

Abstract
Labeling reagents that differ only in their isotopic composition offer a powerful approach to achieve relative quantification between samples by ESI-MS. Heavy and light isotopic forms of cholamine, which contain a positively charged quaternary ammonium group, were synthesized and tested as new labeling reagents for the relative quantification of carboxylic acid-containing metabolites, specifically fatty acids. The positive charge on cholamine ensures that the labeled product is also positively charged under all LC/MS conditions, regardless of mobile phase pH. This leads to high ionization efficiency and correspondingly high detection sensitivity, demonstrated here for the analysis of fatty acids in positive ion mode ESI-MS after reverse-phase separation under acidic conditions. Good accuracy and precision were obtained by mixing heavy- and light-labeled hydrolyzed egg lipid extracts in different known ratios. The relative quantification results for ten observed fatty acids had an average absolute error of 4.6% and an average coefficient of variation (CV) of 2.6%. The labeling strategy yielded a median CV of 6% when employed for fatty acid analysis of eggs from chickens fed various dietary supplements.
INTRODUCTION
Metabolomics involves identifying and quantifying the small molecules present in biological samples. The two most common analysis techniques are NMR and chromatography coupled to mass spectrometry (e.g. GC-MS and LC-MS).1–4 Mass spectrometry offers much greater sensitivity and thereby affords the analysis of numerous low-abundance metabolites, but its quantitative precision is inherently poorer than NMR. Relative quantification of metabolites between two or more samples helps researchers understand biological systems by unveiling interesting differences and through testing hypotheses.1, 5, 6 Many such metabolomic studies use multiple retention-time standards and sophisticated data analysis software in order to achieve reasonable precision for the comparison of samples run separately.7–11 An alternative strategy for quantification of metabolites employs isotopic labeling reagents that react with compounds containing a particular functional group.12–14 Each sample is reacted with a reagent that differs only in its isotopic composition, thereby creating “heavy” and “light” versions of derivatized metabolites, which are easily distinguished by mass spectrometry (Figure 1). Samples are mixed after completion of the labeling reaction and then analyzed by LC-MS. Labeled metabolites co-elute from the LC-column and appear in the mass spectrum as pairs of peaks with a mass-shift equal to the difference in mass of the two isotopic labels. The ratio of peak intensities for each pair yields the relative concentration of each metabolite between the two samples.
Figure 1.
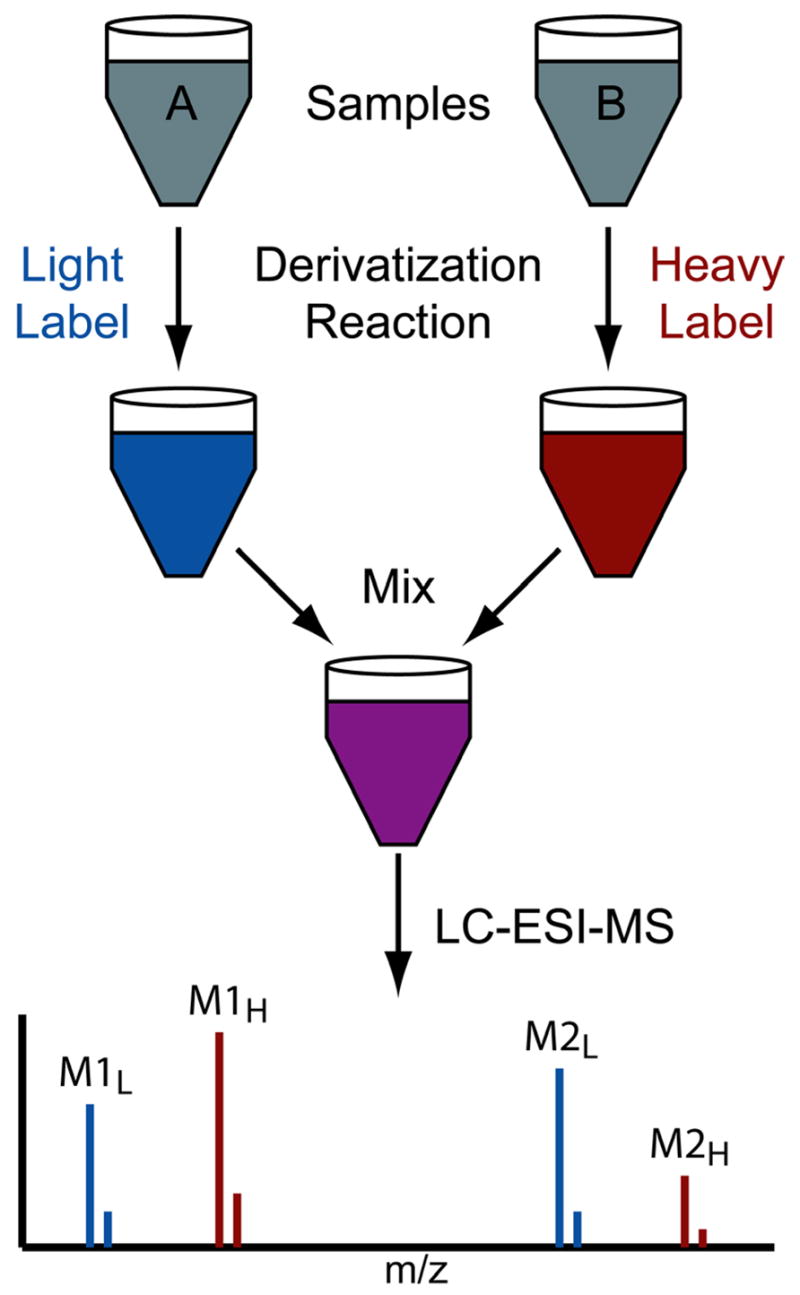
Relative quantification by isotopic labeling. Metabolites containing a certain functional group are derivatized with light- and heavy- isotopic tags prior to mixing the two samples and LC-MS analysis. The ratio of mass spectral peak intensities for each metabolite (e.g. M1L/M1H) provides relative quantification between samples A and B. Note that the two adjacent (large and small) peaks for each compound (e.g. M1L) reflect the natural isotopic distributions of these small molecules.
This isotopic labeling strategy has a number of advantages. First, it improves the precision of relative quantification by minimizing or negating errors associated with run-to-run irreproducibility. Such errors can arise from variations in mass spectrometric detection sensitivity, such as those caused by ionization suppression in electrospray,15–18 or from retention time differences between runs.9, 19 The isotopic pair of labeled compounds, however, co-elute within a single run and therefore have identical retention times and are electrosprayed from identical solution conditions. A second benefit of utilizing a derivatization reagent is that it can help identify a metabolite by indicating the presence of a certain functional group—the one targeted by the reagent. Furthermore, well-designed labeling reagents can improve the chromatographic separation as well as enhance the detection sensitivity. In this paper, we demonstrate relative quantification of carboxylic acid-containing metabolites using isotopic labeling for the analysis of fatty acids.
Fatty acids and other metabolite classes containing carboxylic acids have been analyzed by a number of GC and LC methods. Capillary column GC, often with MS detection, has been used extensively for fatty acid analysis.20–23 The process usually entails hydrolysis of lipids to release free fatty acids, which are then derivatized to methyl, trimethylsilyl, or pentafluorobenzyl esters to increase volatility. Then, just prior to GC-MS analysis, isotopically-labeled internal standards usually are added to the samples. LC-MS methods are becoming more common and have been developed for many metabolite classes that contain carboxylic acids, as reviewed by Johnson.23, 24 An issue in LC-MS of fatty acids is that acidic conditions are preferable for the chromatographic separation, yet basic conditions, which impart a negative charge to the carboxylic acid functional group, yield the greatest detection sensitivity for electrospray ionization mass spectrometry (ESI-MS).23 Several groups have employed derivatization in order to improve detection sensitivity for positive-ion mode ESI-MS under acidic conditions.25–28
We report here a fast and robust derivatization strategy that incorporates both a positively-charged functional group and an isotopic label for improved relative quantification of fatty acids. The carboxylic acid group is converted into an amide by coupling it to cholamine (Scheme 1). The quaternary ammonium group gives the labeled compound a net positive charge, which greatly enhances ionization in positive-ion mode ESI-MS.13, 25, 27, 29 Incorporation of deuterium into the methyl groups provides isotopic variants of the labeling reagent. Separation of the various labeled fatty acids is accomplished with reverse-phase HPLC under acidic conditions. We observe co-elution of the isotopic pairs, as reported by Regnier and coworkers for deuterium labeling on the methyl groups of a quaternary ammonium functional group.30 This co-elution simplifies data analysis and provides precise relative quantification of fatty acids, as demonstrated with egg lipid samples obtained from chickens that were fed diets differing in lipid composition.
Scheme 1.

Reaction of a carboxylic acid metabolite with cholamine to form a product with a quaternary ammonium group, thereby enhancing mass-spectrometric analysis in positive-ion mode.
EXPERIMENTAL
Synthesis of isotopic forms of (2-aminoethyl)trimethylammonium chloride hydrochloride (cholamine-d0 and -d9)
Both cholamine isotopic derivatives were prepared in a similar fashion (Scheme 2) starting from commercially available Boc-protected ethylenediamine (AK Scientific, Inc.) and then reacting with either methyl iodide-d0 or -d3. (The notation dx is used to indicate that the compound contains x deuterium atoms.) The Boc-protected ethylenediamine (164.9 mg, 1.029 mmol) was added to a stirring solution of methanol (Fisher, 20 mL) and KHCO3 (1.0463 g, 12.454 mmol) at room temperature. After moving the reaction to the dark, methyl iodide-d3 (Cambridge Isotope Labs, 2.275 g, 15.69 mmol) was added. After 18 h, the reaction solution was concentrated, resuspended in CHCl3 (Aldrich, 50 mL), and stirred for 1 h before filtering and concentrating it to a viscous oil (~10 mL). The Boc-protected-cholamine-d9 was precipitated by the addition of Et2O (CCI, 70 mL). The supernatant was decanted and the resulting salts washed again with Et2O (40 mL) before drying in vacuo to give 194.6 mg (56%) of Boc-protected-cholamine-d9 intermediate as the iodide salt. Rf = 0.28 (10:2:0.5, DCM:MeOH:NH4OH); 1H NMR (MeOD-d4, 300 MHz) δ 3.63 (m, 2H), 3.56 (m, 2H), 3.40 (s (broad), 1H), 1.54 (s, 9H); 13C NMR (MeOD-d4, 75 MHz) δ 158.11, 80.95, 66.03, 35.85, 28.64 (3C); MS (HRESI-MS) calculated for [C10H14D9N2O2]+ 212.2316, found 212.2319. The Boc-protected-cholamine-d0 intermediate was prepared in a similar manner using methyl iodide-d0. Yield 74%. Rf = 0.28 (10:2:0.5, DCM:MeOH:NH4OH); 1H NMR (MeOD-d4, 300 MHz) δ 3.61 (m, 2H), 3.58 (m, 2H), 3.40 (s (broad), 1H), 3.22 (s, 9H) 1.54 (s, 9H); 13C NMR (MeOD-d4, 75 MHz) δ 158.17, 80.93, 66.30, 54.15 (3C), 35.89, 28.64 (3C); MS (HRESI-MS) calculated for [C10H23N2O2]+ 203.1760, found 203.1765.
Scheme 2.

Synthesis of light(d0)- and heavy(d9)-cholamine.
The Boc-protected-cholamine-d9 intermediate (146.2 mg, 0.4309 mmol) was dissolved in 2 mL of methanol, cooled to 0° C, and then acetyl chloride (Aldrich, 173 μL, 2.43 mmol) was added dropwise. After stirring for 3 h, the reaction was diluted with 50 mL of Et2O causing precipitation of an off-white solid. The Et2O was decanted and the salts were washed iteratively with Et2O (3 × 30 mL) before concentrating in vacuo. Minimal anhydrous acetone was added to dissolve the salts before the addition of 40 mL of Et2O, which again caused the salts to precipitate. The colored mother liquor was decanted and the resulting salts dried in vacuo to give 66.5 mg (84%) of cholamine methyl iodide-d9 as a white chloride salt. Rf = 0.21 (10:4:1, DCM:MeOH:NH4OH); 1H NMR (MeOD-d4, 300 MHz) δ 3.73 (m, 2H), 3.50 (m, 2H), 3.30 (s (broad), 1H); 13C NMR (MeOD-d4, 75 MHz) δ 62.44, 34.21; MS (HRESI-MS) calculated for [C5H6D9N2]+ 112.1800, found 112.1817. Cholamine-d0 was prepared in a similar manner. Yield 91%. Rf = 0.21 (10:4:1, DCM:MeOH:NH4OH); 1H NMR (MeOD-d4, 300 MHz) δ 3.76 (m, 2H), 3.51 (m, 2H), 3.29 (s (broad), 9H); 13C NMR (MeOD-d4, 75 MHz) δ 62.71, 54.30 (3C), 34.27; MS (HRESI-MS) calculated for [C5H15N2]+ 103.1235, found 103.1244.
Animal Protocol and Lipid Extraction from Egg Yolk
Single-comb white Leghorn individually-housed hens each were assigned to one of five dietary regimens: a supplement of 3.5% olive oil (OO), or a supplement of 3.5% safflower oil (SO), or each of these with 0.5% conjugated linoleic acid (OO-CLA and SO-CLA, respectively), or a standard table diet as a control. Following established protocols, egg lipids were extracted and hydrolyzed to free fatty acids.31 Twelve milliliters of 2:1 chloroform:methanol was used to extract lipids from 3 g of egg lipid. A 1 mL aliquot of egg lipid was placed into a 20 mL vial followed by addition of 5 mL of 9:1 acetonitrile:5M HCl. The solution was refluxed until hydrolysis was complete, as visualized by the complete dissolution of the oil layer. Samples were then taken to near dryness on a rotary evaporator, followed by complete drying with a vacuum pump.
Labeling of Chicken Egg Fatty Acid Metabolites with Cholamine
Chicken egg fatty acid extracts were dissolved in 5 mL of dimethyl sulfoxide (DMSO, Sigma). Aliquots (50 μL each) were treated sequentially with: 125 μL of 20 mM 1-Hydroxybenzotriazole (HOBt, Aldrich) in DMSO; 50 μL of 100 mM cholamine-d0 or –d9 in DMSO containing 200 mM triethylamine (TEA, Aldrich); and 125 μL of 20 mM 2-(1H-Benzotriazole-1-yl)-1,1,3,3-tetramethylaminium hexafluorophosphate (HBTU, Novabiochem) in DMSO. The samples were then left to react overnight before being dissolved (1:100) in 75:25 water:acetonitrile with 0.1% formic acid. Heavy and light labeled samples were mixed 1:1 unless otherwise stated. Injection volumes of 2 μL from each mixture were then analyzed by LC-MS as described below.
Labeling Fatty Acid Standards with Cholamine
Myristic acid, oleic acid, and arachidonic acid were purchased from Sigma-Aldrich. A mixture of 0.2 μmol of each of the three acids was treated sequentially with 30 μL of 200 mM HOBt in DMSO, 600 μL of 100 mM cholamine-d0 in DMSO containing 200 mM TEA, and 30 μL of 200 mM HBTU in DMSO. The samples were left to react overnight before being dissolved in 75:25 water:acetonitrile with 0.1% formic acid. A solution of these labeled fatty acids (15 μM each) was analyzed by direct infusion ESI-MS. In addition, these three labeled fatty acids were mixed with the three unlabeled fatty acids (15 μM each) and then a 10 μL injection was analyzed by LC-MS.
LC-MS Analysis
The HPLC system consisted of an LC Packings Famos auto-sampler and UltiMate solvent pump (Dionex Corporation, Sunnyvale, CA). A 150 × 0.300 mm, C18 PepMap 100 capillary column (Dionex Corporation, Sunnyvale, CA) with 3 μm particle size and 100 Å pore-size was used for separation of the analytes. The 38 minute binary gradient elution profile was as follows: t=0, 25% B; t=5, 25% B; t=17, 100% B; t=27, 100% B; t=28, 25% B; and t=38, 25% B. Mobile phase A was 0.1% formic acid (EM, Gibbstown, NJ) in HPLC grade water (Burdick & Jackson, Morristown, NJ) and mobile phase B was 0.1% formic acid in HPLC grade acetonitrile (Burdick & Jackson, Morristown, NJ). The flow rate was 4 μL/min and the LC effluent was directed to the capillary electrospray ionization source of a MicrOTOF time-of-flight mass spectrometer (Bruker Daltonics, Billerica, MA). Positive-ion mode or negative-ion mode electrospray was performed with a potential difference of 4500 V between the spray tip and the inlet using 0.4 bar of N2 as a nebulizer gas and 4.0 L/min of N2 drying gas at 150 °C.
RESULTS AND DISCUSSION
Simulated Relative Quantification of Fatty Acids
Two identical samples of fatty acids from hydrolyzed egg lipid extract were reacted with either the heavy or the light form of cholamine as described in the Experimental Section. The two product solutions were subsequently mixed in ratios of 1:4 and 4:1 to simulate relative quantification. These mixtures were separated by reverse-phase HPLC followed by ESI-MS, and data from the 1:4 mixture are displayed in Figure 2. Extracted ion chromatograms (EICs) were obtained for the masses corresponding to the heavy- and light-labeled versions of ten fatty acids. Chromatographic co-elution of the heavy- and light-labeled fatty acids is clearly demonstrated by the EICs. Example mass spectra are shown for two fatty acids, one with high abundance and the other with low abundance, and these spectra illustrate the 9 Da mass-shift between the heavy and light forms. The ratios of peak intensities were calculated for each pair of heavy- and light-labeled fatty acids from four runs (2 runs each of the 1:4 and 4:1 mixtures); the average ratios are displayed in Table 1. The average experimental ratio for the ten fatty acids was 4.07 compared with an expected ratio of 4.00. The accuracy and precision were very good for this simulated relative quantification experiment: average absolute error was 4.6% and the average CV was 2.6%.
Figure 2.

Extracted ion chromatograms (left) and representative mass spectra (right) of light- and heavy-cholamine labeled fatty acids. For clarity, the EICs are divided between two panels: the upper one shows five more abundant fatty acids and the lower one shows five less abundant fatty acids. Perfect co-elution is observed for the isotopic pairs of peaks (dotted and solid lines for light and heavy labels, respectively). The expected 1:4 intensity ratio and the 9 Da shift are also evident. Note that each compound yields two or three peaks in the mass spectra due to the natural isotopic distribution, but only the tallest (monoisotopic) peak was employed for quantification.
Table 1.
Experimental isotope ratios for ten fatty acids in a simulated relative quantification experiment where the expected ratio was 4.00
| Fatty Acida | Experimental Ratiob | Precision CV (%) | Accuracy|% error| |
|---|---|---|---|
| 16:0 | 4.21 | 3.1 | 5.4 |
| 16:1 | 4.04 | 0.8 | 1.1 |
| 18:0 | 4.30 | 2.3 | 7.5 |
| 18:1 | 4.22 | 2.6 | 5.5 |
| 18:2 | 4.29 | 1.4 | 7.4 |
| 18:3 | 3.98 | 6.3 | 0.6 |
| 20:3 | 3.50 | 4.8 | 12.5 |
| 20:4 | 4.15 | 1.8 | 3.7 |
| 22:5 | 4.04 | 1.7 | 1.0 |
| 22:6 | 3.94 | 1.3 | 1.4 |
| Average | 4.07 | 2.6 | 4.6 |
Fatty acids are referred to by their number of carbons and degree of unsaturation.
A 1:1 mixture was used to normalize ratios for minor variations in reactivity between heavy and light cholamine.
Attributes of Cholamine as an Isotopic Labeling Reagent
Cholamine has a number of advantages as a labeling reagent. First, the product of the reaction between cholamine and a carboxylic acid contains a permanently ionized quaternary ammonium group (Scheme 1). Not only is the ionizability of the acid not destroyed during the coupling reaction, but the mode of ionization switches from negative to positive mode, which facilitates the use of acidic buffers during the LC separation. Second, the reaction conditions are such that no sample clean-up is required. The product-containing solution is simply diluted with LC loading buffer, separated into its components by chromatography, and then mass analyzed. Third, the size and mass of the label is smaller than the targeted analytes; the added mass for the light and heavy labels are 85 Da and 94 Da, respectively. Thus, the chemical structures of the original analytes still contribute significantly to the chromatographic separation, as observed in Figure 2. In other words, the label does not dominate the separation and cause all labeled compounds to elute at the same time, which could increase detection limits because of ion suppression effects,15 and thereby impair observation of some minor constituents. Fourth, the placement of the deuterium on the methyl groups of the quaternary ammonium group, rather than at some more hydrophobic position, ensures co-elution of light- and heavy-isotope derivatives of equivalent metabolites from two different samples, as observed in Figure 2 and reported in the literature.30 In contrast, some deuterium isotope derivatives that are used in relative quantification of peptides produce an undesirable chromatographic shift, which introduces a source of error in calculation of relative abundances.13, 19 Finally, the relatively large 9 Da shift between the light- and heavy-labeled compounds prevents the natural isotope peaks of the light-labeled compound from overlapping with the monoisotopic peak of the heavy-labeled compound.
The sensitivity enhancement produced by cholamine labeling was determined by comparing ESI-MS intensities of labeled and unlabeled fatty acids of various lengths and degrees of unsaturation. Myristic acid (14:0), oleic acid (18:1), and arachidonic acid (20:4) were labeled with cholamine and then diluted to 15 μM in 75:25 water:acetonitrile with 0.1% formic acid. Direct infusion positive-mode ESI-MS of this unpurified mixture, which contained excess labeling reagents, gave signal intensities that were 1.5 to 3 times higher than the three unlabeled acids infused under their optimal conditions (negative-mode ESI-MS at 15 μM in 75:25 water:acetonitrile with 1% ammonium hydroxide). These sensitivity enhancement values represent lower limits and would likely be even greater following LC purification that would remove compounds likely to cause ion suppression (e.g. the excess cholamine and triethylamine). The sample of unlabeled acids did not contain any interfering compounds, but even so it yielded lower signal intensities than the sample of labeled fatty acids.
The improvement offered by labeling fatty acids was further explored by estimating the limits of detection for the labeled fatty acids and for the unlabeled ones under typical acidic LC-MS conditions. An equimolar mixture was prepared of the three cholamine-labeled fatty acids and the three unlabeled fatty acids, where each of the six compounds was 15 μM in a solution of 75:25 water:acetonitrile with 0.1% formic acid. These mixtures were separated by reverse-phase HPLC followed by positive-mode ESI-MS as well as negative-mode ESI-MS (in separate LC runs). The limits of detection for the labeled fatty acids in positive mode were 15 ± 5 fmol, 23 ± 4 fmol, and 30 ± 8 fmol for myristic, oleic and archidonic acids, respectively. The LODs for the unlabeled fatty acids in negative mode were in the picomole range, and as such were 110X, 300X, and 30X higher than for their labeled counterparts in positive mode. The poor LOD values for the unlabeled fatty acids were caused predominantly by the acidic buffer, which limits deprotonation of the carboxylic acid groups. The acidic buffer was employed because it is highly preferred for RPLC of fatty acids,23 which makes the above experiment a fair comparison between cholamine-labeled and unlabeled fatty acids when considering the entire LC-MS analysis method.
Relative Quantification of Fatty Acids from Hydrolyzed Egg Lipid
Dietary lipid composition can have significant effects on the growth and composition of chickens and their eggs. For example, animals fed conjugated linoleic acid (CLA) exhibit improved growth, efficiency of food use, and resistance to certain diseases;32–36 but it also leads to dramatic decreases in egg hatchability. Aydin, et al. have shown that combining other fatty acids with CLA eliminates the hatchability problem.37 These fatty acids in the diet impact the fatty acid composition of the eggs, which affects hatchability. The following experiment used cholamine labeling for relative quantification of fatty acids in egg lipids in order to examine the incorporation of dietary fat into egg yolks.
Results are shown in Figure 3 for the fatty acid analysis of egg lipids after supplementing the standard table diet with olive oil (OO) and safflower oil (SO), with and without CLA. The various supplemented diets are all compared to the standard table diet, and a ratio of 1 in Figure 3a indicates that the supplement had no effect on the amount of that particular fatty acid. Many substantial changes in the fatty acid composition are observed. The error bars in Figure 3 represent the standard deviations obtained from two LC-MS runs for each of two cholamine labeling experiments (i.e. four technical replicates). The median CV across the 12 different fatty acids was 6% (range of 2–20%) for this relative quantification experiment using isotope labels. This precision is quite similar to the 8% value reported for GC-MS of a few metabolites that were quantified by comparison to added isotopic internal standards.38 The labeling approach, however, allows for a more comprehensive metabolite analysis than a targeted approach wherein each isotopic standard has to be purchased, weighed, dissolved, diluted and added to the sample extracts.
Figure 3.
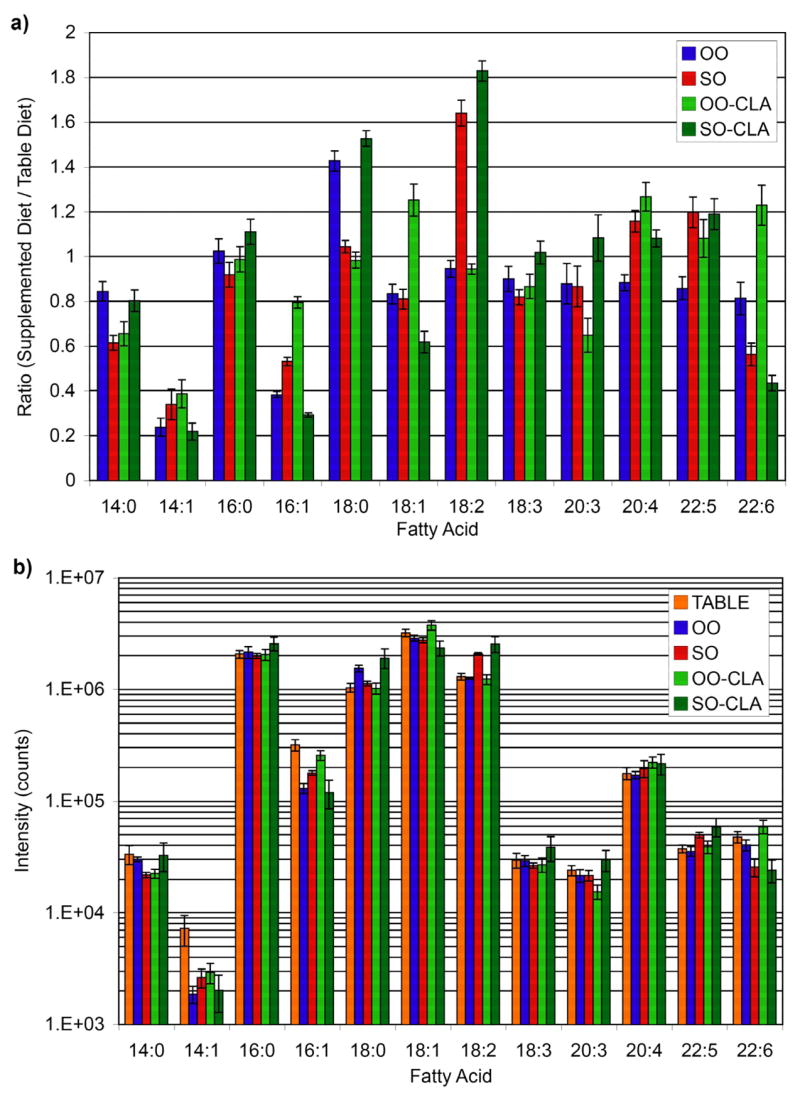
Quantification of fatty acids in hydrolyzed egg lipid extracts from chickens on various diets. (a) The relative quantification results from heavy- and light- isotopic labeling with cholamine are displayed as ratios. (b) This log plot of absolute intensities of labeled fatty acids shows a dynamic range of 3 orders of magnitude.
Quantification by LC-MS without labels or isotopic standards typically involves a simple comparison of peak intensity/area between runs. However, this method is generally less precise due to variations in run-to-run retention times and ionization efficiency.9, 15, 19 This effect is demonstrated by an alternate analysis of the intensity data (Figure 3b). Ratios of peak intensities for each compound between different LC-MS runs were calculated, and the median CV in that case was 12% (range of 2-36%). This variability is similar to the values reported for other metabolite profiling of complex samples that employed either GC-MS (CV = 13.8%)38 or LC-MS (CV = 13.5%)11 without the benefit of an isotopic variant. In contrast, ratioing isotopic peak intensities within a single run using the isotopic labeling approach described herein significantly improves the precision of relative quantification (median CV of only 6%). This example illustrates the improvement that can be expected when employing isotopic labels in a global relative quantification experiment.
CONCLUSIONS
Chemical derivatization with cholamine facilitates LC-MS analysis of fatty acids by greatly enhancing positive mode electrospray ionization after reverse-phase separation under acidic conditions. Metabolite identification did not present a problem for the dozen or so fatty acids of interest in these egg lipid samples, but metabolite database searching for other types of samples could be improved by pairing the knowledge of a carboxylic acid functional group with a metabolite mass. The strategy of isotopic labeling with cholamine assures precise relative quantification without the possibility that matrix effects or run-to-run variations might adversely and unknowingly affect the results. Cholamine labeling is suitable for other carboxylic acid-containing metabolites, such as small polar metabolites, as well as other types of samples, such as lysates from cultured cells. These types of analyses are currently underway in our laboratory.
Acknowledgments
Egg lipid samples were a generous gift from Mark Cook, Shane Huebner and Vanessa Leone. This work was supported by NIH grants 1R21DK070297, 2R01HG002298 and GM065406.
References
- 1.Birkemeyer C, Luedemann A, Wagner C, Erban A, Kopka J. Trends Biotechnol. 2005;23:28–33. doi: 10.1016/j.tibtech.2004.12.001. [DOI] [PubMed] [Google Scholar]
- 2.Rochfort S. J Nat Prod. 2005;68:1813–1820. doi: 10.1021/np050255w. [DOI] [PubMed] [Google Scholar]
- 3.Sumner LW, Mendes P, Dixon RA. Phytochemistry. 2003;62:817–836. doi: 10.1016/s0031-9422(02)00708-2. [DOI] [PubMed] [Google Scholar]
- 4.Weckwerth W, Fiehn O. Curr Opin Biotechnol. 2002;13:156–160. doi: 10.1016/s0958-1669(02)00299-9. [DOI] [PubMed] [Google Scholar]
- 5.Fiehn O, Kopka J, Dormann P, Altmann T, Trethewey RN, Willmitzer L. Nat Biotechnol. 2000;18:1157–1161. doi: 10.1038/81137. [DOI] [PubMed] [Google Scholar]
- 6.Mashego MR, Wu L, Van Dam JC, Ras C, Vinke JL, Van Winden WA, Van Gulik WM, Heijnen JJ. Biotechnol Bioeng. 2004;85:620–628. doi: 10.1002/bit.10907. [DOI] [PubMed] [Google Scholar]
- 7.Wang WX, Zhou HH, Lin H, Roy S, Shaler TA, Hill LR, Norton S, Kumar P, Anderle M, Becker CH. Anal Chem. 2003;75:4818–4826. doi: 10.1021/ac026468x. [DOI] [PubMed] [Google Scholar]
- 8.von Roepenack-Lahaye E, Degenkolb T, Zerjeski M, Franz M, Roth U, Wessjohann L, Schmidt J, Scheel D, Clemens S. Plant Physiol. 2004;134:548–559. doi: 10.1104/pp.103.032714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Smith CA, Want EJ, O’Maille G, Abagyan R, Siuzdak G. Anal Chem. 2006;78:779–787. doi: 10.1021/ac051437y. [DOI] [PubMed] [Google Scholar]
- 10.Nordstrom A, O’Maille G, Qin C, Siuzdak G. Anal Chem. 2006;78:3289–3295. doi: 10.1021/ac060245f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Roy SM, Anderle M, Lin H, Becker CH. Int J Mass Spectrom. 2004;238:163–171. [Google Scholar]
- 12.Shortreed MR, Lamos SM, Frey BL, Phillips MF, Patel M, Belshaw PJ, Smith LM. Anal Chem. 2006;78:6398–6403. doi: 10.1021/ac0607008. [DOI] [PubMed] [Google Scholar]
- 13.Yang WC, Mirzaei H, Liu XP, Regnier FE. Anal Chem. 2006;78:4702–4708. doi: 10.1021/ac0600510. [DOI] [PubMed] [Google Scholar]
- 14.Berry KAZ, Murphy RC. J Lipid Res. 2005;46:1038–1046. doi: 10.1194/jlr.M500014-JLR200. [DOI] [PubMed] [Google Scholar]
- 15.Annesley TM. Clin Chem. 2003;49:1041–1044. doi: 10.1373/49.7.1041. [DOI] [PubMed] [Google Scholar]
- 16.Constantopoulos TL, Jackson GS, Enke CG. J Am Soc Mass Spectrom. 1999;10:625–634. doi: 10.1016/S1044-0305(99)00031-8. [DOI] [PubMed] [Google Scholar]
- 17.Sterner JL, Johnston MV, Nicol GR, Ridge DP. J Mass Spectrom. 2000;35:385–391. doi: 10.1002/(SICI)1096-9888(200003)35:3<385::AID-JMS947>3.0.CO;2-O. [DOI] [PubMed] [Google Scholar]
- 18.Tang L, Kebarle P. Anal Chem. 1993;65:3654–3668. [Google Scholar]
- 19.Pan CL, Kora G, Tabb DL, Pelletier DA, McDonald WH, Hurst GB, Hettich RL, Samatova NF. Anal Chem. 2006;78:7110–7120. doi: 10.1021/ac0606554. [DOI] [PubMed] [Google Scholar]
- 20.Lagerstedt SA, Hinrichs DR, Batt SM, Magera MJ, Rinaldo P, McConnell JP. Mol Genet Metab. 2001;73:38–45. doi: 10.1006/mgme.2001.3170. [DOI] [PubMed] [Google Scholar]
- 21.Eder K. J Chromatogr B Biomed Appl. 1995;671:113–131. doi: 10.1016/0378-4347(95)00142-6. [DOI] [PubMed] [Google Scholar]
- 22.Gutnikov G. J Chromatogr B Biomed Appl. 1995;671:71–89. doi: 10.1016/0378-4347(95)00116-z. [DOI] [PubMed] [Google Scholar]
- 23.Johnson DW. Clin Biochem. 2005;38:351–361. doi: 10.1016/j.clinbiochem.2005.01.007. [DOI] [PubMed] [Google Scholar]
- 24.Saghatelian A, Trauger SA, Want EJ, Hawkins EG, Siuzdak G, Cravatt BF. Biochemistry. 2004;43:14332–14339. doi: 10.1021/bi0480335. [DOI] [PubMed] [Google Scholar]
- 25.Johnson DW. Rapid Commun Mass Spectrom. 2000;14:2019–2024. doi: 10.1002/1097-0231(20001115)14:21<2019::AID-RCM121>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- 26.Yang Y, Griffiths WJ, Lindgren JA, Sjovall J. Rapid Commun Mass Spectrom. 1995;9:289–299. doi: 10.1002/rcm.1290090407. [DOI] [PubMed] [Google Scholar]
- 27.Barry SJ, Carr RM, Lane SJ, Leavens WJ, Monte S, Waterhouse I. Rapid Commun Mass Spectrom. 2003;17:603–620. doi: 10.1002/rcm.957. [DOI] [PubMed] [Google Scholar]
- 28.Cartwright AJ, Jones P, Wolff JC, Evans EH. Rapid Commun Mass Spectrom. 2005;19:1058–1062. doi: 10.1002/rcm.1883. [DOI] [PubMed] [Google Scholar]
- 29.Mirzaei H, Regnier F. Anal Chem. 2006;78:4175–4183. doi: 10.1021/ac0602266. [DOI] [PubMed] [Google Scholar]
- 30.Zhang RJ, Sioma CS, Thompson RA, Xiong L, Regnier FE. Anal Chem. 2002;74:3662–3669. doi: 10.1021/ac025614w. [DOI] [PubMed] [Google Scholar]
- 31.Folch J, Lees M, Sloane Stanley GH. J Biol Chem. 1957:497–509. [PubMed] [Google Scholar]
- 32.Cook ME, DeVoney D, Drake B, Pariza MW, Whigham L, Yang M. In: Advances in Conjugated Linoleic Acid Research. Yurawecz MP, Mossaba MM, Kramer JKG, Pariza MW, Nelson GJ, editors. Vol. 1. AOCS Press; Champaign, IL: 1999. pp. 226–237. [Google Scholar]
- 33.Banni S, Heys SD, Wahle KWJ. In: Advances in Conjugated Linoleic Acid Research. JS, WW, CRA, editors. Vol. 2. AOCS Press; Champaign, IL: 2003. pp. 267–282. [Google Scholar]
- 34.Chin SF, Storkson JM, Albright KJ, Pariza MW. J Nutr. 1994;124:694–701. doi: 10.1093/jn/124.5.694. [DOI] [PubMed] [Google Scholar]
- 35.Pariza MW, YP, MEC Prog Lipid Res. 2001;40:283–298. doi: 10.1016/s0163-7827(01)00008-x. [DOI] [PubMed] [Google Scholar]
- 36.Ostrowska E, Muralitharan M, Cross RF, Bauman DE, Dunshea FR. J Nutr. 1999;129:2037–2042. doi: 10.1093/jn/129.11.2037. [DOI] [PubMed] [Google Scholar]
- 37.Aydin R, Pariza MW, Cook ME. J Nutr. 2001;131:800–806. doi: 10.1093/jn/131.3.800. [DOI] [PubMed] [Google Scholar]
- 38.Gullberg J, Jonsson P, Nordstrom A, Sjostrom M, Moritz T. Anal Biochem. 2004;331:283–295. doi: 10.1016/j.ab.2004.04.037. [DOI] [PubMed] [Google Scholar]