Unpublished. [Google Scholar]
- 95.Zhai H, Han X, Breuker K, McLafferty FW. Consecutive Ion Activation for Top-down Mass Spectrometry: Improved Protein Sequencing by Nozzle-skimmer Dissociation. Anal Chem. 2005;77:5777–5784. doi: 10.1021/ac0580215. [DOI] [PubMed] [Google Scholar]
- 96.Qian K, Rodgers RP, Hendrickson CL, Emmett MR, Marshall AG. Reading Chemical Fine Print: Resolution and Identification of 3000 Nitrogen-Containing Aromatic Compounds from a Single Electrospray Ionization Fourier Transform Ion Cyclotron Resonance Mass Spectrum of Heavy Petroleum Crude Oil. Energy & Fuels. 2001;15:492–498. [Google Scholar]
- 97.Siuti N, Kelleher NL. Decoding Protein Modifications Using Top-down Mass Spectrometry. Nat Methods. 2007;4:817–821. doi: 10.1038/nmeth1097. [DOI] [PMC free article] [PubMed] [Google Scholar]