

Abstract
Yersinia spp, is currently an antibiotic resistance concern and a re-emerging disease. An essential virulence factor YpkA, contains a Ser/Thr kinase domain whose activity modulates pathogenicity. Here we present an approach integrating a machine learning method, homology modeling, and multiple conformational high throughput docking for the discovery of YpkA inhibitors. These first reported inhibitors of YpkA may facilitate studies of pathogenic mechanism of YpkA, and serve as a starting point for development of anti-plague drugs.
Many Gram-negative bacterial pathogens utilize a type III secretion system (TTSS) to inject effector proteins into the cytosol of host cells1. These virulence factors play an important role in bacterial pathogenesis by modulating the host processes that regulate actin cytoskeletal assembly2. With the emergence of antibiotic resistance and the threat of such bacteria being used as biological weapons, targeting virulence proteins for antibiotic design is attractive, as such compounds are unlikely to be cross-resistant or to induce resistance3,4.
Herein, we report our efforts on the discovery of inhibitors for the Yersinia protein kinase A (YpkA). YpkA is an essential virulence determinant in Yersinia spp., which includes the causative agent of plague5. The protein contains a chaperone binding/membrane localization domain6, a Ser/Thr kinase domain, a GDI-like domain that interacts with the Rho-family of small GTPases, and a C-terminal sub-domain responsible in part for actin binding and kinase activation7,8. As the kinase activity of YpkA has been shown to directly correlate to virulence by phosphorylating the small G protein Gαq, inhibition of YpkA could yield new anti-plague therapeutics9,10.
Protein kinase inhibitor design is a challenging problem because of the high similarity and plasticity of the catalytic site11-14. In this study, we applied an approach combining a machine learning method and multiple conformational high throughput docking for the discovery of YpkA inhibitors. The screening strategy employed is illustrated in Figure 1. First, we developed a machine learning support vector machine (SVM) model using a data set of known kinase inhibitors from a diverse kinase collection. The ligand-based SVM model was used as a kinase filter to prioritize the large size of chemical databases and a target-focused library was obtained. Second, we constructed homology models of YpkA based on the MAPK templates, and further performed MD simulations to sample different protein conformations characterized in the catalytic site to account for protein flexibility. Finally, with an ensemble of protein structures and the kinase inhibitor-enriched library, multiple conformational high throughput docking was performed and a number of potent and selective inhibitors of YpkA have been successfully identified.
Figure 1.
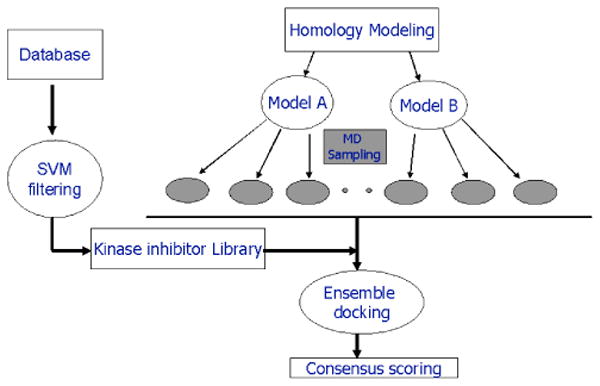
Virtual screening strategy combined machine learning method, homology modeling, and multiple conformational high throughput docking for the discovery of YpkA inhibitors.
In order to develop a general kinase model for database filtering, a data set of known kinase inhibitors was seeded into a randomly selected chemical library serving as the training set. These kinase inhibitors were assembled from the literature and those in complex with a protein target deposited in the protein data bank (PDB). In total, 364 kinase inhibitors were selected covering a diversity of known chemical scaffolds for both Ser/Thr kinases and Tyr kinases. The inactive data set comprising 4220 compounds was randomly selected from the MDDR database (Elsevier MDL, San Leandro, CA). Molecular descriptors were calculated with ADMET/Predictor consisting of 276 descriptors from the 3D structure (SimulationsPlus, Lancaster, CA). The use of ADMET molecular descriptors was anticipated to improve the drug-likeness property of identified compounds, which is a crucial aspect in the late stage of drug development. The SVM model was derived from the molecular descriptors of the training set in distinguishing the active and inactive compounds. As shown in Figure 2, 319 out of 364 inhibitors were classified in the “positive” region, while only 15% of the active compounds were mis-classified as false negative. To validate the model, a testing data set comprising 175 known kinase inhibitors and 669 inactive compounds was applied using the SVM model. 127 out of 175 active compounds were predicted correctly, yielding an enrichment of 70%. This result is promising and comparable to many other machine learning models published recently15,16. Given the high efficiency of the SVM model, we then screened our in-house database collections consisting of more than 2 million compounds, and a kinase-focused library of ∼200,000 compounds was obtained.
Figure 2.
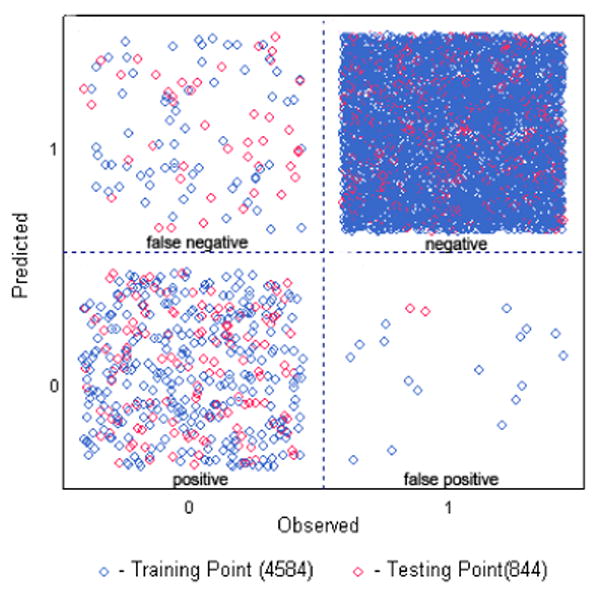
Machine leaning SVM model derived from kinase inhibitors. The “positive” are known kinase inhibitors and the “negative” are randomly selected, inactive compounds. The “false negative” and “false positive” are those that were predicted incorrectly by the SVM model.
Because the structure of YpkA is unavailable, we constructed 3D models based on MAPK. YpkA shares about 20% homology to mammalian Ser/Thr kinases (Figure 3), but considering only the residues near the ATP binding site, the sequence identity to MAPK is 60%. Therefore, there is enough similarity to build a reliable model focused on the catalytic site. Two structural models were constructed based on different templates of MAPK. Model A used the apo structures of p38 (PDB id 1p38 and 1erk), while model B adopted ligand-bound complexes with induced fit at the ATP binding site (PDB id 1a9u and 3erk). As shown in Figure 3, structural differences can be seen within these two models. Model A possesses a more open ATP binding pocket at the Glycine loop (G-loop), while the catalytic site in model B is closed with the G-loop flipping down. As the conformational changes of the G-loop are sensitive to ligand perturbation, both are valid conformations for inhibitor design. We also examined the DFG motif, which is a key element in kinase inhibitor design17. Although in YpkA the corresponding motif is DLG, His293 following the DLGL motif could potentially act in a similar manner to the Phe in mammalian kinases, namely, “His-in” and “His-out” as modeled in A and B.
Figure 3.
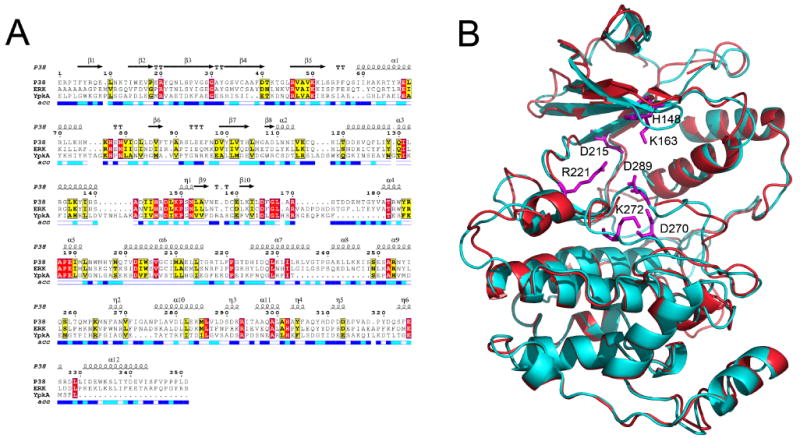
(A) Sequence alignment of YpkA (115-431) with protein kinases P38 and ERK. Strict sequence conservation is shown in red background, and strong sequence conservation in yellow. The solvent-accessibility of each residue in the P38 structure is indicated in the bar at the base of the sequences, with white representing buried residues, dark blue representing solvent-accessible residues, and light blue representing an intermediate value. The secondary structural elements are also indicated according to the structure of P38. (B) Structural alignment of the two homology models of YpkA kinase domain. Model A (red) represents a conformation of YpkA with an open ATP-binding pocket, while model B (cyan) has a closed ATP-binding pocket. The key residues to the ligand binding are shown in magenta.
To further examine the structural features of YpkA, we performed molecular dynamic simulations using both the apo and ATP bound models. The simulations were carried out in vacuo to permit more extended conformational changes. Analysis of the dynamics of the protein at different states revealed a number of active site residues that exhibited high flexibility (Figure 3). In order to sample a good representation of protein conformations for the subsequent ensemble docking, 500 conformers were extracted from 2.0 ns MD simulations and clustered according to a defined residue center at the active site. Five major clusters were obtained with model A and three clusters with model B. From the MD simulations and the docking studies we believe that the conformational changes of the active site residues represent to some extent the plasticity of the ATP binding site upon ligand binding.
With the model of YpkA and the SVM-enriched kinase inhibitor library, we then performed a multiple conformational high throughput docking to search for YpkA inhibitors. The program FlexE was used, which accommodates multiple protein conformations in docking by forming new structural representatives16. A total of eight conformers of YpkA sampled from MD simulations were used in FlexE docking. A kinase focused library consisting of ∼200,000 compounds was subsequently docked into the ensemble of protein structures and ranked according to the FlexX score. To improve the hit selection, we also applied consensus scoring for the FlexX-docked complexes. The top 5% compounds were extracted and re-ranked using X-Score.18 The top 1000 compounds from both the original FlexX score and the X-Score were visually inspected in terms of overall fit, interactions in the binding site, as well as for structural complexity and diversity. A total of 45 compounds were finally selected to experimentally test against YpkA. Seven of the 45 compounds showed complete inhibition at 225 μM to 450 μM, yielding a hit rate of 15%. The IC50 values of these compounds were determined by radiological assay with three compounds exhibiting inhibitory activities at 1.81, 5.87, and 9.72 μM, and the remaining four having IC50 values below 50 μM. Examination of these active compounds revealed a diversity of chemical structure, as represented in Figure 4. Compound 1 possesses a scaffold of indolin-2, which is found in the derivatives of CDK2 inhibitors.19 Compound 2 belongs to the anthraquinone family, potent inhibitors of casein kinase-2.20 The structures of compounds 3 and 4 are quite interesting, as they possess the functional group pyrimidine-2,4,6-trione. As can be seen in figure 5, the binding mode of pyrimidine derivative in YpkA resembles the adenosine moiety of the cofactor, involving in two H-bonding interactions with hinge residues Glu216 and Asp218.
Figure 4.
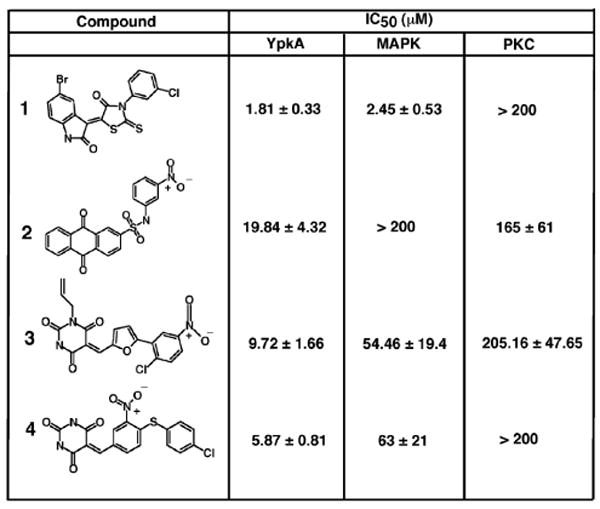
IC50 values of inhibitors from virtual screening with the kinase Homology Model. The chemical structure of each compound is listed along with its IC50 value measured at YpkA concentrations of 0.15μM.
Figure 5.
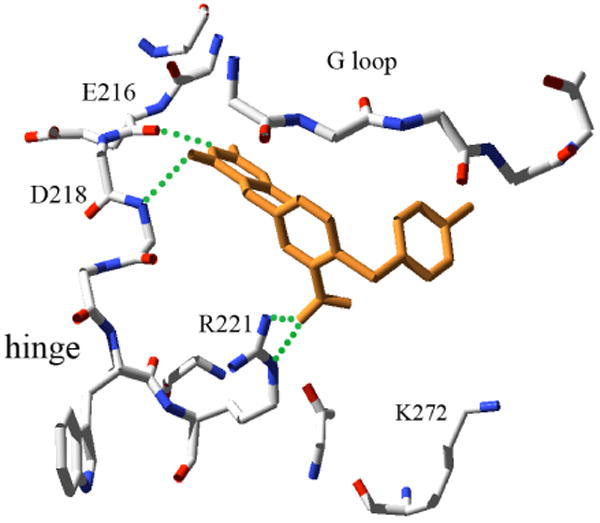
The predicted intermolecular interactions of compound 4 in the YpkA active site. The YpkA inhibitor is shown in orange, hydrogen bonding interactions are shown in green, and the relevant residues of YpkA are colored by atom type and labeled
We further evaluated the selectivity of the YpkA inhibitors by testing against two other kinases, MAPK and protein kinase C (PKC). It is not surprising that some compounds showed comparable inhibitory activities to MAPK, from which the homology models of YpkA were derived. For example, compound 1 showed the best inhibition of YpkA with an IC50 of 1.81 μM, but also exhibited similar activity to MAPK with an IC50 of 2.45 μM. However, compounds 2, 3, 4 are more selective to YpkA over MAPK and PKC with 5 to 10 fold better inhibition (Figure 4). The predicted interactions of compound 4 in the YpkA active site showed that the nitro group forms strong interactions with residue Arg221 at the end of hinge loop (Figure 5). As an Asp or Glu residue is typically present at this position in mammalian serine/threonine kinases, interactions between the basic, positive charged Arg residue and compound 4 may impart selectivity for YpkA. To the best of our knowledge, these are first reported small molecule inhibitors for YpkA, providing a means to investigate the mechanism of YpkA in bacterial pathogenesis, as well as a staring point for the design of potent and selective inhibitors as anti-plague drugs. Although these results are promising, one must consider non-specific inhibition due to the induction of protein aggregation21. Based on our experimental results in figure 4, the reported inhibitors are most likely not acting as aggregation agents and are specifically inhibiting YpkA. This is demonstrated as the compounds have little effect on PKC, but are inhibitory to MAPK, from which our homology model was created.
In summary, we have described an integrated approach combining machine leaning techniques and high throughput docking for the discovery of Yersinia protein kinase A inhibitors. We have made use of the abundant resource of known kinase inhibitors and have developed a SVM model to prioritize these databases. With the construction of homology models and an ensemble of protein structures, we performed multiple conformational high throughput docking on the target-focused library for the search of potent and selective inhibitors of YpkA. The combination of both ligand-based and structure-based knowledge of protein kinases has demonstrated high screening efficiency and reasonable speed, which has allowed us to characterize the first reported inhibitors of Yersinia Protein Kinase A. This integrated approach therefore provides a practical method to account for protein flexibility in a large-scale database for virtual screening of effective inhibitors of therapeutic targets.
Supplementary Material
3D structural models of YpkA. Detailed methods on homology modeling, SVM clustering, MD simulations, virtual screening, YpkA purification, inhibitor data, and experimental details for Figure 4.
Acknowledgments
This work was funded in part by research funds to C.E.S. from the Rockefeller University and PHS grant 1U19AI056510 from the National Institute of Allergy and Infectious Diseases.
Abbreviations
- G-loop
Glycine Loop
- GDI
Guanine Nucleotide Dissociation Inhibitor
- MD
Molecular Dynamics
- PKC
Protein Kinase C
- PDB
Protein Data Bank
- SMV
Support Vector Machine
- SMV
Support Vector Machine
- YpkA
Yersinia Protein Kinase A
References
- 1.Galan JE, Collmer A. Type III secretion machines: bacterial devices for protein delivery into host cells. Science. 1999;284:1322–1328. doi: 10.1126/science.284.5418.1322. [DOI] [PubMed] [Google Scholar]
- 2.Stebbins CE. Structural insights into bacterial modulation of the host cytoskeleton. Curr Opin Struct Biol. 2004;14:731–740. doi: 10.1016/j.sbi.2004.09.011. [DOI] [PubMed] [Google Scholar]
- 3.Henderson DA. The looming threat of bioterrorism. Science. 1999;283:1279–1282. doi: 10.1126/science.283.5406.1279. [DOI] [PubMed] [Google Scholar]
- 4.Marra A. Targeting virulence for antibacterial chemotherapy: identifying and characterising virulence factors for lead discovery. Drugs R D. 2006;7:1–16. doi: 10.2165/00126839-200607010-00001. [DOI] [PubMed] [Google Scholar]
- 5.Perry RD, Fetherston JD. Yersinia pestis--etiologic agent of plague. Clin Microbiol Rev. 1997;10:35–66. doi: 10.1128/cmr.10.1.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Letzelter M, Sorg I, Mota LJ, Meyer S, Stalder J, et al. The discovery of SycO highlights a new function for type III secretion effector chaperones. Embo J. 2006;25:3223–3233. doi: 10.1038/sj.emboj.7601202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Juris SJ, Rudolph AE, Huddler D, Orth K, Dixon JE. A distinctive role for the Yersinia protein kinase: actin binding, kinase activation, and cytoskeleton disruption. Proc Natl Acad Sci U S A. 2000;97:9431–9436. doi: 10.1073/pnas.170281997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Prehna G, Ivanov MI, Bliska JB, Stebbins CE. Yersinia virulence depends on mimicry of host rho-family nucleotide dissociation inhibitors. Cell. 2006;126:869–880. doi: 10.1016/j.cell.2006.06.056. [DOI] [PubMed] [Google Scholar]
- 9.Wiley DJ, Nordfeldth R, Rosenzweig J, DaFonseca CJ, Gustin R, et al. The Ser/Thr kinase activity of the Yersinia protein kinase A (YpkA) is necessary for full virulence in the mouse, mollifying phagocytes, and disrupting the eukaryotic cytoskeleton. Microb Pathog. 2006;40:234–243. doi: 10.1016/j.micpath.2006.02.001. [DOI] [PubMed] [Google Scholar]
- 10.Navarro L, Koller A, Nordfelth R, Wolf-Watz H, Taylor S, et al. Identification of a Molecular Target for the Yersinia Protein Kinase A. Mol Cell. 2007;26:465–477. doi: 10.1016/j.molcel.2007.04.025. [DOI] [PubMed] [Google Scholar]
- 11.Noble ME, Endicott JA, Johnson LN. Protein kinase inhibitors: insights into drug design from structure. Science. 2004;303:1800–1805. doi: 10.1126/science.1095920. [DOI] [PubMed] [Google Scholar]
- 12.Scapin G. Protein kinase inhibition: different approaches to selective inhibitor design. Curr Drug Targets. 2006;7:1443–1454. doi: 10.2174/1389450110607011443. [DOI] [PubMed] [Google Scholar]
- 13.Cavasotto CN, Abagyan RA. Protein flexibility in ligand docking and virtual screening to protein kinases. J Mol Biol. 2004;337:209–225. doi: 10.1016/j.jmb.2004.01.003. [DOI] [PubMed] [Google Scholar]
- 14.Muegge I, Enyedy IJ. Virtual screening for kinase targets. Curr Med Chem. 2004;11:693–707. doi: 10.2174/0929867043455684. [DOI] [PubMed] [Google Scholar]
- 15.Briem H, Gunther J. Classifying “kinase inhibitor-likeness” by using machine-learning methods. Chembiochem. 2005;6:558–566. doi: 10.1002/cbic.200400109. [DOI] [PubMed] [Google Scholar]
- 16.Ford MG, Pitt WR, Whitley DC. Selecting compounds for focused screening using linear discriminant analysis and artificial neural networks. J Mol Graph Model. 2004;22:467–472. doi: 10.1016/j.jmgm.2004.03.006. [DOI] [PubMed] [Google Scholar]
- 17.Nolen B, Taylor S, Ghosh G. Regulation of protein kinases; controlling activity through activation segment conformation. Mol Cell. 2004;15:661–675. doi: 10.1016/j.molcel.2004.08.024. [DOI] [PubMed] [Google Scholar]
- 18.Wang R, Lai L, Wang S. Further development and validation of empirical scoring functions for structure-based binding affinity prediction. J Comput Aided Mol Des. 2002;16:11–26. doi: 10.1023/a:1016357811882. [DOI] [PubMed] [Google Scholar]
- 19.Hardcastle IR, Golding BT, Griffin RJ. Designing inhibitors of cyclin-dependent kinases. Annu Rev Pharmacol Toxicol. 2002;42:325–348. doi: 10.1146/annurev.pharmtox.42.090601.125940. [DOI] [PubMed] [Google Scholar]
- 20.De Moliner E, Moro S, Sarno S, Zagotto G, Zanotti G, et al. Inhibition of protein kinase CK2 by anthraquinone-related compounds. A structural insight. J Biol Chem. 2003;278:1831–1836. doi: 10.1074/jbc.M209367200. [DOI] [PubMed] [Google Scholar]
- 21.McGovern SL, Shoichet BK. Kinase Inhibitors: Not just for kinases anymore. J Med Chem. 2003;46:1478–1483. doi: 10.1021/jm020427b. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
3D structural models of YpkA. Detailed methods on homology modeling, SVM clustering, MD simulations, virtual screening, YpkA purification, inhibitor data, and experimental details for Figure 4.