

Abstract
Mitochondria are widely distributed via regulated transport in neurons, but their sites of biogenesis remain uncertain. Most mitochondrial proteins are encoded in the nuclear genome, and evidence has suggested that mitochondrial DNA (mtDNA) replication occurs mainly or entirely in the cell body. However, it has also become clear that nuclear-encoded mitochondrial proteins can be translated in the axon and that components of the mitochondrial replication machinery reside there as well. We assessed directly whether mtDNA replication can occur in the axons of chick peripheral neurons labeled with 5-bromo-2′-deoxyuridine (BrdU). In axons that were physically separated from the cell body or had disrupted organelle transport between the cell bodies and axons, a significant fraction of mtDNA synthesis continued. We also detected the mitochondrial fission protein Drp1 in neurons by immunofluorescence or expression of GFP-Drp1. Its presence and distribution on the majority of axonal mitochondria indicated that a substantial number had undergone recent division in the axon. Because the morphology of mitochondria is maintained by the balance of fission and fusion events, we either inhibited Drp1 expression by RNAi or overexpressed the fusion protein Mfn1. Both methods resulted in significantly longer mitochondria in axons, including many at a great distance from the cell body. These data indicate that mitochondria can replicate their DNA, divide, and fuse locally within the axon; thus, the biogenesis of mitochondria is not limited to the cell body.
Keywords: mitochondria, axon, biogenesis, mtDNA, fission, fusion
INTRODUCTION
Mitochondria are required for neuronal survival (Nicholls and Budd, 2000), but the different regions of the neuron have varied and changing requirements for mitochondrial functions (Hollenbeck and Saxton, 2005). For example, areas of high ATP demand, such as nodes of Ranvier (Berthold et al., 1993; Waxman and Ritchie, 1993), myelination boundaries (Bristow et al., 2002), active synapses or tracts (Wong-Riley and Welt, 1980; Kageyama and Wong-Riley, 1982; Bindokas et al., 1998; Li et al., 2004), and active growth cones or axonal branches (Morris and Hollenbeck, 1993; Ruthel and Hollenbeck, 2003) attract and accumulate mitochondria. Neurons can redistribute mitochondrial function over time by regulated axonal transport; for example, in growing axons mitochondria move into or out of the distal axon depending on growth cone activity (Morris and Hollenbeck, 1993; Ruthel and Hollenbeck, 2003) and their retention in the distal axon is closely regulated (Hollenbeck and Saxton, 2005).
Mitochondria also turn over throughout the neuronal lifetime; thus, in addition to being redistributed they must also be replaced through organelle biogenesis. One potential challenge to this process is that most mitochondrial proteins are encoded in the nuclear genome. This could limit mitochondrial biogenesis to the cell body and require that all mitochondrial renewal in the distal axon occur by axonal transport. Indeed, studies in PC-12 cells have detected mitochondrial DNA (mtDNA) replication mainly in the perinuclear region (Davis and Clayton, 1996). However, there is also reason to believe that mitochondrial biogenesis could occur within the axon. It is now clear that local protein synthesis occurs in axons (Koenig and Giuditta, 1999; Alvarez et al., 2000; Twiss and van Minnen, 2006), and is essential for axonal functions including guidance (Campbell and Holt, 2001; Ming et al., 2002; Leung et al., 2006), regeneration (Koenig, 1991; Zheng et al., 2001; Verma et al., 2005; Twiss and van Minnen, 2006), and maintenance of mitochondrial membrane polarization (Hillefors et al., 2007). In addition, transcripts for nuclear-encoded mitochondrial proteins are among the species found in axons (Gioio et al., 2001), and mitochondria are found in close proximity to translation sites in the axon (Martin et al., 1998). Finally, components of the mitochondrial replication apparatus have been identified outside the perinuclear region in nonneuronal cells (Larsson et al., 1994; Tiranti et al., 1997; Spelbrink et al., 2000, 2001; Falkenberg et al., 2002; Magnusson et al., 2003) and neuron-like cell lines (Magnusson et al., 2003). In the latter study, evidence of mtDNA replication was observed far from the nucleus, suggesting further that mitochondrial biogenesis might also occur in the axons of bona fide neurons.
Here, we have tested the hypothesis that neuronal mitochondrial biogenesis can occur in distal axons by studying several distinct elements of this process. We have used a sensitive method of 5-bromo-2′-deoxyuridine (BrdU) detection to examine mtDNA replication in axons separated from their cell bodies. We have also localized, inhibited, or overexpressed proteins of mitochondrial fission and fusion and quantified the effects on the state of mitochondria in the distal axon. We find evidence that a portion of neuronal mitochondrial biogenesis does occur in the axon at a significant distance from the cell body.
MATERIALS AND METHODS
Materials
All materials were obtained from Sigma (St. Louis, MO) unless otherwise mentioned. All media were obtained from Mediatech (Herndon, VA). DM1A was purchased from Amersham (Arlington Heights, IL), and Alexa 488-conjugated goat-anti-mouse antibody was from Molecular Probes, (Eugene, OR). Monoclonal antibody against dynamin-related protein 1 (Drp1) and biotinylated anti-BrdU antibody was purchased from BD Biosciences (San Jose, CA). MitoTracker fluorescent dye was obtained from Invitrogen (Carlsbad, CA).
Cell Culture
Dorsal root ganglia (DRG) or sympathetic neurons were isolated from 10- to 11-day-old chicken embryos, dissociated, and cultured as previously described (Hollenbeck, 1993; Olink-Coux and Hollenbeck, 1996; Lee and Hollenbeck, 2003). Coverslips were treated with 1 mg/mL poly-l-lysine and then with 10 μg/mL laminin for 1–2 h at 37°C. Single cells were grown overnight on coated coverslips in C-medium [Leibowitz L-15 medium supplemented with 10% fetal bovine serum (Gibco BRL/Invitrogen), 100 U/mL penicillin and streptomycin (Gibco BRL), 2 mM l-glutamine, 0.6% glucose, 50 ng/mL 2.5-S mouse nerve growth factor (Alomone Labs, Jerusalem, Israel), and 0.5% methyl-cellulose (Dow Chemical Company, Midland, MI)] at 37°C. To prepare axonal halos for axotomy experiments, whole sympathetic ganglia were again isolated from 10- to 11- day-old chicken embryos and placed on coverslips that had been pretreated with short chain poly-l-lysine (Mr30,000–70,000, in 0.1 M boric acid buffer, pH 8.4) overnight at 37°C. The ganglia were grown for 2 days in C-medium containing 0.6 μM Ara-C (cytosine 1-d-arabinofuranoside) (Lee and Hollenbeck, 2003).
BrdU Labeling and Immunocytochemistry
Cultured neurons were treated with BrdU (10 μM) for 3–20 h to reveal the site of DNA synthesis. To control for the background, either BrdU was omitted from the cell culture or it was added in the presence of 100 μM 2′,3′-dideoxycytidine (ddC), an inhibitor of mtDNA polymerization (Simpson et al., 1989; Eriksson et al., 1995). We found that 4–6 h of prior incubation was necessary for ddC to block BrdU incorporation into DNA. To insure that the detected BrdU signals correspond to mtDNA synthesis, the mitochondria were also labeled with 100 nM MitoTracker fluorescent dye (Red CMH2XRos) for 30 min at 37°C. Cells were then washed twice with F+ medium (C-medium without methylcellulose), fixed with 4% paraformaldehyde (PFA, diluted in PBS) at room temperature (RT) for 15 min, and then permeabilized with 0.3% Triton X-100 in PBS for 5 min at RT. Cells were subsequently washed with TBS-Tween 20 (0.05%, TBST), incubated with 2 N HCl (pH 2.0) for 30 min at 45°C to denature mtDNA and expose the antigenic sites to anti-BrdU antibody, and then neutralized with borate buffer (0.1 M, pH 8.3) twice for 5 min. Then cells were washed again with TBST, incubated with 0.5% blocking solution in TBS (Tyramide Signal Amplification kit, PerkinElmer, Boston, MA) for 30 min at RT, and then treated with a 1:50 to 1:100 dilution of biotinylated anti-BrdU antibody in blocking solution for 1 h at 4°C. After antibody incubation, cells were washed with TBST three times for 10 min and treated with 1:200 to 1:300 dilution of Streptavidin-Peroxidase in blocking solution for 1 h at 4°C. Cells were then washed with TBST as before and treated with 1:50 dilution of Biotin-Tyramide in signal amplifying solution for 2 min (RT). After washing again, cells were incubated with Streptavidin-Peroxidase (1:200 to 1:300) for a second time at 4°C for 1 h. Cells were then washed and treated for 2 min with a 1:50 dilution of Fluorescein-Tyramide in signal amplifying solution. Finally, cells were washed and mounted using Citifluor mounting media (Ted Pella, Redding, CA). All samples were observed on a Nikon TE300 microscope with epifluorescence illumination and a 60× planapo objective. MitoTracker and BrdU signals were visualized using Texas-Red and FITC filter sets, respectively, and images were collected using a MicroMax cooled CCD camera (Princeton Instruments) and processed with Metamorph imaging software (Universal imaging, West Chester, PA). This double-precipitation method provided high sensitivity but precluded resolving the location of BrdU+ mtDNA to a particular subregion of a mitochondrion.
Axotomy: Separation of Axons from Their Cell Bodies in Culture
After sympathetic ganglia had been grown in culture for 2 days, their cell body masses were surgically removed using a glass micropipette and micromanipulator as described previously (Olink-Coux and Hollenbeck, 1996; Lee and Hollenbeck, 2003). Ganglia were washed in F+ medium and axons were transected at a distance of 400–500 μm from the cell body mass to insure that no cell bodies were present in the axonal halo after axotomy. The remaining axonal halos were incubated for 2 h at 37°C and then labeled with BrdU for another 3 h. Then they were stained with MitoTracker dye (300 nM), fixed, and BrdU-mtDNA signals were detected using immunocytochemistry as described earlier. For controls, some ganglia were kept intact with their cell bodies still attached and were processed in parallel. To determine the relative contribution of axonal to total cellular mtDNA synthesis, BrdU signals from separated axonal halos were compared with those from axonal halos of the intact ganglia.
Vinblastine Treatment
To depolymerize microtubules (MTs), established cultures were treated with vinblastine as described previously (Morris and Hollenbeck, 1995). Overnight DRG cultures were incubated with 1 μM vinblastine (Calbiochem, La Jolla, CA) in F+ media for 3 h. At the end of treatment, axons depleted of their MTs were examined for their ability to replicate mtDNA using the BrdU assay. In short, treated cells were incubated in F+ medium containing vinblastine (200 nM) for 3 h in the presence or absence of BrdU. Following incubation, cells were stained with MitoTracker dye, washed, fixed with 4% PFA/PBS, and BrdU signals were detected as described earlier. To further analyze the contribution of axonal mtDNA replication to the total mtDNA replication, the level of mtDNA synthesis detected by BrdU was quantified and compared between treated and untreated cells.
Immunofluorescence Staining and Detection
To detect MTs, cells were fixed, permeabilized, and finally blocked following our previous technique (Morris and Hollenbeck, 1995). The absence of MTs in vinblastine-treated cells was confirmed by immunostaining with DM1A anti-tubulin antibody as described earlier (Chada and Hollenbeck, 2003). Endogenous Drp1 was also detected by immunofluorescence using anti-Drp1 antibody. Overnight cultures of isolated DRGs were stained with MitoTracker dye, fixed in 4% PFA/PBS, and then permeabilized with 0.3% Triton X-100. The treatment with detergent reduced the level of cytosolic Drp1, which could have otherwise obscured the small membrane-bound fraction of the protein. Finally, cells were blocked in 3% BSA/PBS for 15 min (RT), stained with mouse anti-Drp1 antibody (1:50), and then with Alexa 488 goat anti-mouse antibody (1:200) for 30–45 min at RT. The fluorescence was viewed using epifluorescent illumination with FITC and Texas Red filter sets. For controls, the primary antibody was either omitted or replaced with DM1A. The percentage of Drp1-positive mitochondria in three different regions of the axons was determined: proximal to the cell body, midaxon, and distal axon (close to the growth cone). In addition, we determined the length of the mitochondria and the position of Drp1-staining puncta on the mitochondria.
Plasmids and Transfections
The expression constructs GFP-Drp1 (Smirnova et al., 2001), pEGFP-Mfn1 (Santel et al., 2003), and small hairpin RNA (shRNA)-Drp1 (Tondera et al., 2005) were generously provided by A. van der Bliek (UCLA, Los Angeles, CA) and A. Santel (University of Geneva, Geneva, Switzerland), respectively. Transient transfection of isolated DRG neurons was performed by electroporation following our previously described protocol (Martinez and Hollenbeck, 2003). After transfection, cells were grown for 48 h, and finally stained with MitoTracker dye and observed using epifluorescent illumination. For RNA interference (RNAi) studies, the shRNA-Drp1expression constructs were cotransfected with pEGFP plasmids at the ratio of 9:1 to detect shRNA-transfected cells. The length of the axonal mitochondria in both shRNA-Drp1 and pEGFP-Mfn1 transfected cells was compared with that in nontransfected cells.
RESULTS
In Situ Detection of mtDNA Replication in Peripheral Axons
Previous studies have detected mtDNA synthesis by labeling cells with the thymidine analog BrdU followed by immunocytochemical detection (Davis and Clayton, 1996; Magnusson et al., 2003). We enhanced the sensitivity of this procedure and developed a double-precipitation method for detection of BrdU incorporated into mtDNA (see Materials and Methods). Our technique was sensitive and robust enough to visualize strong mitochondrial signals throughout the axons of chick peripheral neurons after 3 h or less of BrdU labeling [Fig. 1(A–F)]. This signal was abolished by pre- and coincubation of BrdU-treated cultures with ddC, an inhibitor of the mtDNA polymerase [Fig. 1(G–I)]. The appearance of substantial BrdU incorporation during a short incubation indicates that the rate of mtDNA turnover in peripheral neurons is relatively high. Previous studies have quantified mtDNA turnover in DRG neurons and PC-12 cell lines indirectly by measuring the rate of mtDNA depletion during ddC treatment (Chen et al., 1991; Werth et al., 1994). We carried out BrdU pulse-chase analysis in DRG neurons and found that after a 4 h BrdU pulse, half of the recently replicated mitochondria lost their BrdU signals within 1.5 days (supplemental Fig. S1). Because recently replicated mtDNA was apparent even in the most distal regions of the axons (>1000 μm from cell bodies), we examined the possibility that mtDNA can be replicated outside of the cell body. To insure that the BrdU-marked mitochondria had not initially replicated their DNA in the cell body and then entered the axons by anterograde transport, it was necessary to prevent mitochondrial traffic from cell bodies to axons during exposure to BrdU. That was accomplished in two ways: complete axotomy and MT disruption.
Figure 1.

In situ detection of BrdU-incorporated mtDNA. Chick peripheral neurons were incubated with BrdU and the incorporation into mtDNA was detected by anti-BrdU immunocytochemistry. Phase contrast images (A,D,G), epifluorescence images of MitoTracker-labeled mitochondria (B,E,H), and of BrdU-labeled DNA (C,F,I) are shown here. After 15–20 h of labeling, BrdU signals were readily detected in mitochondria along the length of the axons (A–C). A similar pattern of BrdU localization was obtained even when the labeling period was decreased to 3 h (D–F). However, preincubation with ddC (G–I) resulted in no BrdU signals in the axons even after 15–20 h of BrdU staining and only a low level of background in the cell body (I). Note that due to the amplification with the double-precipitation procedure, the BrdU signals appeared to almost completely overlap with Mitotracker signals rather than being discrete foci within mitochondria. In addition, because of the immediate proximity to the cell body and long duration of BrdU exposure in (A–C), most of the mitochondria seen are BrdU-positive. Scale bar, 10 μm.
mtDNA Replication in Peripheral Axons After Removal of Their Cell Bodies
When whole sympathetic ganglia are cultured for 2–3 days on a poly-l-lysine substratum, they form radial arrays of axons extending outward from the cell body mass with minimal contamination by nonneuronal cells [Fig. 2(A)]. To investigate whether the axons can replicate mtDNA autonomously and independently of their cell bodies, the cell body mass was surgically removed from the ganglion explants, leaving the axonal “halos” on the substratum (Olink-Coux and Hollenbeck, 1996; Lee and Hollenbeck, 2003), 2 h prior to the BrdU pulse [Fig. 2(B)]. Control ganglia were left intact, with their cell body masses still attached. After BrdU incubation, fixation, and immunocytochemical detection, bright BrdU signals were observed not only in intact ganglia [Fig. 2(C–E)] but also in axonal halos without cell bodies [Fig. 2(F–H)]. Thus, even 2 h after cell body removal, axonal mitochondria were still capable of replicating their DNA. This result was consistent with the observations of Magnusson et al. (2003) on enucleated fibroblasts which showed that mtDNA could be synthesized in the absence of the nucleus and that all components of mtDNA synthesis are still present to support replication. To determine the relative contribution of axonal mtDNA synthesis to total neuronal mtDNA synthesis, we quantified BrdU signals from intact ganglia and axonal halos. After a 3 h BrdU labeling period, intact ganglia showed newly synthesized DNA in 12% ± 1.2 (mean ± SEM) of their axonal mitochondria, whereas in axonal halos that had cell bodies removed prior to BrdU incubation, 4.0% ± 0.81 of mitochondria had newly synthesized DNA. In replicate experiments, the absolute level of BrdU labeling of intact ganglia varied, but the ratio of % BrdU-positive mitochondria in axonal halos to that in intact ganglia remained similar, averaging 37% (for halos, n = 2451 mitochondria/7 ganglia; for intact ganglia, n = 2209 mitochondria/6 ganglia). Thus, approximately one-third of the mitochondria with recently replicated DNA present in the axons of these neurons had in fact replicated their DNA within the axons themselves.
Figure 2.
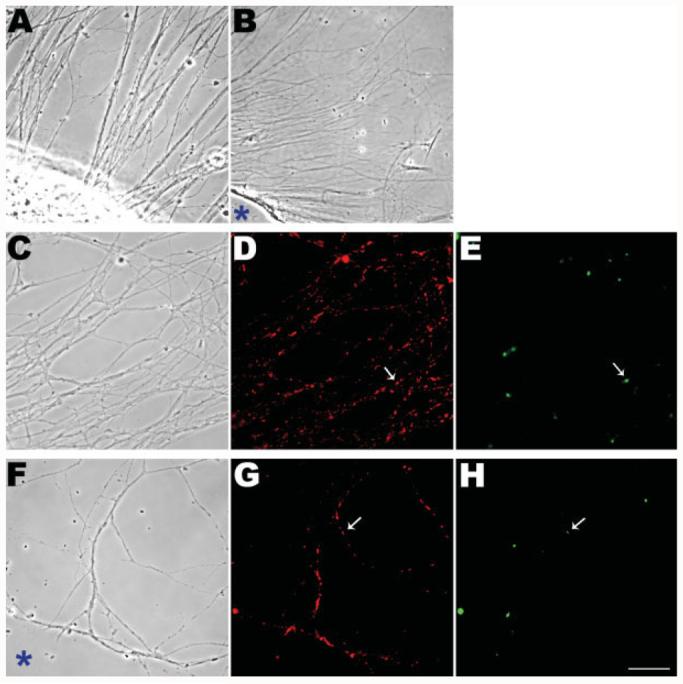
Mitochondria in axons can replicate their DNA in the absence of any connection to their cell bodies. Sympathetic ganglion explants that were grown for 2–3 days formed a radial halo of the axons (A). The cell body mass was surgically removed from ganglia 2 h prior to BrdU exposure (B). Cultures were incubated in BrdU for 3 h followed by fixation and immunocytochemical detection (C–H). BrdU signals were obvious in the axons of both intact ganglia (E) and those without cell bodies (H). Examples of BrdU signals corresponding to MitoTracker-stained mitochondria are shown by arrows in D and G. The sites of the removed cell body masses are shown with asterisks (B,F). Scale bar, 10 μm.
mtDNA Replication in Axons After Depolymerization of Their MTs
To confirm that mtDNA replication can occur in axons, we eliminated MTs from intact DRG neurons using vinblastine treatment [Fig. 3(A–F)]. MTs are the major tracks for long-range movement of mitochondria and their disruption with vinblastine severely limits the transport of mitochondria from the cell body into the axon and results in net retrograde transport (Morris and Hollenbeck, 1995). Following vinblastine treatment, we stained the cells with DM1A anti-tubulin antibody and ensured that their axons were completely depleted of MTs (data not shown). Then drug-treated neurons were incubated with BrdU and their axonal mitochondria were assessed for mtDNA replication. Even after elimination of MTs, mitochondria still incorporated BrdU into their DNA throughout the axons [Fig. 3(F)]. MitoTracker staining again ensured that the BrdU signals were coming from mitochondria [Fig. 3(E)]. Because these axons have few or no MTs, very few or no mitochondria from the cell body could have entered the axons via anterograde transport. Quantification of the BrdU signal showed that after a 3 h incubation period, 43% ± 1.5 of mitochondria present in the axons of control cells had replicated their DNA, whereas 25% ± 2.1 of those in vinblastine-treated cells had. This again indicated that a significant fraction of the replicated mtDNA observed in the axons had actually undergone synthesis there (control cells: n = 546 mitochondria/30 axons, drug-treated cells: n = 506 mitochondria/38 axons; two experiments). Taken together, the results obtained from the axotomy and vinblastine experiments indicated that mitochondria could replicate their DNA directly in the axons.
Figure 3.

Mitochondrial DNA replication in axonal mitochondria after disruption of anterograde traffic by MT depletion. DRG cultures were grown overnight and then treated with vinblastine for 3 h. They were then grown for an additional 3 h in the absence (A–C) or presence of BrdU (D–F) and finally assayed by immunocytochemistry. The phase contrast (A,D), epifluorescence images of MitoTracker (B,E), and BrdU (C,F) staining are shown earlier. BrdU signals were detected along the axon of vinblastine-treated cells (F). No background signal was observed in the control cells which had not been labeled with BrdU (C). Scale bar, 10 μm.
Colocalization of Drp1 Proteins with Axonal Mitochondria
To assess whether mitochondria also divide in the axons, we looked at the expression pattern of the essential mitochondrial fission protein, Dnm1/Drp1/Dlp1 (Bleazard et al., 1999; Labrousse et al., 1999; Sesaki and Jensen, 1999; Smirnova et al., 2001). Drp1 is mainly cytosolic, but a small fraction of the protein is localized to mitochondria, and studies have suggested that it is recruited to the mitochondrial membrane shortly before fission occurs (Labrousse et al., 1999). Following division, Drp1 remains attached to the tips of daughter mitochondria for some time before being released to the cytosolic pool (Labrousse et al., 1999; Smirnova et al., 2001). Therefore, we tracked the appearance and distribution of Drp1 on mitochondria as an indicator of incipient or recently completed fission events.
We first examined the distribution of endogenous Drp1 on axonal mitochondria by immunofluorescence. Consistent with previous studies in nonneuronal cells (Labrousse et al., 1999; Smirnova et al., 2001), Drp1 was concentrated in a punctuate pattern on axonal mitochondria (see Fig. 4). Staining typically appeared to be either distributed along the length or restricted to the tip of mitochondria [Fig. 4(D), arrow head and arrow, respectively]. Quantitative analysis showed that throughout the axons, an average of 88% of the mitochondria had detectable Drp1 puncta associated with them, with a higher fraction positive in the proximal (95%) and distal (92%) regions than in the central region (79%) [Fig. 4(E)]. The presence of Drp1 on axonal mitochondria indicated that these mitochondria were probably capable of dividing, although only a subset of those mitochondria with Drp1 foci will probably undergo fission incipiently (Labrousse et al., 1999; Smirnova et al., 2001). In addition, if recently divided mitochondria in the axon had all undergone fission in the cell body and then entered the axon by anterograde transport, we would have expected a much smaller fraction of axonal mitochondria to be Drp1-positive and their frequency would have decreased toward the distal region of the axon rather than remaining relatively constant and even increasing modestly. Thus, Drp1-mediated fission seems likely not to be limited to cell bodies, but to also occur in axons.
Figure 4.
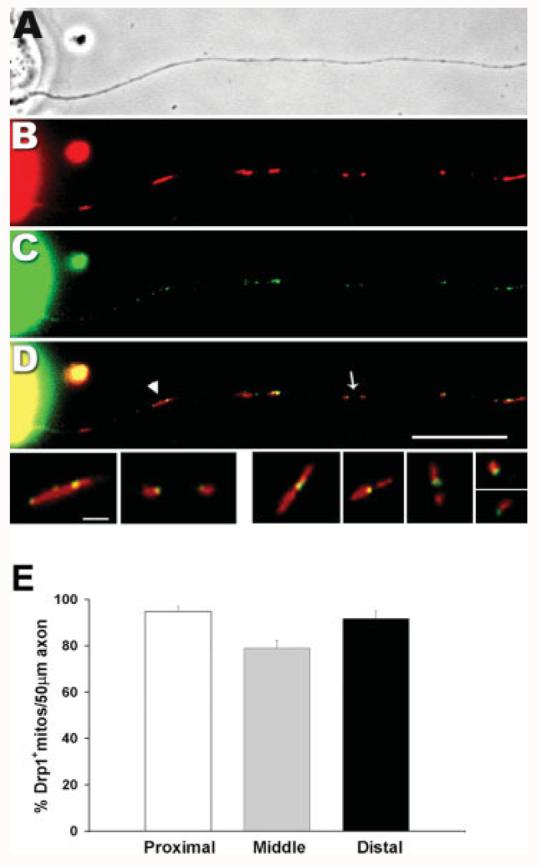
Endogenous Drp1 is colocalized with mitochondria throughout the axon. Overnight DRG cultures were stained with MitoTracker (red, B), fixed, and subjected to anti-Drp1 immunostaining (C). Phase contrast (A) and epifluorescence (B–D) images are shown here. An overlay image (D) shows that Drp1 (in green) is colocalized with mitochondria (in red). Drp1 is concentrated in puncta that are either found along the length of mitochondria (arrow head) or concentrated at the tip of newly divided ones (arrow). Scale bar, 10 μm. The mitochondria marked in (D) are shown enlarged in the bottom row on the left with additional examples on the right. Scale bar for bottom row, 2 μm. Distribution of Drp1-containing mitochondria along the length of the axons (E). The percentage of mitochondria positive for Drp1 was calculated in three different regions of the axon: proximal (0–50 μm from the cell body); distal (0–50 μm from the growth cone); and middle (remainder of the axon shaft). The histogram shows the mean of three experiments ± SEM indicated as error bars (n = 17 axons). The fraction of mitochondria containing Drp1 is similar between proximal (95 ± 2.3%, n = 193 mitochondria) and distal regions (92 ± 3.2%, n = 200) but both are higher than the middle region (79 ± = 3.3%, n = 135) of the axon (p < 0.001, repeated measures ANOVA).
If the presence of Drp1 foci on axonal mitochondria does reflect their role in fission, then we would expect those about to undergo fission to have Drp1 foci near their midpoint, whereas those that have recently divided should have Drp1 at one of their tips. To assess this, we compared the position of Drp1 on axonal mitochondria to mitochondrial length. The results showed that mitochondria with a Drp1 signal concentrated only at one end were less than half the length of those with Drp1 at their middle (mean lengths = 1.26 ± 0.04 μm and 3.3 ± 0.11 μm, respectively; n = 261 mitochondria/22 axons in two experiments).
To confirm these observations in live neurons, we transfected DRG neurons with a GFP-Drp1 expression plasmid and looked at the pattern of exogenous Drp1 expression (see Fig. 5). After 48 h of expression, GFP-Drp1 was also detected in discrete puncta on axonal mitochondria [Fig. 5(C,D)]. Thus, both immunofluorescence detection and exogenous expression studies of Drp1 showed that this fission protein is present on mitochondria in the axons of neurons. Previous studies in nonneuronal cells (Smirnova et al., 2001) have indicated that short mitochondria with Drp1 at their ends have recently divided. We suggest that at least a fraction of those mitochondria with Drp1 puncta near their midpoints are about to divide, whereas those shorter mitochondria with Drp1 puncta present at their ends have recently divided.
Figure 5.
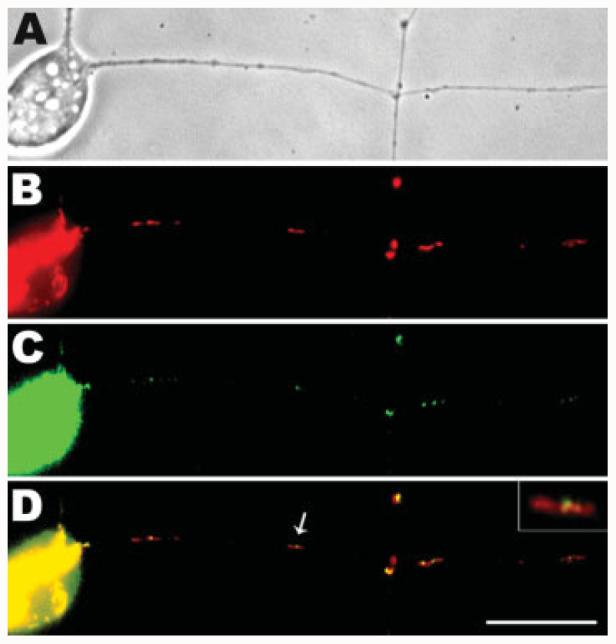
GFP-Drp1 is colocalized with axonal mitochondria in DRG neurons. Isolated DRGs were transfected with a GFP-Drp1 expression plasmid and observed after 48 h. Phase contrast (A) and epifluorescence (B–D) images are shown here. Mitochondria were marked by MitoTracker (red, B). As shown in the merged image (D), the expressed GFP-Drp1 was detected as green puncta (C) on the mitochondria. The mitochondrion indicated by the arrow in D is shown enlarged in the inset. This image shows that the Drp1 fission protein is concentrated near the midpoint of a relatively long mitochondrion. Scale bar, 10 μm.
Reduction of Drp1 Expression Results in Formation of Longer Mitochondria in Distal Axons
To further investigate whether Drp1-mediated fission occurs in axons, we knocked down its expression by shRNA-mediated RNAi, using transient transfection of DRG neurons with an shRNA-Drp1 plasmid that was targeted to Drp1 mRNA (Czauderna et al., 2003; Tondera et al., 2005). The validity and target specificity of this plasmid have been described previously (Tondera et al., 2005). To identify shRNA-Drp1-transfected cells, the shRNA plasmid was cotransfected with a cytoplasmic enhanced green fluorescent protein (EGFP) construct at a 9:1 ratio. When neurons were observed after 48 h, cells transfected with shRNA-Drp1 [Fig. 6(C–H)] had significantly longer axonal mitochondria than did untransfected neurons [Fig. 6(A,B)]. Long mitochondria were detected in transfected neurons from the proximal to the most distal regions of the axons [Fig. 6(D,F,H) respectively]. Indeed, when the lengths of axonal mitochondria in the axons of tranfected and untransfected neurons were measured, the distributions were nearly nonoverlapping [Fig. 6(I)]: 98% of control mitochondria were shorter than 2 μm, whereas in shRNA-Drp1-transfected axons 86% were longer than 2 μm and some even reached lengths of 20–30 μm [Fig. 6(I)]. Transfection with GFP alone had no effect on mitochondrial length (data not shown). The formation of very long mitochondria throughout axons deficient in Drp1 indicates an important role for mitochondrial fission within the axon under normal conditions. In particular, the longest axonal mitochondria are almost completely immobile indicating that they arose in the axons close to where they were observed and that a balance of local mitochondrial fission and fusion occurs in axons.
Figure 6.
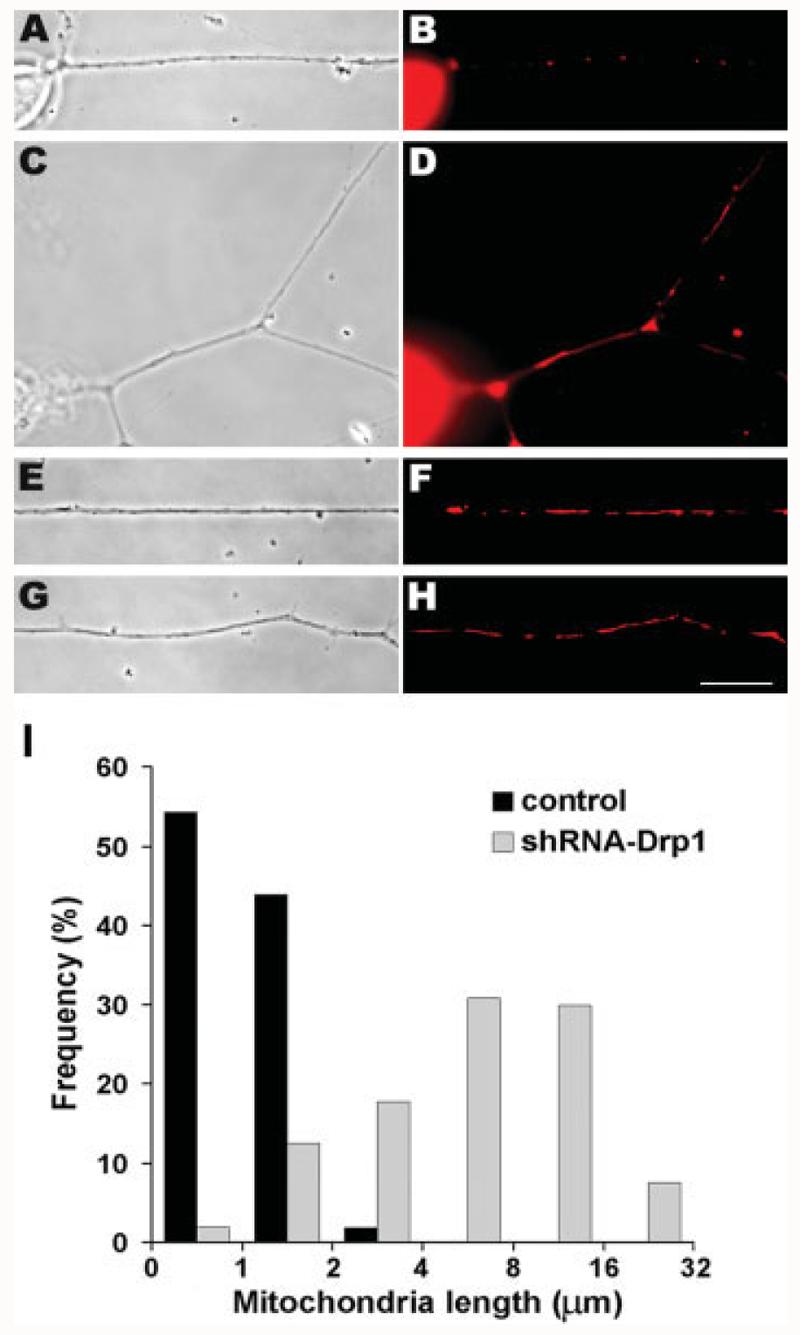
RNAi-mediated knockdown of Drp1 expression results in significantly longer mitochondria in axons. Isolated DRGs were transfected with shRNA-Drp1 and cells were stained with MitoTracker dye to reveal mitochondrial morphology (B,D,F,H). Phase contrast (A,C,E,G) and epifluorescence (B,D,F,H) images show that most mitochondria in transfected cells (C–H) are longer than those in control cells (A,B). Long mitochondria are seen throughout the axons of transfected cells: in the proximal (C,D), middle (E,F), and distal (G,H) regions. Scale bar, 10 μm. Quantification of mitochondrial length change after RNAi knockdown of Drp1 expression (I). The lengths of axonal mitochondria were measured and plotted as a frequency distribution. The mitochondrial length distributions in control and transfected cells were nearly nonoverlapping, differing from each other with p < 0.001 (two-tail t test). In control cells, 98% of mitochondria were ≤2 μm, whereas in transfected cells, 86% of mitochondria were >2 μm long, and a small fraction exceeded 16 μm. Pooled data are shown from three experiments (shRNA: n = 107 mitochondria/13 axons; control: n = 163 mitochondria/12 axons).
Overexpression of Mfn1 Protein Results in Fusion of Axonal Mitochondria
The dramatic increase in mitochondrial length throughout shRNA-Drp1-transfected axons likely resulted from inhibition of fission combined with continued local fusion. To assess the role of fusion in the axon, we overexpressed mitofusin1 (Mfn1) in DRG neurons. Mfn1, the mammalian ortholog of fuzzy onions gene (Fzo1), mediates the fusion of mitochondria and its overexpression in mammalian cell cultures results in the formation of large interconnected reticular mitochondria in perinuclear regions of the cells (Santel et al., 2003). Isolated DRGs were transiently transfected with an EGFP-Mfn1 expression plasmid (Santel et al., 2003) and examined 48 h later, using MitoTracker staining to identify mitochondria (see Fig. 7). Compared to untransfected cells, the axonal mitochondria in transfected cells were very long [Fig. 7(A–C and D–I)], and highly elongated mitochondria were detected from the proximal [Fig. 7(D–F)] to the most distal regions (G–I) of transfected axons. Because Mfn1 proteins are localized to the outer mitochondrial membrane (Santel et al., 2003), we could also visualize mitochondrial morphology using the EGFP signal [Fig. 7(F,I)]. A comparison of the length of the axonal mitochondria between transfected and nontransfected cells gave results similar to Drp1 knockdown [Fig. 7(J)]: Although nearly all mitochondria observed in control axons were ≤2 μm in length, in Mfn1-overexpressing axons the mitochondrial length increased significantly so that 62% of mitochondria became >2 μm and some even reached a length of 30 μm. As in the Drp1 knockdown experiments mentioned earlier, the immobility of these very long mitochondrial in mitofusin overexpression indicates that they must have arisen very close to where they were observed. Thus, the conversion of almost the entire axonal mitochondrial population to extreme length by Mfn1 overexpression indicated that local mitochondrial fusion can occur in DRG axons. Because the maintenance of normal mitochondrial morphology in nonneuronal cells requires a balance of fission and fusion, these data suggest that both events occur in the axon.
Figure 7.
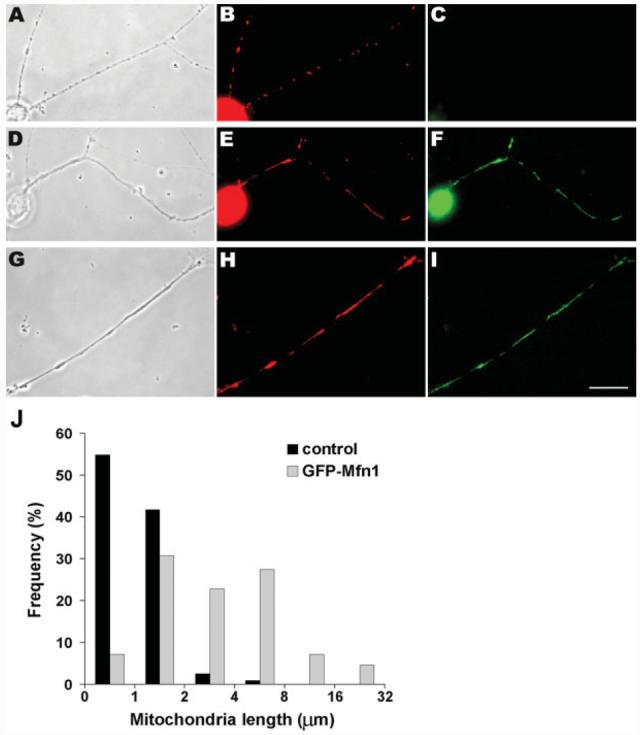
Overexpression of Mfn1 fusion protein induces longer mitochondria throughout DRG axons. Isolated DRGs were transfected with EGFP-Mfn1 expression plasmids and observed 48 h later. Phase contrast (A,D,G) and epifluorescence (B,C,E,F,H,I) images are shown. MitoTracker staining reveals the mitochondrial morphology (B,E,H). Neurons expressing EGFP-Mfn1 (D–F, G–I) had significantly longer mitochondria in all regions of axons than did untransfected cells (A– C). Longer mitochondria are shown in the proximal (E) and distal (H) regions of the axons. Scale bar, 10 μm. Overexpression of Mfn1 protein by transient transfection results in significantly longer mitochondria in axons (J). In transfected cells, the length of the mitochondria was increased very significantly (p < 0.001; two-tail t test); a small percentage (4.6%) of axonal mitochondria exceeded 16 lm in length. Pooled data are shown from two experiments (transfected: n = 124 mitochondria/9 axons, control: n = 115 mitochondria/3 axons).
DISCUSSION
Neurons are large cells that rely profoundly on mitochondrial function (Nicholls and Budd, 2000), and thus the rapid and versatile distribution of mitochondrial activity over time and space is critical (Hollenbeck and Saxton, 2005). They are also typically long-lived cells in which mitochondria must dilute the products of their error-prone DNA replication (Scharpira, 1993) by fission and fusion (Ono et al., 2001; Chan, 2006; Chang and Reynolds, 2006; Hoppins et al., 2007) and renew themselves through biogenesis (Schmitz et al., 1999). The dying-back peripheral neuropathy observed in AIDS patients (Mah et al., 1988; Miller et al., 1988) treated with nucleoside inhibitors of mtDNA replication (Dubinsky et al., 1989; Chen et al., 1991; Pardo et al., 2001; Raff et al., 2002) emphasizes the importance of mitochondrial biogenesis for the integrity of the axon. Other deficiencies in mitochondrial biogenesis, such as those associated with aging, affect neurons more than other cell types (Miquel, 1991; Schmitz et al., 1999) and probably underlie neurodegenerative disorders (Linnane et al., 1989; Schmitz et al., 1999). But where in this large and asymmetric cell does mitochondrial biogenesis occur? Because most mitochondrial proteins are encoded in the nuclear genome (Neupert, 1997), it has been suggested that all mitochondrial biogenesis occurs in the cell body, close to the nucleus (Davis and Clayton, 1996). This was a particularly attractive idea before the demonstration of mRNA localization and protein translation in axons. However, it seems unlikely from first principles that the distal axons of peripheral neurons can depend solely on the replication of mitochondria in the cell body and their subsequent transport. In addition, past studies have focused on mtDNA replication and have not provided evidence excluding mitochondrial fission and fusion from the axon. A body of recent work revealing components of the machinery for mitochondrial replication within axons has refocused our attention on this issue, as has the detection of mtDNA replication in processes of SH-SY5Y neuroblastoma cells (Magnusson et al., 2003). We have addressed the location of three elements of neuronal mitochondrial biogenesis using bona fide peripheral neurons in culture. Our examination of mtDNA replication, fission and fusion indicates that they can occur autonomously within axons.
mtDNA Replication in Axons
We assessed the rate and sites of mtDNA replication in axons of embryonic peripheral neurons, using a BrdU labeling system with high sensitivity. Newly synthesized DNA was detected in mitochondria not only in cell bodies but also throughout the axons (Fig. 1), and pulse-chase experiments with BrdU indicated that axonal mtDNA turned over on a time scale of hours to days, faster than rates derived from radio-labeling of adult brain (Gross et al., 1969; Menzies and Gold, 1971), but comparable to neuron-like cell lines in culture (Chen et al., 1991; Wang et al., 1997). This high rate of turnover implies that in many axons of the peripheral nervous system, mitochondria exiting the cell body would not even reach the synapse before replicating their DNA.
Using axotomy, we demonstrated that mtDNA replication can occur in axons; indeed, quantitative analysis of BrdU signals showed that the % of axonal mitochondria that replicated their DNA during a 3-h period in the absence of any connection to their cell bodies was fully one-third of the % replicated in axons of intact neurons (see Fig. 2). This may represent an upper limit for the level of mtDNA replication in the axon, as mtDNA replication has been reported to increase after nerve axotomy in situ (Korr et al., 1997) at least in proximal axons. Nonetheless, as we also observed in MT-depleted neurons (see Fig. 3), it is clear that mitochondria can replicate their DNA at a significant frequency within axons.
Mitochondrial Fission in Axons
Because mitochondria cannot be made de novo, the fission of preexisting organelles is necessary for generation of the new mitochondria. To address this process in the axon, we focused on Drp1, an essential protein of the mitochondrial fission complex. Mutations in Drp1 prevent fission and result in the formation of large interconnected mitochondria (Bleazard et al., 1999; Labrousse et al., 1999; Sesaki and Jensen, 1999; van der Bliek, 2000; Smirnova et al., 2001). Drp1 is recruited from the cytosol to the mitochondrial membrane before division (Labrousse et al., 1999; Smirnova et al., 2001), and after fission it remains attached to the tip of daughter mitochondria before being eventually released back to the cytosol (Bleazard et al., 1999; Labrousse et al., 1999; van der Bliek, 2000). Thus, the presence of Drp1 on mitochondria indicates an incipient or recent fission. If mitochondria were undergoing fission in the axon, we would expect to find many Drp1-positive mitochondria throughout the length of the axon, with Drp1 puncta at the ends of shorter, recently divided mitochondria, and near the middle of longer ones that are likely to divide soon. This is exactly what we observed: assessed by either immunofluorescent staining or GFP-Drp1 expression, the majority of axonal mitochondria were Drp1-positive. In addition, the fraction of mitochondria that was Drp1-positive was not significantly different between the proximal and most distal region of the axon but both were higher than the central axon shaft. A rise in the percentage of Drp1-positive mitochondria from the central to the most distal region of the axon suggests a possible focus of fission at the growth cone. This might reinforce other mechanisms for increasing mitochondrial activity at active growth cones, including transport and docking (Morris and Hollenbeck, 1993) and local regulation of membrane potential (Verburg, and Hollenbeck, unpublished observations).
The relationship of mitochondrial length to Drp1 position was also consistent with axonal fission. The majority of Drp1-positive axonal mitochondria had Drp1 at one end, and these were on average less than half the length of those that had Drp1 near their middle. If the recently divided mitochondria in the axon were mainly or all derived from fission in the cell body followed by anterograde transport, we would expect a much smaller fraction to be Drp1-positive, and their frequency—especially of short mitochondria with Drp1 still at their ends—would display a proximodistal gradient. Thus, these data suggest that mitochondrial fission events occur regularly outside of the cell body, throughout the axon.
Balance of Fission and Fusion in the Axon
If the full mitochondrial life cycle can unfold in axons, then both fission and fusion should occur there and any disruption of their balance should change the characteristic steady-state distribution of mitochonrial lengths. In fact, when we perturbed this balance in favor of fusion by either inhibiting expression of the fission protein Drp1 (Tondera et al., 2005; Hoppins et al., 2007) or by overexpressing the fusion protein Mfn1 (Santel et al., 2003), axonal mitochondria became very significantly longer, with a length distribution that was essentially nonoverlapping with that of controls (Figs. 6 and 7). Mitochondria as long as 30 μm were found throughout the axons, including in the most distal regions. Because mitochondria of this length are essentially immobile in axons, they must have arisen by fusion, not properly balanced by fission, in the distal axon itself. It seems likely that in the axon, fission and fusion serve the same purposes as they do in the cell body and in nonneuronal cells: fission is the final step in mitochondrial replication, whereas fusion dilutes errors in mtDNA, with both events required to protect mitochondrial integrity and function (Chen et al., 2005; Chang and Reynolds, 2006). However, this balance is probably regulated differently in different regions: the motility of axonal mitochondria may ordinarily require shorter organelles, whereas the formation of longer, interconnected mitochondria in dendrites (Popov et al., 2005) may result in more efficient and coordinated ATP dispersal and ion sequestration over a larger cytoplasmic area, and/or a decrease in energy loss caused by respiration (Skulachev, 2001; Chang and Reynolds, 2006).
Why Do Axons Require Local Mitochondrial Biogenesis?
Our understanding of the neuron as a dynamic metabolic system depends critically upon where in the cell mitochondrial biogenesis occurs. If it is limited to the cell body, then axonal mitochondria are purely visitors in the axon whose lifetime prior to turnover will barely equal the time necessary to reach the distal ends of long peripheral axons. In this case, axons could respond to changes in local need for mitochondrial function only by redistributing or regulating the activity of mitochondria that are already undergoing transport through the axon. However, if a fraction of the neuron's mtDNA replication, fission and fusion occurs in the distal axon, then mitochondrial biogenesis, like protein synthesis, could respond to local events and signals in the axon such as growth cone guidance, synaptic activity, or regeneration after injury. Our data support the second hypothesis and raise the question of which events support and require axonal mitochondria biogenesis.
Supplementary Material
Acknowledgments
The GFP-Drp1 expression plasmid was kindly provided by A. van der Bliek (UCLA). The plasmids shRNA-Drp1 and pEGFP-Mfn1 were generously donated by A. Santel (University of Geneva). The BrdU assay was originally developed by Sunkyung Lee.
Contract grant sponsor: NIH (National Institutes of Neurological Disorders and Stroke); contract grant number: NS027073.
Footnotes
Additional Supporting Information may be found in the online version of this article.
REFERENCES
- Alvarez J, Giuditta A, Koenig E. Protein synthesis in axons and terminals: Significance for maintenance, plasticity and regulation of phenotype. With a critique of slow transport theory. Prog Neurobiol. 2000;62:1–62. doi: 10.1016/s0301-0082(99)00062-3. [DOI] [PubMed] [Google Scholar]
- Berthold CH, Fabricius C, Rydmark M, Andersen B. Axoplasmic organelles at nodes of Ranvier. I. Occurrence and distribution in large myelinated spinal root axons of the adult cat. J Neurocytol. 1993;22:925–940. doi: 10.1007/BF01218351. [DOI] [PubMed] [Google Scholar]
- Bindokas VP, Lee CC, Colmers WF, Miller RJ. Changes in mitochondrial function resulting from synaptic activity in the rat hippocampal slice. J Neurosci. 1998;18:4570–4587. doi: 10.1523/JNEUROSCI.18-12-04570.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bleazard W, McCaffery JM, King EJ, Bale S, Mozdy A, Tieu Q, Nunnari J, et al. The dynamin-related GTPase Dnm1 regulates mitochondrial fission in yeast. Nat Cell Biol. 1999;1:298–304. doi: 10.1038/13014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bristow EA, Griffiths PG, Andrews RM, Johnson MA, Turnbull DM. The distribution of mitochondrial activity in relation to optic nerve structure. Arch Ophthalmol. 2002;120:791–796. doi: 10.1001/archopht.120.6.791. [DOI] [PubMed] [Google Scholar]
- Campbell DS, Holt CE. Chemotropic responses of retinal growth cones mediated by rapid local protein synthesis and degradation. Neuron. 2001;32:1013–1026. doi: 10.1016/s0896-6273(01)00551-7. [DOI] [PubMed] [Google Scholar]
- Chada SR, Hollenbeck PJ. Mitochondrial movement and positioning in axons: The role of growth factor signaling. J Exp Biol. 2003;206:1985–1992. doi: 10.1242/jeb.00263. [DOI] [PubMed] [Google Scholar]
- Chan DC. Mitochondrial fusion and fission in mammals. Annu Rev Cell Dev Biol. 2006;22:79–99. doi: 10.1146/annurev.cellbio.22.010305.104638. [DOI] [PubMed] [Google Scholar]
- Chang DT, Reynolds IJ. Differences in mitochondrial movement and morphology in young and mature primary cortical neurons in culture. Neuroscience. 2006;141:727–736. doi: 10.1016/j.neuroscience.2006.01.034. [DOI] [PubMed] [Google Scholar]
- Chen CH, Vazquez-Padua M, Cheng YC. Effect of anti-human immunodeficiency virus nucleoside analogs on mitochondrial DNA and its implication for delayed toxicity. Mol Pharmacol. 1991;39:625–628. [PubMed] [Google Scholar]
- Chen H, Chomyn A, Chan DC. Disruption of fusion results in mitochondrial heterogeneity and dysfunction. J Biol Chem. 2005;280:26185–26192. doi: 10.1074/jbc.M503062200. [DOI] [PubMed] [Google Scholar]
- Czauderna F, Fechtner M, Aygun H, Arnold W, Klippel A, Giese K, Kaufmann J. Functional studies of the PI(3)-kinase signalling pathway employing synthetic and expressed siRNA. Nucleic Acids Res. 2003;31:670–682. doi: 10.1093/nar/gkg141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis AF, Clayton DA. In situ localization of mitochondrial DNA replication in intact mammalian cells. J Cell Biol. 1996;135:883–893. doi: 10.1083/jcb.135.4.883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubinsky RM, Yarchoan R, Dalakas M, Broder S. Reversible axonal neuropathy from the treatment of AIDS and related disorders with 2′,3′-dideoxycytidine(ddC) Muscle Nerve. 1989;12:856–860. doi: 10.1002/mus.880121012. [DOI] [PubMed] [Google Scholar]
- Eriksson S, Xu B, Clayton DA. Efficient incorporation of anti-HIV deoxynucleotides by recombinant yeast mitochondrial DNA polymerase. J Biol Chem. 1995;270:18929–18934. doi: 10.1074/jbc.270.32.18929. [DOI] [PubMed] [Google Scholar]
- Falkenberg M, Gaspari M, Rantanen A, Trifunovic A, Larsson NG, Gustafsson CM. Mitochondrial transcription factors B1 and B2 activate transcription of human mtDNA. Nat Genet. 2002;31:289–294. doi: 10.1038/ng909. [DOI] [PubMed] [Google Scholar]
- Gioio AE, Eyman M, Zhang H, Lavina ZS, Giuditta A, Kaplan BB. Local synthesis of nuclear-encoded mitochondrial proteins in the presynaptic nerve terminal. J Neurosci Res. 2001;64:447–453. doi: 10.1002/jnr.1096. [DOI] [PubMed] [Google Scholar]
- Gross NJ, Getz GS, Rabinowitz M. Apparent turnover of mitochondrial deoxyribonucleic acid and mitochondrial phospholipids in the tissues of the rat. J Biol Chem. 1969;244:1552–1562. [PubMed] [Google Scholar]
- Hillefors M, Gioio AE, Mameza MG, Kaplan BB. Axon viability and mitochondrial function are dependent on local protein synthesis in sympathetic neurons. Cell Mol Neurobiol. 2007;27:701–716. doi: 10.1007/s10571-007-9148-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollenbeck PJ. Products of endocytosis and autophagy are retrieved from axons by regulated retrograde organelle transport. J Cell Biol. 1993;121:305–315. doi: 10.1083/jcb.121.2.305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollenbeck PJ, Saxton WM. The axonal transport of mitochondria. J Cell Sci. 2005;118:5411–5419. doi: 10.1242/jcs.02745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoppins S, Lackner L, Nunnari J. The machines that divide and fuse mitochondria. Annu Rev Biochem. 2007;76:751–780. doi: 10.1146/annurev.biochem.76.071905.090048. [DOI] [PubMed] [Google Scholar]
- Kageyama GH, Wong-Riley MT. Histochemical localization of cytochrome oxidase in the hippocampus: Correlation with specific neuronal types and afferent pathways. Neuroscience. 1982;7:2337–2361. doi: 10.1016/0306-4522(82)90199-3. [DOI] [PubMed] [Google Scholar]
- Koenig E. Evaluation of local synthesis of axonal proteins in the goldfish Mauthner cell axon and axons of dorsal and ventral roots of the rat in vitro. Mol Cell Neurosci. 1991;2:384–394. doi: 10.1016/1044-7431(91)90025-j. [DOI] [PubMed] [Google Scholar]
- Koenig E, Giuditta A. Protein-synthesizing machinery in the axon compartment. Neuroscience. 1999;89:5–15. doi: 10.1016/s0306-4522(98)00282-6. [DOI] [PubMed] [Google Scholar]
- Korr H, Philippi V, Helg C, Schiefer J, Graeber MB, Kreutzberg GW. Unscheduled DNA synthesis and mitochondrial DNA synthetic rate following injury of the facial nerve. Acta Neuropathol. 1997;94:557–566. doi: 10.1007/s004010050750. [DOI] [PubMed] [Google Scholar]
- Labrousse AM, Zappaterra MD, Rube DA, van der Bliek AM. C. elegans dynamin-related protein DRP-1 controls severing of the mitochondrial outer membrane. Mol Cell. 1999;4:815–826. doi: 10.1016/s1097-2765(00)80391-3. [DOI] [PubMed] [Google Scholar]
- Larsson NG, Oldfors A, Holme E, Clayton DA. Low levels of mitochondrial transcription factor A in mitochondrial DNA depletion. Biochem Biophys Res Commun. 1994;200:1374–1381. doi: 10.1006/bbrc.1994.1603. [DOI] [PubMed] [Google Scholar]
- Lee SK, Hollenbeck PJ. Organization and translation of mRNA in sympathetic axons. J Cell Sci. 2003;116:4467–4478. doi: 10.1242/jcs.00745. [DOI] [PubMed] [Google Scholar]
- Leung KM, van Horck FP, Lin AC, Allison R, Standart N, Holt CE. Asymmetrical β-actin mRNA translation in growth cones mediates attractive turning to netrin-1. Nat Neurosci. 2006;9:1247–1256. doi: 10.1038/nn1775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li YC, Zhai XY, Ohsato K, Futamata H, Shimada O, Atsumi S. Mitochondrial accumulation in the distal part of the initial segment of chicken spinal motoneurons. Brain Res. 2004;1026:235–243. doi: 10.1016/j.brainres.2004.08.016. [DOI] [PubMed] [Google Scholar]
- Linnane AW, Marzuki S, Ozawa T, Tanaka M. Mitochondrial DNA mutations as an important contributor to ageing and degenerative diseases. Lancet. 1989;1:642–645. doi: 10.1016/s0140-6736(89)92145-4. [DOI] [PubMed] [Google Scholar]
- Magnusson J, Orth M, Lestienne P, Taanman JW. Replication of mitochondrial DNA occurs throughout the mitochondria of cultured human cells. Exp Cell Res. 2003;289:133–142. doi: 10.1016/s0014-4827(03)00249-0. [DOI] [PubMed] [Google Scholar]
- Mah V, Vartavarian LM, Akers MA, Vinters HV. Abnormalities of peripheral nerve in patients with human immunodeficiency virus infection. Ann Neurol. 1988;24:713–717. doi: 10.1002/ana.410240604. [DOI] [PubMed] [Google Scholar]
- Martin R, Vaida B, Bleher R, Crispino M, Giuditta A. Protein synthesizing units in presynaptic and postsynaptic domains of squid neurons. J Cell Sci. 1998;111(Part 21):3157–3166. doi: 10.1242/jcs.111.21.3157. [DOI] [PubMed] [Google Scholar]
- Martinez CY, Hollenbeck PJ. Transfection of primary central and peripheral nervous system neurons by electroporation. Methods Cell Biol. 2003;71:339–351. doi: 10.1016/s0091-679x(03)01016-1. [DOI] [PubMed] [Google Scholar]
- Menzies RA, Gold PH. The turnover of mitochondria in a variety of tissues of young adult and aged rats. J Biol Chem. 1971;246:2425–2429. [PubMed] [Google Scholar]
- Miller RG, Parry GJ, Pfaeffl W, Lang W, Lippert R, Kiprov D. The spectrum of peripheral neuropathy associated with ARC and AIDS. Muscle Nerve. 1988;11:857–863. doi: 10.1002/mus.880110810. [DOI] [PubMed] [Google Scholar]
- Ming GL, Wong ST, Henley J, Yuan XB, Song HJ, Spitzer NC, Poo MM. Adaptation in the chemotactic guidance of nerve growth cones. Nature. 2002;417:411–418. doi: 10.1038/nature745. [DOI] [PubMed] [Google Scholar]
- Miquel J. An integrated theory of aging as the result of mitochondrial-DNA mutation in differentiated cells. Arch Gerontol Geriatr. 1991;12:99–117. doi: 10.1016/0167-4943(91)90022-i. [DOI] [PubMed] [Google Scholar]
- Morris R, Hollenbeck P. Axonal transport of mitochondria along microtubules and F-actin in living vertebrate neurons. J Cell Biol. 1995;131:1315–1326. doi: 10.1083/jcb.131.5.1315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris RL, Hollenbeck PJ. The regulation of bidirectional mitochondrial transport is coordinated with axonal outgrowth. J Cell Sci. 1993;104(Part 3):917–927. doi: 10.1242/jcs.104.3.917. [DOI] [PubMed] [Google Scholar]
- Neupert W. Protein import into mitochondria. Annu Rev Biochem. 1997;66:863–917. doi: 10.1146/annurev.biochem.66.1.863. [DOI] [PubMed] [Google Scholar]
- Nicholls DG, Budd SL. Mitochondria and neuronal survival. Physiol Rev. 2000;80:315–360. doi: 10.1152/physrev.2000.80.1.315. [DOI] [PubMed] [Google Scholar]
- Olink-Coux M, Hollenbeck PJ. Localization and active transport of mRNA in axons of sympathetic neurons in culture. J Neurosci. 1996;16:1346–1358. doi: 10.1523/JNEUROSCI.16-04-01346.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ono T, Isobe K, Nakada K, Hayashi JI. Human cells are protected from mitochondrial dysfunction by complementation of DNA products in fused mitochondria. Nat Genet. 2001;28:272–275. doi: 10.1038/90116. [DOI] [PubMed] [Google Scholar]
- Pardo CA, McArthur JC, Griffin JW. HIV neuropathy: Insights in the pathology of HIV peripheral nerve disease. J Peripher Nerv Syst. 2001;6:21–27. doi: 10.1046/j.1529-8027.2001.006001021.x. [DOI] [PubMed] [Google Scholar]
- Popov V, Medvedev NI, Davies HA, Stewart MG. Mitochondria form a filamentous reticular network in hippocampal dendrites but are present as discrete bodies in axons: A three-dimensional ultrastructural study. J Comp Neurol. 2005;492:50–65. doi: 10.1002/cne.20682. [DOI] [PubMed] [Google Scholar]
- Raff MC, Whitmore AV, Finn JT. Axonal self-destruction and neurodegeneration. Science. 2002;296:868–871. doi: 10.1126/science.1068613. [DOI] [PubMed] [Google Scholar]
- Ruthel G, Hollenbeck PJ. Response of mitochondrial traffic to axon determination and differential branch growth. J Neurosci. 2003;23:8618–8624. doi: 10.1523/JNEUROSCI.23-24-08618.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santel A, Frank S, Gaume B, Herrler M, Youle RJ, Fuller MT. Mitofusin-1 protein is a generally expressed mediator of mitochondrial fusion in mammalian cells. J Cell Sci. 2003;116:2763–2774. doi: 10.1242/jcs.00479. [DOI] [PubMed] [Google Scholar]
- Scharpira AHV. Mitochondria cytopathies. Curr Opin Neurobiol. 1993;3:760–767. doi: 10.1016/0959-4388(93)90150-w. [DOI] [PubMed] [Google Scholar]
- Schmitz C, Axmacher B, Zunker U, Korr H. Age-related changes of DNA repair and mitochondrial DNA synthesis in the mouse brain. Acta Neuropathol. 1999;97:71–81. doi: 10.1007/s004010050957. [DOI] [PubMed] [Google Scholar]
- Sesaki H, Jensen RE. Division versus fusion: Dnm1p and Fzo1p antagonistically regulate mitochondrial shape. J Cell Biol. 1999;147:699–706. doi: 10.1083/jcb.147.4.699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simpson MV, Chin CD, Keilbaugh SA, Lin TS, Prusoff WH. Studies on the inhibition of mitochondrial DNA replication by 3′-azido-3′-deoxythymidine and other dideoxynucleoside analogs which inhibit HIV-1 replication. Biochem Pharmacol. 1989;38:1033–1036. doi: 10.1016/0006-2952(89)90245-1. [DOI] [PubMed] [Google Scholar]
- Skulachev VP. Mitochondrial filaments and clusters as intracellular power-transmitting cables. Trends Biochem Sci. 2001;26:23–29. doi: 10.1016/s0968-0004(00)01735-7. [DOI] [PubMed] [Google Scholar]
- Smirnova E, Griparic L, Shurland DL, van der Bliek AM. Dynamin-related protein Drp1 is required for mitochondrial division in mammalian cells. Mol Biol Cell. 2001;12:2245–2256. doi: 10.1091/mbc.12.8.2245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spelbrink JN, Li FY, Tiranti V, Nikali K, Yuan QP, Tariq M, Wanrooij S, et al. Human mitochondrial DNA deletions associated with mutations in the gene encoding Twinkle, a phage T7 gene 4-like protein localized in mitochondria. Nat Genet. 2001;28:223–231. doi: 10.1038/90058. [DOI] [PubMed] [Google Scholar]
- Spelbrink JN, Toivonen JM, Hakkaart GA, Kurkela JM, Cooper HM, Lehtinen SK, Lecrenier N, et al. In vivo functional analysis of the human mitochondrial DNA polymerase POLG expressed in cultured human cells. J Biol Chem. 2000;275:24818–24828. doi: 10.1074/jbc.M000559200. [DOI] [PubMed] [Google Scholar]
- Tiranti V, Savoia A, Forti F, D'Apolito MF, Centra M, Rocchi M, Zeviani M. Identification of the gene encoding the human mitochondrial RNA polymerase (h-mtRPOL) by cyberscreening of the expressed sequence Tags database. Hum Mol Genet. 1997;6:615–625. doi: 10.1093/hmg/6.4.615. [DOI] [PubMed] [Google Scholar]
- Tondera D, Czauderna F, Paulick K, Schwarzer R, Kaufmann J, Santel A. The mitochondrial protein MTP18 contributes to mitochondrial fission in mammalian cells. J Cell Sci. 2005;118:3049–3059. doi: 10.1242/jcs.02415. [DOI] [PubMed] [Google Scholar]
- Twiss JL, van Minnen J. New insights into neuronal regeneration: The role of axonal protein synthesis in pathfinding and axonal extension. J Neurotrauma. 2006;23:295–308. doi: 10.1089/neu.2006.23.295. [DOI] [PubMed] [Google Scholar]
- van der Bliek AM. A mitochondrial division apparatus takes shape. J Cell Biol. 2000;151:F1–F4. doi: 10.1083/jcb.151.2.f1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verma P, Chierzi S, Codd AM, Campbell DS, Meyer RL, Holt CE, Fawcett JW. Axonal protein synthesis and degradation are necessary for efficient growth cone regeneration. J Neurosci. 2005;25:331–342. doi: 10.1523/JNEUROSCI.3073-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang GJ, Nutter LM, Thayer SA. Insensitivity of cultured rat cortical neurons to mitochondrial DNA synthesis inhibitors: Evidence for a slow turnover of mitochondrial DNA. Biochem Pharmacol. 1997;54:181–187. doi: 10.1016/s0006-2952(97)00158-5. [DOI] [PubMed] [Google Scholar]
- Waxman SG, Ritchie JM. Molecular dissection of the myelinated axon. Ann Neurol. 1993;33:121–136. doi: 10.1002/ana.410330202. [DOI] [PubMed] [Google Scholar]
- Werth JL, Zhou B, Nutter LM, Thayer SA. 2′, 3′-Dideoxycytidine alters calcium buffering in cultured dorsal root ganglion neurons. Mol Pharmacol. 1994;45:1119–1124. [PubMed] [Google Scholar]
- Wong-Riley MT, Welt C. Histochemical changes in cytochrome oxidase of cortical barrels after vibrissal removal in neonatal and adult mice. Proc Natl Acad Sci USA. 1980;77:2333–2337. doi: 10.1073/pnas.77.4.2333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng JQ, Kelly TK, Chang B, Ryazantsev S, Rajasekaran AK, Martin KC, Twiss JL. A functional role for intra-axonal protein synthesis during axonal regeneration from adult sensory neurons. J Neurosci. 2001;21:9291–9303. doi: 10.1523/JNEUROSCI.21-23-09291.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.