

Abstract
Background
Pathological cardiac hypertrophy inevitably remodels, leading to functional decompensation. Although modulation of apoptosis-regulating genes occurs in cardiac hypertrophy, a causal role for programmed cardiomyocyte death in left ventricular (LV) remodeling has not been established.
Methods and Results
We targeted the gene for proapoptotic Nix, which is transcriptionally upregulated in pressure overload and Gq-dependent hypertrophies, in the mouse germ-line or specifically in cardiomyocytes (KO) and conditionally overexpressed it in the heart (TG). Conditional forced Nix expression acted synergistically with the pro-hypertrophic Gq transgene to increase cardiomyocyte apoptosis (0.8±0.1% in GqTG vs 7.8±0.6% in GqTG+NixTG, P<0.001) causing lethal cardiomyopathy with LV dilation and depressed systolic function (% fractional shortening (%FS): 39±4 vs 23±4, P=0.042). In the reciprocal experiment, germ-line Nix ablation significantly reduced cardiomyocyte apoptosis (4.8±0.2% in GqTG+NixKO vs 8.4±0.5% in GqTG, P=0.001), which improved %FS (43±3% vs 27±3%, P=0.017), attenuated LV remodeling, and largely prevented lethality in the Gq peripartum model of apoptotic cardiomyopathy. Cardiac-specific (Nkx2.5-Cre) Nix KO mice subjected to transverse aortic constriction (TAC) developed significantly less LV dilation by echocardiography and magnetic resonance imaging, maintained concentric remodeling, and exhibited preserved LVEF (61±2% in TAC cardiac Nix KO vs 36±6% in TAC wild-type, P=0.003) at 9 weeks, with reduced cardiomyocyte apoptosis at day 4 (1.70±0.21% vs 2.73±0.35%, P=0.032).
Conclusions
Nix-induced cardiomyocyte apoptosis is a major determinant of adverse remodeling in pathological hypertrophies, which suggests therapeutic value for apoptosis inhibition to prevent cardiomyopathic decompensation.
Keywords: apoptosis, hypertrophy, cardiomyopathy
Introduction
Cardiac hypertrophy is an independent risk factor for death largely because chronically hypertrophied hearts remodel and dilate, progressing from a stable compensation to dilated cardiomyopathy.1 A better understanding of the mechanisms for functional decompensation of cardiac hypertrophy in response to hemodynamic overload is essential to develop effective preventative measures and therapeutics. A number of candidate pathological events have been identified, including bio-energetically unfavorable changes in contractile protein isoforms,2 the metabolically adverse transition from fatty acid to glucose utilization,3;4 degradation of the cardiac matrix and resulting myocyte slippage,5 and a relative decrease in myocardial vascularization resulting in oxidative stress.6 Each of these factors becomes an additional physiological stressor for hemodynamically overloaded cardiomyocytes, and in combination with the primary stimulus can ultimately overwhelm protective cell survival pathways5 and result in apoptotic cardiomyocyte dropout with replacement fibrosis. Apoptotic loss of myocardium itself can increase hemodynamic stress through ventricular dilation and wall thinning, and is therefore hypothesized to play an important role in the downward functional spiral that ultimately leads to overt heart failure.5;7 While cardiomyocyte apoptosis is commonly observed in pressure overload hypertrophy,7-10 and causes myocardial disease in experimental models wherein it was artificially induced,11-15 the degree to which it contributes to ventricular remodeling in naturally-occurring hypertrophy decompensation remains unclear. Nor is it known whether cardiac myocytes that are programmed to die in pressure overload hypertrophy may nevertheless die a necrotic death if apoptosis is prevented, in which case inhibiting apoptosis would likely prove ineffective in preventing decompensation.
One approach to testing the hypothesis that apoptosis is a critical pathophysiological nodal point for multiple factors that induce hypertrophy decompensation, and for testing the therapeutic efficacy of apoptosis prevention to interrupt the feed-forward cycle of functional deterioration of pressure overload hypertrophy, is pharmacological caspase inhibition.16 This approach is limited, however, by potential toxicity and lack of absolute specificity of these compounds, and has not been reported in pressure overload hypertrophy. We considered that a more selective tactic of identifying pro-apoptotic factor(s) responsible for apoptosis after cardiac pressure overloading and individually manipulating them in the heart could better validate the hypothesis. Previously, we used DNA microarray analysis to identify Nix as a candidate hypertrophy-stimulated pro-apoptotic factor.17 Nix is a nearly ubiquitous member of the Bcl2 family of mitochondrial-localized proteins that is expressed at very low levels in normal hearts, but was strikingly upregulated in cardiac-specific Gq-overexpressing mice, a genetic model that recapitulates the molecular pathways and essential features of pressure overload hypertrophy.18;19 Nix provokes apoptosis in transfected cells and acts synergistically with normal maturational growth to cause apoptotic heart failure in neonatal mice, but its overexpression alone was not sufficient to cause apoptotic cardiac decompensation in normal adult hearts.13;15 It is not known whether Nix is essential for apoptosis that occurs in Gq-mediated or pressure overload hypertrophy, and therefore represents a specific, targetable apoptotic effector of hypertrophy decompensation. To test this, we used cardiac-specific conditional Nix gain- and loss-of-function mouse models in combination with genetic and physiological stress. Our results support a critical role for Nix in remodeling of hemodynamically overloaded hearts and more clearly define the consequences of cardiomyocyte apoptosis in functional decompensation after pressure overload hypertrophy.
Methods
Generation and characterization of genetically modeled mice
Mice with cardiac-specific Gq overexpression or inducible, attenuated expression of Nix were described previously13;18 and were inter-bred. Gq transgenic mice were mated with Nix null mice20 for peripartum cardiomyopathy studies.
To produce cardiac Nix del animals, mice homozygous for floxed Nix alleles (Nixf/f)20 were mated with Nkx-Cre knock-in mice.21 Nixf/f mice were used as wild-type controls. Mice were housed and studied according to procedures approved by the University of Cincinnati Institutional Animal Care and Use Committee. 2D directed M-mode echocardiography, histopathology and TUNEL studies were performed as described previously.13
Expression analysis with quantitative PCR
1 μg total RNA purified from snap-frozen mouse hearts using Trizol (Invitrogen, Carlsbad, CA), was reverse-transcribed using oligo(dT). Amplicons spanning exons 2-4 of Nix (Bnip3L, GenBank NM_009761, nts 298-414) were detected using SYBR Green I during 35 cycles of qPCR (95 C 15 s, 60 C 1 min) using 5′-AAGAGGCAGTTCGCACTGTGACA-3′ (forward) and 5′-TCTACAACTTCTTCTTCTGACTGAGAGCTG-3′ (reverse) primers. TaqMan assays for ANF, SERCA2a, alpha-skeletal actin, and GAPDH were purchased from Applied Biosystems.
Studies with pressure overload modeling
Twelve-week-old mice underwent acute pressure overloading by surgical TAC.19 Periprocedural mortality was ∼25% in both groups. Cardiac MRI studies were performed as described.22 Terminal invasive studies for assessment of transcoarctation gradient and left ventricular hemodynamics were performed as described previously.19
Myocardial histomorphometric analysis
Myocyte cross sectional area was determined using FITC tagged wheat germ agglutinin labeling.23 TUNEL staining used the DeadEnd™ Flourometric TUNEL system (Promega, WI) counterstained with α-sarcomeric actin (Invitrogen, CA) and DAPI (Vector Laboratories) and imaged using a UV transparent 100X oil immersion objective. Only TUNEL positive nuclei within sarcomeric actin stained cardiomyocytes were counted. Ceaved caspase 3 and PARP were analyzed as described.24 Masson's trichrome stained myocardial sections were imaged (200X) and collagen area calculated as percent of total left ventricular myocardial area using NIH Image software.
Statistical Analysis
Results are mean±SEM. Experimental groups were compared using Student t-test for comparision between 2 groups, one-way ANOVA for comparing multiple groups or two-way repeated measures ANOVA for time dependent changes between various groups, followed by Tukey's post-hoc test. Nonparametric testing was employed when data were not normally distributed. Dunn's post-hoc test was employed after ANOVA on ranks. Log rank test was employed for survival analyses. P<0.05 was considered significant.
The authors had full access to and take full responsibility for the integrity of the data. All authors have read and agree to the manuscript as written.
Results
Nix synergizes with Gq-mediated hypertrophy to cause lethal apoptotic heart failure
Nix gene expression increases in human and experimental pressure overload hypertrophy.15;17;24 We previously found that increased expression of Nix alone did not cause functionally-significant cardiomyocyte apoptosis in normal adult mice, but that Nix caused progressive apoptotic cardiac dilation in neonatal mice,13 and that pro-apoptotic effects of Nix were inversely proportional to the rate of maturational cardiomyocyte growth. Here, to define possible synergies between Nix and pathological cardiomyocyte growth,25 we examined the consequences of conditional forced Nix expression in mice with Gq-mediated hypertrophy. Overexpression of Gq is a cardiac myocyte-specific genetic stimulus for pathological hypertrophy that recapitulates many characteristics of compensated pressure overload hypertrophy.18;19
As previously observed,13 cardiac-specific overexpression of Nix beginning in the neonatal period (Figure 1A) increased cardiomyocyte apoptosis to levels (∼4%; Figure 1D) that, over time, significantly diminished echocardiographic left ventricular fractional shortening (%FS, Figure 1C, black bars). Survival to adulthood was not affected (Figure 1B), nor was left ventricular remodeling induced, measured as the ratio of ventricular radius to wall thickness, r/h (Figure 1C, white bars). Gq transgenic mice exhibited characteristic functional impairment (decreased %FS) and mild ventricular remodeling (Figures 1A and 1C), without cardiomyocyte apoptosis (Figure 1D) or mortality (not shown).18;19 Strikingly, although Nix expression was similar in Nix and Nix/Gq compound transgenic hearts (Figure 1D, inset), combined postnatal cardiac overexpression of Nix and Gq proved rapidly lethal (Figure 1B). At six weeks, cardiomyocyte apoptosis was doubled in Nix/Gq mice compared to Nix alone (Figure 1D), with ventricular remodeling (Figure 1A and Figure 1C, white bars) and dramatically depressed %FS (Figures 1A and 1C, black bars). These results demonstrate that Gq-mediated pathological hypertrophy exacerbates apoptosis caused by Nix overexpression, providing additional evidence for synergy between cardiac growth and death pathways.25
Figure 1. Apoptotic synergy between cardiac-expressed Nix and Gq in neonatal mice.
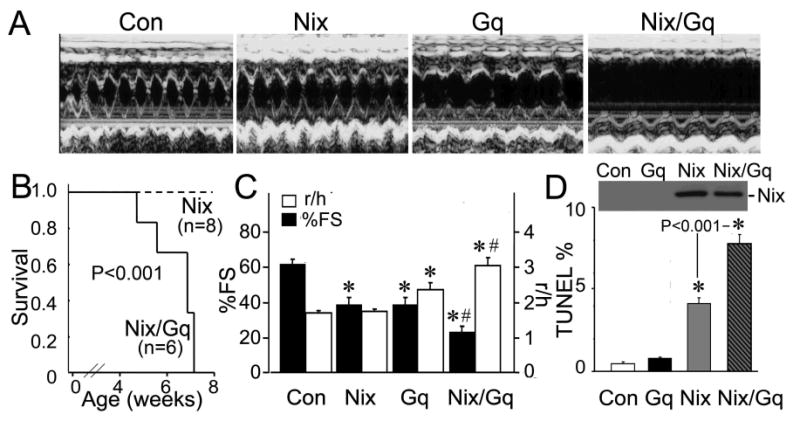
A. Representative left ventricular (LV) M-mode echocardiograms in short axis view at 6 weeks age. B. Survival curves (P value by log-rank test). C. Echocardiographic left ventricular fractional shortening (black, %FS) and remodeling (LV radius/wall thickness, white, r/h; n=4-8/group). D. Representative western blot for Nix (inset) and apoptotic indices by TUNEL analysis (n=3/group). *= P<0.05 vs control, #=P<0.05 for Nix/Gq vs Gq by post-hoc test after one-way ANOVA.
Nix gene ablation prevents Gq-mediated apoptotic peripartum cardiomyopathy
Nix is upregulated in Gq-mediated cardiac hypertrophy,15;17 and the above results show that Gq-mediated hypertrophy contributes to apoptosis caused by Nix expression. A unique manifestation of apoptotic myocardial disease in the Gq mouse is lethal peripartal heart failure.16;23 To demonstrate that Nix-mediated apoptosis contributes to peripartal heart failure in the Gq mouse, we performed a genetic rescue experiment by crossing Gq transgenic mice to mice lacking a functional Nix gene (Nix null). Germ-line Nix null mice exhibit diminished apoptosis during normal erythroblast maturation,20 but have no detectable cardiac abnormalities (unpublished results and Figures 2C and 2D). Crossing α-myosin heavy chain-directed Gq transgenic mice with Nix null mice produces a model in which Gq expression is cardiomyocyte-specific and Nix ablation is systemic. Thus, cardiac phenotypes reflect the cardiac specificity of the hypertrophy stimulus.
Figure 2. Nix gene ablation diminishes apoptosis in Gq-mediated peripartum cardiomyopathy, improving function and minimizing death.
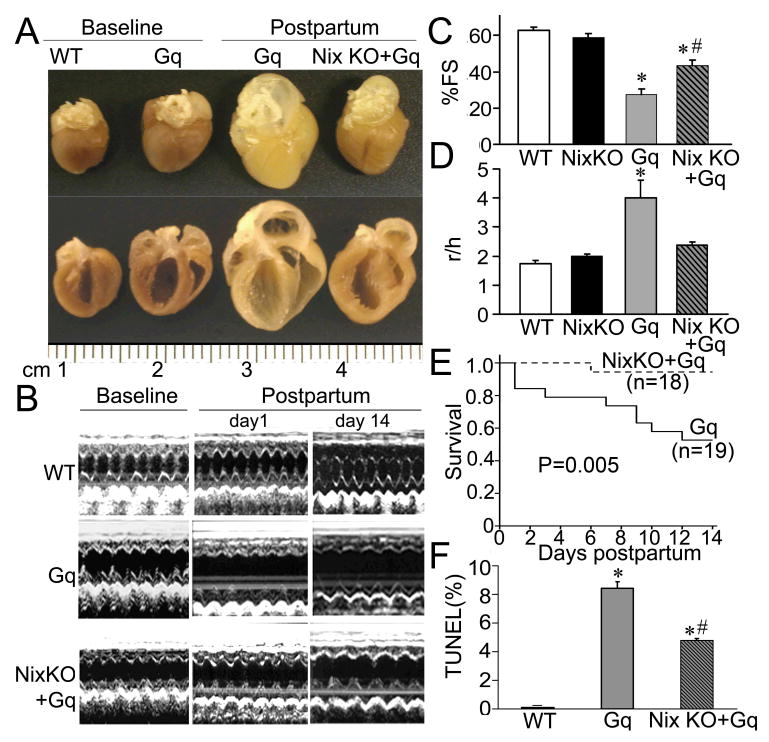
A. Representative non-pregnant (baseline) and post-partum 14 day hearts. B. Representative LV M-mode echocardiograms. C. Echocardiographic LV fractional shortening (%FS) and D. remodeling (LV radius/wall thickness, r/h; n=4-6/group). E. Kaplan Meier survival curves for peripartum Gq expressors, with and without Nix (P value by log-rank test). F. Apoptotic indices at 1 day post-partum (n=4/group), *= P<0.05 vs WT, #=P<0.05 for NixKO+Gq vs Gq by post-hoc test after one-way ANOVA.
There were no cardiac effects of parturition or the peripartum state in Nix null mice (not shown). As previously observed,16;23 Gq mice developed peripartal cardiac enlargement and chamber dilation (Figures 2A and 2B) with striking left ventricular remodeling (increase in r/h; Figure 2D), and a >50% decline in left ventricular fractional shortening (Figures 2B and 2C). Mortality from heart failure in peripartum Gq mice was approximately 50% (Figure 2E), and the rate of cardiomyocyte apoptosis (TUNEL positivity) on the first post-partum day was ∼8% (Figure 2F). By comparison, syngeneic Gq mice lacking a functional Nix gene had reduced peripartal cardiac enlargement and left ventricular dilatation (NixKO+Gq, Figures 2A and B), without remodeling, but with significantly improved ejection performance, and decreased mortality and cardiomyocyte apoptosis (Figures 2C-F). These data establish the importance of Nix-mediated apoptosis in the development of, and mortality that results from, apoptotic cardiomyopathy in peripartum Gq overexpressing mice.
Cardiomyocyte-specific ablation of Nix prevents decompensation of pressure overload hypertrophy
Taken together, the above results show that Nix is necessary and sufficient for apoptotic decompensation of Gq-mediated hypertrophy. To determine whether Nix plays an analogous role in pressure overload hypertrophy (which is also Gq-dependent26;27) in a manner that would not be confounded by extra-cardiac effects of germ-line Nix ablation,20 we ablated the Nix gene specifically in cardiomyocytes using Nkx-2.5-driven Cre21 (Figure 3A). Cardiomyocyte-specific Nix knockout mice (cardiac Nix del) were viable and exhibited no cardiac abnormalities by gross morphometry or echocardiography (Table 1 and data not shown). Compared to wild-type control mice, myocardial Nix mRNA levels measured by real-time quantitative PCR were decreased by 78% at baseline in cardiac Nix del mice, and showed no change four days after imposition of a ∼84 mm Hg trans-aortic gradient by surgical transverse aortic coarctation (TAC), compared to doubling of Nix mRNA after TAC in wild-type mice (Figure 3B). Since Nix mRNA was not detectable using this assay in hearts from germ-line Nix null mice (Figure 3B), Nix mRNA at baseline and after TAC in cardiac Nix del hearts likely represents constitutive Nix gene expression in non-myocyte myocardial cells.28-32
Figure 3. Cardiac-specific Nix ablation prevents functional decompensation after acute transverse aortic coarctation (TAC).
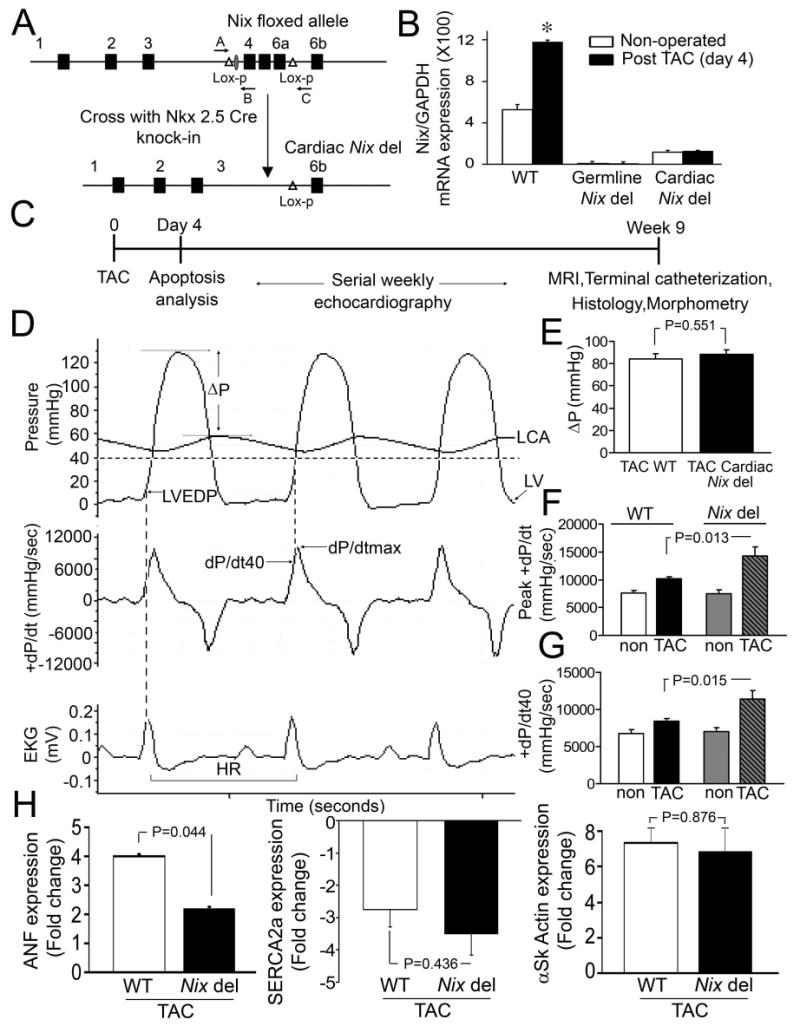
A. Cardiac-specific Nix ablation strategy. B. qPCR of Nix mRNA expression after TAC (n=3/group). *= P<0.05 vs non-operated by t-test. C. Schematic of TAC modeling studies. D. Representative invasively determined hemodynamic tracings after TAC (ΔP:transcoarctation gradient, LVEDP:LV end-diastolic pressure, HR: heart rate). E. Transcoarctation gradient in cardiac Nix del mice (n=11) and controls (n=13). P value reported is by t-test. G. Peak positive dP/dt and H. dP/dt at 40 mm Hg LV pressure in cardiac Nix del mice and Nix floxed controls subjected to TAC (n=4/non-operated group; n=11-13/TAC group; P values reported are by t-test for comparision of TAC groups.) Non operated mice are shown for comparision. H. Change in ANF, SERCA2a, and α-skeletal actin mRNA levels four days after TAC in WT (white bars) and cardiac Nix del (black bars).
Table 1. Morphometric and hemodynamic parameters of cardiac Nix del mice nine weeks after transverse aortic constriction.
| Non-operated WT
(n=6) |
TAC WT
(n=13) |
% change | Non-operated Cardiac Nix del
(n=6) |
TAC Cardiac Nix del
(n=11) |
% change | P value | |
|---|---|---|---|---|---|---|---|
| Heart /body weight (mg/g) | 4.9±0.1 | 8.8±0.9* | 79±16 | 5.3±0.2 | 9.1±0.5* | 71±10 | 0.954 |
| Peak LV pressure (mmHg) | 99±5 | 161±5* | 62±5 | 98±7 | 171±6* | 75±6 | 0.093 |
| Peak positive dP/dt (mmHg/sec) | 7596±441 | 10189±368* | 34±5 | 7507±627 | 14304±1571* | 91±21 | 0.006 |
| Peak negative dP/dt (mmHg/sec) | -8070±576 | -12090±368* | 50±5 | -7740±589 | -15688±1756* | 103±23 | 0.056 |
| dP/dt40 (mmHg/sec) | 6742±569 | 8432±311* | 25±4 | 6990±567 | 11368±1154* | 63±17 | 0.049 |
| LV end-diastolic pressure (mmHg) | 3±2 | 10±1* | 243±45 | 4±2 | 10±2 | 160±60 | 0.301 |
All data are shown as Mean±SEM. P values are for comparision of % change in TAC group between wild type (WT) and cardiac Nix del by Student's t-test.
indicates P<0.05 vs respective non-operated group by Student's t-test.
Cardiac Nix del and wild-type mice underwent surgical TAC,19 producing gradients of 88±4 and 84±4mmHg, respectively (P=NS, Figures 3D top and 3E). The time courses of hypertrophy development (LV mass), functional decompensation (fractional shortening, %FS), and left ventricular remodeling (LV end diastolic dimension [LVEDD] and r/h) were determined by serial echocardiography, followed by terminal invasive hemodynamic studies, cardiac morphometrics, and histological examinations of the myocardium (Figure 3C). Gravimetric cardiac hypertrophy measured nine weeks after TAC, hypertrophy-associated changes in α-skeletal actin and sarcoplasmic reticulum ATPase (SERCA2a) gene expression measured four days after TAC and the rate of left ventricular hypertrophy development determined by weekly echocardiography (Table 1, Figures 3H and 4A), were identical in cardiac Nix del mice and wild-type controls, showing that absence of Nix does not affect the hypertrophic response to pressure overload. In contrast, terminal invasive functional assessments of cardiac Nix del mice revealed enhanced myocardial contractile function late after pressure overloading measured as peak positive LV dP/dt or dP/dt at 40 mmHg LV systolic pressure (Figures 3D, F, and G, and Table 1). Echocardiography confirmed enhanced systolic performance (Figures 4 D-F) and further demonstrated that the differences between TAC wild-type and cardiac Nix del mice in left ventricular diastolic chamber size (Figure 4B), remodeling (r/h; Figure 4C), and systolic performance (Figures 4D-F) were observed by two weeks after TAC, with the trend lines remaining roughly parallel thereafter. These findings, together with a significantly smaller increase in failure-associated ANF mRNA levels (Figure 3H), suggest that the benefits of cardiac myocyte Nix ablation on left ventricular remodeling and ejection performance in pressure overloaded hearts largely accrue early after TAC, when apoptosis is most active.
Figure 4. Cardiac-specific Nix ablation prevents structural remodeling and functional decompensation after TAC.
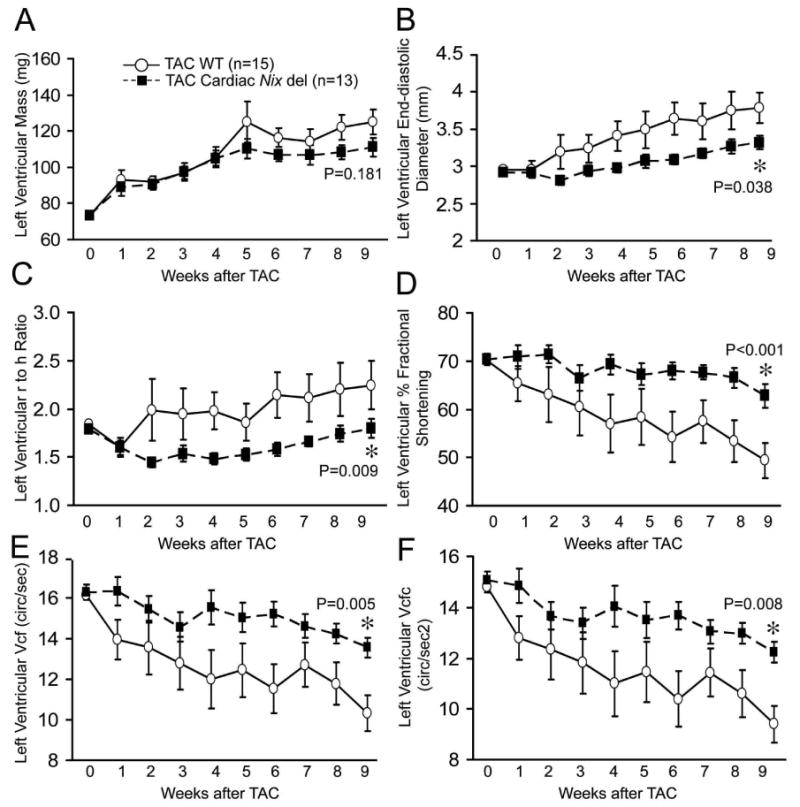
Time-dependent echocardiographic outcomes after TAC in control (Nix floxed, white circles, n=15) and cardiac Nix del (black squares, n=13) mice. A. LV mass. B. LV end-diastolic dimension (LVEDD). C. Ratio of LV radius to wall thickness (r/h). D. LV fractional shortening (%FS). E. Vcf: Velocity of circumferential shortening. F. Vcfc: Vcf corrected for heart rate. P values by post-hoc test vs WT TAC after two-way repeated measures ANOVA.
To better assess the consequences of cardiomyocyte-specific Nix ablation on ventricular remodeling and contractile function after pressure overloading, a cohort of surgically pressure overloaded mice underwent magnetic resonance imaging (MRI).33;34 Compared to non-operated controls, MRI of wild-type mice nine weeks after TAC showed increased left ventricular mass and end-diastolic volume, with reduced sphericity index (increased sphericity), and a ∼50% decline in volumetric ejection fraction (Figures 5A and B and Table 2), representing typical left ventricular remodeling and functional deterioration after acute pressure overloading.35 In contrast, and consistent with the echocardiographic and invasive hemodynamic findings, pressure overloaded cardiac Nix del mice showed no significant left ventricular dilation, and had enhanced left ventricular ejection fraction and a normal left ventricular sphericity index, despite comparable hypertrophy (Figures 5A and B and Table 2). These results reinforce the findings of echocardiographic and terminal studies, that Nix ablation prevents left ventricular remodeling and minimizes left ventricular functional declines in chronically pressure overloaded hearts.
Figure 5. Cardiac-specific Nix ablation abrogates LV remodeling after TAC.
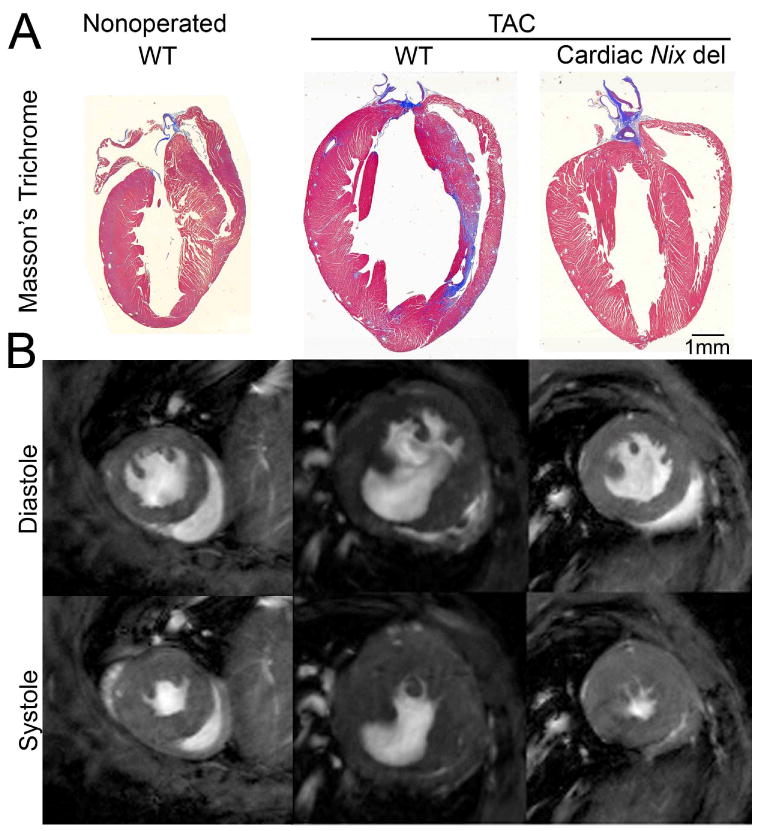
A. Representative Masson's trichrome stained coronal sections. B. Representative magnetic resonance images at 9 weeks after TAC.
Table 2. Magnetic resonance studies of left ventricular structure and function in cardiac Nix del mice nine weeks after transverse aortic constriction.
| Non-operated
(n=4) |
TAC WT
(n=7) |
TAC Cardiac Nix del
(n=5) |
|
|---|---|---|---|
| End-diastolic volume (μl) | 43±4 | 93±9* | 51±4# |
| End-systolic volume (μl) | 13±1 | 62±10* | 20±2 |
| Ejection fraction (%) | 71±2 | 36±5* | 61±2 |
| Mass (mg) | 57±5 | 142±14* | 114±16* |
| Sphericity index | 1.96±0.05 | 1.56±0.06* | 1.84±0.05# |
All data are shown as Mean±SEM. All groups were compared by one-way ANOVA.
indicates P<0.05 vs non-operated WT and
indicates TAC Cardiac Nix del vs TAC WT by post-hoc test (Tukey's).
Cardiomyocyte specific ablation of Nix diminishes myocardial apoptosis in response to pressure overload
Taken together, the above studies demonstrate that cardiac myocyte-specific Nix ablation largely prevents remodeling and functional deterioration in Gq-mediated and pressure overload hypertrophies. In the Gq model, the benefits of Nix ablation and the deleterious effects of Nix overexpression were associated with reciprocal effects on cardiac myocyte apoptosis. To interrogate the effects of Nix ablation on cardiac myocytes in pressure overload hypertrophy, studies of cardiac myocyte hypertrophy, apoptosis, and replacement fibrosis were performed. Cardiac myocyte cross-sectional area, which is increased in pressure overload hypertrophy,36 showed similar ∼35% increases in wild-type and cardiac Nix del mice nine weeks after TAC (Figure 6A), indicating that Nix ablation does not affect cellular hypertrophy.
Figure 6. Cardiac-specific Nix ablation prevents cardiomyocyte apoptosis and myocardial fibrosis after TAC.

Representative myocardial sections showing: A. Cardiomyocyte cross section area (FITC tagged wheat germ agglutinin labeling (200X); B. TUNEL positivity (1000X); C. Cleaved caspase 3 (brown staining; left panels (400X) and immunoblot (right) showing cleaved PARP (top) with Ponceau red loading control (bottom). D. Myocardial fibrosis (Masson's trichrome, (200X) nine weeks after TAC in cardiac Nix del mice and Nix floxed controls. Group data are in bar graphs to the right (n=4/non-operated group and n=6-12/TAC group). P values by t-test (TAC WT vs TAC Nix del). Non operated mice are shown for comparison.
Apoptosis studies in control mice four days after TAC showed a ∼5-fold increase in the number of TUNEL positive cardiomyocytes compared to baseline (Figure 6B), with increased caspase 3 and PARP cleavage (Figure 6C). By comparison, cardiac Nix del hearts after TAC had less TUNEL positivity (Figure 6B) and caspase 3 and PARP cleavage (Figure 6C). Late fibrotic replacement of dead cardiac myocytes37 was also reduced by approximately half in cardiac Nix del subjected to TAC, compared to identically-treated controls (Figure 6D). Since provocation of mitochondrial/intrinsic pathway apoptosis is the only known cellular function of Nix,20 and Nix is specifically upregulated in pressure overload hypertrophy,15;24 these results are consistent with the notion that the geometrical and functional benefits afforded by cardiomyocyte Nix ablation after pressure overload are a consequence of reduced myocardial apoptotic cell loss.
Discussion
The current studies mechanistically link cardiomyocyte apoptosis with ventricular remodeling and functional deterioration during the transition from compensated pressure overload hypertrophy to decompensated heart failure. They further identify induction of Nix gene expression as a critical molecular event causing apoptotic cardiac myocyte drop-out in pressure overload and Gq-mediated hypertrophy. The efficacy of cardiomyocyte salvage by apoptosis inhibition under conditions of where there is no relief from the primary inciting stimulus demonstrates that preventing apoptotic cardiomyocyte death does not simply commit the cell to a necrotic one.
We selected Nix as the candidate effector of apoptosis for our studies of hypertrophy decompensation because Nix transcripts are specifically upregulated in pathological hypertrophy.15;24 Nix protein rapidly localizes to mitochondrial outer membranes via a carboxyl-terminal hydrophobic localization domain, without which Nix lacks apoptotic activity.15;38 Nix protein also undergoes rapid proteolytic degradation,38;39 revealing that apoptosis induced by it is tightly regulated both by rapid “start” and “stop” functions. At the mitochondria, Nix promotes the release of cytochrome c by supporting permeabilization of the mitochondrial outer membrane,20 almost certainly by stimulating oligomerization of the multi-domain pore-forming Bcl-2 proteins Bax and Bak.40 Addition of recombinant Nix to isolated mitochondria does not open permeability transition pores,20 although this does occur in intact cells undergoing Gq-mediated apoptosis.23;41;42 Thus, available evidence indicates that Nix is a hypertrophy-inducible apical regulator of apoptosis mediated strictly via the intrinsic, mitochondrial pathway.43
It is notable that Nix ablation in cardiac myocytes and the germ line did not affect the cardiac response to two hypertrophic stimuli. Thus, the functional and structural benefits of Nix ablation are independent of the magnitude of the hypertrophic response. This observation is not fully consistent with the proposition that “pathological hypertrophy” is intrinsically harmful, or at least fully dispensable, in pressure overload,44-46 and suggests that hypertrophy can be tolerated if its deleterious aspects are neutralized. This notion is also consistent with recent studies demonstrating that micro-ischemia due to inadequate angiogenesis contributes to hypertrophy decompensation, and that enhancing angiogenesis can prevent this.6;47;48 It is interesting to speculate that micro-ischemia in cardiac hypertrophy may actually contribute to hypertrophic Nix gene induction, as hypoxia has been shown to increase Nix transcripts in cultured tumor cell lines,49;50 and in the current studies Nix overexpression alone was not sufficient to cause apoptotic heart failure, whereas the same level of Nix expression in the context of Gq-mediated hypertrophy proved lethal.
Why was hypertrophy-associated apoptosis incompletely suppressed by Nix ablation? First, Nix may not be the only mitochondrial apoptotic factor induced in cardiac hypertrophy. We and others have shown that closely related Bnip3 is strongly induced in cardiomyocyte ischemia.24;51;52 Bnip3 and Nix have nearly identical apoptotic effects in cells,38 and may interact cooperatively in the heart (Dorn and Diwan, unpublished results). Recent evidence that failure of angiogenesis contributes to functional hypertrophy decompensation6;47;53 suggests that cardiomyocyte ischemia may be unavoidable in pressure overload hypertrophy. If so, co-induction of hypertrophic and ischemic apoptotic factors may be the rule, and elimination of apoptosis will not be accomplished by targeting a single gene or gene product.
The second likelihood is that extrinsic, death receptor apoptosis pathways contribute to apoptosis after pressure overload. Certainly, loss of the interleukin-6/gp130 cytokine receptor and its cardiomyocyte survival function leads to dramatic apoptotic decompensation after pressure overloading.12 The pro-inflammatory/pro-apoptotic cytokine tumor necrosis factor-α (TNFα) also contributes to decompensation of pressure overload hypertrophy.54 However, although there is cross-talk between mitochondrial and death receptor apoptosis pathways,55 Nix is not involved. Thus, it is likely that Nix modulates only apoptosis transduced via the intrinsic pathway in decompensating pressure overload hypertrophy, but that death receptor pathways can also contribute.
In conclusion, the current results support the proposition that programmed cardiomyocyte death is at a junction between multiple mechanical and molecular factors that contribute to heart failure, and demonstrate that moderating cardiomyocyte apoptosis can interrupt the cycle of physiological stress leading to myocyte drop-out that, in turn, exacerbates the physiological stress. Retention of cardiac myocytes that were programmed to die is a form of myocardial salvage that helps to maintain normal chamber wall thickness and dimension, and therefore preserves cardiac ejection performance and hemodynamic homeostasis.
Acknowledgments
Funding sources: Supported by NHLBI HL059888, HL077101, and HL080008 (GW Dorn II), American Heart Association (Scientist Development Grant, A Diwan) and the Veterans Administration.
Abbreviations
- TG
transgenic
- KO
knockout
- MRI
magnetic resonance imaging
Footnotes
Conflict of interest disclosures: None
Reference List
- 1.Levy D, Garrison RJ, Savage DD, Kannel WB, Castelli WP. Prognostic implications of echocardiographically determined left ventricular mass in the Framingham Heart Study. N Engl J Med. 1990;322:1561–1566. doi: 10.1056/NEJM199005313222203. [DOI] [PubMed] [Google Scholar]
- 2.Scheuer J, Malhotra A, Hirsch C, Capasso J, Schaible TF. Physiologic cardiac hypertrophy corrects contractile protein abnormalities associated with pathologic hypertrophy in rats. J Clin Invest. 1982;70:1300–1305. doi: 10.1172/JCI110729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dorn GW. The fuzzy logic of physiological cardiac hypertrophy. Hypertension. 2007;49:962–970. doi: 10.1161/HYPERTENSIONAHA.106.079426. [DOI] [PubMed] [Google Scholar]
- 4.Massie BM, Schaefer S, Garcia J, McKirnan MD, Schwartz GG, Wisneski JA, Weiner MW, White FC. Myocardial high-energy phosphate and substrate metabolism in swine with moderate left ventricular hypertrophy. Circulation. 1995;91:1814–1823. doi: 10.1161/01.cir.91.6.1814. [DOI] [PubMed] [Google Scholar]
- 5.Diwan A, Dorn GW. Decompensation of cardiac hypertrophy: cellular mechanisms and novel therapeutic targets. Physiology (Bethesda) 2007;22:56–64. doi: 10.1152/physiol.00033.2006. [DOI] [PubMed] [Google Scholar]
- 6.Dorn GW. Myocardial angiogenesis: its absence makes the growing heart founder. Cell Metab. 2007;5:326–327. doi: 10.1016/j.cmet.2007.04.003. [DOI] [PubMed] [Google Scholar]
- 7.Teiger E, Than VD, Richard L, Wisnewsky C, Tea BS, Gaboury L, Tremblay J, Schwartz K, Hamet P. Apoptosis in pressure overload-induced heart hypertrophy in the rat. J Clin Invest. 1996;97:2891–2897. doi: 10.1172/JCI118747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.De Windt LJ, Lim HW, Taigen T, Wencker D, Condorelli G, Dorn GW, Kitsis RN, Molkentin JD. Calcineurin-mediated hypertrophy protects cardiomyocytes from apoptosis in vitro and in vivo: An apoptosis-independent model of dilated heart failure. Circ Res. 2000;86:255–263. doi: 10.1161/01.res.86.3.255. [DOI] [PubMed] [Google Scholar]
- 9.Sadoshima J, Montagne O, Wang Q, Yang G, Warden J, Liu J, Takagi G, Karoor V, Hong C, Johnson GL, Vatner DE, Vatner SF. The MEKK1-JNK pathway plays a protective role in pressure overload but does not mediate cardiac hypertrophy. J Clin Invest. 2002;110:271–279. doi: 10.1172/JCI14938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.van Empel VP, Bertrand AT, van der N R, Kostin S, Doevendans PA, Crijns HJ, de Wit E, Sluiter W, Ackerman SL, De Windt LJ. Downregulation of apoptosis-inducing factor in harlequin mutant mice sensitizes the myocardium to oxidative stress-related cell death and pressure overload-induced decompensation. Circ Res. 2005;96:e92–e101. doi: 10.1161/01.RES.0000172081.30327.28. [DOI] [PubMed] [Google Scholar]
- 11.Donath S, Li P, Willenbockel C, Al Saadi N, Gross V, Willnow T, Bader M, Martin U, Bauersachs J, Wollert KC, Dietz R, von Harsdorf R. Apoptosis repressor with caspase recruitment domain is required for cardioprotection in response to biomechanical and ischemic stress. Circulation. 2006;113:1203–1212. doi: 10.1161/CIRCULATIONAHA.105.576785. [DOI] [PubMed] [Google Scholar]
- 12.Hirota H, Chen J, Betz UA, Rajewsky K, Gu Y, Ross J, Jr, Muller W, Chien KR. Loss of a gp130 cardiac muscle cell survival pathway is a critical event in the onset of heart failure during biomechanical stress. Cell. 1999;97:189–198. doi: 10.1016/s0092-8674(00)80729-1. [DOI] [PubMed] [Google Scholar]
- 13.Syed F, Odley A, Hahn HS, Brunskill EW, Lynch RA, Marreez Y, Sanbe A, Robbins J, Dorn GW. Physiological growth synergizes with pathological genes in experimental cardiomyopathy. Circ Res. 2004;95:1200–1206. doi: 10.1161/01.RES.0000150366.08972.7f. [DOI] [PubMed] [Google Scholar]
- 14.Wencker D, Chandra M, Nguyen K, Miao W, Garantziotis S, Factor SM, Shirani J, Armstrong RC, Kitsis RN. A mechanistic role for cardiac myocyte apoptosis in heart failure. J Clin Invest. 2003;111:1497–1504. doi: 10.1172/JCI17664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yussman MG, Toyokawa T, Odley A, Lynch RA, Wu G, Colbert MC, Aronow BJ, Lorenz JN, Dorn GW. Mitochondrial death protein Nix is induced in cardiac hypertrophy and triggers apoptotic cardiomyopathy. Nat Med. 2002;8:725–730. doi: 10.1038/nm719. [DOI] [PubMed] [Google Scholar]
- 16.Hayakawa Y, Chandra M, Miao W, Shirani J, Brown JH, Dorn GW, Armstrong RC, Kitsis RN. Inhibition of cardiac myocyte apoptosis improves cardiac function and abolishes mortality in the peripartum cardiomyopathy of Galpha(q) transgenic mice. Circulation. 2003;108:3036–3041. doi: 10.1161/01.CIR.0000101920.72665.58. [DOI] [PubMed] [Google Scholar]
- 17.Aronow BJ, Toyokawa T, Canning A, Haghighi K, Delling U, Kranias E, Molkentin JD, Dorn GW. Divergent transcriptional responses to independent genetic causes of cardiac hypertrophy. Physiol Genomics. 2001;6:19–28. doi: 10.1152/physiolgenomics.2001.6.1.19. [DOI] [PubMed] [Google Scholar]
- 18.D'Angelo DD, Sakata Y, Lorenz JN, Boivin GP, Walsh RA, Liggett SB, Dorn GW. Transgenic Galphaq overexpression induces cardiac contractile failure in mice. Proc Natl Acad Sci U S A. 1997;94:8121–8126. doi: 10.1073/pnas.94.15.8121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sakata Y, Hoit BD, Liggett SB, Walsh RA, Dorn GW. Decompensation of pressure-overload hypertrophy in G alpha q-overexpressing mice. Circulation. 1998;97:1488–1495. doi: 10.1161/01.cir.97.15.1488. [DOI] [PubMed] [Google Scholar]
- 20.Diwan A, Koesters AG, Odley AM, Pushkaran S, Baines CP, Spike BT, Daria D, Jegga AG, Geiger H, Aronow BJ, Molkentin JD, Macleod KF, Kalfa TA, Dorn GW. Unrestrained erythroblast development in Nix-/- mice reveals a mechanism for apoptotic modulation of erythropoiesis. Proc Natl Acad Sci U S A. 2007;104:6794–6799. doi: 10.1073/pnas.0610666104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Moses KA, DeMayo F, Braun RM, Reecy JL, Schwartz RJ. Embryonic expression of an Nkx2-5/Cre gene using ROSA26 reporter mice. Genesis. 2001;31:176–180. doi: 10.1002/gene.10022. [DOI] [PubMed] [Google Scholar]
- 22.Diwan A, Krenz M, Syed FM, Wansapura J, Ren X, Koesters AG, Li H, Kirshenbaum LA, Hahn HS, Robbins J, Jones WK, Dorn GW. Inhibition of ischemic cardiomyocyte apoptosis through targeted ablation of Bnip3 restrains postinfarction remodeling in mice. J Clin Invest. 2007;117:2825–2833. doi: 10.1172/JCI32490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Adams JW, Sakata Y, Davis MG, Sah VP, Wang Y, Liggett SB, Chien KR, Brown JH, Dorn GW. Enhanced Galphaq signaling: a common pathway mediates cardiac hypertrophy and apoptotic heart failure. Proc Natl Acad Sci U S A. 1998;95:10140–10145. doi: 10.1073/pnas.95.17.10140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Galvez AS, Brunskill EW, Marreez Y, Benner BJ, Regula KM, Kirschenbaum LA, Dorn GW. Distinct Pathways Regulate Proapoptotic Nix and BNip3 in Cardiac Stress. J Biol Chem. 2006;281:1442–1448. doi: 10.1074/jbc.M509056200. [DOI] [PubMed] [Google Scholar]
- 25.Dorn GW. Physiologic growth and pathologic genes in cardiac development and cardiomyopathy. Trends Cardiovasc Med. 2005;15:185–189. doi: 10.1016/j.tcm.2005.05.009. [DOI] [PubMed] [Google Scholar]
- 26.Akhter SA, Luttrell LM, Rockman HA, Iaccarino G, Lefkowitz RJ, Koch WJ. Targeting the receptor-Gq interface to inhibit in vivo pressure overload myocardial hypertrophy. Science. 1998;280:574–577. doi: 10.1126/science.280.5363.574. [DOI] [PubMed] [Google Scholar]
- 27.Wettschureck N, Rutten H, Zywietz A, Gehring D, Wilkie TM, Chen J, Chien KR, Offermanns S. Absence of pressure overload induced myocardial hypertrophy after conditional inactivation of Galphaq/Galpha11 in cardiomyocytes. Nat Med. 2001;7:1236–1240. doi: 10.1038/nm1101-1236. [DOI] [PubMed] [Google Scholar]
- 28.Agah R, Frenkel PA, French BA, Michael LH, Overbeek PA, Schneider MD. Gene recombination in postmitotic cells. Targeted expression of Cre recombinase provokes cardiac-restricted, site-specific rearrangement in adult ventricular muscle in vivo. J Clin Invest. 1997;100:169–179. doi: 10.1172/JCI119509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Henderson SA, Goldhaber JI, So JM, Han T, Motter C, Ngo A, Chantawansri C, Ritter MR, Friedlander M, Nicoll DA, Frank JS, Jordan MC, Roos KP, Ross RS, Philipson KD. Functional adult myocardium in the absence of Na+-Ca2+ exchange: cardiac-specific knockout of NCX1. Circ Res. 2004;95:604–611. doi: 10.1161/01.RES.0000142316.08250.68. [DOI] [PubMed] [Google Scholar]
- 30.Jacoby JJ, Kalinowski A, Liu MG, Zhang SS, Gao Q, Chai GX, Ji L, Iwamoto Y, Li E, Schneider M, Russell KS, Fu XY. Cardiomyocyte-restricted knockout of STAT3 results in higher sensitivity to inflammation, cardiac fibrosis, and heart failure with advanced age. Proc Natl Acad Sci U S A. 2003;100:12929–12934. doi: 10.1073/pnas.2134694100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Matkovich SJ, Diwan A, Klanke JL, Hammer DJ, Marreez Y, Odley AM, Brunskill EW, Koch WJ, Schwartz RJ, Dorn GW. Cardiac-specific ablation of G-protein receptor kinase 2 redefines its roles in heart development and beta-adrenergic signaling. Circ Res. 2006;99:996–1003. doi: 10.1161/01.RES.0000247932.71270.2c. [DOI] [PubMed] [Google Scholar]
- 32.Shai SY, Harpf AE, Babbitt CJ, Jordan MC, Fishbein MC, Chen J, Omura M, Leil TA, Becker KD, Jiang M, Smith DJ, Cherry SR, Loftus JC, Ross RS. Cardiac myocyte-specific excision of the beta1 integrin gene results in myocardial fibrosis and cardiac failure. Circ Res. 2002;90:458–464. doi: 10.1161/hh0402.105790. [DOI] [PubMed] [Google Scholar]
- 33.Weiss RG. Imaging the murine cardiovascular system with magnetic resonance. Circ Res. 2001;88:550–551. doi: 10.1161/01.res.88.6.550. [DOI] [PubMed] [Google Scholar]
- 34.Wiesmann F, Ruff J, Hiller KH, Rommel E, Haase A, Neubauer S. Developmental changes of cardiac function and mass assessed with MRI in neonatal, juvenile, and adult mice. Am J Physiol Heart Circ Physiol. 2000;278:H652–H657. doi: 10.1152/ajpheart.2000.278.2.H652. [DOI] [PubMed] [Google Scholar]
- 35.Maslov MY, Chacko VP, Stuber M, Moens AL, Kass DA, Champion HC, Weiss RG. Altered high-energy phosphate metabolism predicts contractile dysfunction and subsequent ventricular remodeling in pressure-overload hypertrophy mice. Am J Physiol Heart Circ Physiol. 2007;292:H387–H391. doi: 10.1152/ajpheart.00737.2006. [DOI] [PubMed] [Google Scholar]
- 36.Dorn GW, Robbins J, Sugden PH. Phenotyping hypertrophy: eschew obfuscation. Circ Res. 2003;92:1171–1175. doi: 10.1161/01.RES.0000077012.11088.BC. [DOI] [PubMed] [Google Scholar]
- 37.Zhang D, Gaussin V, Taffet GE, Belaguli NS, Yamada M, Schwartz RJ, Michael LH, Overbeek PA, Schneider MD. TAK1 is activated in the myocardium after pressure overload and is sufficient to provoke heart failure in transgenic mice. Nat Med. 2000;6:556–563. doi: 10.1038/75037. [DOI] [PubMed] [Google Scholar]
- 38.Chen G, Cizeau J, Vande VC, Park JH, Bozek G, Bolton J, Shi L, Dubik D, Greenberg A. Nix and Nip3 form a subfamily of pro-apoptotic mitochondrial proteins. J Biol Chem. 1999;274:7–10. doi: 10.1074/jbc.274.1.7. [DOI] [PubMed] [Google Scholar]
- 39.Cizeau J, Ray R, Chen G, Gietz RD, Greenberg AH. The C. elegans orthologue ceBNIP3 interacts with CED-9 and CED-3 but kills through a BH3- and caspase-independent mechanism. Oncogene. 2000;19:5453–5463. doi: 10.1038/sj.onc.1203929. [DOI] [PubMed] [Google Scholar]
- 40.Bouillet P, Strasser A. BH3-only proteins - evolutionarily conserved proapoptotic Bcl-2 family members essential for initiating programmed cell death. J Cell Sci. 2002;115:1567–1574. doi: 10.1242/jcs.115.8.1567. [DOI] [PubMed] [Google Scholar]
- 41.Adams JW, Pagel AL, Means CK, Oksenberg D, Armstrong RC, Brown JH. Cardiomyocyte apoptosis induced by Galphaq signaling is mediated by permeability transition pore formation and activation of the mitochondrial death pathway. Circ Res. 2000;87:1180–1187. doi: 10.1161/01.res.87.12.1180. [DOI] [PubMed] [Google Scholar]
- 42.Miyamoto S, Howes AL, Adams JW, Dorn GW, Brown JH. Ca2+ dysregulation induces mitochondrial depolarization and apoptosis: role of Na+/Ca2+ exchanger and AKT. J Biol Chem. 2005;280:38505–38512. doi: 10.1074/jbc.M505223200. [DOI] [PubMed] [Google Scholar]
- 43.Crow MT, Mani K, Nam YJ, Kitsis RN. The mitochondrial death pathway and cardiac myocyte apoptosis. Circ Res. 2004;95:957–970. doi: 10.1161/01.RES.0000148632.35500.d9. [DOI] [PubMed] [Google Scholar]
- 44.Esposito G, Rapacciuolo A, Naga Prasad SV, Takaoka H, Thomas SA, Koch WJ, Rockman HA. Genetic alterations that inhibit in vivo pressure-overload hypertrophy prevent cardiac dysfunction despite increased wall stress. Circulation. 2002;105:85–92. doi: 10.1161/hc0102.101365. [DOI] [PubMed] [Google Scholar]
- 45.Hill JA, Karimi M, Kutschke W, Davisson RL, Zimmerman K, Wang Z, Kerber RE, Weiss RM. Cardiac hypertrophy is not a required compensatory response to short-term pressure overload. Circulation. 2000;101:2863–2869. doi: 10.1161/01.cir.101.24.2863. [DOI] [PubMed] [Google Scholar]
- 46.Sano M, Schneider MD. Still stressed out but doing fine: normalization of wall stress is superfluous to maintaining cardiac function in chronic pressure overload. Circulation. 2002;105:8–10. [PubMed] [Google Scholar]
- 47.Sano M, Minamino T, Toko H, Miyauchi H, Orimo M, Qin Y, Akazawa H, Tateno K, Kayama Y, Harada M, Shimizu I, Asahara T, Hamada H, Tomita S, Molkentin JD, Zou Y, Komuro I. p53-induced inhibition of Hif-1 causes cardiac dysfunction during pressure overload. Nature. 2007;446:444–448. doi: 10.1038/nature05602. [DOI] [PubMed] [Google Scholar]
- 48.Shiojima I, Sato K, Izumiya Y, Schiekofer S, Ito M, Liao R, Colucci WS, Walsh K. Disruption of coordinated cardiac hypertrophy and angiogenesis contributes to the transition to heart failure. J Clin Invest. 2005;115:2108–2118. doi: 10.1172/JCI24682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sowter HM, Ratcliffe PJ, Watson P, Greenberg AH, Harris AL. HIF-1-dependent regulation of hypoxic induction of the cell death factors BNIP3 and NIX in human tumors. Cancer Res. 2001;61:6669–6673. [PubMed] [Google Scholar]
- 50.Sowter HM, Ferguson M, Pym C, Watson P, Fox SB, Han C, Harris AL. Expression of the cell death genes BNip3 and NIX in ductal carcinoma in situ of the breast; correlation of BNip3 levels with necrosis and grade. J Pathol. 2003;201:573–580. doi: 10.1002/path.1486. [DOI] [PubMed] [Google Scholar]
- 51.Hamacher-Brady A, Brady NR, Logue SE, Sayen MR, Jinno M, Kirshenbaum LA, Gottlieb RA, Gustafsson AB. Response to myocardial ischemia/reperfusion injury involves Bnip3 and autophagy. Cell Death Differ. 2007;14:146–157. doi: 10.1038/sj.cdd.4401936. [DOI] [PubMed] [Google Scholar]
- 52.Regula KM, Ens K, Kirshenbaum LA. Inducible expression of BNIP3 provokes mitochondrial defects and hypoxia-mediated cell death of ventricular myocytes. Circ Res. 2002;91:226–231. doi: 10.1161/01.res.0000029232.42227.16. [DOI] [PubMed] [Google Scholar]
- 53.Shiojima I, Walsh K. Regulation of cardiac growth and coronary angiogenesis by the Akt/PKB signaling pathway. Genes Dev. 2006;20:3347–3365. doi: 10.1101/gad.1492806. [DOI] [PubMed] [Google Scholar]
- 54.Sun M, Chen M, Dawood F, Zurawska U, Li JY, Parker T, Kassiri Z, Kirshenbaum LA, Arnold M, Khokha R, Liu PP. Tumor necrosis factor-alpha mediates cardiac remodeling and ventricular dysfunction after pressure overload state. Circulation. 2007;115:1398–1407. doi: 10.1161/CIRCULATIONAHA.106.643585. [DOI] [PubMed] [Google Scholar]
- 55.Yin XM. Signal transduction mediated by Bid, a pro-death Bcl-2 family proteins, connects the death receptor and mitochondria apoptosis pathways. Cell Res. 2000;10:161–167. doi: 10.1038/sj.cr.7290045. [DOI] [PubMed] [Google Scholar]