

Abstract
We describe the design, synthesis, and properties of DNA-like molecules in which the base pairs are expanded by benzo homologation. The resulting size-expanded genetic helices are called xDNA (“expanded DNA”) and yDNA (“wide DNA”). The large component bases are fluorescent, and they display high stacking affinity. When singly substituted into natural DNA, they are destabilizing because the benzo-expanded base pair size is too large for the natural helix. However, when all base pairs are expanded, xDNA and yDNA form highly stable, sequence-selective double helices. The size-expanded DNAs are candidates for components of new, functioning genetic systems. In addition, the fluorescence of expanded DNA bases makes them potentially useful in probing nucleic acids.
Keywords: benzoadenine, base pair, DNA polymerase, fluorescence, stacking
I. Introduction
Over the past decade there has been a substantial research effort focused on designing replacements for the bases in DNA. Until recently, DNA base replacements have been studied mainly for their effects in the context of natural DNA. For example, studies are often aimed at testing local effects of new bases and base pairs on the stability of the helix, or on local biomolecular interactions with individual bases and pairs in natural DNA.1–8
With the present project we are taking a different approach: we are asking whether all of the pairs of DNA can be replaced with a new geometry. Our strategy has been to design new base pairs that are larger than natural ones, and assemble them to make enlarged DNA-like helices, called xDNA and yDNA. The aim of this Account is to outline progress made in assembling and studying these helices, and to point out some of our future aims.
One of the long-term goals of research on expanded DNAs is the design and exploration of the properties of a nonnatural genetic system. This line of research could be classified as “biomimetic chemistry”9 or “synthetic biology”,10,11 both of which involve assembly of systems from smaller components (atoms and molecules, biochemical pathways) with the purpose of behaving like natural biomolecules, natural pathways and ultimately, living organisms. If chemists are to design a new genetic system, this might well start with a design of a molecule analogous to DNA. However, DNA alone is not a genetic system in itself, but is rather the information-encoding part of the genetic system. Thus, to make a functioning system one would need, at a minimum, not only an analogue of DNA, but also its monomeric building blocks and polymerase enzymes that replicate it.
By working to recapitulate the functions of Nature’s genetic system, we can test our knowledge of how it operates, and how tightly constrained the natural design is. A second goal of our research in size-expanded DNAs is the development of tools for use in biology and medicine. For example, a set of expanded-size nucleotides might be useful as probes of steric effects in enzymes that recognize the natural nucleotides,12 In addition, size-expanded fluorescent oligomers might have applied utility in genetic reporting and imaging.
II. Background and Related Studies
The formalism of Watson and Crick, who noted the analogous structures of hydrogen bonded purine-pyrimidine pairs in 1953,13 had a strong influence on the DNA chemistry field in its first three or four decades. The main examples of non-AT/GC pairs prior to the mid-1990s were cases that still adhered to the paradigmatic purine-pyrimidine rule (Fig. 1). Among the most important early examples in that vein were the designed purine-pyrimidine base pairs of Benner and of Rappaport.14,15
Figure 1.
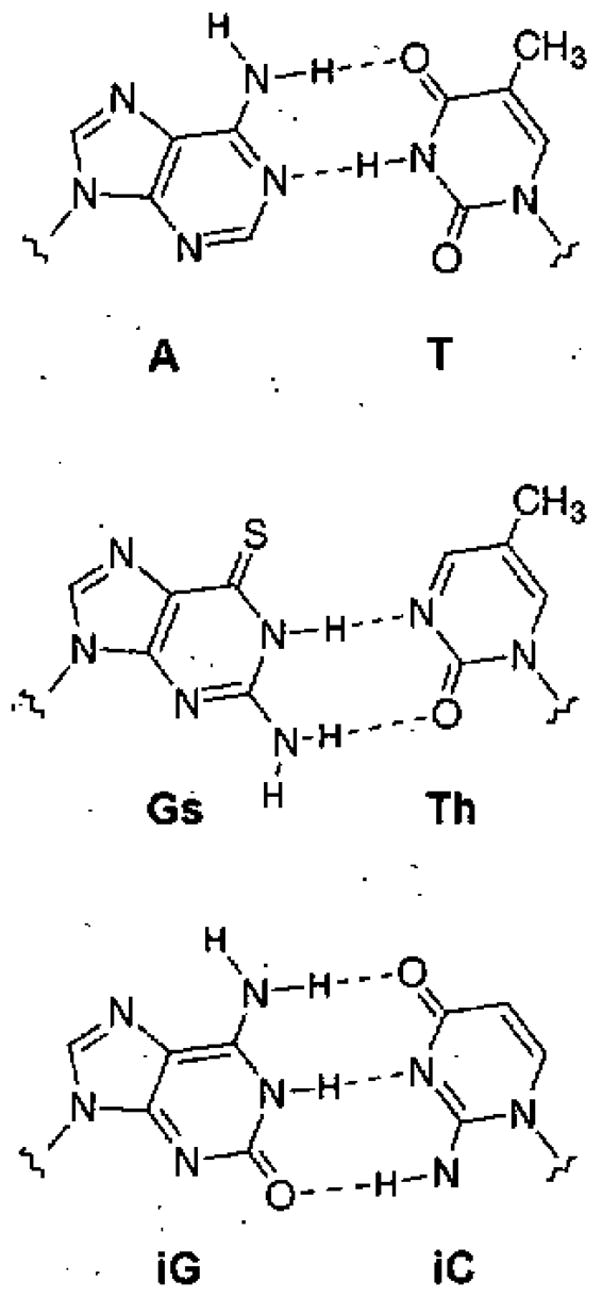
Examples of natural and nonnatural pur-pyr pairs: A-T, Gs-Th,16 isoG-isoC.15 All of these conform to Watson and Crick’s purine-pyrimidine structural paradigm for the DNA double helix.
The concept of nucleotides having increased size originated with Leonard, who in 1974 synthesized an adenine ribonucleoside analogue (1) in which the base was “stretched out” by benzo fusion.16 The benzoadenosine nucleotide was studied as a substrate for several ATP-dependent enzymes, including firefly luciferase and a cyclic AMP-dependent protein kinase.12 Subsequently, a similar GTP analogue was also synthesized,17 and a 2′-deoxy variant of benzoA was later prepared from the ribonucleoside.18 At the time of Leonard’s work, it was not practical to synthesize oligonucleotides. However, he predicted that benzoA was too large for a natural DNA or RNA helix.19
Only a few examples exist of studies in DNA involving bases larger, than natural ones. Benzo-fused DNA bases were prepared by Moreau and by Saito;20,21 however, their point of attachment was at the natural position; thus base pairs with the natural partner would adopt the normal rather than stretched geometry. The same is true of a large cytosirie derivative reported by Matteucci.22 In studies more closely related to our strategy, Seley prepared a thiophene-fused adenine-like nucleobase (2);23 this molecule was expanded in its potential pairing size, but it has not been incorporated into DNA to test this possibility. Finally, Matsuda has prepared DNA bases with extra rings (see 3), and has examined their pairing properties.24 Some of these were intended to increase the number of hydrogen bonds to four per pair, without changing the overall DNA geometry; however, a recent report describes base pairs (inserted into the natural DNA context) that could take on a stretched glycosidic distance.25
It should be noted that other laboratories are also using different approaches to pursuing the development of designed genetic sets.26–32 The approaches include use of modified sugars rather than modified bases, or the development of replicable base pairs that are meant to function in the context of the natural genetic system rather than orthogonally to the natural system.
III. Benzo-Fused Molecular Designs
xDNA
At the start of this work we sought a benzo-fused design of pyrimidines that would match the geometric expansion of purines conceived by Leonard.12 A generalized design would, in principle, allow for the combination of expanded versions of A, G, C, and T with the natural base complements and might allow them to form a regular helix. This concept led to the new deoxyribosides dxT and dxC, and to the complete set of expanded DNA (xDNA) nucleosides (4–7). All of the expanded bases have their pairing edges shifted outward by 2.4 Å (the width of benzene), with similar vectors for this extension. Preliminary AMI calculations predicted the structural stability and tautomeric preferences of the xDNA bases,33 and later high-level calculations by Fuentes-Cabrera et at. added support.34
The base pair designs for xDNA (Figure 2) are in some respects closely analogous to those of DNA, but are also different in important ways. Natural DNA sequences are composed of one of four letters at each position, and involve purines paired with pyrimidines. In xDNA, benzopurines are paired with pyrimidines and benzopyrimidines with purines, and there are eight components of xDNA,35 with four types of ring systems.
Figure 2.
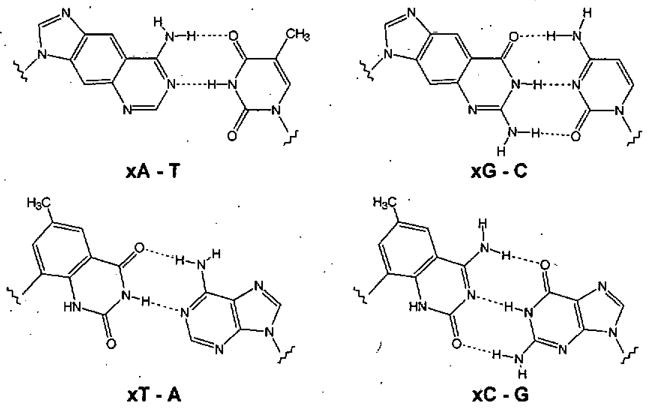
The four base pairing schemes of xDNA. xDNA is comprised of eight bases, while natural DNA has four bases with two pairing schemes.
Pairing selectivity in xDNA comes (in principle) not only from complementary hydrogen bonding, but also complementary size. For example, adenine paired with thymine is a mismatch in xDNA, because (although it has complementary hydrogen bonding groups) the pair is too small for the xDNA helix by approximately 2.4 Å. For that reason, the 8-base xDNA genetic set and the natural DNA genetic set are “orthogonal”, in that they are not expected to be able to interact with one another (Fig. 3). However, it also occurred to us that a subset of xDNA designs might allow the modified xDNA bases to recognize natural DNA or RNA. In that subset, all the xDNA bases would be segregated onto one strand, entirely composed of xA, xG, xT, xC.35 Such size-expanded strands, in the proper sequence, might bind natural nucleic acids and still form a large helix (Fig. 3B). In principle, one might use this recognition, and the inherent fluorescence of xDNA bases (see below), to sense natural DNAs or RNAs.
Figure 3.
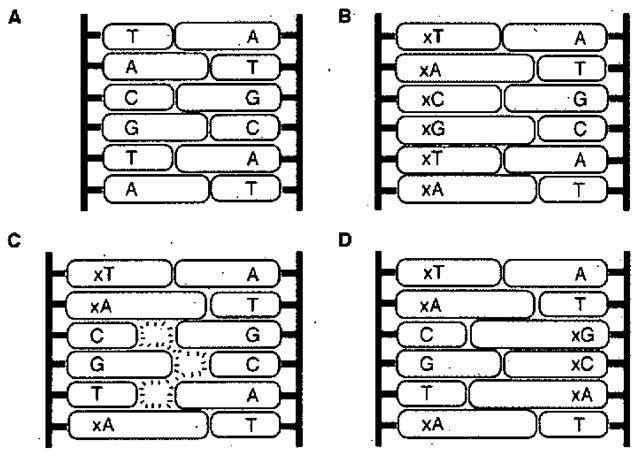
Illustration of nucleobase size differences that lead to two classes of expanded DNA helices. (A) Watson-Crick DNA strands are complementary in size. (B) An all-expanded xDNA strand is size complementary to natural DNA. (C) Mixing expanded and non-expanded bases on a strand (8-base xDNA) results in a size mismatch (orthogonality) with natural DNA. (D) 8-base xDNA can be complementary to another 8-base xDNA strand.
yDNA
Modeling studies suggested that there was more than one way to increase the size of a base pair by benzo fusion. This led to the “yDNA” (an abbreviation of ‘wide DNA’) design.36–38 Four benzo-widened hucleoside components of yDNA are shown below (8–11). Simple analysis of base pair geometry suggested that yDNA would have about the same degree of helix expansion as xDNA, and analysis of possible base stacking geometries (assuming a right-handed helix) suggested that yDNA is at least as favorable in area of overlap as xDNA.
A genetic pairing system based on yDNA design would (aside from the geometry of extension) be analogous to xDNA. However, AM1 calculations of the yDNA bases suggested that tautomeric preferences might be more complex than for the xDNA cases. The yT and yA bases were predicted to prefer the desired tautomers, but yC and yG were calculated to prefer different tautomers than needed for pairing as in the original design.38,39 Of course, aqueous solvation might change the relative tautomeric preferences. As a result of these calculations we focused our initial synthetic efforts only on yT, yC, and yA, and postponed studies aimed at yG.
IV. Synthesis of size-expanded nucleosides
The syntheses of seven xDNA and yDNA deoxynucleosides have been completed to date; in general, the expanded pyrimidines (dxT, dxC, dyT, dyC) are readily prepared in good yields, while the expanded purines (dxA, dxG, dyA) are more challenging.
The synthesis of dxA started with the approach of Leonard,40 and then deviated to complete the synthesis in fewer steps and higher yields (Scheme I).41 The deoxyribonucleoside 4 was subsequently converted to an amidine-protected phosphoramidite (Scheme 1) which can be used to make synthetic DNA oligonucleotides on an automated synthesizer. The synthesis of dxG42 began with intermediate 12 from the dxA synthesis. Starting from 12, the dxG phosphoramidite was synthesized in twelve steps and 4.6% overall yield, and is the most challenging of the expanded nucleosides to prepare.
Scheme 1.
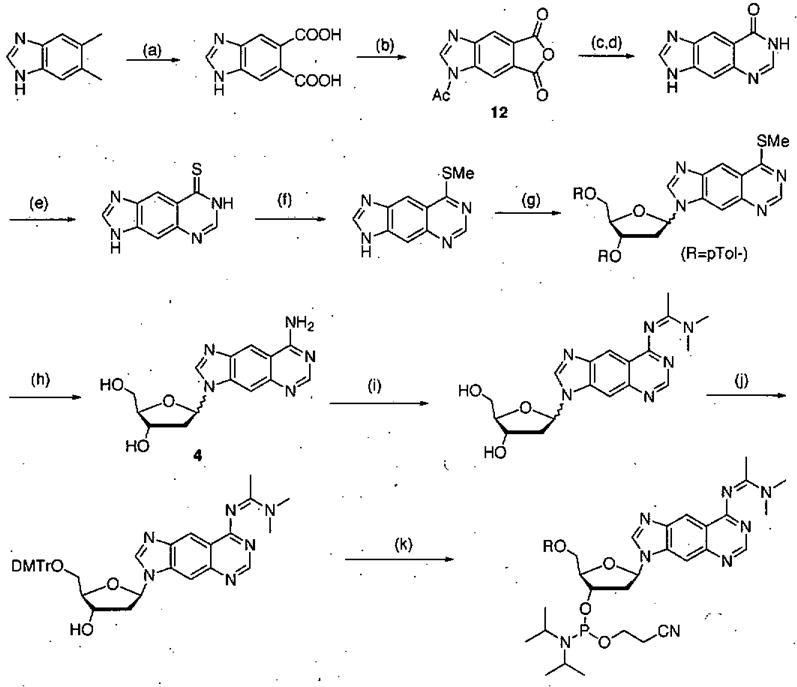
Synthetic route for preparation of dxA phosphoramidite derivative. Conditions: (a) KMnO4, tBuOH, H2O, 70 °C, 1.5 h, 60%; (b) Ac2O 155 °C, 3 h, 90%; (c) TMSN3, 90–95 °C, 3 h; (d) formamidine acetate, DMF, 155 °C, 3hr, 55% (two steps); (e) P2S5, Py, 140 °C, 36 h, 100%; (f) CH3I, KOH, 1 h, 64%; (g) NaH, CH3CN, Hoffer’s chlorosugar, 6 h,.37%; (h) NH3, EtOH, 140 °C, 36 h, 61%; (i) N,N-dimethylacetamide dimethylacetal, MeOH, 80 °C, 72 h, 69%; (j) DMTrCl, DIPEA, Py, 23 °C 6 h, 54%; (k) N, N-diisopropyl-cyanoethyl-chlorophosphoramidite, CH2Cl2, DIPEA, 23 °C, 5 h, 90%. (Reprinted with permission of J. Am. Chem. Soc.)
In contrast to the expanded purines, the dxT and dxC deoxyribonucleosides were made much more efficiently, in up to 34% overall yield.41,42 The key step for these C-glycosides was glycoside bond formation (Scheme 2); in the presence of Pd(OAc)2 and AsPh3, the iodinated base analogues were added via Heck reaction onto a silyl-protected dihydrofuran.43
Scheme 2.
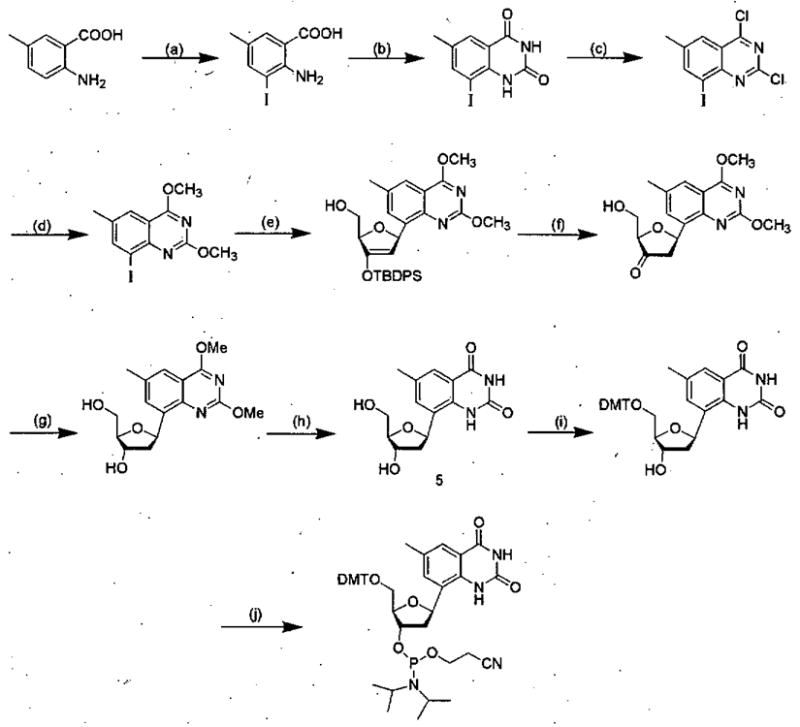
Synthetic route for preparation of dxT phosphoramidite derivative. Conditions: a). ICl, HCl(aq.), 96%; b) urea, 150 °C, 92%; c) POCl3, N, N-diethylaniline, reflux, 90%; d) NaOCH3/CH3OH, reflux, 87%; e) 1,2-dehydro-3-O-(t-butyldiphenylsilyl)-5-hydroxymethyl-furan, Pd(OAc) 2, AsPh3, N(Bu) 3, 75 °C, 64%; f) TBAF, THF, 0 °C, 84%; g) NaB(OAc)3H, THF, AcOH, 15 °C, 93%; h) NaI, AcOH, 60 °C, 100%; i) 4,4′-dimethoxytrityl chloride, DIPEA, pyridine, 84%; j) N,N-diisopropylammonium tetrazolide, 2-cyanoethyl tetraisopropylphosphoramidite, CH2Cl2, 82%. (Reprinted with permission of J. Am. Chem, Soc.)
The syntheses of three yDNA nucleosides have also been reported recently.36,38 The preparation of the expanded deoxyadenosine analogue, dyA, was achieved in ten steps and 5.7% overall yield. The yC and yT variants were prepared in good yields in-a manner closely analogous to the earlier syntheses of their xDNA analogues (see Scheme 2).40
V. Fluorescence of Expanded DNA Bases
The additional conjugation rendered by the benzo-fusion in the expanded DNA bases makes them inherently fluorescent, a useful property that is practically absent in the native DNA bases. In conjunction with the abilities of xDNA and yDNA to pair and assemble (below), this “built-in” fluorescence lends itself to possible applications in probing DNA/RNA hybridization and protein-DNA interactions.
The expanded DNA bases fluoresce in the blue-violet to violet range with conveniently large Stokes shifts of 50–80 nm (Fig. 4).36,38,41,42 They are efficient fluorophores, with quantum yields between 0.3 and 0.6. Studies are currently underway to evaluate interaction of x,yDNA bases in the context of common fluorescence assays involving static fluorescence, time-resolved fluorescence, energy transfer, quenching, etc. Thus it appears that expanded DNA bases will join other recent fluorescent DNA base analogues that can report on biophysical and biochemical phenomena that occur inside the helix.21,44–52
Figure 4.
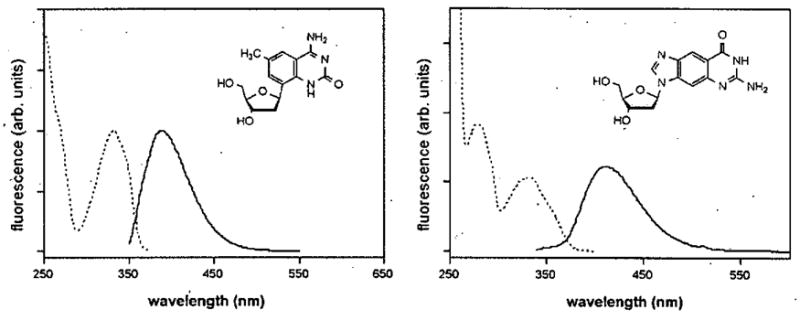
Fluorescence spectra of two xDNA nucleosides (dxC and dxG) in methanol.41,42 Emission spectra are shown with solid lines, and excitation spectra with dashed lines. All xDNA and yDNA nucleobases synthesized to date are efficient fluorophores.
VI. Base Pairing and Stacking in Natural DNA
Before studying fully expanded helices; we tested the effects of benzo-expanding a single pair in otherwise natural DNA. Expanded-size analogues were incorporated into short double helices and their stability was measured (by thermal, denaturation) relative to fully natural helices. The results showed consistently that a single expanded pair in the middle of a natural DNA duplex was destabilizing to the double helix. This is not too surprising considering the size mismatch of the expanded base pair in natural DNA, which would impose strain on the backbone. For the xDNA analogues, the destabilization penalty ranged from 0.3–1.7 kcal/mol relative to natural base pairs.53 For yDNA pairs, the range was 0,6–2.2 kcal/mol.36,38 Interestingly, while single expanded DNA analogues decreased thermal stability of the duplex, they were still selective for their Watson-Crick partners. For example, xA formed a more stable duplex paired opposite T than it did when paired opposite C, G, A, or a sugar lacking a base.53
Stacking interactions between the aromatic DNA bases play a dominant role in duplex stability,54,55 and these interactions appear to be driven largely by van der Waals forces and, for hydrophobic molecules, by solvophobic effects.55,56 Through experiments with xDNA bases dangling at the ends of duplex DNAs, we observed strong stacking of the benzo-fused bases (Fig, 5). In comparison with the natural bases as overhanging moieties, xA and xT are better stackers by 1.2 and 1.0 kcal per base.53 For the expanded analogues xG and xG the advantage in stacking over the natural bases was 0.5 and 0.7 kcal per base, respectively.42 Subsequent studies with the yDNA bases showed that most stack nearly as strongly as the xDNA bases.36,38 These exceptionally strong stacking abilities apparently result from a larger surface area (lending greater polarizability) and greater hydrophobicity. Similar observations with bases having increased aromatic surfaces have been noted by a number of laboratories.24,57–60
Figure 5.
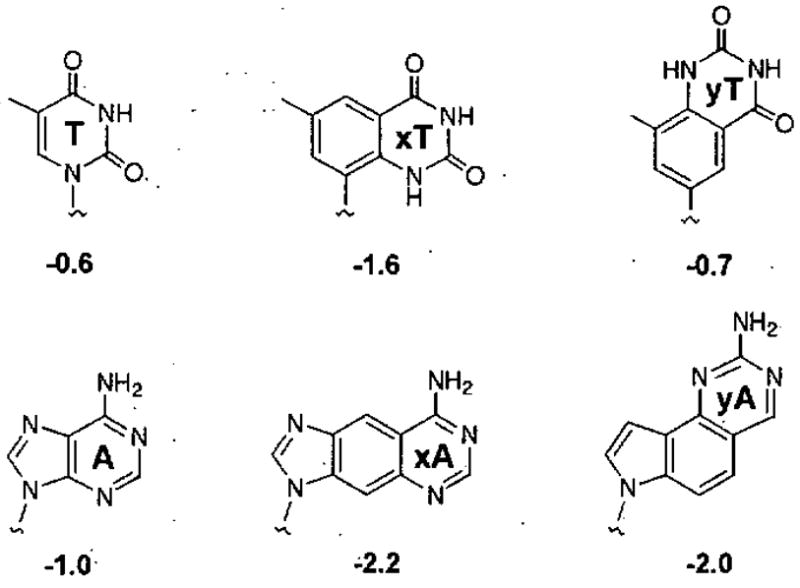
High stacking proficiency of xDNA and yDNA bases.36,38,42,58 Free energies for stacking (kcal/mol, 37 °C) were measured at the end of a short DNA duplex. The strong stacking of the expanded bases is likely due both to stronger van der Waals forces and the hydrophobic effect.62
VII. Size-expanded Double Helices
In a functioning genetic system, not only must isolated bases pair correctly, but they must also recognize their pairing partners when strung together in information-encoding sequences. In natural DNA, there are four monomeric components forming four pair orientations (A-T, T-A, C-G, G-C), which are assembled by polymerases (or by a DNA synthesizer). With xDNA, by contrast, there exist eight pairing possibilities; thus the assembly of xDNA could be considerably more complex.
As mentioned above, a single expanded base pair is destabilizing when incorporated into natural DNA, However, one can envision a double helix composed entirely of expanded-size base pairs. By accommodating this size in all pairs, the backbone distortion created by a lone expanded pair might, in principle, be relieved.
Base pairing in xDNA
In 2003 our lab demonstrated that a self-complementary oligomer composed entirely of xA-T expanded base pairs (a 10-mer) showed a cooperative, sigmoidal transition in its UV absorbance as the temperature was raised, a property characteristic of double-stranded DNA.61 Interestingly, this complex denatured at a temperature higher than a natural duplex of identical sequence (56 °C vs. 21 °C (Fig. 6)), which might be explained by the expanded bases’ enhanced stacking.
Figure 6.
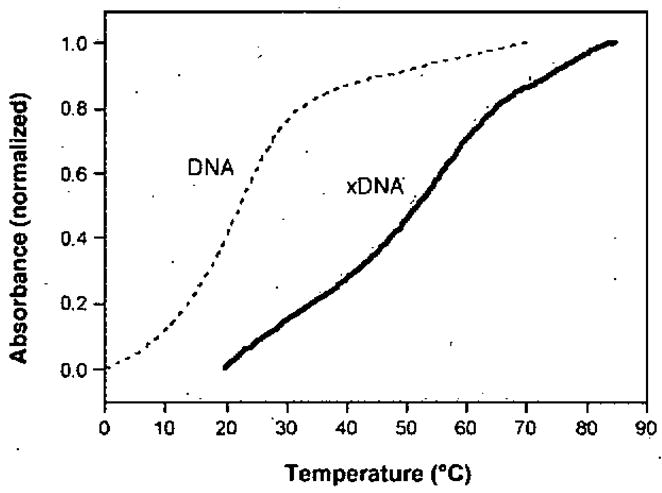
Thermal denaturation curves showing the high stability of an xDNA helix compared to DNA of the same sequence.61
Subsequently we determined a high-resolution NMR solution structure of this complex consisting of xA-T and T-xA base pairs.62 xDNA, like native B-DNA, exhibits a right-handed turn, Watson-Crick hydrogen bonding patterns, anti glycosidic bond conformations and standard 2′-endo deoxyribose sugar conformations (Fig. 7). However, the duplex diameter is larger by 3.0 Å, contributing to a smaller helical twist requiring more base pairs per turn (12, vs. 10 in natural DNA). In addition, the major and minor grooves are wider than those of natural DNA by 2.5 Å and 2.2 Å, respectively,(Fig. 7A).
Figure 7.
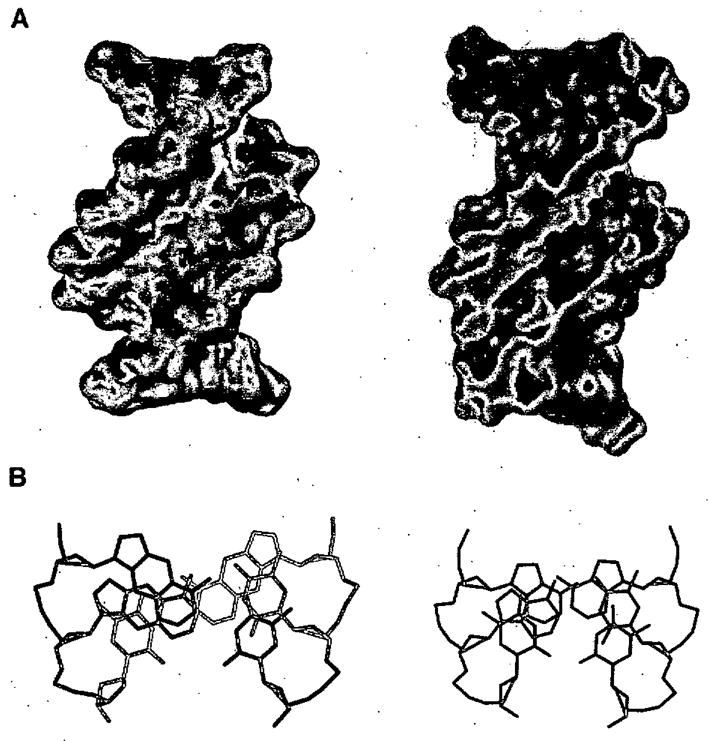
Structures of xDNA measured by NMR, with comparison to DNA (right).62 (A) Space-filling models showing wider grooves of xDNA, but similar right-handed helical turn. (B) View of three adjacent xDNA pairs, showing large surface area of overlap compared to DNA.
One of the most important hallmarks of the natural genetic system is that DNA hybridizes not only with high affinity, but also with robust sequence selectivity. We tested this with xDNA strands that contained a single mismatch. These expanded duplexes also exhibited high mismatch selectivity for the expected hydrogen bonding partners, with a selectivity profile (Fig. 8) remarkably analogous to that of natural DNA.35 This reinforces the possibility that an artificial genetic system other than the natural one may one day be used to selectively encode information. On a practical level, such selectivity is also important if xDNA is to be applied in hybridization-based assays with RNA or DNA.
Figure 8.
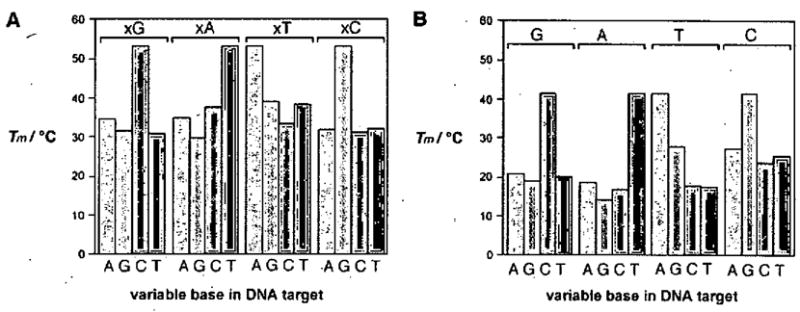
Graphs showing base pairing selectivity of xDNA (A) compared to DNA (B), as measured by melting temperatures of short duplexes containing a single mismatched pair as shown. (Reprinted with permission of Angew. Chem. Int. Ed.)
Pairing in yDNA
The development of yDNA as part of a second designed genetic system is also furthered by data on its mismatch selectivity towards complementary hydrogen bonding partners.36,38 We demonstrated that short oligomers composed entirely of yC/yT bases exhibited greater selectivity against mismatched targets than did natural DNA of analogous sequence. For example, for mismatches opposite yT, Tm values dropped 15–27 °C with a single mismatch, representing sizeable selectivity. By, comparison, natiiralDNA showed smaller Tm drops of 9–16 °C.
XIII. Early Studies of Polymerase Replication of Expanded DNAs
As was discussed in the introduction, one of the long-term goals of this work is to determine whether size-expanded DNAs can mimic the biochemical functions of the natural genetic system. One of the most important functions of natural DNA is its ability to template its own synthesis, catalyzed by DNA polymerase enzymes. Thus we ask the same question of xDNA and yDNA: is it possible to find, modify, design or evolve an enzyme to replicate these expanded helix-forming polymers?
We expect that discovery or development of such an enzyme - an xDNA or yDNA replicase - will not be a simple task, because of the highly specialized functions of natural DNA polymerase enzymes. Many DNA polymerases work efficiently (up to hundreds of nucleotides per second) and with high specificity, making an error in nucleotide selection only once per 10,000 to 100,000 events.63 Data suggest that this selectivity is governed chiefly by the enzyme tightly surrounding the base pair being synthesized, using steric effects to reject incorrect nucleotides.64 Even small steric differences can cause large kinetic effects.65 For this reason, we might expect most DNA polymerases to accept xDNA base pairs reluctantly.
Early experiments with xDNA bases in template strands of DNA add some support to this expectation (Fig. 9). DNA polymerase I from E. coli (Klenow fragment) inserts natural DNA nucleotides opposite xDNA bases relatively poorly, with efficiency ca. 1000-fold lower than natural pairs.66 However, the fact that there is any activity at all could be considered promising, and interestingly, some selectivity remains in this activity. For example, thymine (rather than C, G, or A) is preferentially inserted opposite benzoadenine (Fig. 9B), and similar selectivity is also seen with other expanded bases.66
Figure 9.
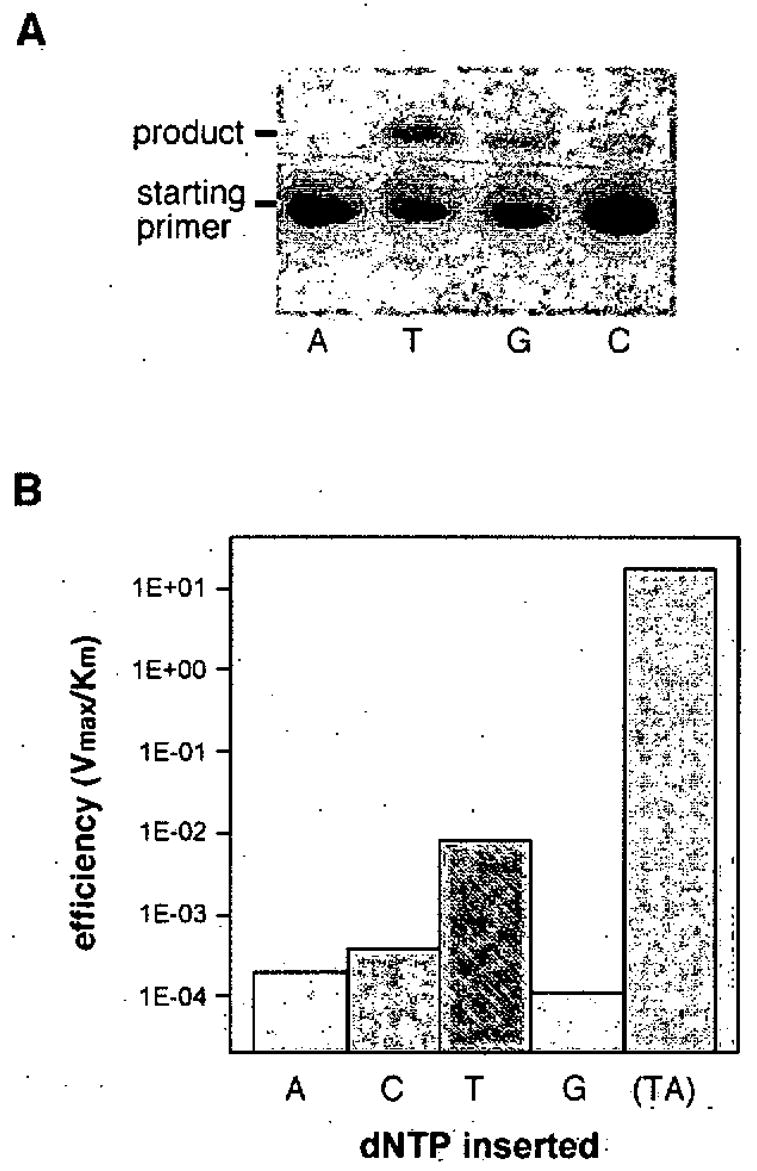
Preliminary data showing the ability of DNA polymerase I (Klenow fragment) to function with the xA base.66 (A) Polyacrylamide gel showing addition of dxA to the end of a primer opposite A, T, G, or C; (B) histogram showing relative efficiency of insertion of dTTP opposite xA. Note log scale of efficiency; natural TA base pair efficiency is given for comparison at right.
Addition of a single nucleotide is not replication, so there remains much work to be done in finding replicases for expanded DNA. The longer-term goals will require the synthesis of entire complementary xDNA or yDNA strands, and demonstration that two complementary strands can template enzymatic synthesis. Possible strategies for this might include searching for naturally occurring enzymes that are more forgiving in their substrate specificity,67 or mutagenesis of existing enzymes. A combination of such approaches may eventually yield enzymes capable of full replication of expanded DNAs.
IX. Is Further Expansion Possible? The Naphtho-fusion Approach
The self-assembly of the xDNA and yDNA systems leads naturally to a new question: how far can base pairs be expanded and still allow for this assembly? A homologous way to test this would be to add yet another benzene ring to the pairs. This would yield 4.8Å of expansion, giving hypothetical xxDNA and yyDNA designs (Fig. 10).68
Figure 10.
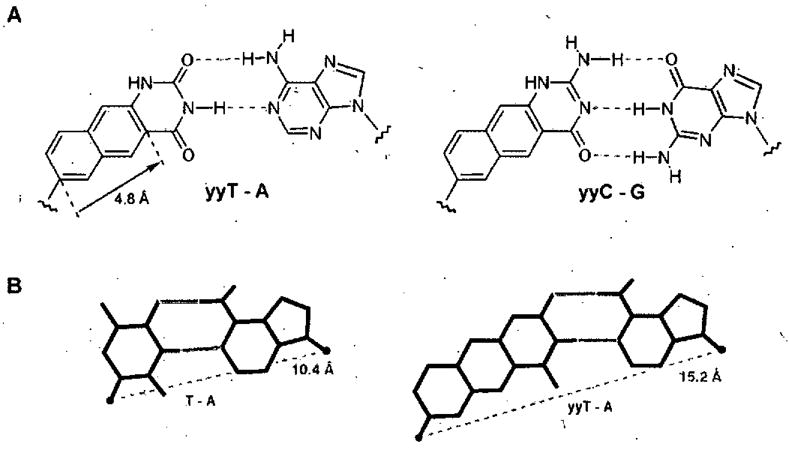
Pairing scheme of doublewide DNA (yyDNA). The proposed double helix has an expected diameter nearly 5Å wider than natural DNA. (A) Expected base pair. structures; (B) comparison of DNA pair size and geometry with yyDNA pair.
A recent study began testing this question in the yyDNA (“doublewide DNA”) system. It proved straightforward to synthesize the two naphtho-T and -C analogues (dyyT (13) and dyyC (14)).68 They are both fluorescent (like their smaller yDNA analogues), but with red-shifted absorption and emission, as expected for the more greatly conjugated π systems. A preliminary series of helix assembly studies has been carried out in the yyDNA system. Results showed complex melting behavior consistent with strong stacking of the component bases.68 Mixing plots, CD and fluorescence studies all gave evidence for double-stranded helix formation. Quenching and fluorescence resonance energy transfer experiments also pointed to double-stranded complexes of yyDNA. Further studies will be needed to evaluate the structure and stability of these complexes.
There are a number of possible ways to assemble naphtho-expanded systems. For example, it may be possible to pair xDNA bases with other xDNA bases, and one could imagine a similar yyDNA design as well. Thus there is more work to be done in testing the limits of expanded helices.
X. Conclusions and Future Prospects
Our experiments confirm that natural DNA is not the only base-pairing framework that allows for stable and selective helix assembly. Size-expanded DNA sequences can spontaneously assemble themselves into right-handed, double-stranded helices with antiparallel orientation. They form base pairs with sequence selectivity and stability at least as high as natural DNA. Thus, it is clear that new base pair structures and geometries can be grafted onto the natural DNA backbone, with retention of helix assembly properties.
The research to date also suggests that xDNAs and yDNAs may have useful applications. The fluorescent monomers may be useful probes of DNA-modifying enzymes and nucleotide-dependent enzymes in general. When expanded DNA monomers are assembled into oligomeric strands, such strands can bind natural DNAs or RNAs sequence selectively, forming expanded helices. This binding occurs with high affinity, suggesting uses of xDNAs or yDNAs in binding natural RNAs or DNAs even where they are partially blocked or folded in existing structures.
These experiments also give us new perspectives about the evolved design of the natural genetic system. Clearly, DNA base pairs did not evolve their structure because that was the only framework that could properly function in self-assembly. Our data show that size-expanded base pair designs appear to function as well. This suggests that it is more important to have a regular helix with consistent base pair geometries, than it is to conserve the absolute size of DNA. Perhaps the most convincing reason for the evolution of DNA’s native structure (rather than something like xDNA) is its chemical simplicity. The natural DNA bases are smaller and have fewer carbon-carbon bonds than our expanded designs, which makes their prebiotic synthesis easier. Although the greater structural complexity does not prevent chemists from making molecules such as xDNA, it does represent an advantage for natural DNA in a primordial soup where base assembly first occurred.
There remains much to be discovered with size-expanded genetic sets. In the biological realm, how far can the biomimicry be taken: can expanded DNAs be replicated? Can they be transcribed? Can they evolve? Can they function inside native or modified cells? And beyond the basic biological implications, are there other useful applications of expanded DNAs in biotechnology or nanotechnology? Such questions will keep us and others in the field of base pair design occupied for some time to come.
Acknowledgments
We thank the US National Institutes of Health (GM63587) for support. A. H. F. L. acknowledges a fellowship from the Croucher Foundation.
Biographies
Andrew T. Krueger - Andrew Krueger received his B.S. in chemistry from the University of Wisconsin-Madison in 2003. While at Wisconsin, he was engaged in research under Prof. Howard E. Zimmerman where he worked on the synthesis and photochemistry of substituted 4,4-diphenylcyclohexenones. He then moved to Stanford University to pursue graduate work under Professor Eric Kool. His research interests include the synthesis, evaluation, structural studies of novel DNA base analogues.
Haige Lu - Haige Lu received his B.Sc. degree from Peking University in 2001. He then moved to Stanford University, where he is carrying out his Ph.D. study under the supervision of Professor Eric Kool. His research concentrates on the synthesis, structure and DNA polymerase kinetics of yDNA (wide DNA).
Alex H. F. Lee - Alex Lee was born and brought up in Hong Kong. He pursued his B.Sc. and M.Phil, degrees from The Chinese University of Hong Kong, and obtained his Ph.D. from The Hong Kong Polytechnic University in 2003 with Professor Albert S, C. Chan. Then he was awarded the Croucher Foundation Fellowship and undertook his postdoctoral training with Professor Eric T. Kool at Stanford University (2003–2006). His research interests include anti tumor antibiotics, free radical-mediated DNA cleavage mechanistic studies; modified nucleosides, expanded DNA and novel nucleic acids research and development.
Eric T. Kool - Eric Kool is the George and Hilda Daubert Professor in the Department of Chemistry at Stanford University. He received his undergraduate degree from Miami University (Ohio) in 1982. He pursued graduate studies as an NSF Fellow at Columbia University with Ronald Breslow, and received his postdoctoral training at Caltech with Peter Dervan. Kool started his independent research career in 1990 as Assistant Professor at the University of Rochester. He moved to Stanford in 1999, where he teaches and directs his research group. His research interests include the design of molecular mimics for biological molecules and pathways, and development of new chemical approaches for biosensing and bioimaging.
References
- 1.McLaughlin LW, Wilson M, Ha SB. Use of Nucleoside Analogues to Probe Biochemical Processes. In: Barton D, Nakanishi K, editors. Comprehensive Natural Products Chemistry. Elsevier; Oxford, UK: 1999. pp. 251–284. [Google Scholar]
- 2.Kool ET, Morales JC, Guckian KM. Mimicking the Structures and Functions of DNA: Insights into DNA Stability and Replication. Angew Chem Int Ed. 2000;39:990–1009. doi: 10.1002/(sici)1521-3773(20000317)39:6<990::aid-anie990>3.0.co;2-0. [DOI] [PubMed] [Google Scholar]
- 3.Horlacher J, Hottiger M, Podust VN, Hubscher U, Benner SA. Recognition by Viral and Cellular DNA Polymerases of Nucleosides Bearing Bases with Nonstandard Hydrogen Bonding Patterns. Proc Natl Acad Sci USA. 1995;92:6329–6333. doi: 10.1073/pnas.92.14.6329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Henry AA, Romesberg FE, Beyond ACGT. Augmenting Nature’s Alphabet. Curr Opin Chem Biol. 2003;7:727–33. doi: 10.1016/j.cbpa.2003.10.011. [DOI] [PubMed] [Google Scholar]
- 5.Johnson SC, Sherrill CB, Marshall DJ, Moser MJ, Prudent JR. A Third Base Pair for the Polymerase Chain Reaction: Inserting IsoC and IsoG. Nucleic Acids Res. 2004;32:1937–1941. doi: 10.1093/nar/gkh522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Paul N, Nashine VC, Hoops G, Zhang P, Zhou J, Bergstrom DE, Davisson VJ. DNA Polymerase Template Interactions Probed by Degenerate Isosteric Nucleobase Analogs. Chem Biol. 2003;10:815–825. doi: 10.1016/j.chembiol.2003.08.008. [DOI] [PubMed] [Google Scholar]
- 7.Nichols R, Andrews PC, Zhang P, Bergstrom DE. A Universal Nucleoside for Use at Ambiguous Sites in DNA Primers. Nature. 1994;369:492–493. doi: 10.1038/369492a0. [DOI] [PubMed] [Google Scholar]
- 8.Parsch J, Engels JW. Stacking and Stability of RNA Duplexes Containing Fluorobenzene and Fluorobenzimidazole Nucleosides. Nucleosides Nucleotides Nucleic Acids. 2001;20:815–818. doi: 10.1081/NCN-100002436. [DOI] [PubMed] [Google Scholar]
- 9.Breslow R. Studies in Biomimetic Chemistry. Pure Appl Chem. 1998;70:267–270. [Google Scholar]
- 10.Benner SA. Synthetic Biology: Act Natural. Nature. 2003;421:118. doi: 10.1038/421118a. [DOI] [PubMed] [Google Scholar]
- 11.Rawls R. ‘Synthetic Biology’ Makes Its Debut. Chem Eng News. 2000 April 24;:49–53. [Google Scholar]
- 12.Leonard NJ. Dimensional Probes Of Enzyme-Coenzyme Binding-Sites. Acc Chem Res. 1982;15:128–135. [Google Scholar]
- 13.Watson JD, Crick FHC. The Structure of DNA. Cold Spr Harbor Symp Quant Biol. 1953;18:123–131. doi: 10.1101/sqb.1953.018.01.020. [DOI] [PubMed] [Google Scholar]
- 14.Piccirilli JA, Krauch T, Moroney SE, Benner SA. Enzymatic Incorporation of a New Base pair into DNA and RNA Extends the Genetic Alphabet. Nature. 1990;343:33. doi: 10.1038/343033a0. [DOI] [PubMed] [Google Scholar]
- 15.Rappaport HP. Replication of the Base Pair 6-thioguanine/5-methyl-2-pyrimidine with the Large Klenow Fragment of Escherichia coli DNA Polymerase I. Biochemistry. 1993;32:3047. doi: 10.1021/bi00063a016. [DOI] [PubMed] [Google Scholar]
- 16.Scopes DI, Barrio JR, Leonard NJ. Defined Dimensional Changes in Enzyme Cofactors: Fluorescent “Stretched-out” Analogs of Adenine Nucleotides. Science. 1977;195:296–298. doi: 10.1126/science.188137. [DOI] [PubMed] [Google Scholar]
- 17.Leonard NJ, Keyser GE. Synthesis of Fluorescent Nucleotide Analogues: 5′-Mono-, Di-, and Triphosphates of Linear-benzoguanosine, Linear-benzoinosine, and Linear-benzoxanthosine. Proc Natl Acad Sci USA. 1979;76:4262–4. doi: 10.1073/pnas.76.9.4262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lessor RA, Gibson KJ, Leonard NJ. Synthesis and Biochemical Evaluation of 2′-deoxy-lin-benzoadenosine Phosphates. Biochemistry. 1984;23:3868–3873. doi: 10.1021/bi00312a012. [DOI] [PubMed] [Google Scholar]
- 19.Czarnik AW, Leonard NJ. Foreshortened Nucleotide Analogs as Potential Base-pairing Complements for lin-benzoadenosine. J Am Chem Soc. 1982;104:2624–2631. [Google Scholar]
- 20.Godde F, Toulme JJ, Moreau S. Biochemistry. 1998;37:13765. doi: 10.1021/bi9811967. [DOI] [PubMed] [Google Scholar]
- 21.Okamoto A, Tainaka K, Saito I. J Am Chem Soc. 2003;125:4972–4973. doi: 10.1021/ja034090u. [DOI] [PubMed] [Google Scholar]
- 22.Matteucci MD, von Krosigk U. Hybridization Properties of Oligonucleotides Bearing a Tricyclic 2′-Deoxycytidine Analog Based on a Carbazole Ring System. Tetrahedron Lett. 1996;37:5057–5060. [Google Scholar]
- 23.Seley KL, Januszczyk P, Hagos A, Zhang L, Dransfield DT. Synthesis and Antitumor Activity of Thieno-Separated Tricyclic Purines. J Med Chem. 2000;43:4877–4883. doi: 10.1021/jm000326i. [DOI] [PubMed] [Google Scholar]
- 24.Minakawa N, Kojima N, Hikishima S, Sasaki T, Kiyosue A, Atsumi N, Ueno Y, Matsuda A. New Base Pairing Motifs. J Am Chem Soc. 2003;125:9970–82. doi: 10.1021/ja0347686. [DOI] [PubMed] [Google Scholar]
- 25.Hikishima S, Minakawa N, Kuramoto K, Fujisawa Y, Ogawa M, Matsuda A. Angew Chem Int Ed. 2005;44:596–598. doi: 10.1002/anie.200461857. [DOI] [PubMed] [Google Scholar]
- 26.Switzer CY, Moroney SE, Benner SA. Enzymatic Recognition of the Base Pair Between Isocytidine and Isoguanosine. Biochemistry. 1993;32:10489–10496. doi: 10.1021/bi00090a027. [DOI] [PubMed] [Google Scholar]
- 27.Leconte AM, Matsuda S, Hwang GT, Romesberg FE. Efforts towards Expansion of the Genetic Alphabet: Pyridone and Methyl Pyridone Nucleobases. Angew Chem Int Ed. 2006;45:4326–4329. doi: 10.1002/anie.200601272. [DOI] [PubMed] [Google Scholar]
- 28.Hirao I, Harada Y, Kimoto M, Mitsui T, Fujiwara T, Yokoyama S. A Two-unnatural-base-pair system toward the Expansion of the Genetic Code. J Am Chem Soc. 2004;126:13298–13305. doi: 10.1021/ja047201d. [DOI] [PubMed] [Google Scholar]
- 29.Chaput JC, Szostak JW. TNA synthesis by DNA polymerases. J Am Chem Soc. 2003;125:9274–9275. doi: 10.1021/ja035917n. [DOI] [PubMed] [Google Scholar]
- 30.Jung KH, Marx A. Nucleotide Analogues as Probes for DNA Polymerases. Cell Mol Life Sci. 2005;62:2080–2091. doi: 10.1007/s00018-005-5117-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tanaka K, Tengeiji A, Kato T, Toyama N, Shiro M, Shionoya M. Efficient incorporation of a copper hydroxypyridone base pair in DNA. J Am Chem Soc. 2002;124:12494–12498. doi: 10.1021/ja027175o. [DOI] [PubMed] [Google Scholar]
- 32.Zimmermann N, Meggers E, Schultz PG. A novel silver(I)-mediated DNA base pair. J Am Chem Soc. 2002;124:13684–13685. doi: 10.1021/ja0279951. [DOI] [PubMed] [Google Scholar]
- 33.Liu H, Gao J, Maynard L, Saito YD, Kool ET. Toward a New Genetic System with Expanded Dimensions: Size-Expanded Analogs of Deoxyadenosine and Thymidine. J Am Chem Soc. 2004;126:1102–1109. doi: 10.1021/ja038384r. [DOI] [PubMed] [Google Scholar]
- 34.Fuentes-Cabrera M, Sumpter BG, Wells JC. Size-Expanded Bases: An Ab Initio Study of Their Structural and Electronic Properties. J Phys Chem B. 2005;109:21135–21139. doi: 10.1021/jp055210i. [DOI] [PubMed] [Google Scholar]
- 35.Gao J, Liu H, Kool ET. Assembly of the Complete Eight-base Artificial Genetic Helix, xDNA, and its Interaction with the Natural Genetic System. Angew Chem Int Ed. 2005;44:3118–3122. doi: 10.1002/anie.200500069. [DOI] [PubMed] [Google Scholar]
- 36.Lu H, He K, Kool ET. yDNA: A New Geometry for Size-Expanded Base Pairs. Angew Chem Int Ed. 2004;43:5834–5836. doi: 10.1002/anie.200461036. [DOI] [PubMed] [Google Scholar]
- 37.Lee AHF, Kool ET. A New Four-Base Genetic Helix, yDNA, Composed of Widened Benzopyrimidine-Purine Pairs. J Am Chem Soc. 2005;127:3332–3338. doi: 10.1021/ja0430604. [DOI] [PubMed] [Google Scholar]
- 38.Lee AHF, Kool ET. Novel Benzopyrimidines as Widened Analogues of DNA Bases. J Org Chem. 2005;70:132–40. doi: 10.1021/jo0483973. [DOI] [PubMed] [Google Scholar]
- 39.Fuentes-Cabrera M, Sumpter BG, Lipkowski P, Wells JC. Size-Expanded yDNA Bases: An Ab Initio Study. J Phys Chem B Condens Matter Mater Surf Interfaces Biophys. 2006;110:6379–84. doi: 10.1021/jp057356n. [DOI] [PubMed] [Google Scholar]
- 40.Leonard NJ, Sprecker MA, Morrice AG. Defined Dimensional Changes in Enzyme Substrates and Cofactors. Synthesis of lin-Benzoadenosine and Enzymatic Evaluation of Derivatives of the Benzopurines. J Am Chem Soc. 1976;98:3987–3994. doi: 10.1021/ja00429a040. [DOI] [PubMed] [Google Scholar]
- 41.Liu H, Gao J, Maynard L, Saito YD, Kool ET. Toward a New Genetic System with Expanded Dimensions: Size-Expanded Analogues of Deoxyadenosine and Thymine. J Am Chem Soc. 2004;126:1102–1109. doi: 10.1021/ja038384r. [DOI] [PubMed] [Google Scholar]
- 42.Liu H, Gao J, Kool ET. Size-Expanded Analogues of dG and dC: Synthesis and Pairing Properties in DNA. J Org Chem. 2005;70:639–647. doi: 10.1021/jo048357z. [DOI] [PubMed] [Google Scholar]
- 43.Zhang HC, Daves GD., Jr Syntheses of 2′-Deoxypseudouridine, 2′-Deoxyformycin B, and 2′3′-Dideoxyformycin B by Palladium-Mediated Glycal-Aglycon Coupling. J Org Chem. 1992;57:4690–4696. [Google Scholar]
- 44.Sandin P, Wilhelmsson LM, Lincoln P, Powers VEC, Brown T, Albinsson B. Fluorescent Properties of DNA Base Analogue tC upon Incorporation into DNA – Negligible Influence of Neighboring Bases on Fluorescence Quantum Yield. Nucleic Acids Res. 2005;33:5019–5025. doi: 10.1093/nar/gki790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ward DC, Reich E, Stryer L. Fluorescence Studies of Nucleotides and Polynucleotides. I. Formycin, 2-aminopurine Riboside, 2, 6-diaminopurine Riboside, and their Derivatives. J Biol Chem. 1969;244:1228–1237. [PubMed] [Google Scholar]
- 46.Charubala R, Maurinsh J, Rosler A, Melguizo M, Jungmann O, Gottlieb M, Lehbauer J, Hawkins M, Pfleiderer W. Pteridine Nucleosides: New Versatile Building Blocks in Oligonucleotide Synthesis. Nucleosides Nucleotides. 1997;16:1369. [Google Scholar]
- 47.Greco NJ, Tor Y. Simple Fluorescent Pyrimidine Analogues Detect the Presence of DNA Abasic Sites. J Am Chem Soc. 2005;127:10784–10785. doi: 10.1021/ja052000a. [DOI] [PubMed] [Google Scholar]
- 48.Coleman RS, Madaras ML. Synthesis of a Novel Coumarin C-Riboside as a Photophysical Probe of Oligonucleotide Dynamics. J Org Chem. 1998;63:5700. [Google Scholar]
- 49.Saito Y, Miyauchi Y, Okamoto A, Saito I. Base-discriminating Fluorescent (BDF) Nucleoside: Distinction of Thymine by Fluorescence Quenching. Chem Commun. 2004;7:1704–1705. doi: 10.1039/b405832a. [DOI] [PubMed] [Google Scholar]
- 50.Beuck C, Singh L, Bhattacharya A, Hecker W, Parmar VS, Seitz O, Weinhold E. Polycyclic Aromatic DNA-base Surrogates: High-affinity Binding to an Adenine-specific Base-flipping DNA Methyltransferase. Angew Chem Int Ed. 2003;42:3958–3960. doi: 10.1002/anie.200219972. [DOI] [PubMed] [Google Scholar]
- 51.Jiang YL, Stivers JT, Song F. Base-flipping Mutations of Uracil DNA Glycosylase: Substrate Rescue using a Pyrene Nucleotide Wedge. Biochemistry. 2002;41:11248–11254. doi: 10.1021/bi026227j. [DOI] [PubMed] [Google Scholar]
- 52.Valis L, Amann N, Wagenknecht HA. Detection of Single Base Mismatches and Abasic Sites using Phenanthridinium as an Artificial DNA Base and Charge Donor. Org Biomol Chem. 2005;7:36–38. doi: 10.1039/b414672g. [DOI] [PubMed] [Google Scholar]
- 53.Gao J, Liu H, Kool ET. Expanded-Size Bases in Naturally-Sized DNA: Evaluation of Steric Effects in Watson-Crick Pairing. J Am Chem Soc. 2004;126:11826–11831. doi: 10.1021/ja048499a. [DOI] [PubMed] [Google Scholar]
- 54.Petersheim M, Turner DH. Base-stacking and Base-pairing Contributions to Helix Stability: Thermodynamics of Double-helix Formation with CCGG, CCGGp, CCGGAp, ACCGGp, CCGGUp, and ACCGGUp. Biochemistry. 1983;22:256–263. doi: 10.1021/bi00271a004. [DOI] [PubMed] [Google Scholar]
- 55.Guckian K, Schweitzer BA, Ren RX-F, Sheils CJ, Paris PL, Tahmassebi DC, Kool ET. Experimental Measurement of Aromatic Stacking Affinities in the Context of Duplex DNA. J Am Chem Soc. 1996;118:8182–8183. doi: 10.1021/ja961733f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lai JS, Qu J, Kool ET. Fluorinated DNA Bases as Probes of Electrostatic Effects in DNA Base Stacking. Angew Chem Int Ed. 2003;42:5973–5977. doi: 10.1002/anie.200352531. [DOI] [PubMed] [Google Scholar]
- 57.Lin KY, Jones RJ, Matteucci MJ. Tricyclic 2′-Deoxycytidine Analogs: Syntheses and Incorporation into Oligodeoxynucleotides which have Enhanced Binding to Complementary RNA. Am Chem Soc. 1995;117:3873–3874. [Google Scholar]
- 58.Sando S, Nakatani K, Saito I. New Method for Scanning of Single-Nucleotide Polymorphisms (SNPs): Recognition of Guanine-Guanine Mismatches by Dimeric Naphthyridine. Nucleic Acids Symp Ser. 2000;44:119–120. doi: 10.1093/nass/44.1.119. [DOI] [PubMed] [Google Scholar]
- 59.Uotani Y, Nakano S, Nakashima S, Anno Y, Fujii M, Sugimoto N. Large Stacking Stability of a Base Pair-mimic Nucleotide on the DNA Duplex. Nucleic Acids Res Suppl. 2003;3:79–80. doi: 10.1093/nass/3.1.79. [DOI] [PubMed] [Google Scholar]
- 60.Dogan Z, Paulini R, Rojas Stutz JA, Narayanan S, Richert C. 5′-Tethered Stilbene Derivatives as Fidelity- and Affinity-enhancing Modulators of DNA Duplex Stability. J Am Chem Soc. 2004;126:4762–4763. doi: 10.1021/ja0394434. [DOI] [PubMed] [Google Scholar]
- 61.Liu H, Gao J, Lynch SR, Maynard L, Saito D, Kool ET. A Four Base Paired Genetic Helix with Expanded Size. Science. 2003;302:868–871. doi: 10.1126/science.1088334. [DOI] [PubMed] [Google Scholar]
- 62.Liu H, Lynch SR, Kool ET. Solution Structure of xDNA: A Paired Genetic Helix with Increased Diameter. J Am Chem Soc. 2004;126:6900–6905. doi: 10.1021/ja0497835. [DOI] [PubMed] [Google Scholar]
- 63.Kunkel TA, Bebenek K. DNA Replication Fidelity. Ann Rev Biochem. 2000;69:497–529. doi: 10.1146/annurev.biochem.69.1.497. [DOI] [PubMed] [Google Scholar]
- 64.Kool ET. Active Site Tightness and Substrate Fit in DNA Replication. Ann Rev Biochem. 2002;71:191–219. doi: 10.1146/annurev.biochem.71.110601.135453. [DOI] [PubMed] [Google Scholar]
- 65.Kim TW, Delaney JC, Essigmann JM, Kool ET. Probing the Active Site Tightness of DNA Polymerase in Sub-Angstrom Increments. Proc Natl Acad Sci USA. 2005;102:15803–15808. doi: 10.1073/pnas.0505113102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Liu H. PhD Thesis. Stanford University; 2004. [Google Scholar]
- 67.Mizukami S, Kim TW, Helquist SA, Kool ET. Varying DNA Base Pair Size in Sub-Angstrom Increments: Evidence for a Loose, Not Large, Active Site in Low-fidelity Dpo4 Polymefase. Biochemistry. 2006;45:2772–2778. doi: 10.1021/bi051961z. [DOI] [PubMed] [Google Scholar]
- 68.Lee AHF, Kool ET. Exploring the Limits of DNA Base Pair Size: Naphthohomologous DNA Bases and Pairs. J Am Chem Soc. 2006;128:9219–9230. doi: 10.1021/ja0619004. [DOI] [PMC free article] [PubMed] [Google Scholar]