

Abstract
The medial nucleus of the trapezoid body (MNTB) is specialized for high frequency firing by expression of Kv3 channels, which minimize action potential (AP) duration, and Kv1 channels, which suppress multiple AP firing, during each calyceal giant EPSC. However, the outward K+ current in MNTB neurons is dominated by another unidentified delayed rectifier. It has slow kinetics and a peak conductance of ∼37 nS; it is half-activated at −9.2 ± 2.1 mV and half-inactivated at −35.9 ± 1.5 mV. It is blocked by several non-specific potassium channel antagonists including quinine (100 μm) and high concentrations of extracellular tetraethylammonium (TEA; IC50 = 11.8 mm), but no specific antagonists were found. These characteristics are similar to recombinant Kv2-mediated currents. Quantitative RT-PCR showed that Kv2.2 mRNA was much more prevalent than Kv2.1 in the MNTB. A Kv2.2 antibody showed specific staining and Western blots confirmed that it recognized a protein ∼110 kDa which was absent in brainstem tissue from a Kv2.2 knockout mouse. Confocal imaging showed that Kv2.2 was highly expressed in axon initial segments of MNTB neurons. In the absence of a specific antagonist, Hodgkin–Huxley modelling of voltage-gated conductances showed that Kv2.2 has a minor role during single APs (due to its slow activation) but assists recovery of voltage-gated sodium channels (Nav) from inactivation by hyperpolarizing interspike potentials during repetitive AP firing. Current-clamp recordings during high frequency firing and characterization of Nav inactivation confirmed this hypothesis. We conclude that Kv2.2-containing channels have a distinctive initial segment location and crucial function in maintaining AP amplitude by regulating the interspike potential during high frequency firing.
Potassium currents have multiple and diverse roles in shaping electrical signalling, with different suites of voltage-gated and rectifying non-gated channels setting neuronal membrane potentials, firing threshold, action potential waveform and firing patterns. Identification of the channel family and subunits contributing to these functions in native neurons is complicated by their heterogeneous subunit composition, their particular functional localization to the plasma membrane and by the absence of specific antagonists for some families. For these reasons the full complement of the K+ channels and subunits underlying native K+ currents are still not known for any identified central neuron. We have focused on studying the potassium currents of a ‘simple’ neuron within the medial nucleus of the trapezoid body (MNTB), which serves as a relay in the binaural circuits involved in sound source localization.
These neurons receive the glutamatergic calyx of Held giant synapse, which reliably triggers postsynaptic APs with large, well-timed EPSCs which have a magnitude of around 30 times firing threshold. In vivo recordings show the calyceal input fires spontaneously at frequencies ranging between 0 and 100 Hz (Kopp-Scheinpflug et al. 2003) and sound-evoked firing frequencies in the MNTB neuron range from these spontaneous rates to several hundred hertz and even up to 800 Hz for short periods with electrically evoked firing (Taschenberger & von Gersdorff, 2000). Although the interspike membrane potential is elevated during stimulus trains, each EPSP triggers only one postsynaptic AP. Voltage-gated K+ channels shape AP waveform and firing frequency in the presynaptic terminal and postsynaptic MNTB neuron. Postsynaptic Kv1 channels suppress AP firing during the EPSP decay so that the output of the MNTB neuron precisely follows the timing and pattern of the presynaptic incoming AP train (Brew & Forsythe, 1995; Dodson et al. 2002; Brew et al. 2003; Gittelman & Tempel, 2006). Kv3 channels speed AP repolarization, promoting short APs and high frequency firing (Wang et al. 1998; Rudy et al. 1999; Dodson & Forsythe, 2004). A-type potassium currents (IA) mediated by Kv4 channels are also present in mouse MNTB neurons (Johnston et al. 2008).
Under voltage clamp, the greater part of the MNTB outward current is resistant to Kv1 and Kv3 antagonists, indicating that another K+ current must also play a role in MNTB neuron excitability. In this paper we use biophysical, immunohistochemical, molecular and modelling methods to characterize this voltage-gated K+ conductance. It is a slow-gating delayed rectifier that generates large currents on depolarization and is mediated by Kv2.2-containing channels. The slow kinetics of this Kv2 conductance limits its contribution to the repolarization of a single AP. However, during high frequency repetitive stimulation, cumulative activation of this conductance causes hyperpolarization of the interspike potential, promoting recovery of voltage-gated Na+ channels from inactivation and so enhancing AP firing.
Methods
Brain slice preparation
Brain slices were prepared as previously described (Barnes-Davies & Forsythe, 1995; Wong et al. 2006). Briefly, CBA/Ca mice aged P10–P19 (postnatal days) were decapitated in accordance with the UK Animals (Scientific Procedures) Act 1986 and the brain was removed in a slush of iced artificial cerebrospinal fluid (aCSF) of composition (in mm): 250 sucrose, 2.5 KCl, 10 glucose, 1.25 NaH2PO4, 0.5 ascorbic acid, 26 NaHCO3, 4 MgCl2, 0.1 CaCl2, gassed with 95% O2–5% CO2 (pH 7.4). The brain was placed ventral side up and the brainstem removed with a cut at the level of the pons and angled ∼20 deg toward the cerebellum. The rostral surface was then mounted on the stage of an Integraslice 7550 mm (Campden Instruments, Loughborough, UK). Four to five transverse slices 120–150 μm thick were cut from the area containing the MNTB. Slices were then transferred to aCSF containing (in mm): 125 NaCl, 2.5 KCl, 10 glucose, 1.25 NaH2PO4, 2 sodium pyruvate, 3 myo-inositol, 0.5 ascorbic acid, 26 NaHCO3, 2.5 MgCl2, 0.5 CaCl2, gassed with 95% O2–5% CO2 (pH 7.4), incubated at 37°C for 1 h and then stored at room temperature until required.
Electrophysiology
For recording, one slice was transferred to a Peltier controlled environmental chamber on the stage of an upright E600FN microscope (Nikon, Tokyo, Japan) and continuously perfused with aCSF (∼1 ml min−1 at 25–27°C, or 35–37°C for current clamp and Nav recordings). Patch pipettes were pulled from thick-walled borosilicate glass (GC150F-7.5, Harvard Apparatus, UK) and whole-cell patch recordings were made from visually identified neurons in the MNTB using an Optopatch amplifier (Cairn, Faversham, UK).
For studying K+ currents the pipette solution contained (in mm): 97.5 potassium gluconate, 32.5 KCl, 10 Hepes, 5 EGTA (or 10 BAPTA) 0.01 ZD 7288 (to block Ih) and 1 MgCl2 (pH 7.2 with KOH) and had series resistances of 4–12 MΩ. The perfusing aCSF contained 1 μm tetrodotoxin (TTX) to block INa, and usually contained 3 mm TEA and 10 nm dendrotoxin-I (DTx-I) unless otherwise stated. For current-clamp recording pipette solutions were identical but with ZD 7288 omitted and the perfusing aCSF contained no ion channel blockers.
For studying Na+ currents the pipette solution contained (in mm): 120 CsCl, 10 TEA-Cl, 5 4-aminopyridine, 5 EGTA, 10 Hepes, 5 NaCl (pH 7.2 with NaOH) and had series resistances of 3–8 MΩ. The perfusing aCSF contained 20 μm DNQX, 10 μm bicuculline, 1 μm strychnine, 100 μm CdCl2, 3 mm TEA and 1 mm CsCl. Leao et al. (2005) demonstrated that, when recording Nav currents from MNTB slices, space clamp and voltage error only induced minor inaccuracy; however, we blocked around half of the sodium current by including 5 nm TTX in the aCSF (IC50 of the MNTB subunits Nav1.1 and 1.6 is ∼5 nm, Catterall et al. 2003) to further reduce voltage-clamp error.
The average series resistance from 28 recordings was 6 ± 0.4 MΩ. All series resistances were compensated by at least 75% with 10 μs lag. The holding potential in voltage clamp was −77 mV; current-clamped neurons were held at ∼ −70 mV. Data were excluded from analysis if more than 250 pA was required to hold at −70 mV.
Data acquisition and analysis
Data were acquired using pCLAMP 9.2 and a Digidata 1322A interface (Molecular Devices) filtered and digitized at 2 kHz and 10–50 kHz, respectively, for K+ currents. For Na+ currents, data were filtered at 5 kHz and digitized at 50 kHz.
For K+ currents: no leak subtraction was performed on the K+ currents. K+-mediated leak is non-linear (outwardly rectifying) as described by the Goldman–Hodgkin–Katz (GHK) current equation; conventional leak subtraction (which is linear) fails to subtract all the leak. Therefore the residual current in Fig. 2D, when fitted by a GHK current equation of the form: I = Pz2(VF2/RT) × (([K+]in − [K+]out × exp(−zFV/RT))/1 – exp(−zFV/RT)) (Hille, 2001) is consistent with complete block of the voltage-activated current components. For example, the DTx-I-sensitive current (shown in online Supplemental material, Fig. S1) 40 ms into each pulse was corrected for non-linearity of the single channel current using a modified GHK equation of the form: i = α(F2V/RT) × (([K+]in − [K+]out × exp(−FV/RT))/1 − exp(−FV/RT)), where the terms have their usual meaning and α is a normalization factor (Clay, 2000). For estimating the IC50 for TEA block of the Kv2 current, concentrations of 3, 10, 30 and 100 mm were all measured relative to 1 mm TEA (which was already present to block Kv3 currents). A Hill equation of the form: Proportion of channels occupied (Pocc) = [TEA]/IC50 + [TEA] was then fitted to the data, with the Pocc value for 1 mm TEA varied to give the best fit (which gave a Pocc of 0.05 for 1 mm). Junction potentials of −7 mV have been added to stated voltages for the K+-based pipette solution. For Na+ currents: voltages were corrected for −4 mV junction potentials and leak subtraction was performed using the scaled pulse method to minimize capacitive artifacts (this method was not employed on the K+ current, see above).
Figure 2. Kinetics of the slow high voltage-activated current.

A, in the presence of TEA (3 mm) and DTx-I (10 nm) an initial depolarization from −97 to −37 mV activates, and then inactivates the A-current. After 20 ms further voltage steps were applied to activate the remaining voltage-gated current. B, the voltage dependence of activation is fitted by a single exponential; the mean time constant is plotted against voltage and shows an e-fold increase with 17.7 mV depolarization (n = 6) grey trace. C, enlargement of the dash-boxed area in A; an activation curve was measured from these tail currents, measured at the latency indicated by the filled diamond. D, a sustained conditioning voltage step (8 s) over voltages ranging from −107 to +43 mV showed the isolated delayed rectifier to have slow inactivation, which was assessed by measuring the current on stepping to +43 mV (filled triangle). E, a Boltzmann function was fitted to the magnitude of activation tail currents (filled diamonds) and another Boltzmann function was fitted to the inactivation data (filled triangles) from the data shown in C and D (see text for mean data). F, deactivation rates were measured after maximally activating the current by stepping to +53 mV for 15 ms and then stepping to the indicated voltages. Single exponentials fitted the tail currents, with the averaged data plotted in G. Mean data are plotted for 6 neurons. Deactivation accelerated e-fold with 30.6 mV hyperpolarization (grey trace, n = 6; NB, fits close to zero current were omitted as being too small for reasonable accuracy).
Activation and inactivation parameters were determined by a Boltzmann function of the form 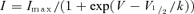 with variables
with variables 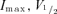 and k (the slope factor). All fits were performed using Clampfit 9.2 (Molecular Devices) or Excel (Microsoft) with least squares minimization. Data are presented as mean ± s.e.m. and either a two-tailed Student's t test (paired or unpaired) or an ANOVA was used to assess significance, which is indicated by * for 95% confidence and ** for 99%.
and k (the slope factor). All fits were performed using Clampfit 9.2 (Molecular Devices) or Excel (Microsoft) with least squares minimization. Data are presented as mean ± s.e.m. and either a two-tailed Student's t test (paired or unpaired) or an ANOVA was used to assess significance, which is indicated by * for 95% confidence and ** for 99%.
Quantitative RT-PCR (QRT-PCR)
RNA extraction (each homogenate consisted of MNTBs pooled from 8 mice) was carried out using RNeasy lipid tissue mini kit (Qiagen) and samples were DNase treated (DNAfree, Ambion). RT–PCR was performed on 1–2 μg of the RNA sample with SuperScript III first strand synthesis kit (Invitrogen) using the oligo-dT primers. The resulting cDNA was then used for fluorescence PCR (Power SYBR Green PCR Master Mix, Applied Biosystems and ABI PRISM 7000 thermal cycler). Primer sequences were: Kv2.1 forward TGGAGAAGCCCAACTCATC, reverse CAATGGTGGAGAGGACAATG; Kv2.2 forward CACCTGGCTTGAACAGAAAG, reverse TTGCTTCGGATAATGTCCAC; Kv3.1 forward TGAACAAGACCGAAATCGAG, reverse CGAAGGTGAACCAGACCAC; actin forward TGCTCCTCCTGAGCGCAAGTACTC reverse CGGACTCATCGTACTCCTGCTTGC. Amplification occurred under the following conditions: 50°C 2 min, 95°C 10 min, followed by 95°C 15 s and 60°C 60 s for 40 cycles. Primer efficiency was > 95% assessed by serial dilutions and construction of a standard curve. Relative expression levels were determined using the comparative CT method (User Bulletin 2, ABI PRISM Sequence Detection System, pp. 11–15, 1997, PE Applied Biosystems).
Immunohistochemistry
P13 CBA/Ca mice brainstems were frozen in Tissue Tek (Sakura) using hexane and dry ice. Cryostat sections (20 μm) were mounted on polylysine-coated slides and fixed in 4% paraformaldehyde/PBS for 25 min at 4°C. After washing 3 × 5 min in 100 mm PBS with 0.1% Triton X-100 (PBS-T) the slides were treated with citrate buffer (pH 6.0) for 20 min at 95°C (antigen retrieval). After re-washing in PBS-T, sections were blocked with 1% bovine serum albumin, 1% goat serum in PBS-T for 1 h at 20°C and then incubated with the primary anti-Kv2.2 (Alomone, Israel; 1: 1000) and in some cases also anti-Kv1.2 (NeuroMab, 1: 1000) in blocking buffer overnight at 4°C. After washing with PBS-T, the secondary antibody (goat anti-rabbit Alexa-fluor 488, 1: 1000, and for anti-Kv1.2 goat anti-mouse Alexa-fluor 568, 1: 500, Molecular Probes) was applied for 2 h at 20°C, then mounted with Vectashield (Vector Laboratories). DAPI was included to visualize cell nuclei. Images were obtained using a Zeiss LSM 510 Meta confocal microscope. Control sections underwent identical procedures, but were pre-incubated with the immunizing peptide. Fluorescence intensity was measured in grey levels using ImageJ 1.38 (NIH, USA)
Solutions and drugs
All chemicals were obtained from Sigma except: TTX (Latoxan), dendrotoxin-I and recombinant (r)-stromatoxin-1 (Alomone), linopiridine, DNQX, ZD 7288 and bicuculline (Tocris Cookson) and E4031 (Calbiochem).
Immunoblotting
Proteins were extracted from Kv2.2 knockout (Lexicon, TF1551) and wild-type (WT) mouse brain tissues using a manual homogenizer and NP-40 lysis buffer (20 mm Tris-HCl, pH 7.4, 137 mm NaCl, 2.0 mm EDTA, 1% NP-40) freshly supplemented with phosphatase and protease inhibitors, followed by brief sonication and centrifugation. Samples (40 μg of proteins) were diluted into SDS-PAGE sample buffer and resolved by SDS-PAGE. Immunoblotting was performed using anti-Kv2.2 primary antibody (Alomone) and chemiluminescence detection (ECL, GE Healthcare, UK).
Neuron modelling
A Hodgkin–Huxley-type model of the Kv2.2 conductance of the form:
| (1) |
was implemented using the NEURON simulation software (Hines & Carnevale, 2001). P dictated the magnitude of the conductance and was adjusted to give a current magnitude equal to the measured values, GHK(v) specified the Goldman–Hodgkin–Katz dependence on voltage, m was the activation variable and h was the inactivation variable. The steady-state values of m and h were given by the Boltzman functions in Fig. 2E. Time constants (in ms) for changes in m were derived from least squares fits to the measured activation and deactivation time constants (Model Fig. 5A) and were specified as
| (2) |
Time constants for changes in h were fixed at 900 ms, independent of voltage.
Figure 5. A Hodgkin–Huxley model of an MNTB neuron shows that Kv2 regulates the interspike potential and affects availability of Nav channels during high-frequency firing.

A, time constants used to generate the model Kv2.2 were obtained from a fit (dashed line) to the activation and deactivation time constants obtained from voltage-clamp experiments (see Fig. 2). B, the magnitudes of the ionic conductances present in the implementation of the single compartment MNTB model adapted from Macica et al. (2003). C, the membrane potential of the model MNTB neuron in response to synaptic trains at 50 Hz (left) and 200 Hz (right) in the presence (black) and absence (grey) of the Kv2.2 conductance; arrows indicate AP peak and interspike potential at the end of each train, respectively. D, the magnitude of the Kv2.2 current passing during the APs in the traces shown above in C. Note that the Kv2.2 current dramatically increases in magnitude at the start of the 200 Hz train (right) but is smaller and relatively stable in the 50 Hz train (left). Note the difference in interspike potential (black arrow) and action potential height (grey arrow).
The single-compartment MNTB model described by Macica et al. (2003) was manipulated using the NEURON simulation software (Hines & Carnevale, 2001). Conductances for the different channels incorporated in this model are given in Fig. 5B. The reversal potential of the leak current was set to −70 mV. Synaptic currents were constructed using a reversal potential of 0 mV and were of the form:
| (3) |
where v was voltage, G was conductance and B (and C) were given by,
| (4) |
where z was a scaling factor set to normalize the maximum of X to 1.
Results
Under voltage clamp most of the outward current in MNTB neurons is resistant to block by perfusion of Kv3 and Kv1 channel antagonists, 3 mm TEA and 10 nm DTx-I, respectively (Fig. 1). The current–voltage (I–V) relationships for outward current recorded under control conditions (Fig. 1A) and for that measured after antagonist perfusion (Fig. 1B) were measured at a latency of 40 ms (dashed vertical line) and the result plotted in Fig. 1C. The recordings were made from animals aged P10–P14. The TEA and DTx-I-insensitive current has two distinct components, as shown in Fig. 1B: a fast activating transient A-type current (arrow) which has been characterized previously (Johnston et al. 2008) and a slowly activating delayed rectifier.
Figure 1. A slow delayed rectifier in MNTB neurons.

A, control outward K+ currents evoked by step commands from a pre-pulse voltage of −97 mV to the indicated voltages, in 10 mV increments under voltage-clamp conditions. B, repeat of the same voltage-clamp protocol in the same neuron following perfusion of DTx-I (10 nm) and TEA (3 mm). Arrow indicates unmasking of a small A-type current. C, the mean current–voltage (I–V) relationship for control (black, n = 18) and following DTx and TEA perfusion (grey, n = 18). Note that more than half of the outward current is insensitive to DTx and TEA; this current is therefore not mediated by Kv1 or Kv3 channels.
The slowly activating delayed rectifier
Isolation of a slowly activating outward current was achieved by pharmacological block of the Kv1 and Kv3 components and inactivation of the A-current by a pre-pulse to −37 mV for 20 ms (Fig. 2A). Increasing step depolarizations generated a slowly activating and deactivating current (see Fig. 2A and F). The rising phase of the current was well fitted by a single exponential, with a time constant which varied with voltage (Fig. 2B). Activation rates were slow at negative voltages (τ = 39.6 ± 5.2 ms at −27 mV, n = 6) and accelerated e-fold with 17.7 mV depolarization, the curve forming an asymptote at ∼2.3 ms by +43 mV. A Boltzmann function fitted to tail currents (Fig. 2C) gave a half-activation, 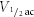 , of −9.2 ± 2.1 mV and kac of −9.0 ± 0.4 (Fig. 2E, diamonds, n = 6), indicating that the current activates from ∼ −30 mV and is maximally activated by +20 mV.
, of −9.2 ± 2.1 mV and kac of −9.0 ± 0.4 (Fig. 2E, diamonds, n = 6), indicating that the current activates from ∼ −30 mV and is maximally activated by +20 mV.
Steady-state inactivation was assessed using the protocol in Fig. 2D and fitting the current evoked at +43 mV with a Boltzmann function. This gave a V½in of −35.9 ± 1.5 mV and kin of 10.8 ± 0.8 (Fig. 2E, triangles, n = 7) showing that this current exhibits little steady-state inactivation at resting potentials. Following activation, the conductance slowly inactivated (Fig. 2D) but neither single nor double exponentials gave reliable fits over a range of voltages. The fitting problem is most likely caused by changes in voltage error associated with the declining current passing across the residual series resistance as the current inactivates, therefore quantification was limited to measuring the time to half-inactivation, which at −7 mV (where complete inactivation occurs) was 669 ± 32 ms (n = 5). Following a train of brief depolarizing pulses, the rate of inactivation was similar to that during a sustained depolarization (data not shown) which is consistent with the absence of ‘U’-type inactivation (Klemic et al. 1998). The slow deactivation of this tail current (Fig. 2F) also exhibits voltage dependence. The current was maximally activated by stepping to +53 mV for 15 ms (> 5τ) prior to stepping back to test voltages. Decays were fitted with single exponentials and mean time constants are plotted in Fig. 2G, showing an e-fold increase with 30.6 mV hyperpolarization.
Which K+ channel families do not contribute to this slow delayed rectifier?
We have made use of well-characterized pharmacological tools to identify Kv1 and Kv3 conductances in MNTB neurons, but many other pharmacological agents were tested in order to distinguish between the voltage-gated families which might underlie the slow delayed rectifier. As above, the Kv1 subfamily was eliminated with DTx-I (Dodson et al. 2002; Brew et al. 2003; Gittelman & Tempel, 2006); 10 nm was a maximally effective dose because 100 nm had no further effect. No DTX-I-insensitive low-voltage-activated currents were observed in the MNTB and potential contributions from Kv1.3 and Kv1.4 channels are unlikely since CP339 818 (Nguyen et al. 1996; Jager et al. 1998) had no effect on mouse MNTB currents (5–10 μm, n = 3, data not shown) as previously observed in the rat (Dodson et al. 2002). We eliminated the Kv3 family by perfusion of 1–3 mm TEA (Brew & Forsythe, 1995; Wang et al. 1998; Coetzee et al. 1999; Song et al. 2005) which at these concentrations also blocks BKCa. There is already evidence against the presence of large calcium-sensitive K+ currents in the MNTB (Brew & Forsythe, 1995); nevertheless, we conducted voltage-clamp experiments with low external calcium ([Ca2+]o/[Mg2+]o = 0.5: 2.5 mm) and with high concentrations of internal calcium buffer (5 mm EGTA or 10 mm BAPTA) so this delayed rectifier is not a BK or SK current. Although many of the Kv7 channels (KCNQ/M-current) mediate slowly activating currents, they are all sensitive to low concentrations of linopirdine (Robbins, 2001; Lawrence et al. 2006; Vervaeke et al. 2006; Hu et al. 2007) and the slow outward current was unaffected by 50 μm linopirdine (n = 2, data not shown). The Kv4 family underlies A-type currents (Birnbaum et al. 2004) and Kv4.3 is responsible for the A-current observed here (Johnston et al. 2008) but A-currents were removed by voltage-dependent inactivation (see Fig. 2A). EAG/ERG/ELK (Kv10, 11 and 12) subfamilies were excluded from the present study as addition of E4031 (1 μm) to the internal patch solution and left the current unaffected (Gessner & Heinemann, 2003; Royer et al. 2005; Lamarca et al. 2006; Furlan et al. 2007). Contributions from HCN channels were routinely blocked by inclusion of ZD 7288 (10 μm) in the patch solution. We could find no evidence for significant activation of KNa channels in the MNTB under these physiological recording conditions. We were unable to induce significantly increased outward currents on adding 23 mm [Na+] to internal solutions. Furthermore, addition of 2 mm [ATP]i and reduction of [Cl−]o to 7 mm (n = 3, data not shown) had no influence on the measured outward currents. Taken together these results exclude all of the known K+ channel families (Coetzee et al. 1999): Kv1, Kv3, Kv4, EAG, KCNQ, BK, KNa, SK, HCN, with the exception of Kv2.
Pharmacological agents acting on the slow delayed rectifier
This putative Kv2 conductance was largely insensitive to 3 mm [Ba2+]o (n = 5, data not shown), but perfusion of 100 μm quinine caused complete block of the voltage-gated component. The remaining current was fitted by the GHK current equation (Fig. 3A), consistent with mediation by leak channels (see Methods). Unfortunately, quinine is not specific and will block a range of K+ channel families and voltage-gated Na+ channels (data not shown) so it cannot be used to assess physiological significance in current-clamp recordings. One report suggests that a purified tarantula toxin called stromatoxin-1 modifies the gating of Kv2 channels and has a high affinity for Kv2.1 subunits (Escoubas et al. 2002). Recent reports using a recombinant stromatoxin-1 in neocortical pyramidal neurons confirmed that it reversibly blocks native Kv2.1 currents but with a much reduced affinity (Guan et al. 2007). We tested r-stromatoxin-1 (Alomone) using concentrations in the range of 100 nm to 5 μm (n = 5, three different lots, data not shown) but it had no effect on the slow delayed rectifier.
Figure 3. There were no specific blockers of this slow delayed rectifier.

A, in the presence of TEA and DTx-I (black trace) quinine (100 μm) blocked the remaining sustained voltage-gated current (grey trace). The GHK current equation fits the quinine-insensitive current showing that it is not voltage gated, but reflects the K+ leak currents. Inset traces show the respective control (black) and test (grey) traces from the same cell (without inactivation of the A-current; pre-pulse potential −97 mV). B, 4-aminopyridine (4-AP, 5–10 mm, n = 4) gives a partial block of the current; inset shows control and test (grey) current traces from the same cell (pre-pulse potential −77 mV). The 4-AP-insensitive current is not fitted by the GHK current equation, indicating that voltage-gated current remains. C, high concentrations of TEA blocked the slow-delayed rectifier. A dose–response curve to TEA was conducted and data pooled from multiple cells. Each point is the mean current from 3–4 neurons. The grey curve is a fit to the Hill equation and gives an IC50 of 11.8 mm. The open triangle was a free parameter to allow for block by 1 mm TEA which was present in all solutions to block Kv3 currents (see Methods). Inset: representative traces are shown from one cell at +13 mV.
The classic K+ channel antagonist 4-aminopyridine (4-AP) is non-specific, blocking Kv1, Kv2, Kv3 and Kv4 currents (Coetzee et al. 1999) as well as this slow delayed rectifier (Fig. 3B). Although low concentrations of external TEA (1–3 mm) are relatively specific for Kv3 channels, higher concentrations will also block many K+ channels. The MNTB slow delayed rectifier was sensitive to high concentrations of TEA, showing a dose–response curve with an IC50 of 11.8 mm (Fig. 3C). The current was insensitive to low concentrations of TEA applied internally (2 mm, n = 2, data not shown), which points to an incompatibility with Kv2.1 subunits (Taglialatela et al. 1991). The pharmacological profiles from studies of recombinant Kv2 channels in expression systems show greater sensitivity to TEA and 4-AP than observed here, but the pharmacology can be influenced by the incorporation of subunits from the ‘electrically silent’ families (Post et al. 1996; Robertson, 1997) and since both Kv5.1 and Kv6.4 are expressed in the MNTB (Lein et al. 2007) it is possible that the native Kv2 channel is not a Kv2.2 homomultimer.
We performed QRT-PCR on tissue dissected from MNTB nuclei to determine which Kv2 subunits are expressed. Figure 4A shows that Kv2.2 transcripts were present at considerably higher levels than Kv2.1 (n = 3). The above pharmacology, lack of ‘U’-type inactivation and the absence of immunostaining for Kv2.1 in the mouse MNTB (R. E. W. Fyffe, personal communication) are consistent with this current being mediated by a Kv2.2 rather than a Kv2.1 channel.
Figure 4. Kv2.2 mRNA and protein are present in MNTB principal neurons.

A, QRT-PCR of Kv3.1, 2.1 and 2.2 mRNA, expressed relative to Kv3.1. Each value is the mean of 3 separate reactions normalized to actin transcripts. B, Western blots with the Kv2.2 antibody in brain tissue from CBA mice (WT, centre right) and following pre-incubation with the blocking peptide (BP) shows a band at around 110 kDa. The left blots using the same Kv2.2 antibody in brain tissue obtained from homozygous Kv2.2 knockout mice (Lexicon, TF1551) and control littermates (WT, centre left). C, Kv2.2 immunoreactivity (green) is present in the MNTB and VNTB of the superior olivary complex and at lower levels across the auditory brainstem. Bright ‘sparks’ of fluorescence can be seen in the MNTB. D, at higher magnification MNTB principal neurons and the fluorescent sparks are clear. A plot of intensity along the dashed white line shows the incidence of the highly fluorescent sparks (red dashed lines) often alongside a neuronal profile (*). Inset (top) shows blocking peptide negative control. E, a confocal projection of one MNTB neuron double-labelled for Kv2.2 and DAPI (blue nucleus) showing a highly stained putative initial segment region (arrow). F, a confocal projection of a triple-labelled MNTB neuron showing co-localization of Kv2.2 (green) with Kv1.2 (red) in adjacent parts of the axon initial segment (AIS) and DAPI (blue).
The specificity of an anti-Kv2.2 antibody was verified by Western blot (Fig. 4B); the antibody stained a single band at the appropriate molecular weight for mouse Kv2.2 (accession number XP_984002) that was absent in the presence of the blocking (immunizing) peptide (Fig. 4D, n = 3) and not detected in brains from homozygous Kv2.2 knockout mice. We found that anti-Kv2.2 broadly labelled most neurons in the auditory brainstem; however, neurons in the MNTB and the ventral nucleus of the trapezoid body (VNTB) showed particularly strong focal labelling (Fig. 4C and D, n = 5) and some labelling of blood vessels was observed. Similar staining was observed in rat MNTB (n = 2, data not shown). In addition to the somatic labelling of the MNTB, which was predominantly cytoplasmic, intensely stained spots and rod-like structures were observed of up to 10 μm in length. Figure 4D shows a higher magnification image of one MNTB nucleus. These high intensity areas are clearly seen in a line-plot of the fluorescence intensity measured across the medio-lateral axis of the MNTB (dashed white line and plotted as grey scale level above blocking peptide control). Two distinct levels of staining above background were seen, an intermediate level in the MNTB neuronal somata (20–50 arbitrary units (a.u.), stars) and the very bright spots and rods (above 100 a.u., indicated by red dashed lines). The bright regions were always peripheral to cytoplasmic staining and often adjacent to somatic profiles.
Kv2.2 channels are concentrated in the axon initial segment
Confocal imaging showed that these intensely labelled regions were associated with individual MNTB neurons (Fig. 4E, arrow). We observed diffuse Kv2.2 immunostaining in the cytoplasm of principal MNTB neurons (consistent with protein synthesis, trafficking or presence in other internal organelles). The most intense labelling was observed in processes either immediately adjacent to or near principal neuron somata; the 4 μm-thick confocal projection in Fig. 4E shows a single MNTB neuron with green cytoplasmic staining of Kv2.2, a blue DAPI-stained nucleus and an intense green band which appears to form part of the axon initial segment (AIS). To test this hypothesis we used co-immunolocalization with Kv1.2 antibodies known to be present in MNTB AIS (Dodson et al. 2002), and elsewhere in the brain. The co-labelling with Kv1.2 (red) and Kv2.2 (green) shows clear association of both Kv1 and Kv2 K+ channels with the initial part of the MNTB neuron axon (Fig. 4F). This is consistent with evidence of interaction between the measured Kv1 and Kv2 currents, as indicated by the non-parallel and overlapping I–V curves in control and following DTx-I application (see Supplemental Fig. S1A and B). DTx-I blocks Kv1 current at negative voltages but the outward current at positive voltages (mediated by Kv2 channels) increases in amplitude and occasionally surpasses control levels (Supplemental Fig. S1A, black and purple arrows, respectively); implying that the (unblocked) Kv1 conductance reduces voltage-clamp quality for positive voltage steps, consistent with location of Kv1 and Kv2 channels either in the same or succeeding electrotonic compartments. In support of this conclusion Kv2 current was not observed in any excised outside-out patches from MNTB somata (Supplemental Fig. S2, n = 8) but was present in all whole-cell recordings (n > 67). These electrophysiological observations, combined with the Kv2.2 immunostaining, are consistent with the hypothesis that initial segment Kv2.2 channels mediate the slow delayed rectifier current recorded from the soma.
Modelling of the Kv2.2 conductance
In the absence of a specific antagonist to Kv2.2 we investigated the physiological function of this slow delayed rectifier by using a Hodgkin–Huxley-type model of an MNTB neuron. The model was based on the published MNTB neuron model of Macica et al. (2003) containing: Nav, KLVA (Kv1), KHVA (Kv3) and Ileak (Fig. 5B) to which a Kv2.2 conductance was added. The A-current was not included in the model for simplicity, as it has been shown to have minimal effects in trains of action potentials (Johnston et al. 2008). APs were elicited by suprathreshold ‘calyceal synaptic’ inputs (Fig. 5C) in the presence or absence of the model Kv2.2 conductance. At low firing frequencies (50 Hz) the presence of the Kv2.2 current (shown in Fig. 5C and D) had little effect. The interspike interval was slightly depolarized and AP amplitudes slightly reduced when the Kv2.2 was excluded from the model. However, at higher AP firing rates the Kv2.2 current did not completely deactivate during the shorter interspike interval, enabling accumulation of the current during the early stages of a 200 Hz train (Fig. 5D, right). The elevated Kv2.2 conductance during the interspike interval opposed the summating synaptic conductances, resulting in a more hyperpolarized interspike potential (compare with arrow marking interspike potential in the absence of the Kv2.2 current; Fig. 5C, right). In the absence of this hyperpolarizing drive from the Kv2.2 current, the AP amplitudes were reduced (grey arrow, Fig. 5C) and this attenuation was due to increased Nav inactivation caused by the more depolarized interspike intervals.
Hyperpolarization of the interspike potential
The rapidly gating high voltage-activated Kv3 current is known to make a major contribution to action potential repolarization in MNTB neurons and other fast-spiking cells (Rudy et al. 1999). So the slow activation of Kv2 channels means that Kv2 conductance is unlikely to contribute to single APs when AP half-widths are around 0.5 ms, which is consistent with the model. Both Kv1 and Kv3 currents turn off relatively rapidly (Brew & Forsythe, 1995) with their activity essentially following the AP waveform (Klug & Trussell, 2006), and there is little evidence for their cumulative activation during repetitive stimulation until the highest frequencies are reached (e.g. 600 Hz; Klug & Trussell, 2006). The model shows that activation of Kv2.2 conductance can accumulate at high firing frequencies (200 Hz and above) and hyperpolarizes the interspike potential. To test this prediction in native MNTB neurons we evoked trains of 60 APs under current-clamp conditions, using direct current injection of simulated synaptic currents without short-term depression. This method of stimulation avoids variability caused by quantal release and short-term depression during repetitive stimulation. The AP waveforms are similar to those evoked by orthodromic synaptic stimulation (Dodson et al. 2003; Postlethwaite et al. 2007) and are more physiological than step current injection.
The interspike potential was measured under current-clamp conditions during 50 Hz and 200 Hz stimulus trains. The experimental responses are shown in Fig. 6A (50 Hz black, 200 Hz grey traces). At 50 Hz the interspike potential showed a small decline of 1.9 ± 0.5 mV by the 60th spike (compare dashed line versus arrow in Fig. 6A black trace, n = 7, P = 0.014). In contrast the interspike potential during a 200 Hz train in the same cell (Fig. 6A, grey trace) declined by 4.7 ± 0.7 mV (Fig. 6C, n = 4, P = 0.0075). Similar results were seen in two further cells, but these were excluded from averaged data because the first stimulus generated two APs; multiple firing of MNTB neurons is a common finding in mice (Brew et al. 2003). This cumulative and frequency-dependent hyperpolarization was also seen in four cells patched with 10 mm BAPTA in place of EGTA in the pipette, ruling out any affects of Ca2+-activated K+ channels. Furthermore, no ATP was included in the patch pipette ruling out any effects from the Na+,K+-ATPase. Note that the fast after-hyperpolarization (AHP) remains constant since it is mediated by Kv3 channels, whose activation follows the AP waveform (Klug & Trussell, 2006). The frequency-dependent decrease in interspike potential confirms the predicted effect of Kv2.2 from the MNTB model (Fig. 5C and D).
Figure 6. Frequency-dependent hyperpolarization of the interspike potential under physiological conditions.

A, example 50 Hz (black) and 200 Hz (grey) trains of APs in the same MNTB neuron evoked with identical current trains at the two frequencies. Note the different time scales. Dashed lines indicate the peak of the 1st interspike potential and arrows show the peak of the last. Note that AP amplitudes remain fairly constant throughout, but the hyperpolarization of the interspike interval is greater with higher frequency stimulation. The 1st and 60th action potentials from 50 and 200 Hz overlain (black and grey, respectively) show that only the interspike voltage changes. C, plot of the peak interspike potential for the cell in A.
Sensitivity of the Nav current to the interspike potential
The Kv2-MNTB model also predicted effects on the availability of Nav channels during a train. However, the measured interspike hyperpolarization during a high frequency AP train is only a few millivolts. We therefore examined the MNTB Na+ current inactivation in greater detail. The steady-state inactivation of the Nav current was measured under voltage clamp as shown in Fig. 7A (inset) with the peak Na+ current evoked on voltage steps to −4 mV plotted against the pre-pulse conditioning voltage. The curve was fitted with a Boltzmann function giving a V½in of −55.4 ± 1.7 mV and a slope (kin) of 6.3 ± 0.1 (Fig. 7A, n = 5). These data confirm that MNTB Nav channels exhibit little steady-state inactivation at resting membrane potentials; however, during repetitive stimulation the interspike potential occurs over the steepest part of the inactivation voltage range. Therefore a K+ current need only induce small changes in the interspike potential to have a pronounced effect on the recovery of Nav channels from inactivation during high frequency stimulation.
Figure 7. Sensitivity of INa to the interspike potential.

A, the steady-state inactivation of INa was determined by plotting the peak current measured at −4 mV against the voltage of the 500 ms pre-pulse, which was fitted with a Boltzmann distribution giving a V½in of −55.4 ± 1.7 mV and a kin of 6.3 ± 0.1 (n = 5); inset shows example traces. B, the protocol used to asses the sensitivity of INa to changes in interspike potential (Vis); a 500 ms −94 mV pre-pulse (to remove all steady-state inactivation) was followed by test pulses to −4 mV interleaved with 5 ms intervals, simulating the time between spikes in a 200 Hz train, as indicated by the overlaid AP (red). C, example traces (all from one cell) generated from the protocol in B, show the sensitivity of INa to Vis. Black, Vis hyperpolarized from −55 to −59 mV (identical to Fig. 5D). Blue, Vis is kept constant at −55 mV. Green, Vis depolarized from −55 to −51 mV, mimicking mild summation of the interspike potential. D, average INa amplitudes during the trains shown in C; data normalized to 2nd spike to show change from the initial level of inactivation. Asterisks indicate onset of statistical significance.
To further demonstrate the influence of the interspike potential on Nav channel inactivation we used a voltage-clamp protocol designed to maximally activate the INa in a repetitive fashion (Fig. 7B, VTest). Each test pulse was followed by a 5 ms ‘interspike period’ reproducing the interspike interval for a 200 Hz AP train (see the protocol in Fig. 7B with an AP overlaid in red). A 1 ms ‘repolarizing’ step to −67 mV (VAHP, Fig. 7B) mimicked the AHP mediated by fast Kv3 currents, then the remaining 4 ms interspike voltage (Vis) was stepped over a range of potentials from −55 mV to ± 5 mV (Fig. 7B), matching the observed range of the interspike potential during AP trains.
The first INa in each train was the largest, since there was little resting inactivation at these voltages, with the second depolarizing step following the 5 ms interspike period being reduced by 40.5 ± 5.8% (n = 7, P ≤ 0.001). This incomplete recovery from inactivation was a constant and measurements were made relative to this second pulse in the train. A train of 60 repeats showed a 17 ± 14.4% increase in INa (n = 7, P = 0.004 measured at the 60th spike, compared to the 2nd) when Vis was matched to the measured values during an action potential train (voltages in Fig. 6C were used). Sodium current magnitude stabilized around the 15th repetition (Fig. 7C and D, black). In contrast, when Vis was maintained constant at −55 mV, INa decreased by 25.1 ± 8.6% (n = 8, P = 0.02, measured at the 60th VTest) and was still declining by the 60th repeat (Fig. 7C and D, blue trace).
The model showed that Kv2 channels caused a more hyperpolarized interspike potential (Fig. 5D). So to test what effect the absence of Kv2.2 would have on the sodium current's inactivation we set Vis to depolarize, giving mild summation of the interspike potential (Fig. 7C, final Vis = −50.6 mV). Under these conditions we observed a 49.9 ± 4.0% decline in INa from the 2nd Vtest (Fig. 7C and D, green trace, n = 5, P = 0.007). These results further validate the predictions of the MNTB model, suggesting that Kv2 channels function to hyperpolarize the interspike potential during high frequency stimulation, promoting recovery of Nav channel inactivation and ultimately maintaining AP amplitude during repetitive stimulation.
Kv2.2 current increases with development and has a tonotopic gradient
The Kv2 current magnitude increased with postnatal age (assessed in the presence of 3 mm TEA and 10 nm DTx-I to isolate Kv2.2). A significant increase in current magnitude was observed across the age range of P10–P14 (P < 0.0001, ANOVA) and additional data from P18–P19 animals showed a further increase with the same level of significance, as shown in Fig. 8A. Note that absolute current amplitude is plotted; since the Kv2 channels are not uniformly distributed, normalization to cell capacitance is of little relevance. But for comparison purposes this normalization was performed on pooled data from the P10–P14 animals and compared with the P18–P19 group. This gave Kv2 current amplitudes of 63.16 ± 10.77 (n = 6) and 148.85 ± 20.26 pA pF−1 (n = 5), respectively, and are significantly different (P = 0.003). The V½ac for Kv2.2 was unchanged with age (P18–P19 versus P10–P14).
Figure 8. Age and tonotopic gradients of Kv2.2 current.

A, the current measured 40 ms into a +13 mV step depolarization (peak activation) is plotted against postnatal age. A significant increase in the current magnitude is observed with development (statistical significance assessed by ANOVA, P < 0.001). B, the MNTB nucleus was divided into 3 parts, medial (M), intermediate (I) and lateral (L). C, the current measured 40 ms into a +13 mV step depolarization is plotted against location for P12 animals. A significant trend to larger currents in lateral neurons was seen across the tonotopic axis. NB, all data recorded in the presence of 10 nm DTx and 3 mm TEA.
Both Kv1 and Kv3 channels show a functional tonotopic distribution along the medio-lateral axis of the MNTB (Brew & Forsythe, 2005) with higher magnitude currents in the medial versus the lateral neurons. To test for> a tonotopic gradient of Kv2.2 current, the MNTB was divided into 3 parts; medial, intermediate and lateral (Fig. 8B). The Kv2.2 current magnitude was plotted against location for data from P12 mice. Use of data from other ages was avoided to minimize developmental variation (see Fig. 8A). A significant gradient of current magnitudes was detected (P = 0.03, Fig. 8C) with a tendency for larger current magnitude in lateral compared to medial neurons. This is opposite to the gradient for the Kv3 and Kv1 channels (Li et al. 2001; Brew & Forsythe, 2005), but similar to that of a previously reported slowly deactivating current of the rat MNTB (Brew & Forsythe, 2005).
Discussion
Kv1, Kv3 and Kv4 potassium currents have well-recognized roles in maintaining high fidelity transmission and generating fast action potential waveforms in the MNTB. Here we identify and characterize the fourth voltage-gated K+ conductance mediating a delayed rectifier in the MNTB. Although there are no specific blockers for this current, we have studied it by conducting voltage-clamp experiments under conditions where other known conductances are blocked. Our electrophysiology, pharmacology, PCR and immunohistochemical data provide strong evidence that this slow-activating conductance is mediated by Kv2.2-containing channels located in the axon initial segment. It acts during high frequency action potential firing, contributing to hyperpolarization of the interspike potential and enhancing recovery of sodium channels from inactivation.
Voltage-gated conductances of MNTB neurons
It is well established that a low voltage-activated current in the MNTB is mediated by Kv1 channels, while a high voltage-activated current is mediated by Kv3 channels. In the presence of Kv1 and Kv3 blockers, more than half (6 nA at +30 mV) of the high voltage-activated outward current remains and we have shown that this is mediated by a Kv2 channel. The A-current mediated by Kv4 channels also makes a small contribution in mouse (Brew et al. 2003; Johnston et al. 2008) but it is absent in rat MNTB neurons (Dodson et al. 2002). This A-current is largely inactivated at resting potentials and is completely inactivated by the voltage protocols used in this study. Together with previous publications, these data show that mouse MNTB neurons express voltage-gated potassium channels from each of the four classic voltage-gated K+ channel gene families. Their distinct locations and functions suggest general rules for the role of these gene families in regulating neuronal excitability: Kv1 regulates AP firing threshold, Kv2 assists recovery of sodium channels from inactivation, Kv3 enables fast action potential repolarization and Kv4 channels further influence AP waveform and interspike interval, conditional on the previous voltage history.
The high voltage-activated slow delayed rectifier is a Kv2.2 channel
Our whole-cell patch data showing slow kinetics and positive voltage activation of the antagonist-insensitive current are compatible with recombinant Kv2 channels (Coetzee et al. 1999). The general pharmacology of this current is also consistent with this interpretation: it is insensitive to the Kv1 antagonist dendrotoxin-I and is insensitive to low concentrations of TEA (1–3 mm, which block Kv3 and BK channels), but is blocked by higher concentrations showing an IC50 of 11.8 mm TEA (see Fig. 3). It is also partially sensitive to 4-aminopyridine (which blocks Kv1, Kv3, Kv2 and Kv4 channels) and insensitive to E4031, an antagonist of EAG and related channels, and the Kv7/KCNQ antagonist linopirdine. It is blocked by micromolar levels of quinine (Schmalz et al. 1998). The biophysical data and pharmacological profile are consistent with the MNTB delayed rectifier being of the Kv2 family, and the immunohistochemical and molecular evidence shows that the current is mediated by channels containing Kv2.2 subunits.
Early immunostaining data reported high Kv2 expression in the cerebellum and brainstem (Hwang et al. 1993) with little subcellular overlap in expression of Kv2.1 and Kv2.2. Recent in situ hybridization studies show no evidence of Kv2.1 and low levels of Kv2.2 mRNA in the MNTB (Lein et al. 2007). Our PCR data confirm that Kv2.2 is expressed in the MNTB while Kv2.1 mRNA levels were negligible. Immunohistochemistry confirmed broad expression of Kv2.2 in brainstem neurons, accompanied by intense immunofluorescence of MNTB neuron axon initial segments. We also demonstrate functional restriction of the slow current to this compartment (see Supplemental Fig. S1). The axonal location of Kv2.2 is also observed in other brain areas, e.g. cortical inhibitory neurons and perforant pathway axons in the dentate gyrus (Hwang et al. 1993). The Kv2.2 staining is in marked contrast to the focal Kv2.1 aggregates associated with synaptic profiles in spinal motoneurons (Muennich & Fyffe, 2004) and hippocampal neurons (Misonou et al. 2004), indeed, the mouse MNTB shows no detectable expression of Kv2.1 by immunohistochemistry (R. E. W. Fyffe, personal communication).
Other recent evidence has suggested that the MNTB nucleus expresses sodium-dependent K+ channels (Yang et al. 2007). In contrast to this report our recordings made in the absence of added [Na+]i show large sustained outward K+ currents (routinely > 6 nA). Under our recording conditions the outward currents changed little on adding [Na+] to the patch solution, and they showed little chloride dependence, nor were they blocked by ATP. One difference between these studies is that in order to make substitutions of [Na+]i while maintaining the [K+] electrochemical gradient, hyperosmotic solutions were used by Yang et al. in their whole-cell recordings. Our data (collected at normal osmolarity) showed large voltage-gated outward K+ currents without adding internal [Na+] and both immunohistochemical and molecular data are consistent with the dominant slow outward K+ conductance being Kv2.2.
The presence of Kv2.2 mRNA and the intense immunolabelling in the MNTB strongly support the functional expression of Kv2-containing channels, but it is unclear whether the channels are homo- or heteromeric. Heteromers of Kv2.1 and Kv2.2 subunits are unlikely due to lack of, or weak expression of, Kv2.1 in the MNTB and specific evidence that these subunits do not associate in other systems (Malin & Nerbonne, 2002). It is well established that both Kv2.1 and Kv2.2 subunits form functional channels with members of the electrically silent families (i.e. Kv5, 6, 8 and 9) resulting in alterations in activation ranges and pharmacological sensitivities (Post et al. 1996; Robertson, 1997; Salinas et al. 1997). Certainly mRNA expression for ‘silent’ subunits has been detected in the MNTB, e.g. Kv5.1 and Kv6.4 (Lein et al. 2007) so coexpression with Kv2.2 is theoretically possible. It is currently unclear whether our observed insensitivity to recombinant ScTx-1 reflects subunit heterogeneity or the recombinant origin of the toxin. However, as each subunit contributes 1/4 to the selectivity filter (which is close to the binding sites of 4-AP and TEA) a Kv2.2 heteromeric channel would readily explain the decreased sensitivity to these channel blockers.
A physiological role for Kv2.2 channels in the MNTB
The slow activation rate and depolarized activation range of the Kv2.2-mediated current means that it can make little contribution to single APs in the MNTB, particularly because MNTB APs are so fast, with half-widths of less than 0.5 ms. This contrasts with other studies of Kv2 channels showing that Kv2.1 contributes to AP repolarization (Blaine & Ribera, 2001; Malin & Nerbonne, 2002). However, in these areas (superior cervical ganglion and Xenopus neurons) AP half-widths range between 2.5 and 3.5 ms, so allowing more time for Kv2 activation during one AP. The unusual speed of the MNTB action potential is achieved by expression of Nav1.6 sodium channels (Leao et al. 2005) combined with the high expression of Kv3 potassium channels (Wang et al. 1998; Li et al. 2001). The AP waveform is dominated by the fast activating and deactivating Kv1 and Kv3 channels (Brew & Forsythe, 1995), their activity follows the AP waveform, turning off with the falling phase of the AP (Klug & Trussell, 2006). It is in the period between spikes where the slower Kv2.2 channels are free to exert their influence. Despite the fact that only a fraction of the Kv2.2 channels turn on with a single action potential their slow deactivation allows the activity to accumulate, particularly at higher frequencies. A frequency-dependent role for Kv2 (Shab) currents has also been proposed at the Drosophila neuromuscular junction (Ueda & Wu, 2006).
In the absence of specific antagonists for native Kv2.2 currents, our proposal, based on the properties of voltage-gated Na+ currents and modelling of the Na+ and K+ currents, is that Kv2 channels contribute to maintaining high frequency firing by assisting recovery of Na+ channels from inactivation, thus ensuring that more Na+ channels are able to contribute during trains of APs.
As the interspike potential falls on the steepest part of the inactivation range for the sodium current, small changes in interspike voltage will have a large effect on the availability of Nav channels. We show that after the 1st spike in a 200 Hz train around 60% of the sodium current is able to reactivate, but with the cumulative membrane hyperpolarization this amount increased by ∼17%. More importantly the available INa did not decline with spike number, which would result in eventual failure of transmission. Kv2.2 channels are open during the interspike potential; open K+ channels hyperpolarize the membrane potential. So to support the proposed role of Kv2.2 channels we simulated their absence by either depolarizing the interspike potential or maintaining it at the initial level. In both cases we observed a significant reduction in the available Nav channels throughout a 200 Hz train. Together, these findings support the conclusion that the Kv2.2-mediated hyperpolarization will sustain firing by preventing cumulative inactivation of INa during high frequency trains. In addition, the magnitude of this Kv2.2 conductance increases with maturation, similar to many other properties of this synapse which together facilitate higher frequency transmission (Taschenberger & von Gersdorff, 2000; Joshi & Wang, 2002; Joshi et al. 2004).
Further work is required to determine the subunit stoichiometry of Kv2 channels but heteromers with electrically silent subunits have been demonstrated in expression systems (Post et al. 1996; Robertson, 1997; Salinas et al. 1997). Such precision has yet to be achieved in native neurons, but with further knowledge of the spectrum of subunits expressed in the MNTB principal neuron, the MNTB could provide the first answer to this problem.
Acknowledgments
This work was funded by the MRC. J.J. was an MRC PhD scholar.
Supplementary material
Online supplemental material for this paper can be accessed at: http://jp.physoc.org/cgi/content/full/jphysiol.2008.153734/DC1 and http://www.blackwell-synergy.com/doi/suppl/10.1113/jphysiol.2008.153734
References
- Barnes-Davies M, Forsythe ID. Pre- and postsynaptic glutamate receptors at a giant excitatory synapse in rat auditory brainstem slices. J Physiol. 1995;488:387–406. doi: 10.1113/jphysiol.1995.sp020974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birnbaum SG, Varga AW, Yuan L-L, Anderson AE, Sweatt JD, Schrader LA. Structure and function of Kv4-family transient potassium channels. Physiol Rev. 2004;84:803–833. doi: 10.1152/physrev.00039.2003. [DOI] [PubMed] [Google Scholar]
- Blaine JT, Ribera AB. Kv2 channels form delayed-rectifier potassium channels in situ. J Neurosci. 2001;21:1473–1480. doi: 10.1523/JNEUROSCI.21-05-01473.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brew HM, Forsythe ID. Two voltage-dependent K+ conductances with complementary functions in postsynaptic integration at a central auditory synapse. J Neurosci. 1995;15:8011–8022. doi: 10.1523/JNEUROSCI.15-12-08011.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brew HM, Forsythe ID. Systematic variation of potassium current amplitudes across the tonotopic axis of the rat medial nucleus of the trapezoid body. Hear Res. 2005;206:116–132. doi: 10.1016/j.heares.2004.12.012. [DOI] [PubMed] [Google Scholar]
- Brew HM, Hallows JL, Tempel BL. Hyperexcitability and reduced low threshold potassium currents in auditory neurons of mice lacking the channel subunit Kv1.1. J Physiol. 2003;548:1–20. doi: 10.1113/jphysiol.2002.035568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catterall WA, Goldin AL, Waxman SG. International Union of Pharmacology. XXXIX. Compendium of voltage-gated ion channels: sodium channels. Pharmacol Rev. 2003;55:575–578. doi: 10.1124/pr.55.4.7. [DOI] [PubMed] [Google Scholar]
- Clay JR. Determining K+ channel activation curves from K+ channel currents. Eur Biophys J. 2000;29:555–557. doi: 10.1007/s002490000091. [DOI] [PubMed] [Google Scholar]
- Coetzee WA, Amarillo Y, Chiu J, Chow A, Lau D, McCormack T, Moreno H, Nadal MS, Ozaita A, Pountney D, Saganich M, Vega-Saenz de Miera E, Rudy B. Molecular diversity of K+ channels. Ann N Y Acad Sci. 1999;868:233–285. doi: 10.1111/j.1749-6632.1999.tb11293.x. [DOI] [PubMed] [Google Scholar]
- Dodson PD, Barker MC, Forsythe ID. Two heteromeric Kv1 potassium channels differentially regulate action potential firing. J Neurosci. 2002;22:6953–6961. doi: 10.1523/JNEUROSCI.22-16-06953.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dodson PD, Billups B, Rusznak Z, Szucs G, Barker MC, Forsythe ID. Presynaptic rat Kv1.2 channels suppress synaptic terminal hyperexcitability following action potential invasion. J Physiol. 2003;550:27–33. doi: 10.1113/jphysiol.2003.046250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dodson PD, Forsythe ID. Presynaptic K+ channels: electrifying regulators of synaptic terminal excitability. Trends Neurosci. 2004;27:210–217. doi: 10.1016/j.tins.2004.02.012. [DOI] [PubMed] [Google Scholar]
- Escoubas P, Diochot S, Celerier M-L, Nakajima T, Lazdunski M. Novel tarantula toxins for subtypes of voltage-dependent potassium channels in the Kv2 and Kv4 subfamilies. Mol Pharmacol. 2002;62:48–57. doi: 10.1124/mol.62.1.48. [DOI] [PubMed] [Google Scholar]
- Furlan F, Taccola G, Grandolfo M, Guasti L, Arcangeli A, Nistri A, Ballerini L. ERG conductance expression modulates the excitability of ventral horn GABAergic interneurons that control rhythmic oscillations in the developing mouse spinal cord. J Neurosci. 2007;27:919–928. doi: 10.1523/JNEUROSCI.4035-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gessner G, Heinemann SH. Inhibition of hEAG1 and hERG1 potassium channels by clofilium and its tertiary analogue LY97241. Br J Pharmacol. 2003;138:161–171. doi: 10.1038/sj.bjp.0705025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gittelman JX, Tempel BL. Kv1.1 containing channels are critical for temporal precision during spike initiation. J Neurophysiol. 2006;96:1203–1214. doi: 10.1152/jn.00092.2005. [DOI] [PubMed] [Google Scholar]
- Guan D, Tkatch T, Surmeier DJ, Armstrong WE, Foehring RC. Kv2 subunits underlie slowly inactivating potassium current in neocortical pyramidal neurons. J Physiol. 2007;581:941–960. doi: 10.1113/jphysiol.2007.128454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hille B. Ion Channels of Excitable Membranes. Sunderland: Sinauer Associates Inc. MA USA; 2001. [Google Scholar]
- Hines ML, Carnevale NT. NEURON: a tool for neuroscientists. Neuroscientist. 2001;7:123–135. doi: 10.1177/107385840100700207. [DOI] [PubMed] [Google Scholar]
- Hu H, Vervaeke K, Storm JF. M-channels (Kv7/KCNQ channels) that regulate synaptic integration, excitability, and spike pattern of CA1 pyramidal cells are located in the perisomatic region. J Neurosci. 2007;27:1853–1867. doi: 10.1523/JNEUROSCI.4463-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang P, Fotuhi M, Bredt D, Cunningham A, Snyder S. Contrasting immunohistochemical localizations in rat brain of two novel K+ channels of the Shab subfamily. J Neurosci. 1993;13:1569–1576. doi: 10.1523/JNEUROSCI.13-04-01569.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jager H, Rauer H, Nguyen AN, Aiyar J, Chandy KG, Grissmer S. Regulation of mammalian Shaker-related K+ channels: evidence for non-conducting closed and non-conducting inactivated states. J Physiol. 1998;506:291–301. doi: 10.1111/j.1469-7793.1998.291bw.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnston J, Griffin SJ, Baker C, Forsythe ID. Kv4 (A-type) potassium currents in the mouse medial nucleus of the trapezoid body. Eur J Neurosci. 2008;27:1391–1399. doi: 10.1111/j.1460-9568.2008.06116.x. [DOI] [PubMed] [Google Scholar]
- Joshi I, Shokralla S, Titis P, Wang LY. The role of AMPA receptor gating in the development of high-fidelity neurotransmission at the calyx of Held synapse. J Neurosci. 2004;24:183–196. doi: 10.1523/JNEUROSCI.1074-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joshi I, Wang LY. Developmental profiles of glutamate receptors and synaptic transmission at a single synapse in the mouse auditory brainstem. J Physiol. 2002;540:861–873. doi: 10.1113/jphysiol.2001.013506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klemic KG, Shieh C-C, Kirsch GE, Jones SW. Inactivation of Kv2.1 potassium channels. Biophys J. 1998;74:1779–1789. doi: 10.1016/S0006-3495(98)77888-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klug A, Trussell LO. Activation and deactivation of voltage-dependent K+ channels during synaptically driven action potentials in the MNTB. J Neurophysiol. 2006;96:1547–1555. doi: 10.1152/jn.01381.2005. [DOI] [PubMed] [Google Scholar]
- Kopp-Scheinpflug C, Lippe WR, Dorrscheidt GJ, Rubsamen R. The medial nucleus of the trapezoid body in the gerbil is more than a relay: comparison of pre- and postsynaptic activity. J Assoc Res Otolaryngol. 2003;4:1–23. doi: 10.1007/s10162-002-2010-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamarca V, Grasa L, Fagundes DS, Arruebo MP, Plaza MA, Murillo MD. K+ channels involved in contractility of rabbit small intestine. J Physiol Biochem. 2006;62:227–236. doi: 10.1007/BF03165751. [DOI] [PubMed] [Google Scholar]
- Lawrence JJ, Saraga F, Churchill JF, Statland JM, Travis KE, Skinner FK, McBain CJ. Somatodendritic Kv7/KCNQ/M channels control interspike interval in hippocampal interneurons. J Neurosci. 2006;26:12325–12338. doi: 10.1523/JNEUROSCI.3521-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leao RM, Kushmerick C, Pinaud R, Renden R, Li G-L, Taschenberger H, Spirou G, Levinson SR, von Gersdorff H. Presynaptic Na+ channels: locus, development, and recovery from inactivation at a high-fidelity synapse. J Neurosci. 2005;25:3724–3738. doi: 10.1523/JNEUROSCI.3983-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lein ES, Hawrylycz MJ, Ao N, Ayres M, Bensinger A, Bernard A, et al. Genome-wide atlas of gene expression in the adult mouse brain. Nature. 2007;445:168–176. doi: 10.1038/nature05453. [DOI] [PubMed] [Google Scholar]
- Li W, Kaczmarek LK, Perney TM. Localization of two high-threshold potassium channel subunits in the rat central auditory system. J Comp Neurol. 2001;437:196–218. doi: 10.1002/cne.1279. [DOI] [PubMed] [Google Scholar]
- Macica CM, von Hehn CAA, Wang L-Y, Ho C-S, Yokoyama S, Joho RH, Kaczmarek LK. Modulation of the Kv3.1b potassium channel isoform adjusts the fidelity of the firing pattern of auditory neurons. J Neurosci. 2003;23:1133–1141. doi: 10.1523/JNEUROSCI.23-04-01133.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malin SA, Nerbonne JM. Delayed rectifier K+ currents, IK, are encoded by Kv2α-subunits and regulate tonic firing in mammalian sympathetic neurons. J Neurosci. 2002;22:10094–10105. doi: 10.1523/JNEUROSCI.22-23-10094.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Misonou H, Mohapatra DP, Park EW, Leung V, Zhen D, Misonou K, Anderson AE, Trimmer JS. Regulation of ion channel localization and phosphorylation by neuronal activity. Nat Neurosci. 2004;7:711–718. doi: 10.1038/nn1260. [DOI] [PubMed] [Google Scholar]
- Muennich EA, Fyffe RE. Focal aggregation of voltage-gated, Kv2.1 subunit-containing, potassium channels at synaptic sites in rat spinal motoneurones. J Physiol. 2004;554:673–685. doi: 10.1113/jphysiol.2003.056192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen A, Kath J, Hanson D, Biggers M, Canniff P, Donovan C, Mather R, Bruns M, Rauer H, Aiyar J, Lepple-Wienhues A, Gutman G, Grissmer S, Cahalan M, Chandy K. Novel nonpeptide agents potently block the C-type inactivated conformation of Kv1.3 and suppress T cell activation. Mol Pharmacol. 1996;50:1672–1679. [PubMed] [Google Scholar]
- Post MA, Kirsch GE, Brown AM. Kv2.1 and electrically silent Kv6.1 potassium channel subunits combine and express a novel current. FEBS Lett. 1996;399:177–182. doi: 10.1016/s0014-5793(96)01316-6. [DOI] [PubMed] [Google Scholar]
- Postlethwaite M, Hennig MH, Steinert JR, Graham BP, Forsythe ID. Acceleration of AMPA receptor kinetics underlies temperature-dependent changes in synaptic strength at the rat calyx of Held. J Physiol. 2007;579:69–84. doi: 10.1113/jphysiol.2006.123612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robbins J. KCNQ potassium channels: physiology, pathophysiology, and pharmacology. Pharmacol Ther. 2001;90:1–19. doi: 10.1016/s0163-7258(01)00116-4. [DOI] [PubMed] [Google Scholar]
- Robertson B. The real life of voltage-gated K+ channels: more than model behaviour. Trends Pharmacol Sci. 1997;18:474–483. doi: 10.1016/s0165-6147(97)01140-1. [DOI] [PubMed] [Google Scholar]
- Royer A, Demolombe S, El Harchi A, Le Quang K, Piron J, Toumaniantz G, Mazurais D, Bellocq C, Lande G, Terrenoire C, Motoike HK, Chevallier JC, Loussouarn G, Clancy CE, Escande D, Charpentier F. Expression of human ERG K+ channels in the mouse heart exerts anti-arrhythmic activity. Cardiovascular Res. 2005;65:128–137. doi: 10.1016/j.cardiores.2004.09.030. [DOI] [PubMed] [Google Scholar]
- Rudy B, Chow A, Lau D, Amarillo Y, Ozaita A, Saganich M, Moreno H, Nadal MS, Hernandez-Pineda R, Hernandez-Cruz A, Erisir A, Leonard C, Vega-Saenz de Miera E. Contributions of Kv3 channels to neuronal excitability. Ann N Y Acad Sci. 1999;868:304–343. doi: 10.1111/j.1749-6632.1999.tb11295.x. [DOI] [PubMed] [Google Scholar]
- Salinas M, Duprat F, Heurteaux C, Hugnot J-P, Lazdunski M. New modulatory α subunits for mammalian Shab K+ channels. J Biol Chem. 1997;272:24371–24379. doi: 10.1074/jbc.272.39.24371. [DOI] [PubMed] [Google Scholar]
- Schmalz F, Kinsella J, Koh SD, Vogalis F, Schneider A, Flynn ERM, Kenyon JL, Horowitz B. Molecular identification of a component of delayed rectifier current in gastrointestinal smooth muscles. Am J Physiol Gastrointest Liver Physiol. 1998;274:G901–G911. doi: 10.1152/ajpgi.1998.274.5.G901. [DOI] [PubMed] [Google Scholar]
- Song P, Yang Y, Barnes-Davies M, Bhattacharjee A, Hamann M, Forsythe ID, Oliver DL, Kaczmarek LK. Acoustic environment determines phosphorylation state of the Kv3.1 potassium channel in auditory neurons. Nat Neurosci. 2005;8:1335–1342. doi: 10.1038/nn1533. [DOI] [PubMed] [Google Scholar]
- Taglialatela M, Vandongen AM, Drewe JA, Joho RH, Brown AM, Kirsch GE. Patterns of internal and external tetraethylammonium block in four homologous K+ channels. Mol Pharmacol. 1991;40:299–307. [PubMed] [Google Scholar]
- Taschenberger H, von Gersdorff H. Fine-tuning an auditory synapse for speed and fidelity: developmental changes in presynaptic waveform, EPSC kinetics, and synaptic plasticity. J Neurosci. 2000;20:9162–9173. doi: 10.1523/JNEUROSCI.20-24-09162.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ueda A, Wu C-F. Distinct frequency-dependent regulation of nerve terminal excitability and synaptic transmission by IA and IK potassium channels revealed by Drosophila Shaker and Shab mutations. J Neurosci. 2006;26:6238–6248. doi: 10.1523/JNEUROSCI.0862-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vervaeke K, Gu N, Agdestein C, Hu H, Storm JF. Kv7/KCNQ/M-channels in rat glutamatergic hippocampal axons and their role in regulation of excitability and transmitter release. J Physiol. 2006;576:235–256. doi: 10.1113/jphysiol.2006.111336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Gan L, Forsythe ID, Kaczmarek LK. Contribution of the Kv3.1 potassium channel to high-frequency firing in mouse auditory neurones. J Physiol. 1998;509:183–194. doi: 10.1111/j.1469-7793.1998.183bo.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong AY, Billups B, Johnston J, Evans RJ, Forsythe ID. Endogenous activation of adenosine A1 receptors, but not P2X receptors, during high-frequency synaptic transmission at the calyx of Held. J Neurophysiol. 2006;95:3336–3342. doi: 10.1152/jn.00694.2005. [DOI] [PubMed] [Google Scholar]
- Yang B, Desai R, Kaczmarek LK. Slack and Slick KNa channels regulate the accuracy of timing of auditory neurons. J Neurosci. 2007;27:2617–2627. doi: 10.1523/JNEUROSCI.5308-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.