

Abstract
Growth factor deprivation of endothelial cells induces apoptosis, which is characterized by membrane blebbing, cell rounding, and subsequent loss of cell–matrix and cell–cell contacts. In this study, we show that initiation of endothelial apoptosis correlates with cleavage and disassembly of intracellular and extracellular components of adherens junctions. β-Catenin and plakoglobin, which form intracellular links between vascular endothelial cadherin (VE-cadherin) and actin-binding α-catenin in adherens junctions, are cleaved in apoptotic cells. In vitro incubations of cell lysates and immunoprecipitates with recombinant caspases indicate that CPP32 and Mch2 are involved, possibly by initiating proteolytic processing. Cleaved β-catenin from lysates of apoptotic cells does not bind to endogenous α-catenin, whereas plakoglobin retains its binding capacity. The extracellular portion of the adherens junctions is also altered during apoptosis because VE-cadherin, which mediates endothelial cell–cell interactions, dramatically decreases on the surface of cells. An extracellular fragment of VE-cadherin can be detected in the conditioned medium, and this “shedding” of VE-cadherin can be blocked by an inhibitor of metalloproteinases. Thus, cleavage of β-catenin and plakoglobin and shedding of VE-cadherin may act in concert to disrupt structural and signaling properties of adherens junctions and may actively interrupt extracellular signals required for endothelial cell survival.
INTRODUCTION
Programmed cell death, or apoptosis, is fundamental to development and disease processes (Carson and Ribeiro, 1993; Thompson, 1995). Anchorage of cells to the extracellular matrix through integrins (Frisch and Francis, 1994; Re et al., 1994), as well as cadherin-mediated homeotypic cell–cell interactions (Hermiston and Gordon, 1995), is thought to play a crucial role in cell survival. These adhesive interactions involve a variety of transmembrane receptors that are linked via junctional plaque proteins to the cytoskeleton. Cytoskeletal organization, rather than engagement or clustering of cell adhesion molecules, appears to be the critical determinant of life or death for endothelial cells (Chen et al., 1997). During apoptosis, a number of molecules involved in cytoskeletal regulation and signaling, such as α-fodrin, gelsolin, growth arrest-specific gene 2 (Gas2), extracellular-regulated kinase kinase-1 (MEKK-1), PKC, and p21-activated kinase (PAK2), have been shown to be substrates for proteolytic cleavage by caspases, the family of death proteases (Brancolini et al., 1995; Martin et al., 1995; Cryns et al., 1996; Kothakota et al., 1997; Nicholson and Thornberry, 1997; Rudel and Bokoch, 1997). The caspases are a family of highly conserved aspartate-specific cysteine-proteases related to the mammalian interleukin-1β–converting enzyme and the product of the Caenorhabditis elegans ced-3 gene (reviewed by Alnemri, 1997) that are involved in the final execution phase of apoptosis.1
Endothelial cells undergo apoptosis in response to removal of growth factors and exhibit classical biochemical and morphologic changes associated with apoptosis (Hase et al., 1994). The first morphologic changes observed after growth factor removal are cell retraction and membrane blebbing, with subsequent loss of cell–cell and cell–matrix contacts, resulting in cell detachment. These observations suggest that cytoskeletal disruption occurs with eventual loss of cell–cell contacts during apoptosis. An important element involved in endothelial cell–cell adhesion is the adherens junction, in which an endothelial-specific cadherin, vascular endothelial (VE)-cadherin, anchors the cytoskeleton of neighboring cells via the catenins. β-Catenin and plakoglobin (γ-catenin) are two related molecules, which connect the intracellular domain of VE-cadherin to α-catenin, a vinculin homolog that binds actin (Lampugnani et al., 1995).
In this study, we examined modifications to adherens junction components that occur during growth factor deprivation-induced apoptosis of human umbilical vein endothelial cells (HUVEC). We demonstrate cleavage of β-catenin and plakoglobin in vitro, which appears to be mediated in part by caspases. Cleavage of β-catenin in apoptotic endothelial cells, but not plakoglobin, inhibits its interaction with the cytoskeleton through α-catenin. In addition, we present data for metalloproteinase-mediated shedding of VE-cadherin after growth factor deprivation. Dissolution of the adherens junction and loss of cell–cell contacts may be a critical component of the apoptotic program.
MATERIALS AND METHODS
Antibodies and Reagents
Monoclonal antibodies to α-catenin, β-catenin, and plakoglobin were purchased from Transduction Laboratories (Lexington, KY); polyclonal anti-β-catenin from Sigma (St. Louis, MO); monoclonal anti-VE-cadherin from Chemicon (Temecula, CA); and monoclonal antibodies to adenomatous polyposis coli (APC) (Ab-1 and Ab-5) from Calbiochem (San Diego, CA). HRP-labeled anti-mouse and anti-rabbit antibodies were from Vector Laboratories (Burlingame, CA), and FITC-conjugated goat anti-mouse antibody was from Cappel (Durham, NC). Dr. Vishva Dixit, Genentech (South San Francisco, CA), and Dr. Kim Orth, University of Michigan, kindly provided recombinant CPP32, ICE LAP3, and Mch2. Dr. Roy Black, Immunex (Seattle, WA), provided the metalloproteinase inhibitor N-{d,l-[2-(hydroxyaminocarbonyl)methyl]-4-methylpentanoyl}l-3-(2′naphthyl)-alanyl-l-alanine, 2-aminoethyl amide (TAPI).
Cell Culture and Induction of Apoptosis
HUVEC were isolated as described (Jaffe et al., 1973) and cultured in RPMI (Life Technologies, Chagrin Falls OH), supplemented with 15% calf serum (Life Technologies), 3% endothelial growth supplement prepared as described previously (Gospodarowicz et al., 1983), with 0.05 mg/ml heparin, and with penicillin and streptomycin. Apoptosis was induced in confluent monolayers by incubation with RPMI without any supplements for 14–16 h. Apoptotic cells, which appeared as “floaters” in the culture medium, were harvested by centrifugation at 1,000 × g for 5 min. Adherent, viable cells remaining on the culture dish and control cells (cultured in normal growth medium) were scraped off the culture dish and centrifuged before lysis. For experiments with the metalloproteinase inhibitor, cells were exposed to 50 μM TAPI in RPMI without supplements. After 8 h, floaters and adherent cells were harvested as described above. After removal of the apoptotic cells, the supernatants were concentrated approximately 10-fold in a Centriprep-30 concentrator (Amicon, Beverly, MA).
Preparation of Cell Lysates and Western Blot Analysis
Cells were lysed in 50 mM Tris-HCl, pH 7.4, 250 mM NaCl, 0.5% NP-40, 10% glycerol, 5 mM EDTA, 50 mM NaF, 0.5 mM Na3VO4, 10 mM β-glycerophosphate, PMSF, leupeptin, and aprotinin. Total protein concentration was determined by use of the BCA assay (Pierce, Rockford, IL). Samples were prepared in reducing loading buffer and then separated on 7.5% SDS-PAGE. Proteins were transferred to Immobilon (Millipore, Bedford, MA) for Western blot analysis. Membranes were blocked with 5% nonfat dry milk in PBS for 30 min at room temperature (RT). Primary antibody was incubated in PBS with 0.5% nonfat dry milk for 1 h at 37°C. After washing four times for 10 min, membranes were incubated with peroxidase-labeled second antibody for 1 h at RT. After four more washes, the blots were developed with ECL (Amersham, Arlington Heights, IL) and exposed to film for varying times, usually 15 s to 5 min.
For Western blot analysis of adenomatous polyposis coli (APC), a 3% low melt NuSieve agarose gel (FMC BioProducts, Rockland, ME) in Tris-borate-EDTA/0.1% SDS was cast after pouring a 15% polyacrylamide plug and prewarming the gel apparatus to 37°C. The molten agarose was poured while at 65°C–70°C. The agarose gel was run without a stacking gel in Laemmli running buffer (25 mM Tris base, 192 mM glycine, and 0.1% SDS) at 15 V/cm until the proteins in the 40- to 60-kDa range had migrated off the bottom of the gel. Proteins were then transferred to Immobilon by capillary transfer with TBS/0.04% SDS. After transfer, the membrane was blocked with 10% nonfat dry milk in TBST (20 mM Tris base, 137 mM NaCl, pH 7.6, with HCl, O.1% Tween-20) for 2 h at RT. Membranes were incubated with 1 μg/ml anti-APC (Ab-1) in 5% nonfat dry milk in TBST for 1 h at 37°C, followed by three washes with TBST, and finally incubated with HRP-conjugated anti-mouse IgG in 5% nonfat dry milk in TBST at RT for 1 h. After six washes with TBST, the membranes were developed with the ECL system (Amersham).
Immunoprecipitations
For immunoprecipitations, 250 μg total protein were incubated overnight with 1–2 μg of antibody in 50 μl at 4°C. Rabbit anti-mouse IgG antibody was added to immunoprecipitations with monoclonal antibodies. After 30 min incubation at 4°C, 20 μl of a 1:1 slurry of protein A sepharose (Bio-Rad, Richmond, CA) was added to each immunoprecipitate. After another 30 min at 4°C, the samples were centrifuged and the pellets washed six times in lysis buffer. Proteins were eluted into 2× reducing loading buffer by boiling for 10 min. After centrifugation, samples were subjected to Western blot analysis as described above.
Caspase-mediated Cleavage
For in vitro cleavage of β-catenin and plakoglobin by recombinant caspases in whole cell lysates, control cells were washed with PBS before lysis, and then lysed in caspase reaction buffer consisting of 10 mM HEPES/KOH, pH 7.4, 2 mM EDTA, 5 mM dithiothreitol, 1% NP-40, and the protease inhibitors leupeptin and aprotinin as previously described (Casciola-Rosen et al., 1996). Cell lysates were cleared by centrifugation at 27,000 × g for 5 min. Cell lysate (10 μg) was incubated with or without 100 ng recombinant CPP32/apopain or Mch2 in a total volume of 10 μl for 45 min at 37°C. Reactions were stopped by the addition of 4× sample buffer. The proteins were separated on SDS-PAGE and analyzed by Western blotting as described above.
For analysis of direct cleavage of β-catenin and plakoglobin by CPP32/apopain and Mch2, β-catenin and plakoglobin were immunoprecipitated from control cell lysates as described above. Beads were washed twice in lysis buffer and once in caspase reaction buffer. The reaction was performed in a total volume of 30 μl, and 33 ng/ml recombinant caspase for 1.5 h at 37°C. The reaction was stopped by addition of 15 μl 4× sample buffer, and the samples were analyzed as described above.
Cell Fractionation
Cell fractionation was performed using digitonin to gently solubilize the plasma membrane (Boyle et al., 1984). Apoptotic cells were harvested by centrifugation and control cells by trypsin EDTA treatment and centrifugation. Cell pellets were resuspended in 300 μl PBS and then diluted into 5 ml ice-cold nuclear buffer (150 mM NaCl, 150 mM sucrose, 20 mM HEPES, pH 7.4, 5 mM KCl, 2 mM dithiothreitol, 1 mM MgCl2, 0.5 mM CaCl2, 0.1 mM PMSF, and protease inhibitors with 0.1% digitonin). The suspension was gently mixed with a pipet on ice for 3 min and then centrifuged at 500 × g for 10 min to pellet the nuclei. Then the remaining cytosolic supernatant was centrifuged for 60 min at 100,000 × g at 4°C. For immunoblot analysis, the equivalent of 200,000 cells/lane was used. Unmodified p21Cip1/Waf1 and proliferating cell nuclear antigen are detected only in the nuclear extracts, whereas vinculin is observed only in cytoplasmic fractions (Levkau et al., 1998a,b), demonstrating a low level of cross-contamination of subcellular fractions.
Immunofluorescence
Cells were seeded on eight-well permanox chamber slides (Nunc, Naperville, IL) previously coated with 2% gelatin, at 25,000 cells/well, and grown at 37°C in a humid incubator. Two days after plating, cells were serum starved for 4 h, and fixed with 3.7% paraformaldehyde for 20 min at RT. Cells were washed two times for 10 min with PBS before permeabilization with 0.5% Triton X-100 for 10 min. After two more washes with PBS, quenching was performed by three 5-min washes with 50 mM NH4Ac in PBS. The samples were washed twice for 10 min with PBS and blocked with 1% BSA in PBS for 30 min. Primary antibodies were diluted in 0.1% BSA in PBS and incubated for 1 h. After three washes with PBS, cells were incubated with FITC-labeled goat anti-mouse antibody (Cappel) for 30 min and washed again before they were mounted with Vectashield mounting medium (Vector Labs). Analysis was performed by confocal microscopy (Bio-Rad).
RESULTS
β-Catenin and Plakoglobin, but Not α-Catenin, Are Cleaved during HUVEC Apoptosis
Growth factor deprivation of HUVEC induces apoptosis with characteristic morphologic and biochemical features associated with apoptosis (membrane blebbing, nuclear condensation and fragmentation, DNA laddering, caspase activation, and substrate cleavage; Levkau et al., 1998b). The first morphologic changes observed after growth factor removal are retraction, membrane blebbing, and loss of cell–cell and cell–matrix contacts, which suggest dramatic cytoskeletal changes that ultimately result in cell detachment and the appearance of floating apoptotic cells in the culture medium. To study changes in adherens junctions during apoptosis, we compared control cells in growth factor-supplemented medium with HUVEC deprived of growth factors for 14–16 h and evaluated expression of three components of the adherens junction—α- and β-catenin and plakoglobin (Figure 1). After 14–16 h, approximately 45% of the HUVEC are apoptotic and floating in the medium (100% apoptotic by biochemical and morphological criteria; Levkau et al., 1998b). The remaining adherent (viable) cells will survive and proliferate if returned to growth factor-supplemented medium. In lysates of apoptotic cells, we find that β-catenin and plakoglobin, but not α-catenin, undergo distinct proteolytic changes with time and that the native proteins completely disappear in the pure apoptotic population (Figure 1a).
Figure 1.

β-Catenin and plakoglobin, but not α-catenin, are cleaved during HUVEC apoptosis. (a) HUVEC were deprived of growth factors for 12–16 h, and apoptotic cells (A) which appear as “floaters” were harvested by centrifugation of the medium. The remaining adherent and viable cell population (V) and control cells (C) cultured in full growth medium were harvested with a cell scraper and centrifuged before cell lysis. Cell lysates (25 μg) were analyzed by separation on a 7.5% SDS-PAGE, by transfer to Immobilon membrane, and by Western blotting with monoclonal antibodies to α-catenin, β-catenin, and plakoglobin (γ-catenin). (b and c) HUVEC were deprived of growth factors for 0, 2, 4, 8, and 12 h, and floaters and viable cells were pooled, lysed, and analyzed by SDS-PAGE and Western blotting with monoclonal antibodies to β-catenin (b) and plakoglobin (c). A lysate of pure apoptotic cells (A) was also included for comparison. Letters and Roman numerals with arrows indicate the fragments observed, some of which are only observed with very short exposure times (fragment A of β-catenin in panel b).
For both β-catenin and plakoglobin, the kinetics of appearance of proteolytic products are different for individual fragments (Figure 1, b and c). Five different proteolytic fragments of β-catenin are observed (Figure 1b): 2 and 4 h after growth factor deprivation, fragments A (only seen in the 1-s exposure), B, and C are detected, with fragment C increasing at 4 h; fragment D appears only after 8 h; fragment E is detected only at 12 h, but may also be present at earlier time points and may be undetectable because of adjacent bands; and in the pure apoptotic population, fragments C and D are the main proteolytic fragments with small amounts of fragment B present as well. The initial plakoglobin fragment observed at 2 h is fragment I, with fragments II and III increasing with time, and fragment IV appearing after 8 h. None of the plakoglobin fragments disappear during apoptosis, which suggests that they represent independent and final products.
Two Recombinant Caspases, CPP32 and Mch2, Cleave Endogenous β-Catenin and Plakoglobin in HUVEC Lysates, Whereas Only CPP32 Cleaves the Immunoprecipitated Proteins
Because activation of caspases and caspase-mediated cleavage of intracellular proteins are crucial steps in apoptosis, we evaluated the amino acid sequence of β-catenin for potential consensus cleavage sites (Talanian et al., 1997; Thornberry et al., 1997) for CPP32-like caspases (DXXD) or Mch2 (L/V/IXXD). Five potential Mch2 and two potential CPP32 cleavage sites are present in β-catenin (Figure 2a), including a CPP32 cleavage site that is located at the far C terminus of the molecule (residue 764). To test whether the C terminus of β-catenin is lost in the apoptotic fragments of β-catenin, we used a polyclonal antibody to the C-terminal peptide (amino acids 768–781), which immediately follows the putative C-terminal CPP32 cleavage site (Figure 2a). Western blot analysis of control, viable, and apoptotic HUVEC demonstrates that this antibody recognizes intact β-catenin in control and viable cells, but none of the proteolytic β-catenin fragments from the apoptotic cell population (Figure 2b).
Figure 2.

Possible involvement of the caspases in the cleavage of β-catenin in HUVEC apoptosis. (a) A schematic diagram of β-catenin shows possible caspase cleavage sites: CPP32-like sites (black arrows) and Mch2-like sites (white arrows) indicating consensus cleavage sites with amino acid sequences of DXXD and (L/I/V)XXD, respectively. The α-catenin–binding site and regions important for down-regulation of β-catenin are illustrated with a line, and the region of armadillo repeats is indicated in gray. The epitopes recognized by the antibodies used for analysis are also shown. (b) Control (C), viable (V), and apoptotic (A) cell lysates were analyzed by SDS-PAGE and Western blotting with either a monoclonal antibody (571–781 C) or a polyclonal antibody (768–781 C) to the indicated C-terminal peptides of β-catenin. The polyclonal antibody to amino acids 768–781 of β-catenin does not recognize the apoptotic fragments C and D, which indicates that the C terminus of the molecule is lost. (c) Lysates from control HUVEC were incubated with recombinant caspases CPP32, ICE LAP3, and Mch2 or reaction buffer for 30 and 90 min, subjected to 7.5% SDS-PAGE, and analyzed by Western blotting with antibody 768–781 C-β-catenin. After just 30 min incubation with CPP32, no native β-catenin remains, and fragments A and B (comparable to those observed in the time course of endothelial apoptosis, Figure 1b) are prominent. After 90 min, fragment A is no longer detectable and fragment C becomes apparent; while even after a 90-min incubation with ICE LAP3, no significant cleavage is observed; primarily, fragment B is observed after Mch2 incubation. (d) Lysates from control HUVEC were incubated with recombinant CPP32 and Mch2 for 45 min, subjected to 7.5% SDS-PAGE, and analyzed by Western blotting with antibody 571–781 C-β-catenin. Lysates from control (C) and apoptotic cells (A) were included for comparison. In control lysates, β-catenin is cleaved by both CPP32 and Mch2, but the final in vitro cleavage products (fragments B and C with CPP32 and only fragment B with Mch2) differ from fragments of β-catenin in apoptotic HUVEC (fragments C and D). (e) Immunoprecipitated β-catenin was incubated with recombinant CPP32, recombinant Mch2, or no enzyme (buffer), and compared with control (C) and apoptotic (A) cell lysates after SDS-PAGE and Western analysis with antibodies 768–781 C or 571–781 C to the indicated C-terminal peptides of β-catenin. Antibody 768–781 C does not recognize immunoprecipitated β-catenin that was cleaved by CPP32. This finding and the minor band shift from cleavage suggest that CPP32 cleaves at the most proximal DXXD located at the C terminus (764 in panel a).
To test whether recombinant caspases can cleave β-catenin in vitro, we incubated lysates of control HUVEC, as a source of endogenous β-catenin, with recombinant CPP32, ICE LAP3, or Mch2. After incubation for 30 min with CPP32, no native β-catenin remains and two fragments (A and B), similar to proteolytic products observed early in apoptotic cells, are detected (Figure 2c). After 90 min incubation with CPP32, the predominant cleavage product is fragment C (Figure 2b). Incubation with ICE LAP3 does not significantly alter the levels of native β-catenin. The primary cleavage fragment observed at both time points with Mch2 is fragment B (Figure 2d), a proteolytic product observed at earlier time points in apoptotic HUVEC but only a minor component of β-catenin fragments present in pure apoptotic cells (Figure 1b).
Since caspases are part of a proteolytic cascade in which one caspase can activate other caspases, it is possible that recombinant CPP32 and Mch2 do not directly cleave β-catenin, but rather activate other caspases present in control cell lysates that are responsible for the observed cleavage fragments. Therefore, to determine whether β-catenin is a direct substrate of CPP32 and Mch2, we immunoprecipitated β-catenin from control cell lysates and subsequently incubated the immunoprecipitates with either CPP32 or Mch2. This approach has been successfully used to show direct cleavage of other intracellular proteins (Levkau et al., 1998a,b). As shown in Figure 2e, CPP32 cleaves immunoprecipitated β-catenin to fragments A and E but does not result in the smaller fragments, B, C, and D, observed in apoptotic cells. Consistent with cleavage at the most C-terminal CPP32 putative cleavage site of β-catenin, the antibody to the β-catenin sequence immediately following the most C-terminal CPP32 cleavage site does not recognize either fragment A or E, which suggests cleavage at the sequence DLMD764G. Mch2, on the other hand, fails to cleave immunoprecipitated β-catenin, which suggests that β-catenin is not a direct substrate for Mch2. Thus, additional factors present in cell lysates appear to be necessary to achieve the complete cleavage pattern observed in apoptotic cells.
Analysis of plakoglobin in control cell lysates and incubation with recombinant Mch2 demonstrates that significant amounts of only the highest molecular weight cleavage fragment observed in apoptotic HUVEC, fragment I, are detected after 45 min (Figure 3a). In contrast, recombinant CPP32 generates all of the cleavage products present in apoptotic cells (Figure 3a), and no native plakoglobin remains after a 45-min incubation with CPP32. When direct cleavage is analyzed with immunoprecipitated plakoglobin (Figure 3b), Mch2 is ineffective, while CPP32 generates fragment I, which comigrates with the primary and highest molecular weight band in apoptotic HUVEC, suggesting that CPP32 is responsible for the initial cleavage of native plakoglobin.
Figure 3.

In vitro cleavage of plakoglobin by recombinant caspases. (a) Lysates from control HUVEC were incubated with recombinant CPP32 and Mch2 for 45 min. The reaction products, as well as lysates from control (C) and apoptotic (A) cells, were subjected to 7.5% SDS-PAGE, followed by Western blotting with a monoclonal antibody to plakoglobin (553–738 C). Mch2 generates a cleavage fragment I, which comigrates with the highest molecular weight cleavage product in apoptotic cells, whereas CPP32 cleavage results in all of the fragments observed in apoptotic cells. (b) Plakoglobin was immunoprecipitated from 250 μg lysate of control HUVEC and incubated with recombinant CPP32, Mch2, or reaction buffer (buffer), with lysates from control (C) or apoptotic (A) cells included in the analysis for comparison. CPP32 cleavage of immunoprecipitated plakoglobin results in fragment I observed in apoptotic cells, whereas Mch2 has no effect.
Cleaved β-Catenin, but Not Plakoglobin, Displays Reduced α-Catenin Binding
To examine whether cleavage of β-catenin and plakoglobin in apoptotic cells alters their structural properties as part of the adherens junction, we asked whether the cleaved proteins retain their ability to bind to α-catenin. As shown in Figure 4a, fragment B (a minor fragment of cleaved β-catenin in apoptotic cells) and fragment C can be coimmunoprecipitated with α-catenin. The faster migrating cleavage fragment D is not detectable, which suggests that it has lost the α-catenin binding site (Aberle et al., 1994, 1996; Barth et al., 1997). In contrast, at least three of the four cleavage products of plakoglobin coimmunoprecipitate with α-catenin, which suggests that cleavage occurs near the C terminus rather than the N terminus of the protein known to bind α-catenin (Sacco et al., 1995; Aberle et al., 1996). The lowest molecular weight fragment of plakoglobin, fragment IV, cannot be detected because it comigrates with the 50-kDa heavy chain IgG band. In accord with the analysis of α-catenin immunoprecipitates, significantly less α-catenin is present in β-catenin immunoprecipitates from apoptotic cells than in immunoprecipitates from viable cells, whereas the association of α-catenin with plakoglobin appears unchanged (Figure 4b).
Figure 4.

Cleavage of β-catenin, but not plakoglobin, impairs association with α-catenin. α-Catenin, β-catenin, and plakoglobin were immunoprecipitated from 250 μg of lysates prepared from control (C), viable (V), or apoptotic (A) HUVEC, and analyzed by SDS-PAGE and Western blotting. In panel a, samples immunoprecipitated with the anti-α-catenin antibody were analyzed by use of the anti-β-catenin (571–781 C) or plakoglobin antibodies. Direct lysates (D) of apoptotic cells are shown for comparison. Fragments B and C, but not D, of β-catenin can still associate with α-catenin, whereas all apoptotic fragments of plakoglobin retain their ability to bind to α-catenin. (b) Samples immunoprecipitated with antibody 571–781 C to β-catenin or anti-plakoglobin antibodies were analyzed by Western blot with anti-α-catenin or anti-β-catenin (571–781 C) antibody. In apoptotic cells, β-catenin, but not plakoglobin, displays reduced binding of α-catenin, even though all of the fragments of β-catenin are immunoprecipitated.
APC Disappears in Apoptotic Cells
Since at least one of the cleavage products of β-catenin, fragment D, in apoptotic cells appears to have lost the N terminus, which contains both the α-catenin–binding site and sites critical for down-regulation (Aberle et al., 1996; Barth et al., 1997) of β-catenin (Figure 2a), we asked whether changes in apoptotic cells led to alterations in β-catenin interaction with APC, a protein known to regulate β-catenin degradation (Munemitsu et al., 1995). As shown in Figure 5a, β-catenin coimmunoprecipitates with APC in control and viable cells, whereas it is absent in APC immunoprecipitates of the apoptotic cell population. As this could be due to an absence of APC in apoptotic HUVEC, we tested whether APC is present in the apoptotic cell population. As shown in Figure 5b, APC is undectable in the apoptotic cells, and, thus, the loss of association between β-catenin and APC in apoptotic HUVEC appears to be primarily a function of the loss of APC.
Figure 5.
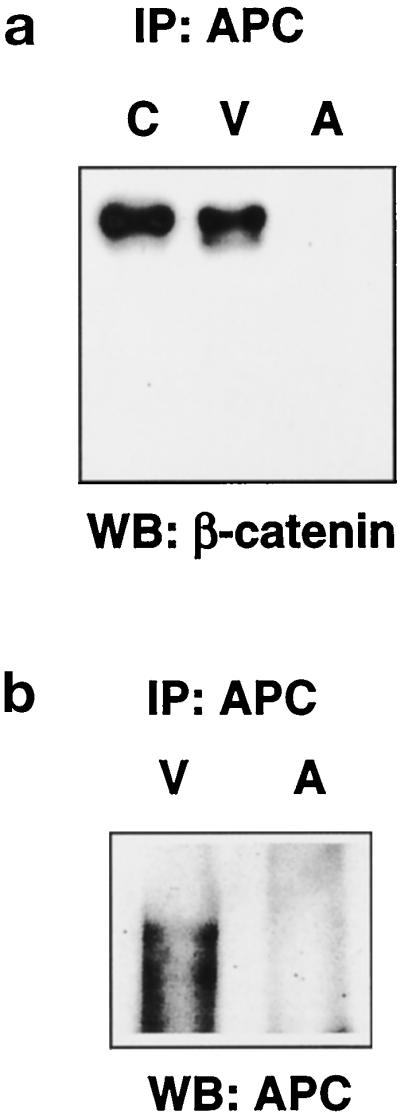
APC disappears in apoptotic HUVEC. (a) APC was immunoprecipitated from 250 μg lysate, and the immunoprecipitates from control (C), viable (V), and apoptotic (A) HUVEC were analyzed by Western blotting with the monoclonal antibody to β-catenin (571–781 C). Association of β-catenin and APC is abolished in apoptotic cells. (b) For analysis of the relative levels of APC under the different conditions, APC was immunoprecipitated from 750 μg of lysate from viable (V) and apoptotic (A) HUVEC with APC antibody (Ab-5), and the samples were separated on a 3% vertical agarose gel with Tris-borate-EDTA/0.1% SDS, transferred by inverse capillary transfer, and blotted with APC antibody Ab-1. APC is absent in apoptotic HUVEC.
Localization of β-Catenin after Growth Factor Withdrawal and Apoptosis
Immunolocalization of β-catenin in growth factor-deprived HUVEC 4 h after growth factor removal by confocal microscopy demonstrates increased staining for β-catenin at the cell periphery in areas lacking cell–cell contact (Figure 6c). In contrast, β-catenin staining is primarily localized to adherens junctions in control cells (Figure 6a). After growth factor removal, punctate staining is also observed within the cells when the bottom level of the cells is analyzed. However, the most striking difference from control cells after growth factor removal is observed in an optical section 4 μm above the bottom level where there is a redistribution of β-catenin to perinuclear/nuclear regions, especially in retracting cells (compare Figures 6b and 6d). To further evaluate possible changes in the intracellular localization of β-catenin, we analyzed cytoplasmic and nuclear extracts from control cells and cells 5 h after growth factor withdrawal. Although cleaved β-catenin is present in both nuclear and cytoplasmic extracts of control cells, uncleaved β-catenin is preferentially lost from the cytoplasmic fraction, and fragment E is only observed in the nuclear fraction in growth factor-deprived cells (Figure 6e).
Figure 6.

Changes in β-catenin distribution between nuclear and cytosolic compartments after growth factor removal (a–d). Control (a and b) and viable HUVEC (c and d) were stained with anti-β-catenin antibody (571–781 C) and analyzed by confocal microscopy. In control cells, staining at the bottom level of the cells can be detected in adherens junctions (arrows in a). In contrast, after growth factor removal (c), increased peripheral staining is observed, despite the lack of cell–cell contact (arrows), and an increase in punctate cytosolic staining is observed as well (arrowheads). Images 4 μm above the bottom level (b and d) are shown for the same cells analyzed in panels a and c. In control cells (b), some of the adherens junction staining (arrows) is still visible. In contrast, after growth factor removal (d), perinuclear and nuclear staining is prominent (arrowheads). (e) Cytosolic (cyt) and nuclear (nuc) extracts of HUVEC grown in full growth medium (control) and of pooled HUVEC (apoptotic and viable cells) 5 h after growth factor removal were prepared as described in MATERIALS AND METHODS. The equivalent of 200,000 cells/fraction was separated on 7.5% SDS-PAGE, transferred to Immobilon, and analyzed for the presence of β-catenin with the anti-β-catenin antibody 571–781 C. Growth factor removal leads to a selective reduction of native β-catenin in the cytosol, while the cleaved apoptotic fragments of β-catenin are found in both the cytosol and the nucleus, except for fragment E, which appears only in the nuclear fraction.
The Extracellular Domain of VE-Cadherin Is Shed from the Cell Surface during Apoptosis, and Shedding Can Be Abolished by an Inhibitor of Metalloproteinases
VE-cadherin, the endothelial cell-specific cadherin that mediates homeotypic cell–cell interaction, is not detectable in lysates of apoptotic cells with an antibody to the extracellular domain (Figure 7a). A reduction in the intensity of the VE-cadherin band and the appearance of an additional band, fragment A, are observed as early as 2 h after removal of growth factors (Figure 7b). To test whether the extracellular domain is shed into the culture medium during apoptosis, we performed Western blot analysis on the concentrated conditioned medium from growth factor-deprived HUVEC. A VE-cadherin band of approximately 90 kDa, fragment B, can be detected in the conditioned medium, which indicates that a portion of the VE-cadherin extracellular domain is shed from the cell surface during apoptosis (Figure 7c). Both fragments of VE-cadherin, fragments A and B, are detected in viable cell lysates.
Figure 7.

VE-cadherin is shed from the cell surface during HUVEC apoptosis. (a) An antibody that recognizes the extracellular domain of VE-cadherin was used to evaluate control (C), viable (V), and apoptotic (A) cells. The extracellular epitope of VE-cadherin recognized by the antibody is absent in apoptotic cells. (b) After growth factor removal from HUVEC for 0, 2, 4, 8, and 12 h, pools of viable and apoptotic cells show a time-dependent loss of the extracellular epitope of VE-cadherin by Western analysis. A proteolytic fragment (A) is observed at 2 h and also decreases with time. (c) Analysis of supernatants (concentrated 10-fold) collected from apoptotic HUVEC cultures after 16 h without growth factors demonstrates that VE-cadherin is shed into the medium. Two fragments (A and B) are detected in viable and apoptotic cell lysates, and the approximately 90-kDa fragment (B) is also found in the supernatant.
To determine whether shedding of VE-cadherin during HUVEC apoptosis is mediated by a metalloproteinase, as shown for a number of adhesion molecules and cytokine receptors (reviewed by Ehlers and Riordan, 1991; Hooper et al., 1997), cells were deprived of growth factors in the presence or absence of the metalloproteinase inhibitor TAPI, used to identify the tumor necrosis factor-α (TNF-α) convertase (Black et al., 1997). Fewer floaters accumulate in the supernatant of HUVEC cultures deprived of growth factors in the presence of TAPI. In addition, fewer cells with membrane blebbing are visible, compared with cells without the inhibitor (our unpublished observations). Western blot analysis reveals that native VE-cadherin and fragment A are present on viable cells with or without the inhibitor, but there is a dramatic increase in both VE-cadherin and fragment A in the presence of TAPI. Only very low levels of fragment A and no native VE-cadherin are detectable in the floaters in the absence of the inhibitor, whereas substantial levels of both native VE-cadherin and fragment A are detectable in apoptotic cells in the presence of TAPI. The extracellular domain of VE-cadherin is detectable in the conditioned medium only in the absence of TAPI but not in the presence of the metalloproteinase inhibitor (Figure 8a). Thus, although proteolytic processing of VE-cadherin is not inhibited by TAPI, shedding of the extracellular domain is prevented. Both native and cleaved VE-cadherin accumulate on viable and apoptotic cells. β-Catenin cleavage is not affected by inhibition of metalloproteinases, which indicates that it is independent of VE-cadherin shedding (Figure 8b).
Figure 8.

Metalloproteinase-mediated shedding of VE-cadherin in apoptotic HUVEC. HUVEC were cultured without growth factors for 8 h in the presence or absence of 50 mM of a metalloproteinase inhibitor, TAPI, and viable (V) and apoptotic (A) cells and supernatants (sup) were harvested separately and analyzed by SDS-PAGE and Western blotting with an antibody to (a) VE-cadherin or (b) β-catenin (571–781 C). The relative levels of native VE-cadherin and fragment A increase in TAPI-treated cells, but TAPI does not prevent formation of fragment A. However, the soluble VE-cadherin fragment (B) is only detectable in the absence of TAPI but not in its presence, consistent with VE-cadherin shedding during HUVEC apoptosis mediated by a metalloproteinase.
DISCUSSION
β-Catenin and Plakoglobin Cleavage by Recombinanat Caspases In Vitro Generates Fragments Corresponding to Those Observed during Endothelial Cell Apoptosis
Both β-catenin and plakoglobin are cleaved in human endothelial cells undergoing apoptosis after growth factor removal, and the cleavages appear to be partially mediated by caspases. In contrast to β-catenin and plakoglobin, α-catenin remains intact, despite the presence of multiple potential caspase cleavage sites. Primarily, two fragments of β-catenin are detected in apoptotic cells, whereas four fragments of plakoglobin are observed. Although antibodies to other epitopes may identify additional cleavage fragments, the generation of a small number of fragments, despite the presence of a large number of additional putative caspase cleavage sites (Figure 2a), suggests specific and limited cleavage of each molecule.
Analysis of the cleavage of β-catenin suggests that it occurs in multiple steps. In control cell lysates, recombinant CPP32 and Mch2 are able to generate the primary, but not all, cleavage fragments seen in apoptotic cells. Cleavage of immunoprecipitated β-catenin by recombinant caspases is even more limited, with only CPP32 able to cleave at the most C-terminal DXXD site. A likely explanation for differences in fragments generated by recombinant caspases incubated with control cell lysates and immunoprecipitated β-catenin is the ability of individual caspases to activate other caspases present in cell lysates that are then able to cleave β-catenin, as shown for the indirect cleavage of PARP by Mch2 through intermediate activation of CPP32 (Orth et al., 1996). Alternatively, caspases may activate other proteolytic activities during apoptosis. Because fragments B and C in apoptotic cell lysates can still associate with α-catenin, the N-terminal α-catenin–binding site (Aberle et al., 1994, 1996; Barth et al., 1997) appears to remain intact, which suggests that cleavage may occur approximately 20 kDa upstream of the C terminus. The smallest fragment, fragment D, which loses its ability to bind to α-catenin, must be cleaved additionally at an N-terminal site.
Another component of adherens junctions, plakoglobin, is also cleaved during apoptosis. In whole cell lysates, CPP32 cleavage appears to generate all of the fragments present in apoptotic cells, whereas Mch2 cleavage efficiently generates only the main fragment. Cleavage of immunoprecipitates of plakoglobin with recombinant CPP32 results in more limited cleavage than whole cell lysates, and only fragment A, the first fragment to appear during apoptosis, is detected. These data suggest that CPP32 is responsible for the initial cleavage during apoptosis, and it may activate other caspases responsible for the other cleavage products. In contrast, Mch2 cannot cleave immunoprecipitated plakoglobin, which suggests that all of its activity in control cell lysates is mediated by other enzymes.
Implications of β-Catenin Cleavage for Adherens Junctions and Signaling
One of the two most prominent forms of cleaved β-catenin in apoptotic cells, fragment D, is unable to associate with α-catenin, which suggests that the cleavage generating this fragment is occurring at the N terminus, and separates the α-catenin–binding site in β-catenin from the rest of the molecule. The reduced association between β- and α-catenin could facilitate retraction and cytoskeletal disruption in cells undergoing apoptosis (Figure 9). In theory, plakoglobin could compensate for this decrease in β-catenin binding to α-catenin because most of the cleavage fragments of plakoglobin retain their ability to bind to α-catenin. However, recruitment of β-catenin and plakoglobin to adherens junctions follows a different time course (Lampugnani et al., 1995), which suggests that they play different roles in assembly and disassembly of adherens junctions.
Figure 9.

Caspases and metalloproteinases may contribute to dissolution of adherens junctions during endothelial apoptosis. Initiation of apoptosis by growth factor deprivation of HUVEC leads to rapid dissolution of adherens junctions that may be mediated by metalloproteinase-dependent shedding of the extracellular domain of VE-cadherin and cleavage of β-catenin and plakoglobin that appear to involve caspases. A cleavage fragment of β-catenin (β′), but not plakoglobin (γ′), loses its ability to bind α-catenin, and, therefore, the intracellular connection of the adherens junction to the actin cytoskeleton is altered. These rapid intracellular and extracellular modifications of the adherens junctions impair cell–cell contact and result in intracellular disruption of the cytoskeleton.
In addition to its structural role in adherens junctions, β-catenin forms a complex with the LEF-1 family of transcription factors that enters the nucleus (Behrens et al., 1996; Huber et al., 1996). The primary structure of β-catenin consists of an N-terminal portion of approximately 130 amino acids, a central region of 550 amino acids that contains 12 sequence repeats of 42 amino acids known as armadillo repeats, and a C-terminal region of 100 amino acids (Figure 2a). Deletion mutations have mapped (Figure 2) the binding of cadherins, APC, and the LEF-1 transcription factors to the armadillo repeat region (Hulsken et al., 1994; Rubinfeld et al., 1995; Behrens et al., 1996; Molenaar et al., 1996), whereas the α-catenin–binding site spans the N-terminal domain and the first armadillo repeat. Transactivator function required for gene activation by the β-catenin–LEF-1 complex appears to be localized to the C-terminal region (van de Wetering et al., 1997). Although our studies have not mapped the exact cleavage sites of β-catenin associated with apoptosis of HUVEC, we present evidence consistent with significant cleavage (removal of approximately 20 kDa) from both the N and C termini of β-catenin. These cleavages, as well as the loss of APC, could significantly affect β-catenin turnover and transcriptional activity. Constitutive transcriptional activation of the LEF-1–β-catenin complex has been described for cells that lack APC or contain APC mutants lacking the β-catenin–binding site (Korinek et al., 1997; Rubinfeld et al., 1997). Given the ability of β-catenin–LEF-1 complexes to bind and regulate E-cadherin transcription (Huber et al., 1996), it will be interesting to determine levels of VE-cadherin transcription after growth factor removal and possible implications for maintenance of adherens junctions during apoptosis.
Shedding of VE-Cadherin by a Metalloproteinase during Apoptosis
A number of cell surface proteins, among them cytokine precursors, growth factors, cytokine receptors, and cell adhesion molecules, are shed from the cell surface in a regulated manner (reviewed by Ehlers and Riordan, 1991; Hooper et al., 1997). In a number of cases, shedding can be inhibited by metalloproteinase inhibitors, which suggests that some cell surface sheddases are metalloproteinases (Gearing et al., 1994; McGeehan et al., 1994; Mohler et al., 1994; Crowe et al., 1995; Kayagaki et al., 1995; Müllberg et al., 1995; Arribas et al., 1997). Cloning of the first sheddase, TNF-α–converting enzyme (TACE), demonstrated that it belongs to the family of metalloproteinase disintegrins (MDCs or ADAMs) and cleaves the surface-bound precursor of TNF-α (Black et al., 1997; Moss et al., 1997). Kuzbanian is another metalloproteinase disintegrin involved in the cleavage and activation of a transmembrane protein, Notch (Pan and Rubin, 1997), which has homologs expressed in endothelial cells (Uyttendaele et al., 1996). Several other MDCs have metalloproteinase domains with an active site consensus sequence, among them MDC15, which was cloned from endothelial cells (Herren et al., 1997) and also may exhibit sheddase activity.
Regulation of VE-cadherin surface expression by shedding has not been reported previously. However, other cell adhesion molecules, such as N-cadherin, are shed from the cell surface in a regulated manner. N-Cadherin is down-regulated during retinal development by metalloproteinase-sensitive shedding from the cell surface (Roark et al., 1992; Paradies and Grunwald, 1993). In these studies, we provide the first evidence that, in endothelial cells undergoing apoptosis after growth factor removal, the extracellular domain of VE-cadherin is lost from the cell surface by a metalloproteinase-sensitive mechanism. Our data also demonstrate metalloproteinase-independent processing of VE-cadherin, which gives rise to two membrane-bound fragments, one of which comigrates with the metalloproteinase-shed extracellular domain of VE-cadherin. This fragment may remain bound to the transmembrane cleavage product similar to other cleaved transmembrane molecules, such as the protein tyrosine phosphatase LAR (Streuli et al., 1992). Interestingly, LAR is localized in adherens junctions and directly associates with β-catenin and plakoglobin (Aicher et al., 1997).
In the presence of the metalloproteinase inhibitor TAPI, the culture medium of serum-deprived HUVEC contains few apoptotic cells, all of which express native VE-cadherin or a cleavage fragment of VE-cadherin that remains cell associated. Loss of VE-cadherin by specific shedding may facilitate detachment and clearance of apoptotic endothelial cells (Figure 9). As has been shown for other adhesion molecules, shedding of VE-cadherin may play a role not only in apoptosis but also in other biological settings that involve reorganization of adherens junctions.
Endothelial Cell Function and Disruption of Adherens Junctions
Disruption of the VE-cadherin–catenin complex in adherens junctions has been demonstrated after binding of neutrophils (or neutrophil membranes) to activated endothelium (Del Maschio et al., 1996; Allport et al., 1997). Disorganization of the adherens junction after neutrophil binding did not result in endothelial cell retraction or injury and was specific for the VE-cadherin complex. Similar to our study, Allport et al. observed degradation of the VE-cadherin complex, including loss of VE-cadherin and proteolytic modification of β-catenin and plakoglobin, with no change in α-catenin. We do not know whether the cleavage products observed after neutrophil adhesion to endothelium are the same as those observed during apoptosis. However, proteolytic cleavage of adherens junction components that transiently disrupt the adherens junction have been suggested by others to be critical for neutrophil transmigration across the endothelial cell layer and for induction of permeability changes in endothelium. Similarly, thrombin treatment of endothelial cells increases endothelial cell permeability, disrupts endothelial adherens junction organization (Rabiet et al., 1996), and truncates the cytoplasmic domain of VE-cadherin, which prevents catenin binding and increases monolayer permeability (Navarro et al., 1995). Thus, shedding of VE-cadherin and/or disruption of the adherens junctions by proteolytic modification of β-catenin and plakoglobin may be important for regulation of endothelial permeability.
Our studies demonstrate that during endothelial apoptosis there is disruption of the adherens junction complex, including metalloproteinase-mediated shedding of VE-cadherin and caspase-mediated cleavage of β-catenin and plakoglobin (Figure 9). We also observe caspase-mediated cleavage of focal adhesion kinase (FAK), an important component of the focal adhesion complex, in growth factor-deprived HUVEC (Levkau et al., 1998a). Cleavage of FAK precedes endothelial cell detachment and coincides with loss of FAK and paxillin from focal adhesion sites and their redistribution into characteristic membrane blebs of dying cells. FAK cleavage prevents association of FAK with normal downstream targets, such as paxillin, and may actively interrupt survival signals and propagate the cell death program. A recent screen of cDNA pools for caspase substrates identified gelsolin as a substrate readily cleaved by CPP32 (Kothakota et al., 1997). Expression of the gelsolin cleavage product in multiple cell types caused the cells to round up, detach from the plate, and undergo nuclear fragmentation. Further, neutrophils isolated from mice lacking gelsolin have delayed onset of apoptosis. Thus, our studies add components of the adherens junction (specifically, β-catenin, plakoglobin, and VE-cadherin) to the list of proteins associated with regulation of the cytoskeleton that are cleaved during apoptosis and promote cell shape change.
Prolonged disruption of the adherens junction during endothelial cell apoptosis may be critical to the apoptotic process by providing a cell geometry permissive for subsequent irreversible apoptotic events. Studies by Chen et al. demonstrate that endothelial cells, plated on different patterns on microfabricated surfaces to alter the extent of cell spreading while maintaining a constant cell–matrix interaction area, show a higher apoptotic index when the endothelial cells are more rounded (Chen et al., 1997). Thus, the rapid dissolution of components of the adherens junctions we observe in apoptotic HUVEC may be important for the activation of apoptotic signaling pathways and/or for maintenance of endothelial cells in an “apoptosis-permissive geometry” until the final committment to the irreversible stages of the apoptotic process.
ACKNOWLEDGMENTS
The authors thank Bonnie Ashleman for expert technical assistance with the confocal microscopy and Barbara Droker for editorial assistance; Drs. Vishva Dixit (Genentech) and Kim Orth (University of Michigan) for providing recombinant caspases; and Dr. Roy Black (Immunex, Seattle) for providing TAPI. This work was supported by National Institutes of Health grant HL-18645 (R.R., E.W.R.). B.L. is a recipient of a training research scholarship from the Deutsche Forschungsgemeinschaft, Germany.
Footnotes
Abbreviations used: ADAM, a disintegrin and metalloprotease domain; APC, adenomatous polyposis coli; BCA, bicinchoninic acid; caspases, cysteinyl aspartate-specific proteinases; CED-3, Caenorhabditis elegans death gene-3; CPP32, Caspase 3, apopain; Yama, DNA-PK DNA-dependent protein kinase; E-cadherin, uvomorulin; FAK, focal adhesion kinase; Gas2, growth arrest-specific gene 2; HUVEC, human umbilical vein endothelial cell(s); LEF-1, lymphoid enhancer factor-1; Mch2, caspase 6; MDC, metalloproteinase-like, disintegrin-like, cysteine-rich protein; MEKK-1, extracellular-regulated kinase kinase-1; N-cadherin, neural cadherin; PAK2, p21-activated kinase; RPMI, RPMI media; RT, room temperature; TACE, TNF-α-converting enzyme; TAPI, N-{d,l-[2-(hydroxyaminocarbonyl)methyl]-4-methylpentanoyl}l-3-(2′naphthyl)-alanyl-l-alanine, 2-aminoethyl amide; TBST, 20 mM Tris base, 137 mM NaCl, pH 7.6, with HCl, O.1% Tween-20; TNF-α, tumor necrosis factor α; VE-cadherin, vascular endothelial cadherin.
REFERENCES
- Aberle H, Butz S, Stappert J, Weissig H, Kemler R, Hoschuetzky H. Assembly of the cadherin-catenin complex in vitro with recombinant proteins. J Cell Sci. 1994;107:3655–3663. doi: 10.1242/jcs.107.12.3655. [DOI] [PubMed] [Google Scholar]
- Aberle H, Schwartz H, Hoschuetzky H, Kemler R. Single amino acid substitutions in proteins of the armadillo gene family abolish their binding to α-catenin. J Biol Chem. 1996;271:1520–1526. doi: 10.1074/jbc.271.3.1520. [DOI] [PubMed] [Google Scholar]
- Aicher B, Lerch MM, Muller T, Schilling J, Ullrich A. Cellular redistribution of protein tyrosine phosphatases LAR and PTPsigma by inducible proteolytic processing. J Cell Biol. 1997;138:681–696. doi: 10.1083/jcb.138.3.681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allport JR, Ding H, Collins T, Gerritsen ME, Luscinskas FW. Endothelial-dependent mechanisms regulate leukocyte transmigration: a process involving the proteasome and disruption of the vascular endothelial-cadherin complex at endothelial cell-to-cell junctions. J Exp Med. 1997;186:517–527. doi: 10.1084/jem.186.4.517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alnemri ES. Mammalian cell death proteases: a family of highly conserved aspartate specific cysteine proteases. J Cell Biochem. 1997;64:33–42. doi: 10.1002/(sici)1097-4644(199701)64:1<33::aid-jcb6>3.0.co;2-0. [DOI] [PubMed] [Google Scholar]
- Arribas J, Coodly L, Vollmer P, Kishimoto TK, Rose-John S, Massagué J. Diverse cell surface protein ectodomains are shed by a system sensitive to metalloproteinase inhibitors. J Biol Chem. 1997;271:11376–11382. doi: 10.1074/jbc.271.19.11376. [DOI] [PubMed] [Google Scholar]
- Barth AI M, Pollack AL, Altschuler Y, Mostov KE, Nelson WJ. NH2-terminal deletion of β-catenin results in stable colocalization of mutant β-catenin with adenomatous polyposis coli protein and altered MDCK cell adhesion. J Cell Biol. 1997;136:693–706. doi: 10.1083/jcb.136.3.693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behrens J, von Kries JP, Kühl M, Bruhn L, Wedlich D, Grosschedl R, Birchmeyer W. Functional interaction of β-catenin with the transcription factor LEF-1. Nature. 1996;382:638–642. doi: 10.1038/382638a0. [DOI] [PubMed] [Google Scholar]
- Black RA, et al. A metalloproteinase disintegrin that releases tumor-necrosis factor-α from cells. Nature. 1997;385:729–733. doi: 10.1038/385729a0. [DOI] [PubMed] [Google Scholar]
- Boyle WJ, Lampert MA, Lipsick JS, Baluda MA. Avian myeloblastosis virus and E26 virus oncogene products are nuclear proteins. Proc Natl Acad Sci USA. 1984;81:4265–4269. doi: 10.1073/pnas.81.14.4265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brancolini C, Benedetti M, Schneider C. Microfilament reorganization during apoptosis: the role of Gas2, a possible substrate for ICE-like proteases. EMBO J. 1995;14:5179–5190. doi: 10.1002/j.1460-2075.1995.tb00202.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carson DA, Ribeiro JM. Apoptosis and disease. Lancet. 1993;341:1251–1254. doi: 10.1016/0140-6736(93)91154-e. [DOI] [PubMed] [Google Scholar]
- Casciola-Rosen L, Nicholson DW, Chong T, Rowan KR, Thornberry NA, Miller DK, Rosen A. Apopain/CPP 32 cleaves proteins that are essential for cellular repair: a fundamental principle of apoptotic death. J Exp Med. 1996;183:1957–1964. doi: 10.1084/jem.183.5.1957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen CS, Mrksich M, Huang S, Whitesides GM, Ingber DE. Geometric control of cell life and death. Science. 1997;276:1425–1428. doi: 10.1126/science.276.5317.1425. [DOI] [PubMed] [Google Scholar]
- Crowe PD, Walter BN, Mohler KM, Otten-Evans C, Black RA, Ware CF. A metalloprotease inhibitor blocks shedding of the 80-kD TNF receptor and TNF processing in T lymphocytes. J Exp Med. 1995;181:1205–1210. doi: 10.1084/jem.181.3.1205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cryns VL, Bergeron L, Zhu H, Li H, Yuan J. Specific cleavage of α-fodrin during Fas- and tumor necrosis factor-induced apoptosis is mediated by an interleukin-1β-converting enzyme/CED-3 protease distinct from the poly(ADP-ribose) polymerase protease. J Biol Chem. 1996;271:31277–31282. doi: 10.1074/jbc.271.49.31277. [DOI] [PubMed] [Google Scholar]
- Del Maschio A, Zanetti A, Corada M, Rival Y, Ruco L, Lampugnani MG, Dejana E. Polymorphonuclear leukocyte adhesion triggers the disorganization of endothelial cell-to-cell adherens junctions. J Cell Biol. 1996;135:497–510. doi: 10.1083/jcb.135.2.497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehlers MRW, Riordan JF. Membrane proteins with soluble counterparts: Role of proteolysis in the release of transmembrane proteins. Biochemistry. 1991;30:10065–10074. doi: 10.1021/bi00106a001. [DOI] [PubMed] [Google Scholar]
- Frisch SM, Francis H. Disruption of epithelial cell-matrix interactions induces apoptosis. J Cell Biol. 1994;124:619–626. doi: 10.1083/jcb.124.4.619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gearing AJH, et al. Processing of tumor necrosis factor-α precursor by metalloproteinases. Nature. 1994;370:555–557. doi: 10.1038/370555a0. [DOI] [PubMed] [Google Scholar]
- Gospodarowicz D, Cheng J, Lirette M. Bovine brain and pituitary fibroblast growth factors: comparison of their abilities to support the proliferation of human and bovine vascular endothelial cells. J Cell Biol. 1983;97:1677–1685. doi: 10.1083/jcb.97.6.1677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hase M, Araki S, Kaji K, Hayashi H. Classification of signals for blocking apoptosis in vascular endothelial cells. J Biochem. 1994;116:905–909. doi: 10.1093/oxfordjournals.jbchem.a124614. [DOI] [PubMed] [Google Scholar]
- Hermiston ML, Gordon JI. In vivo analysis of cadherin function in the mouse intestinal epithelium: essential roles in adhesion, maintenance of differentiation, and regulation of programmed cell death. J Cell Biol. 1995;129:489–506. doi: 10.1083/jcb.129.2.489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herren B, Raines EW, Ross R. Expression of a disintegrin-like protein in cultured vascular cells and in vivo. FASEB J. 1997;11:173–180. doi: 10.1096/fasebj.11.2.9039960. [DOI] [PubMed] [Google Scholar]
- Hooper NM, Karran EH, Turner AJ. Membrane protein secretases. Biochem J. 1997;321:265–279. doi: 10.1042/bj3210265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huber O, Korn R, McLaughlin J, Ohsugi M, Herrmann BG, Kemler R. Nuclear localization of β-catenin by interaction with transcription factor LEF-1. Mech Dev. 1996;59:3–10. doi: 10.1016/0925-4773(96)00597-7. [DOI] [PubMed] [Google Scholar]
- Hülsken J, Birchmeier W, Behrens J. E-cadherin and APC compete for the interaction with β-catenin and the cytoskeleton. J Cell Biol. 1994;127:2061–2069. doi: 10.1083/jcb.127.6.2061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaffe EA, Nachman RL, Becker CG, Minick RC. Culture of human endothelial cells derived from umbilical veins: identification by morphologic and immunologic criteria. J Clin Invest. 1973;52:2745–2756. doi: 10.1172/JCI107470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kayagaki N, Kawasaki A, Ebata T, Ohmoto H, Ikeda S, Inoue S, Yoshino K, Okumura K, Yagita H. Metalloproteinase-mediated release of human Fas ligand. J Exp Med. 1995;182:1777–1783. doi: 10.1084/jem.182.6.1777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korinek V, Barker N, Morin PJ, van Wichen D, de Weger R, Kinzler KW, Vogelstein B, Clevers H. Constitutive transcriptional activation by a β-catenin-Tcf complex in APC-/- colon carcinoma. Science. 1997;275:1784–1787. doi: 10.1126/science.275.5307.1784. [DOI] [PubMed] [Google Scholar]
- Kothakota S, et al. Caspase-3-generated fragment of gelsolin: effector of morphological change in apoptosis. Science. 1997;278:294–298. doi: 10.1126/science.278.5336.294. [DOI] [PubMed] [Google Scholar]
- Lampugnani MG, Corada M, Caveda L, Breviario F, Ayalon O, Geiger B, Dejana E. The molecular organization of endothelial cell to cell junctions: Differential association of plakoglobin, β-catenin, and α-catenin with vascular endothelial cadherin (VE-cadherin) J Cell Biol. 1995;129:203–217. doi: 10.1083/jcb.129.1.203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levkau B, Herren B, Koyama H, Ross R, Raines EW. Caspase-mediated cleavage of pp125FAK and disassembly of focal adhesions in human endothelial cell apoptosis. J Exp Med. 1998a;187:579–586. doi: 10.1084/jem.187.4.579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levkau B, Koyama H, Raines EW, Clurman BE, Herren B, Orth K, Roberts JM, Ross R. Cleavage of p21Cip1/Waf1 and p27Kip1 mediates apoptosis in endothelial cells through activation of Cdk2: role of a CPP 32-like caspase cascade. Mol Cell. 1998b;1:553–563. doi: 10.1016/s1097-2765(00)80055-6. [DOI] [PubMed] [Google Scholar]
- Martin SJ, O’Brien GA, Nishioka WK, McGahon AJ, Mahboubi A, Saido TC, Green DR. Proteolysis of fodrin (non-erythroid spectrin) during apoptosis. J Biol Chem. 1995;270:6425–6428. doi: 10.1074/jbc.270.12.6425. [DOI] [PubMed] [Google Scholar]
- McGeehan GM, et al. Regulation of tumor necrosis factor-α processing by a metalloproteinase inhibitor. Nature. 1994;370:558–561. doi: 10.1038/370558a0. [DOI] [PubMed] [Google Scholar]
- Mohler KM, et al. Protection against a lethal dose of endotoxin by an inhibitor of tumor necrosis factor processing. Nature. 1994;370:218–220. doi: 10.1038/370218a0. [DOI] [PubMed] [Google Scholar]
- Molenaar M, van de Wetering M, Oosterwegel M, Peterson-Maduro J, Godsave S, Korinek V, Roose J, Destrée O, Clevers H. XTcf-3 transcription factor mediates β-catenin-induced axis formation in Xenopus embryos. Cell. 1996;86:391–399. doi: 10.1016/s0092-8674(00)80112-9. [DOI] [PubMed] [Google Scholar]
- Moss ML, et al. Cloning of a disintegrin-metalloproteinase that processes precursor tumor-necrosis factor-α. Nature. 1997;385:733–736. doi: 10.1038/385733a0. [DOI] [PubMed] [Google Scholar]
- Müllberg J, Durie FH, Otten-Evans C, Alderson MR, Rose-John S, Cosman D, Black RA, Mohler KM. A metalloprotease inhibitor blocks shedding of the IL-6 receptor and the p60 TNF receptor. J Immunol. 1995;155:5198–5205. [PubMed] [Google Scholar]
- Munemitsu S, Albert I, Souza B, Rubinfeld B, Polakis P. Regulation of intracellular β-catenin levels by the adenomatous polyposis coli (APC) tumor-supressor protein. Proc Natl Acad Sci USA. 1995;92:3046–3050. doi: 10.1073/pnas.92.7.3046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navarro P, Caveda L, Breviario F, Mândoteanu I, Lampugnani MG, Dejana E. Catenin-dependent and -independent functions of vascular endothelial cadherin. J Biol Chem. 1995;270:30965–30972. doi: 10.1074/jbc.270.52.30965. [DOI] [PubMed] [Google Scholar]
- Nicholson DW, Thornberry NA. Caspases: killer proteases. Trends Biochem Sci. 1997;22:299–306. doi: 10.1016/s0968-0004(97)01085-2. [DOI] [PubMed] [Google Scholar]
- Orth K, O’Rourke K, Salvesen GS, Dixit VM. Molecular ordering of apoptotic mammalian CED-3/ICE-like proteases. J Biol Chem. 1996;271:20977–20980. doi: 10.1074/jbc.271.35.20977. [DOI] [PubMed] [Google Scholar]
- Pan D, Rubin GM. Kuzbanian controls proteolytic processing of Notch and mediates lateral inhibition during Drosophila and vertebrate neurogenesis. Cell. 1997;90:271–280. doi: 10.1016/s0092-8674(00)80335-9. [DOI] [PubMed] [Google Scholar]
- Paradies NE, Grunwald GB. Purification and characterization of NCAD90, a soluble endogenous form of N-Cadherin, which is generated by proteolysis during retinal development and retains adhesive and neurite-promoting function. J Neurosci Res. 1993;36:33–45. doi: 10.1002/jnr.490360105. [DOI] [PubMed] [Google Scholar]
- Rabiet MJ, Plantier JL, Rival Y, Genoux Y, Lampugnani M G, Dejana E. Thrombin-induced increase in endothelial permeability is associated with changes in cell-to-cell junction organization. Arterioscler Thromb Vasc Biol. 1996;16:488–496. doi: 10.1161/01.atv.16.3.488. [DOI] [PubMed] [Google Scholar]
- Re F, Zanetti A, Sironi M, Polentarutti N, Lanfrancone L, Dejana E, Colotta F. Inhibition of anchorage-dependent cell spreading triggers apoptosis in cultured human endothelial cells. J Cell Biol. 1994;127:537–546. doi: 10.1083/jcb.127.2.537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roark EF, Paradies NE, Lagunowich LA, Grunwald GB. Evidence for endogenous proteases, mRNA level and insulin as multiple mechanisms of N-cadherin down-regulation during retinal development. Development. 1992;114:973–984. doi: 10.1242/dev.114.4.973. [DOI] [PubMed] [Google Scholar]
- Rubinfeld B, Souza B, Albert I, Munemitsu S, Polakis P. The APC protein and E-cadherin form similar but independent complexes with α-catenin, β-catenin and plakoglobin. J Biol Chem. 1995;270:5549–5555. doi: 10.1074/jbc.270.10.5549. [DOI] [PubMed] [Google Scholar]
- Rubinfeld B, Robbins P, El-Gamil M, Albert I, Porfiri E, Polakis P. Stabilization of β-catenin by genetic defects in melanoma cell lines. Science. 1997;275:1790–1792. doi: 10.1126/science.275.5307.1790. [DOI] [PubMed] [Google Scholar]
- Rudel T, Bokoch GM. Membrane and morphological changes in apoptotic cells regulated by caspase-mediated activation of PAK2. Science. 1997;276:1571–1574. doi: 10.1126/science.276.5318.1571. [DOI] [PubMed] [Google Scholar]
- Sacco PA, McGranahan TM, Wheelock MJ, Johnson KR. Identification of plakoglobin domains required for association with N-cadherin and α-catenin. J Biol Chem. 1995;270:20201–20206. doi: 10.1074/jbc.270.34.20201. [DOI] [PubMed] [Google Scholar]
- Streuli M, Krueger NX, Ariniello PD, Tang M, Munro JM, Blattler JM, Adler DA, Disteche CM, Saito H. Expression of the receptor-linked protein tyrosine phosphatase LAR: proteolytic cleavage and shedding of the CAM-like extracellular region. EMBO J. 1992;11:897–907. doi: 10.1002/j.1460-2075.1992.tb05128.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talanian RV, Quinlan C, Trautz S, Hackett MC, Mankovich JA, Banach D, Ghayur T, Brady KD, Wong WW. Substrate specificities of caspase family proteases. J Biol Chem. 1997;272:9677–9682. doi: 10.1074/jbc.272.15.9677. [DOI] [PubMed] [Google Scholar]
- Thompson CB. Apoptosis in pathogenesis and treatment of disease. Science. 1995;267:1456–1462. doi: 10.1126/science.7878464. [DOI] [PubMed] [Google Scholar]
- Thornberry NA, et al. A combinatorial approach defines specificities of members of the caspase family and granzyme B. Functional relationships established for key mediators of apoptosis. J Biol Chem. 1997;272:17907–17911. doi: 10.1074/jbc.272.29.17907. [DOI] [PubMed] [Google Scholar]
- Uyttendaele H, Marazzi G, Wu G, Yan Q, Sassoon D, Kitajewski J. Notch4/int-3, a mammary proto-oncogene, is an endothelial cell-specific mammalian Notch gene. Development. 1996;122:2251–2259. doi: 10.1242/dev.122.7.2251. [DOI] [PubMed] [Google Scholar]
- van de Wetering M, et al. Armadillo coactivates transcription driven by the product of the Drosophila segment polarity gene dTCF. Cell. 1997;88:789–799. doi: 10.1016/s0092-8674(00)81925-x. [DOI] [PubMed] [Google Scholar]