

Abstract
A study of the structure–activity relationships (SAR) of 5′-O-[N-(Salicyl)sulfamoyl]adenosine (6), a potent inhibitor of the bifunctional enzyme salicyl-AMP ligase (MbtA, encoded by the gene Rv2384) in Mycobacterium tuberculosis, is described, targeting the salicyl moiety. A systematic series of analogues was prepared exploring the importance of substitution at the C-2 position revealing that a hydroxy group is required for optimal activity. Examination of a series of substituted salicyl derivatives indicated that substitution at C-4 was tolerated. Consequently, a series of analogues at this position provided 4-fluoro derivative, which displayed an impressive MIC99 of 0.098 μM against whole-cell M. tuberculosis under iron-limiting conditions. Examination of other heterocyclic, cycloalkyl, alkyl, and aminoacyl replacements of the salicyl moiety demonstrated that these nonconserative modifications were poorly tolerated, a result consistent with the fairly strict substrate specificities of related non-ribosomal peptide synthetase (NRPS) adenylation enzymes.
Keywords: Mycobacterium tuberculosis, tuberculosis, adenylation inhibitor, siderophore biosynthesis, mycobactin, nonribosomal peptide synthetase
Introduction
Tuberculosis (TBa) is the leading cause of infectious disease mortality by a bacterial pathogen.1 TB is extremely difficult to treat since the etiological agent, Mycobacterium tuberculosis, is slow growing, capable of switching its metabolism to a latent or non-replicating state, and possesses a nearly impenetrable cell-wall, which provides a permeability barrier that limits the uptake of many antibiotics. As a result of these factors, effective therapy requires prolonged treatment with 3–4 antibiotics. A combination of poor patient compliance and the inevitable evolution of drug resistance has resulted in the development of multidrug resistant tuberculosis (MDR-TB) defined as resistance to both the first-line agents isoniazid and rifampin. Further, co-infection with HIV/AIDS is a deadly combination and over one-third of HIV-related deaths are due to TB or related mycobacterial infections. Despite the tremendous advances in TB chemotherapy made in the 20th century, TB has now reemerged as one of the most significant threats to global public health, thus there is a great demand for new drugs to combat TB. The Global Alliance for TB Drug Development (http://www.tballiance.org) recommends that an ideal new TB drug should shorten the duration of effective treatment, improve the treatment of MDR-TB, and provide more effective treatment of latent TB infection. Additionally, drugs that are specific antitubercular agents as opposed to broad-spectrum antibiotics may be advantageous to prevent disturbance to beneficial commensal bacteria as well as minimize the potential development of cross-resistance.
Iron is an essential micronutrient for almost all known organisms including M. tuberculosis where it serves as an obligate cofactor for numerous metalloproteins.2 A particular exception is Borrelia burgdorferi, the causative agent of Lyme disease, which apparently has evolved to utilize manganese instead of iron.3 Notwithstanding this anomaly, iron acquisition is critical for bacterial pathogenesis and bacteria have evolved a variety of mechanisms to obtain this vital nutrient. The most common mechanism involves the synthesis, secretion, and reuptake of small molecule iron-chelators termed siderophores.2 In a mammalian host, siderophore production is crucial since the concentration of free iron is approximately 10−24 M, which is far too low to support bacterial colonization and growth.4 Thus, inhibition of siderophore biosynthesis represents a logical strategy for the development of a new class of antibiotics.5 An alternate strategy pioneered by Miller and co-workers involves antagonism of siderophore function using synthetic siderophore analogues that likely cannot be taken up by bacterial siderophore transport system.32
M. tuberculosis produces two series of structurally related siderophores, collectively known as the mycobactins, that are critical for virulence and growth (Figure 1).6, 7 Mycobactin biosynthesis is initiated by MbtA, an adenylate-forming enzyme that catalyzes a two-step reaction and is responsible for incorporating salicylic acid into the mycobactins (Figure 1).8 MbtA first binds its substrates salicylic acid and ATP then catalyzes their condensation to afford acyladenylate 2 and pyrophosphate. The acyladenylate remains tightly bound whereas pyrophosphate dissociates. Next, MbtA binds the N-terminal aryl carrier domain of MbtB and catalyzes the transfer of the salicyl moiety onto the nucleophilic sulfur atom of the phosphopantetheinyl cofactor of MbtB to afford thioester tethered-MbtB 3 that is elaborated to the mycobactins by a mixed nonribosomal peptide synthetase polyketide synthase (NRPS-PKS) assembly line.8
Figure 1.

Biosynthesis of the mycobactins and carboxymycobactins.6 The depicted lipid side chain is a representative as both 4 and 5 are isolated as a suite of compounds with various length lipid residues.
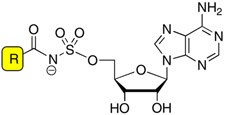
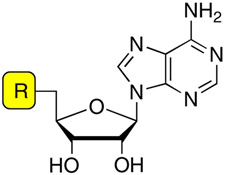
Acyladenylates have been shown to bind several orders of magnitude more tightly than the substrate acids since they simultaneously occupy both substrate binding pockets.9, 10 Thus acyladenylate analogues that incorporate a stabile linker as a bioisostere of the labile acylphosphate function provide potent adenylation enzyme inhibitors. The general inhibitor scaffold is comprised of four domains (aryl, linker, glycosyl, and base) as depicted in Figure 2. The most crucial portion of the inhibitor scaffold is the linker domain since this must be metabolically stable and appropriately position both the aryl and nucleoside moieties in their respective binding pockets. We have previously explored both the molecular geometry and polarity of the linker pharmacophore with the preparation of β-ketophosphonate, acylsulfamate, acylsulfamide, sulfamate, β-ketosulfonamide, α,α-difluoro-β-ketosulfonamide, acyltriazole, and vinylsulfonamide linkages as surrogates for the labile acylphosphate linkage.11–13 Inhibitors incorporating the acylsulfamate and acylsulfamide linkages were found to be the most potent with low nanomolar apparent inhibition constants and possessed submicromolar antitubercular activity against whole-cell M. tuberculosis rivaling the first-line agent isoniazid.11, 14 Next, we systematically examined the glycosyl domain and found that both the 3′-hydroxy and 4′-ribofuranose ring oxygen were dispensable for bioactivity while modifications making the sugar either more or less flexible were detrimental.15 In this article, we explore the importance of the aryl ring of the bisubstrate inhibitor scaffold.
Figure 2.
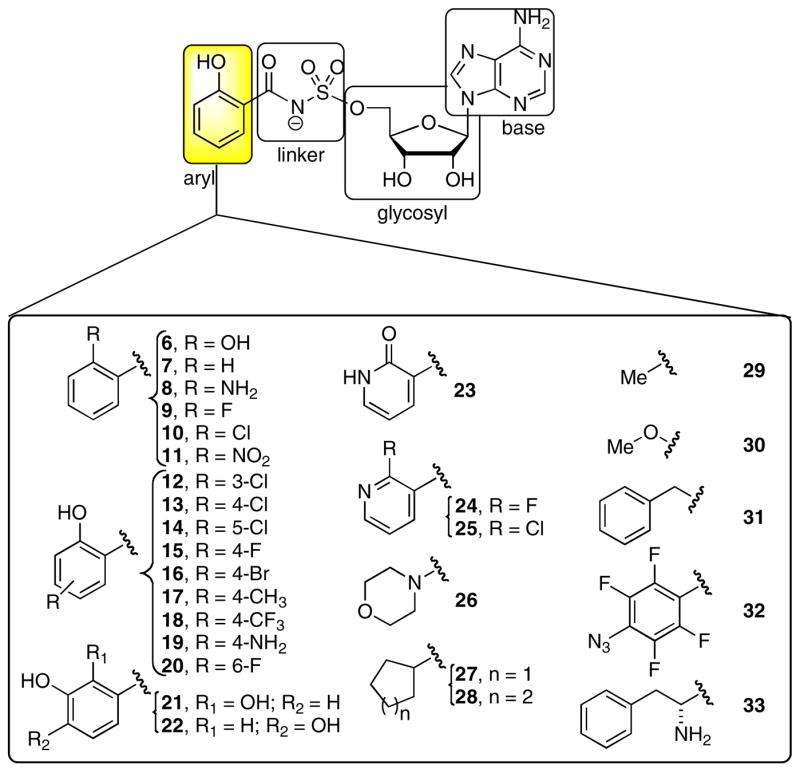
Bisubstrate inhibitors of MbtA. The expanded portion of the figure shows the Ar modifications described herein.
Results and Discussion
Chemistry
Since NRPS adenylation domains exhibit a fairly strict substrate specificity, bisubstrate inhibitors containing a number of conservative aryl modifications were prepared to explore the importance of the ortho-hydroxy group, to define the steric requirements of the shallow aryl acid binding pocket of MbtA, and to assess the potential for interacting with specific residues within the active site such as C240. Additionally, a number of heteroaryl, cycloalkyl, alkyl, and aminoalkyl modifications were targeted. The syntheses of 611, 14, 16, 711, 16, 811, 916, 2116, and 2217 were disclosed previously, but 8 and 21 were synthesized by alternate routes in improved overall yields. The other twenty-two inhibitors were synthesized through three related synthetic routes as described below.
Salicyl acid derivatives 12–20 and 23 were prepared according to the method described by Tan, Quadri and co-workers from the corresponding acids 34a–j by direct activation with CDI to afford intermediate cyclic anhydrides 35a–j, which were not isolated but subsequently reacted with either protected 5′-O-(sulfamoyl)adenosine derivatives 36 or 37 employing either DBU or Cs2CO3 as base to afford 38a–j (Scheme 1).14 Catalytic hydrogenation of 38h provided p-aminosalicyl derivative 39. Final deprotection of the isopropylidene acetal (80% aq TFA) or TBS ethers (TBAF) furnished bisubstrate inhibitors 12–20 and 23. Anthranoyl analogue 8 was prepared analogously from isatoic anhydride and 36 (not shown, see Experimental Section). The tert-butyldimethylsilyl (TBS) protected nucleosides were preferred for compounds, such as nicotinyl derivative 23 that decomposed under acidic conditions. Compounds 15–18 and 23, which were initially isolated as the tetrabutylammonium salts, were converted to the corresponding sodium salts by ion-exchange with a strong cation exchange resin in the sodium form.
Scheme 1a.

aReaction conditions: (a) CDI, MeCN, 60 °C; (b) 36 or 37, DBU or Cs2CO3, DMF; (c) 80% aq TFA, 0 °C; or TBAF, THF; (d) H2, Pd/C.
Direct CDI activation of aryl acids lacking a 2-hydroxy group was much less efficient. In these cases the acyl imidazolidate that forms in situ is substantially less reactive than the cyclic anhydride species (see compound 35, Scheme 1) formed with salicylic acid derivatives. For most other aryl-, heteoaryl-, and alkyl carboxylic acids (40a–k) the corresponding preformed N-hydroxysuccinimide (NHS) esters provided superior coupling yields. Thus, coupling of NHS esters 41a–k to 36 or 37 mediated by either DBU or Cs2CO3 afforded acylsulfamate derivatives 42a–k (Scheme 2).18 Catalytic hydrogenation of 42c provided 2,3-dihydroxybenzoyl derivative 43. In general Cs2CO3 as base is favored since in a few instances, such as 42a, DBU salts of the coupled products were obtained – the DBU could not removed by an acid wash, ion-exchange, or by chromatography (co-eluting with 1% Et3N). Deprotection of the isopropylidene acetal (80% aq TFA) or the TBS ethers (TBAF) of 42a–b, 42d–k, and 43 yielded the final bisubstrate inhibitors 10, 11, 21, 24, 25, 27–29, and 31–33. Compounds 27–29, and 31, which were initially isolated as tetrabutylammonium salts, were converted to the corresponding sodium salts by ion-exchange chromatography. The acetonide protected nucleosides were preferred for compounds, such as 4-azido-2,3,5,6-tetrafluorphenyl derivative 32 that decomposed under the fluoride mediated conditions (TBAF or HF·pyr) required for TBS deprotection.
Scheme 2a.

aReaction conditions: (a) DCC, NHS, THF; (a) 36 or 37, DBU or Cs2CO3, DMF; (c) 80% aq TFA or TBAF, THF; (d) H2, Pd/C.
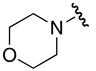
Attempts to prepare a morpholine derivative from the corresponding NHS ester 44 and 5′-O-sulfamoyladenosine derivative 37 did not afford the anticipated product 45, but rather 46 that was deprotected with TBAF to provide 47 in 56% overall yield from 44 (Scheme 3). The β-alanine fragment in 47 was confirmed by HRMS and NMR.19 The β-alanine moiety of compound 46 is hypothesized to arise from a sequential series of reactions as depicted in Scheme 3 below. Initial nucleophilic attack of sulfamate 37 onto the succinimidyl carbonyl of 44 followed by opening of the imide affords acylhydroxamate intermediate 48, which in turn undergoes Lössen rearrangement to provide isocyanate 49 and expulsion of the carbamic acid of morpholine. Decarboxylation of the carbamic acid affords morpholine that condenses with isocyanate 49 to provide the observed β-alanine urea derivative 46. Precedent exists for opening of the succinimide by a nucleophile.20 Failure to obtain the desired product arises from the attenuated electrophilicity of the N-(morpholinyl)carbonyl group, which favors addition at the more reactive imide carbonyls.
Scheme 3a.

Based on the above synthetic considerations, morpholinyl analogue 26 was successfully prepared by simply interchanging the electrophile. Thus, treatment of chlorosulfonylisocyanate 51 with morpholine 50 provided chlorosulfonylurea 52 (Scheme 4). This intermediate was not isolated, but condensed with 2′,3′-O-isopropylideneadenosine 53 to afford 54, subsequent acetonide deprotection (80% aq TFA) and ion-exchange to the sodium salt afforded 26.
Scheme 4a.
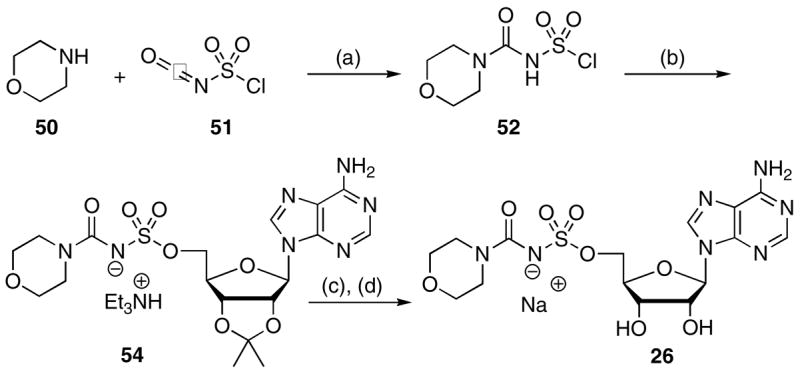
aReaction conditions: (a) CH2Cl2, pyr; (b) 53, MeCN; (c) 80% aq TFA; (d) ion-exchange, 4% overall.
Treatment of 5′-O-(sulfamoyl)adenosine derivative 36 with carbonyl diimidazole afforded [(imidazolyl)carbonyl]sulfamate 55 (Scheme 5). This intermediate was not isolated but treated with methanol and triethylamine, subsequent acetonide deprotection (80% aq TFA) and ion-exchange to the sodium salt afforded 30.
Scheme 5a.
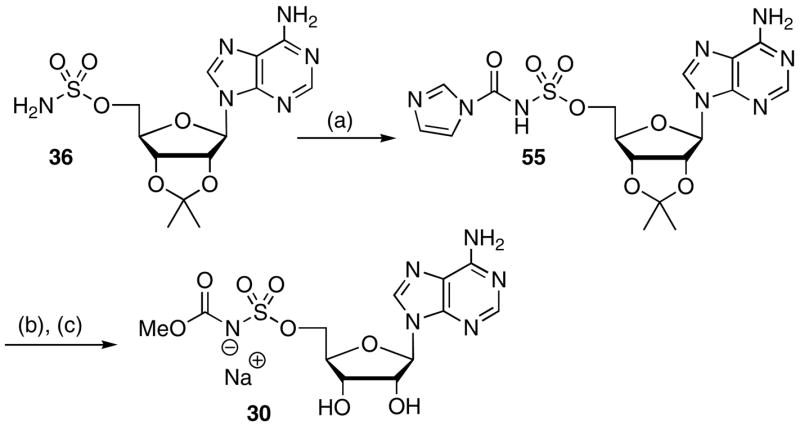
aReaction conditions: (a) MeCN, CDI; (b) MeOH, Et3N; (c) 80% aq TFA; (d) ion-exchange, 64% overall.
Bicyclic chromone 58 was prepared as both structural and modeling studies suggested that the ortho-hydroxy group of 6 internally hydrogen bonds with the sulfamate nitrogen (Figure 4).12, 21 This analogue simultaneously explores modifications to both the aryl and linker domains of the inhibitor scaffold and was synthesized from 2′,3′-O-isopropylideneadenosine 53 by interconversion of the 5′-alcohol to an azide, Boc protection of the N6-amine of adenine, and hydrogenation to afford 56 (Scheme 6).22 CDI mediated coupling of chromone-3-carboxylic acid provided 57 that was deprotected with aqueous TFA to furnish 58.
Scheme 6a.
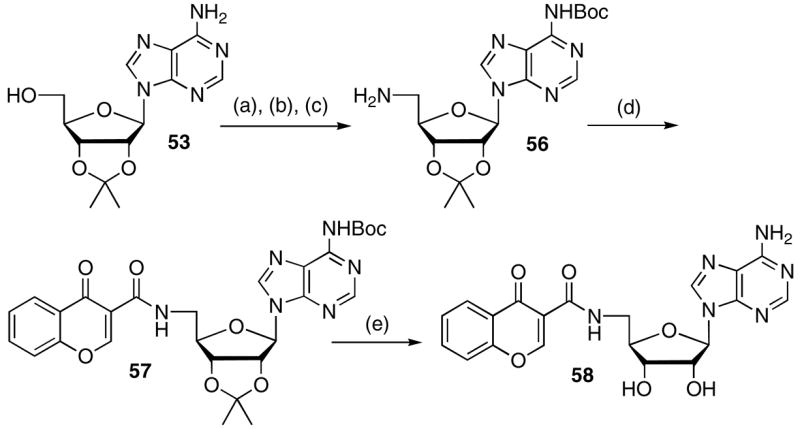
aReaction conditions: (a) ref. 21, 92%; (b) Boc2O, NaH, DMF/THF (4:1), 16 h, 76%; (c) H2 (1 atm), Pd/C, MeOH, 95%; (d) i) CDI, DBU, DMF ii) chromone-3-carboxylic acid; (e) 80% aq TFA, 20% (2 steps).
Finally, acylurea analogue 63 was prepared in order to examine the importance of the sulfonyl group of the linker domain of 6 (Figure 2). Acylurea 63 was prepared from 2-benzyloxybenzamide 59 by treatment with oxalyl chloride to provide intermediate acylisocyanate 60 (Scheme 7). After complete removal of the solvent and excess oxalyl chloride, a solution of 5-aminoadenosine derivative 56 in acetonitrile was added to provide 61, which was sequentially deprotected with aqueous TFA to 62 and hydrogenated to afford acylurea 63.
Scheme 7a.
aReaction conditions: (a) (COCl)2, CH2Cl2, 50 °C, 3 h; (b) 52, MeCN, 16 h, 88%; (c) H2, Pd/C, 51%; (d) 80% aq TFA, 70%.
Enzyme Assay
Enzyme assays were performed at 37 °C with recombinant MbtA expressed in E. coli in a buffer of 75 mM Tris-HCl, pH 7.5, 10 mM MgCl2, 2 mM DTT, 250 μM salicylic acid, 10 mM ATP, and 1 mM PPi.15 The initial rates of pyrophosphate exchange (≤ 10% reaction) were monitored using an enzyme concentration (typically 5–10 nM) by measuring the amount of [32P]ATP formed after addition of [32P]PPi. The enzyme concentration was determined by active-site titration with inhibitor 6. The apparent inhibition constants (Kiapp) were determined by fitting the concentration-response plots either to the Hill equation (eq 1, see Experimental Section) or to the Morrison equation (eq 2, see Experimental Section). The Morrison equation was employed in cases where the inhibitors exhibited tight-binding behavior (Kiapp ≤ 100·[E]). This class of inhibitors has been shown to exhibit reversible and competitive inhibition toward MbtA with respect to both salicylic acid and ATP.15 Attempts to determine the true inhibition constants (Kiapp) have been hampered by the tight-binding nature of the inhibitors, which has precluded assessment of the Ki by traditional steady-state kinetic methods.23 The Ki of the parent inhibitor 6 is 6.6 nM (measured at 250 μM salicylic acid, 10 mM ATP). If one accounts for the competitive substrate salicylic acid, which is held at 250 μM or 120·KM, then the Ki is 55 pM. However, this analysis still neglects the effect of the nonvaried substrate ATP, which is held at 10 mM, or 55·KM, thus the true Ki for 6 is estimated at approximately 1 pM, a value that has been independently confirmed by isothermal calorimetry (Daniel Wilson, Thomas Anderson, unpublished results). All of the Kiapp values reported are uncorrected for substrate concentrations and represent an upper limit of the true dissociation constant. Although the Kiapp reported herein are not a measure of the true inhibitor potency, the differences are reflective of free energy differences associated with inhibitor binding to MbtA presuming equivalent modalities of inhibition.
Aryl Ring SAR Study
To explore the significance of the ortho-hydroxy group, a systematic series of analogues was prepared bearing substitution at the 2-position. Several important trends emerged from this series. Deletion of the hydroxy afforded benzoyl analogue 7 and resulted in a concomitant 14-fold loss of binding affinity (Table 1). Substitution of the ortho-hydroxy group reduced potency in all cases but by widely varying amounts: by 6-fold for 2-fluoro 9, 117-fold for 2-amino 8, 1100 fold for 2-nitro 11, and 2700-fold for 2-chloro 10. Docking studies of the parent ligand 6 into a homology model of MbtA as well as quantum mechanics calculations free from the constraints of the active site show that 6 adopts a coplanar conformation that is stabilized by an internal hydrogen bond where the phenol acts as a hydrogen bond donor to the sulfamate nitrogen (pKa ~ 1), which is deprotonated (Figure 3).11 The X-ray cocrystal structure of DhbE with adenylated 2,3-dihydroxybenzoic acid also shows a similar coplanar arrangement.21 Molecular mechanics simulations of 10 and 11 show that the bulky nitro- and chloro-substituents disfavor the required coplanar conformation and additionally are unable to form an internal hydrogen bond. The 117-fold loss of activity when the aryl hydroxy is replaced by an amino group suggests that the carboxamide side chain of N235 in MbtA must present the amino group to the inhibitor (for ease of interpretation using the DhbE X-ray structure, residues are numbered as in DhbE). The 2-fluoro analog, whch notably lacks the metabolically labile phenol function, displayed the smallest destabilization of binding, consistent with its ability to adopt a coplanar arrangement in quantum calculations. Unfortunately, the bicyclic chromone analogue 58 that mimics the postulated planar conformation of 6 was inactive. The related acyclic acylurea derivative 63 exhibited a profound 329-fold loss in potency relative to acylsulfamide 64 (Table 2). Modeling suggests that twisting of the urea functionality of 63 is required for binding, partially explaining its decreased activity relative to 6. Cyclization as in 58 imposes even greater energy penalties for this twisting, which likely explains the further loss of potency of this compound.
Table 1.
 | ||||||||
|---|---|---|---|---|---|---|---|---|
| Compound, R = | KIapp (μM)a | MIC99 (μM)b (iron-deficient) | MIC99 (μM)c (iron-rich) | Compound, R = | KIapp (μM)a | MIC99 (μM)b (iron-deficient) | MIC99 (μM)c (iron-rich) | |
|
0.0066 ± 0.00015d | 0.39 | 1.56 |
|
||||
| 0.092 ± 0.007 | 12.5 | 50 | 2.9 ± 0.4 | > 200 | > 200 | |||
| 0.77 ± 0.12 | >200 | > 200 | 0.175 ± 0.019 | 12.5 | 50 | |||
| 0.038 ± 0.008 | 12.5 | 50 |
 26 26
|
|||||
| 18.1 ± 1.7 | >200 | > 200 | ||||||
| 7.32 ± 0.9 | >200 | > 200 | > 100 | > 200 | > 200 | |||
|
0.061 ± 0.003 | 50 | > 200 |
|
||||
| 0.012 ± 0.0006 | 12.5 | 50 | 49.8 ± 2.0 | > 200 | > 200 | |||
| 0.020 ± 0.0014 | 12.5 | 50 | 17.3 ± 2.8 | > 200 | > 200 | |||
| 0.012 ± 0.0016 | 0.098 | 0.39 |
 29 29
|
|||||
| 0.021 ± 0.001 | 6.25–12.5 | 50 | > 100 | > 200 | > 200 | |||
| 0.014 ± 0.0007 | 1.56 | 25 | ||||||
| 4.38 ± 0.44 | >200 | > 200 |
|
|||||
| 0.040 ± 0.004 | 1.56 | 25 | ||||||
| 0.0073 ± 0.0006 | 0.78 | 3.13 | 2.5 ± 0.4 | > 200 | > 200 | |||
 21 21
|
0.137 ± 0.012 | >200 | > 200 |
|
> 100 | > 200 | > 200 | |
 22 22
|
> 100 | >200 | > 200 |
 32 32
|
> 100 | > 200 | > 200 | |
 23 23
|
3.7 ± 0.5 | >200 | > 200 |
 33 33
|
36.5 ± 2.2 | > 200 | > 200 | |
Value ± std. error;
grown in GAST media without added Fe3+;
grown in GAST media supplemented with Fe3+;
see ref. 15.
Figure 3.
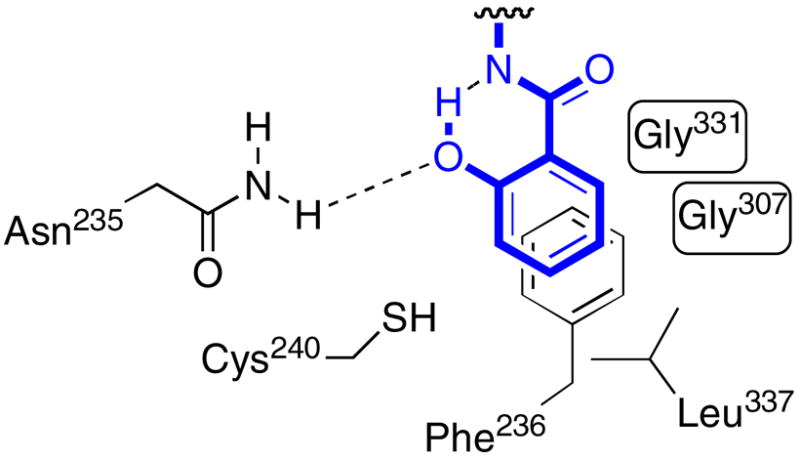
Schematic diagram of key nucleoside/protein interactions for analog 7 based on docking.
Table 2.
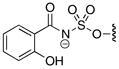 1
1
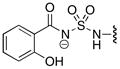 64
64
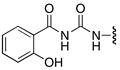 63
63
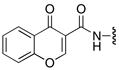 58
58
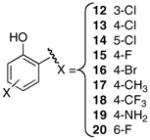
Analogues 12–14 were prepared to define the steric requirements of the shallow binding pocket and to identify potential sites for further modification. A homology model of MbtA reveals a shallow hydrophobic binding pocket for salicylic acid consisting of F236, C240, G306, V329, G331, and L337. The chlorine atom is isosteric with a methyl group and importantly can be modified by standard palladium-mediated coupling reactions. The SAR from this chloro-scan showed that substitution at the 4-position of the aryl ring was most tolerated resulting in a modest 2-fold decrease in potency relative to 6 whereas substitution at the 3- and 5-positions reduced inhibitor potency 3- and 9-fold respectively (Table 1). Since the optimal position was found to be the 4-position, several additional substituents (F, Br, CH3, CF3, and NH2) were examined at this position. None of the analogues in this series (15–19) exceeded the potency of 4-Cl 13 and strongly electron-withdrawing substituents were strongly disfavored. Thus, 4-CF3 18 exhibited a remarkable 313-fold loss of potency relative to the 4-CH3 17. Additionally, modeling suggested that a 6-substituent might be able to accept a poor-geometry hydrogen bond from the backbone amide of G307. The only 6-substituted compound in the series, 6-fluorosalicyl 20, showed activity nearly identical to the parent compound.
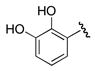
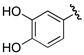
Since many siderophores contain an aryl-capped residue derived from 2,3-dihydroxybenzoic acid, inhibitor 21 was evaluated against MbtA and found to possess a 20-fold loss in potency relative to the parent compound 6. Bacillus subtilis uses the adenylating enzyme DhbE in the synthesis of the 2,3-dihydroxybenzoic acid-capped siderophore bacillibactin and compounds 6 and 21 were shown to possess KIapp values toward DhbE of 106 and 85 nM respectively; however, this analysis neglected the tight-binding nature of the inhibitors and thus underestimated both the inhibitor potency as well as selectivity.16, 24 MbtA shares 42% overall sequence identity to DhbE, but significantly higher homology in the ligand binding site. Of the 21 residues contacting the adenylate ligand in the X-ray cocrystal structure of DhbE (within 4 Å), 16 are identical in MbtA and the remaining 5 residues represent conservative changes (Y236F, S240C, A308S, V337L, T411S).21 In DhbE, the 3-hydroxy hydrogen bonds to S240, which is a cysteine in MbtA and therefore a weaker hydrogen-bonding partner. In addition, the nearby V337L substitution reduces the space available for the meta-hydroxyl. Therefore residues S240 and V337 probably contribute to the specificity differences between DhbE and MbtA. Bacillus anthracis incorporates native substrate 3,4-dihydroxybenzoic acid to make the siderophore petrobactin.25 Accordingly, compound 22 was prepared to probe the MbtA active site compatibility for 3,4-dihydroxybenzoic acid. 3,4-Dihydroxybenzoyl analogue 22 was a modest nanomolar inhibitor of AsbC (IC50 = 250 nM17), but displayed no activity toward MbtA indicating important active site difference between the two adenylation enzymes.
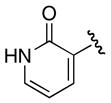
Incorporation of a nitrogen at the 3-position in pyridyl analogue 23–25 was explored because of the S240C and V337L substitutions. A nitrogen internal to the aryl ring would avoid any steric issues present with a 3-hydroxyl group, and molecular modeling of 24 and 25 showed that C240 can potentially donate a hydrogen bond to the 3-nitrogen. The carbon to nitrogen substitutions in 2-Cl pyridyl 25 provided an 103-fold increase in potency relative to 2-Cl phenyl 10, while 2-F pyridyl 24 decreased activity 76-fold relative to 2-F phenyl 9. Additionally, the 2-OH pyridyl 23 suffered a 560-fold loss in potency relative to 6, a result that can be partially reconciled since 23 exists exclusively as the keto tautomer. The lack of any apparent trend with 24 and 25 suggests that multiple effects are responsible for the observed activity.
In order to explore the tolerance for nonaromatic groups, a small set of alkyl derivatives 27–29 were prepared. The trend from this small series shows that larger alkyl substituents led to an increase activity. This series was not further explored systematically since the best compound 28 exhibited a 2600-fold loss in potency relative to 6. In docking studies, all low energy conformations of 28 showed unfavorable contacts between the cyclohexyl hydrogens and residues Phe236 and Gly331, which are on opposite faces of the aryl binding pocket, suggesting the pocket is not tall enough to accommodate saturated ring systems. However, methoxy analogue 30 was found to exhibit a greater than 40-fold increase in activity relative to methyl analogue 29 and this unexpected increase in activity warrants further investigation.
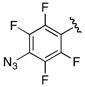
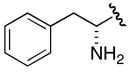
Incorporation of a methylene spacer between the linkage carbonyl and the phenyl group in benzyl analogue 31 led to complete loss of activity (>1000-fold decrease in potency relative to 7). 4-Azido-2,3,5,6-tetrafluorophenyl derivative 32 was prepared as a potential photoaffinity probe, but found to be completely inactive.26 Finally, the phenylalanine derived inhibitor 33 was evaluated to probe the selectivity of MbtA toward aminoacyl adenylate inhibitors. Surprisingly, compound 33 exhibited moderate micromolar activity suggesting that the amino group of 33 is able to favorably interact with MbtA.
Inhibition of M. tuberculosis H37Rv
Compounds 6–33, 58, and 63–64 were evaluated for whole-cell activity against M. tuberculosis H37Rv under iron-limiting and iron-rich conditions. The minimum inhibitory concentrations (MIC99) that inhibited >99% of cell growth are shown in tables 1–2. The MIC99 for 6–8 and 64 have been previously reported under iron-deficient conditions, but the MIC99 under iron-rich conditions have not yet been disclosed.11 In general, the MIC99 values were approximately 100-fold greater than the corresponding KIapp values. The strong correlation between in vitro enzyme inhibition and whole-cell biological activity provides support for the designed mechanism of action. 4-Fluorosalicyl derivative 15 displayed the most potent activity with an impressive MIC99 of 0.098 μM under iron-deficient conditions, which is 4-fold more potent that the parent compound 6. Although the 2-hydroxy group is required for optimal activity, the finding that this can be replaced with a fluoro in analogue 9 and still maintain respectable biological activity may be useful to increase the metabolic stability due to potential glucuronidation of the phenol. Interestingly, chromone derivative 58 displayed modest activity with an MIC99 of 12.5–25 μM under iron deficient conditions despite no apparent enzyme inhibition. However, it should be recognized that the inhibition studies were conducted under supersaturating concentrations of substrates, which underestimate compound potency by more than 1000-fold..
Another consideration is inhibitor selectivity since siderophore production is not essential under iron-rich conditions, suggesting that activity under iron-rich conditions is due to off-target binding and subsequent inhibition of alternate biochemical pathways.14 The parent inhibitor 6 has an MIC99 of 0.39 μM under iron deficient conditions and 1.56 μM under iron-rich conditions. The ratio of MIC99s of iron-rich/iron-deficient (MIC99Fe+/MIC99Fe−) is a measure of the inhibitor selectivity and the selectivity ratio for 6 is only 4. By contrast, the first-line antitubercular agent isoniazid, which disrupts cell-well biosynthesis, was found to be equipotent under iron-rich and iron-deficient conditions (MIC99Fe+ = MIC99Fe− = 0.19 μM). p-Aminosalicyl derivative 19 displayed improved selectivity showing a 16-fold increase in potency under iron-deficient conditions, suggesting that this modification reduced off-target binding. Compound 6 was previously shown to exhibit moderate activity against Yerinia pseudotuberculosis with an MIC99 of 20 μM and > 400 μM under iron–deficient and iron–rich conditions respectively.15 Bioinformatic analysis of M. tuberculosis and Y. pseudotuberculosis suggests that the observed phenotypic difference between these organisms elicited by 6 may be due to the presence of over thirty functionally related fatty acid adenylating (FAAD) enzymes found in M. tuberculosis, but not Y. pseudotuberculosis.27 Many of these FAAD enzymes catalyze essential reactions in the biosynthesis M. tuberculosis lipids and may represent potential off-target proteins inhibited by this class of adenylation inhibitors.28
Conclusion
A series of 5′-O-[(N-acyl)sulfamoyl]adenosines was prepared and evaluated for inhibition of MbtA and activity against whole-cell M. tuberculosis under iron-deficient and iron-rich conditions. A comprehensive and systematic evaluation of the acyl group revealed that a benzoyl substituent is required for antitubercular activity and potent enzyme inhibition. Modifications of the benzoyl group indicated that a hydroxy or fluoro group at C-2 is necessary, and that small substituents at C-4 are tolerated. 4-Fluorosalicyl 15 was found to exhibit the most antitubercular activity while p-aminosalicyl 19 was the first analogue that exhibited improved selectivity against M. tuberculosis under iron-rich conditions. Overall, the collective SAR demonstrated that the aryl domain is poorly tolerant to modification in accord with the fairly strict specificity of NRPS adenylation enzymes; however minor modifications were found to provide incremental enhancement in both potency and selectivity of this new class of adenylation inhibitors.
Experimental Section
General Procedures
All commercial reagents (Sigma-Aldrich, Acros), 4-bromosalicylic acid (ABCR), 4-trifluormethylsalicylic acid (TCI America) were used as provided unless otherwise indicated. An anhydrous solvent dispensing system (J. C. Meyer) using two packed columns of neutral alumina was used for drying THF, Et2O, and CH2Cl2 while two packed columns of molecular sieves were used to dry DMF and the solvents were dispensed under argon. Anhydrous grade DME, MeOH, and MeCN were purchased from Aldrich. Pyridine was freshly distilled from KOH, Et3N was distilled from CaH2. Flash chromatography was performed with Silia P grade silica gel 60 (Silicycle) with the indicated solvent system. All reactions were performed under an inert atmosphere of dry Ar or N2 in oven-dried (150 °C) glassware. 1H and 13C NMR spectra were recorded on a Varian 600 MHz spectrometer. Proton chemical shifts are reported in ppm from an internal standard of residual chloroform (7.26 ppm) or methanol (3.31 ppm), and carbon chemical shifts are reported using an internal standard of residual chloroform (77.3 ppm) or methanol (49.1 ppm). Proton chemical data are reported as follows: chemical shift, multiplicity (s = singlet, d = doublet, t = triplet, q = quartet, p = pentet, m = multiplet, br = broad), coupling constant, integration. High resolution mass spectra were obtained on Agilent TOF II TOF/MS instrument equipped with either an ESI or APCI interface. High resolution mass spectra were obtained on Agilent TOF II TOF/MS instrument equipped with either an ESI or APCI interface. Analytical HPLC were obtained on a Agilent 1100 Series HPLC system with a PDA detector. Optical rotations were measured on Rudolph Autopol III polarimeter. Melting points were measured on electrothermal Mel-Temp manual melting point apparatus and are uncorrected.
General Procedure for CDI Mediated Acylation.14, 18
A solution of 34a–j (3.0 mmol, 3.0 equiv) and CDI (3.6 mmol, 3.6 equiv) in DMF (10 mL) was stirred at 60 °C for 2 h. The solution was cooled to rt and a mixture of either 3611 or 3714 (1.0 mmol, 1.0 equiv) and either DBU or Cs2CO3 (1.5 mmol, 1.5 equiv) was then added and the reaction stirred a further 3 h at rt. The reaction mixture was concentrated in vacuo and purified by flash chromatography to afford the title compound.
General Procedure for Synthesis of NHS Esters
Method A: To a solution of carboxylic acid 40a–e and 40j-k, (1.0 mmol, 1.0 equiv) in THF (10 mL) at 0 °C was added N-hydroxysuccinimide (1.0 mmol, 1.0 equiv) and DCC (1.0 mmol, 1.0 equiv). The resulting mixture was stirred for 30 min at 0 °C then 2 h at rt. The reaction mixture was filtered to remove the DCU precipitate and the filtrate was concentrated under reduced pressure. Purification by flash chromatography afforded the desired N-hydroxysuccinimdyl aroyl ester. Method B: To a solution of acid chloride/anhydride 40f–i (1.0 mmol, 1.0 equiv) in THF (10 mL) at 0 °C was added N-hydroxysuccinimide (1.0 mmol, 1.0 equiv) and pyridine (1.0 mmol, 1.0 equiv). The resulting mixture was stirred for 30 min at 0 °C then 2 h at rt. The reaction mixture was filtered and the filtrate was concentrated under reduced pressure. The product was used without further purification.
General Procedure for NHS-Ester Mediated Acylation.11, 18
To a solution of N-hydroxysuccinimdyl ester 41a–k (1.0 mmol) in DMF (10 mL) at 0 °C was added 36 or 37 (1.5 mmol, 1.5 equiv) and DBU or Cs2CO3 (2.0 mmol, 2.0 equiv). The reaction mixture was warmed to rt and stirred 16 h. The reaction was concentrated under reduced pressure and the residue taken up in EtOAc and filtered. The solids were washed with additional EtOAc (100 mL) and the combined filtrate was concentrated. Purification by flash chromatography (EtOAc/MeOH/Et3N) afforded the title compound.
General Procedure for TFA deprotection
To a solution of 5′-O-[N-acyl(sulfamoyl)]-2′,3′-O-isopropylideneadenosine triethylammonium salt (0.2 mmol) was added 80% aq TFA (2.5 mL). The resulting solution was stirred for 30 min at 0 °C then concentrated under reduced pressure. Purification by flash chromatography (EtOAc/MeOH/Et3N) afforded the title compound.
General Procedure for TBS deprotection
To a solution of 5′-O-[N-acyl(sulfamoyl)]-2′,3′-O-di-(tert-butyldimethylsilyl)adenosine triethylammonium salt (0.1 mmol, 1 equiv) in THF (5 mL) at rt was added TBAF (1.0 M in THF, 0.25 mmol, 2.5 equiv). The reaction mixture was stirred 30 min then concentrated in vacuo. Purification by flash chromatography (EtOAc/MeOH/Et3N) afforded the title compound.
General Procedure for Ion-Exchange
A solution of a nucleoside tetrabutylammonium or triethylammonium salt (0.25 mmol, 1 equiv) in H2O (0.5 mL) was added to a short column (10 × 50 mm, Dowex 50WX8-100-Na+) and incubated for 10 min before eluting with H2O (20 mL). The fractions containing the product were lyophilized to afford the sodium salt as a flocculant white solid. The Dowex cation exchange resin was converted to the sodium form by sequentially washing the column with MeOH (50 mL), H2O (50 mL), 1 N aq NaOH (25 mL), and H2O (100 mL).
5′-O-[N-(2-Aminobenzoyl)sulfamoyl]-2′,3′-O-isopropylideneadenosine triethylammonium salt
To a solution of isatoic anhydride (47 mg, 0.284 mmol, 1.1 equiv) in DMF (1.0 mL) at rt was added 36 (100 mg, 0.26 mmol, 1.0 equiv) and Cs2CO3 (169 mg, 0.52 mmol, 2.0 equiv). The reaction was stirred 12 h at rt then concentrated in vacuo. Purification by flash chromatography (100:25:1 EtOAc/MeOH/TEA) afforded the title compound (135 mg, 86%). 1H NMR, 13C NMR, and HRMS agreed with literature values.11 This compound was deprotected as described.11
N-Hydroxysuccinimdyl 2-chlorobenzoate (41a)
This was prepared from 2-chlorobenzoic acid (780 mg, 5.0 mmol) using the general procedure for NHS ester synthesis. Purification by flash chromatography (1:1 Hexanes/EtOAc) afforded the title compound (880 mg, 70%): Rf 0.5 (1:1 Hexanes/EtOAc); 1H NMR (600 MHz, CDCl3) δ 2.91 (s, 4H), 7.39 (ddd, J = 7.8, 6.0, 1.8 Hz, 1H), 7.53–7.57 (m, 2H), 8.10 (dd, J = 7.8, 1.8 Hz, 1H); 13C NMR (150 MHz, CDCl3) δ 25.9, 124.8, 127.1, 131.8, 132.6, 134.8, 135.8, 160.4, 169.2; MS (ESI+) calcd for C11H9ClNO4 [M+H]+ 254.0, found 254.0.
5′-O-[N-(2-Chlorobenzoyl)sulfamoyl]-2′,3′-O-isopropylideneadenosine (42a)
This was prepared from 41a (126 mg, 0.50 mmol) using the general procedure for NHS-ester mediated acylation. Purification by flash chromatography (100:10:1 EtOAc/MeOH/TEA) afforded the title compound (58 mg, 22%): Rf 0.2 (100:10:1 EtOAc/MeOH/TEA); [α]D21 − 102 (c 2.79, MeOH); 1H NMR (600 MHz, CD3OD) δ 1.36 (s, 3H), 1.59 (s, 3H), 4.35 (d, J = 1.8 Hz, 2H), 4.58 (d, J = 1.8 Hz, 1H), 5.20 (d, J = 6.0 Hz, 1H), 5.37 (dd, J = 6.0, 3.0 Hz, 1H), 6.24 (d, J = 3.0 Hz, 1H), 7.24 (t, J = 7.2 Hz, 1H), 7.28 (t, J = 7.2 Hz, 1H), 7.33 (d, J = 7.8 Hz, 1H), 7.46 (d, J = 7.8 Hz, 1H), 8.18 (s, 1H), 8.46 (s, 1H); 13C NMR (150 MHz, CD3OD) δ 24.4, 26.3, 68.9, 82.1, 84.5, 84.6, 90.7, 114.1, 118.9, 126.4, 128.9, 129.6, 129.8, 130.7, 139.5, 140.3, 149.3, 152.8, 156.1, 174.7; HRMS (APCI+) calcd for C20H22ClN6O7S [M+H]+ 525.0959, found 525.0953 (error 1.1 ppm).
5′-O-[N-(2-Chlorobenzoyl)sulfamoyl]adenosine (10)
This was prepared from 42a (29 mg, 0.050 mmol) using the general procedure for TFA deprotection. Purification by flash chromatography afforded the title compound (22 mg, 93%): Rf 0.18 (100:15:2 MeOH/EtOAc/TEA); [α]D21 −18.7 (c 0.83, MeOH); 1H NMR (600 MHz, CD3OD) δ 4.37 (br s, 1H), 4.49 (t, J = 4.8 Hz, 1H), 4.62–4.70 (m, 3H), 6.11 (d, J = 4.8 Hz, 1H), 7.34–7.37 (m, 1H), 7.44–7.47 (m, 3H), 8.36 (s, 1H), 8.51 (s, 1H); 13C NMR (150 MHz, CD3OD) δ 70.3, 71.5, 74.5, 82.4, 89.3, 119.3, 127.1, 128.9, 130.2, 130.9, 132.4, 133.5, 142.7, 144.3, 148.9, 150.8, 165.7; HRMS (ESI+) calcd for C17H18ClN6O7S [M+H]+ 485.0641, found 485.0649 (error 0.2 ppm).
N-Hydroxysuccinimdyl 2-nitrobenzoate (41b)
This was prepared from 2-nitrobenzoic acid (835 mg, 5.0 mmol) using the general procedure for NHS ester synthesis. Purification by flash chromatography (6:4 Hexanes/EtOAc) afforded the title compound (1.10 g, 85%): Rf 0.2 (6:4 Hexanes/EtOAc); 1H NMR (600 MHz, CDCl3) δ 2.89 (s, 4H), 7.76–7.79 (m, 2H), 7.92–7.95 (m, 1H), 8.06–8.08 (m, 1H); 13C NMR (150 MHz, CDCl3) δ 25.9, 123.2, 124.8, 130.9, 133.6, 133.7, 147.9, 161.2, 168.7; MS (ESI+) calcd for C11H9N2O6 [M+H]+ 265.0, found 264.9.
2′,3′-O-Isopropylidene-5′-O-[N-(2-nitrobenzoyl)sulfamoyl]adenosine triethylammonium salt (42b)
This was prepared from 41b (133 mg, 0.50 mmol) using the general procedure for NHS-ester mediated acylation. Purification by flash chromatography (100:10:1 EtOAc/MeOH/TEA) afforded the title compound (75 mg, 28%): Rf 0.17 (100:10:1 EtOAc/MeOH/TEA); [α]D21 −142 (c 2.38, MeOH); 1H NMR (600 MHz, CD3OD) δ 1.23 (t, J = 7.2 Hz, 9H), 1.36 (s, 3H), 1.59 (s, 3H), 3.13 (q, J = 7.2 Hz, 6H), 4.32–4.35 (m, 2H), 4.58 (br s, 1H), 5.18 (d, J = 6.0, 1H), 5.37–5.39 (m, 1H), 6.23 (d, J = 2.4, 1H), 7.52 (t, J = 7.2 Hz, 1H), 7.62 (t, J = 8.4 Hz, 1H), 7.66 (d, J = 7.2 Hz, 1H), 7.82 (d, J = 8.4 Hz, 1H), 8.18 (s, 1H), 8.45 (s, 1H); 13C NMR (150 MHz, CD3OD) δ 8.0, 24.4, 26.3, 46.7, 68.8, 82.2, 84.5, 84.6, 90.7, 114.1, 118.9, 123.4, 129.3, 129.7, 132.6, 135.5, 140.2, 148.1, 149.3, 152.8, 156.1, 172.6; HRMS (ESI+) calcd for C20H22N7O9S [M+H]+ 536.1199, found 536.1201 (error 0.4 ppm).
5′-O-[N-(2-Nitrobenzoyl)sulfamoyl]adenosine triethylammonium salt (11)
This was prepared from 42b (55 mg, 1.0 mmol) using the general procedure for TFA deprotection. Purification by flash chromatography (100:25:2.5 EtOAc/MeOH/TEA) afforded the title compound (11 mg, 22%): Rf 0.15 (100:25:2.5 EtOAc/MeOH/TEA); [α]D21 −99.7 (c 0.290, MeOH); 1H NMR (600 MHz, CD3OD) δ 1.24 (t, J = 7.8 Hz, 9H), 3.14 (q, J = 7.8 Hz, 6H), 4.31–4.34 (m, 1H), 4.40 (t, J = 3.6 Hz, 2H), 4.45 (dd, J = 3.6 Hz, 1H), 4.72 (t, J = 6.0 Hz, 1H), 6.09 (d, J = 6.0 Hz, 1H), 7.52 (t, J = 8.4, 1H), 7.61 (t, J = 7.2 Hz, 1H), 7.68 (d, J = 7.2 Hz, 1H), 7.83 (d, J = 8.4 Hz, 1H), 8.18 (s, 1H), 8.49 (s, 1H); 13C NMR (150 MHz, CD3OD) δ 8.0, 46.7, 68.5, 71.3, 74.9, 83.5, 87.9, 118.9, 123.4, 129.3, 129.5, 132.7, 135.7, 139.8, 147.9, 149.7, 152.7, 156.1, 172.7; HRMS (APCI+) calcd for C17H18N7O9S [M+H]+ 496.0876, found 496.0904 (error 5.6 ppm).
5′-O-[N-(3-Chloro-2-hydroxybenzoyl)sulfamoyl]-2′,3′-O-isopropylideneadenosine (38a)
This was prepared from 3-chlorosalicylic acid (170 mg, 1.0 mmol) using the general procedure for CDI mediated acylation. Purification by flash chromatography (100:5:1 EtOAc/MeOH/TEA) afforded the title compound (83 mg, 15%): Rf 0.15 (100:5:1 EtOAc/MeOH/TEA); [α]D21 −32.3 (c 2.00, MeOH); 1H NMR (600 MHz, CD3OD) δ 1.31 (s, 3H), 1.55 (s, 3H), 4.32–4.34 (m, 2H), 4.54 (br s, 1H), 5.09–5.11 (m, 1H), 5.34 (dd, J = 6.0, 3.0 Hz, 1H), 6.20 (d, J = 3.0 Hz, 1H), 6.71 (t, J = 7.8 Hz, 1H), 7.35 (d, J = 7.8 Hz, 1H), 7.83 (d, J = 7.8 Hz, 1H), 8.14 (s, 1H), 8.39 (s, 1H); 13C NMR (150 MHz, CD3OD) δ 24.3, 26.2, 68.9, 82.1, 84.5, 84.6, 90.8, 114.1, 118.0, 119.0, 120.8, 121.3, 128.7, 133.3, 140.2, 149.2, 152.7, 156.1, 156.8, 172.9; HRMS (ESI+) calcd for C20H22ClN6O8S [M+H]+ 541.0903, found 541.0903 (error 0 ppm).
5′-O-[N-(3-Chloro-2-hydroxybenzoyl)sulfamoyl]adenosine triethylammonium salt (12)
This was prepared form 38a (30 mg, 0.060 mmol) using the general procedure for TFA deprotection. Purification by flash chromatography (100:15:1 EtOAc/MeOH/TEA) afforded the title compound (20 mg, 65%): Rf 0.17 (100:15:1 EtOAc/MeOH/TEA); [α]D21 −32.8 (c 0.570, MeOH); 1H NMR (600 MHz, CD3OD) δ 1.28 (t, J = 7.2 Hz, 9H), 3.18 (q, J = 7.2 Hz, 6H), 4.34–4.35 (m, 1H), 4.39–4.46 (m, 3H), 4.74 (t, J = 5.4 Hz, 1H), 6.11 (d, J = 6.0 Hz, 1H), 6.76 (t, J = 7.8 Hz, 1H), 7.40 (dd, J = 7.8, 1.2 Hz, 1H), 7.91 (dd, J = 7.8, 1.2 Hz, 1H), 8.20 (s, 1H), 8.53 (s, 1H); 13C NMR (150 MHz, CD3OD) δ 8.1, 46.6, 68.5, 71.1, 74.8, 83.3, 87.9, 117.9, 118.9, 120.9, 121.3, 128.7, 133.1, 139.8, 149.6, 152.6, 156.0, 156.7, 172.6; HRMS (ESI+) calcd for C17H18ClN6O8S [M+H]+ 501.0595, found 501.0585 (error 2.0 ppm).
5′-O-[N-(4-Chloro-2-hydroxybenzoyl)sulfamoyl]-2′,3′-O-isopropylideneadenosine triethylammonium salt (38b)
This was prepared from 4-chlorosalicylic acid (43 mg, 0.25 mmol) using the general procedure for CDI mediated acylation. Purification by flash chromatography (100:10:1 EtOAc/MeOH/TEA) afforded the title compound (94 mg, 70%): Rf 0.15 (100:10:1 EtOAc/MeOH/TEA); [α]D21 −88.4 (c 1.86, MeOH); 1H NMR (600 MHz, CD3OD) δ 1.24 (t, J = 7.8 Hz, 9H), 1.34 (s, 3H), 1.58 (s, 3H), 3.09 (q, J = 7.8 Hz, 6H), 4.29 (dd, J = 10.8, 4.2 Hz, 1H), 4.32 (dd, J = 10.8, 3.6 Hz, 1H), 4.53–4.54 (m, 1H), 5.11 (dd, J = 6.0, 1.8 Hz, 1H), 5.37 (dd, J = 6.0, 3.0 Hz, 1H), 6.21 (d, J = 3.0 Hz, 1H), 6.72–6.80 (m, 2H), 7.83 (d, J = 9.0 Hz, 1H), 8.14 (s, 1H), 8.41 (s, 1H); 13C NMR (150 MHz, CD3OD) δ 8.3, 24.2, 26.2, 46.6, 68.8, 82.2, 84.51, 84.53, 90.8, 114.1, 115.9, 116.6, 118.3, 118.4, 131.6, 138.3, 140.2, 149.2, 152.8, 156.1, 161.7, 172.6; MS (ESI+) calcd for C20H22ClN6O8S [M+H]+ 541.09, found 541.08.
5′-O-[N-(4-Chloro-2-hydroxybenzoyl)sulfamoyl]adenosine triethylammonium salt (13)
This was prepared from 38b (35 mg, 0.060 mmol) using the general procedure for TFA deprotection. Purification by flash chromatography (100:15:1 EtOAc/MeOH/TEA) afforded the title compound (19 mg, 60%): Rf 0.14 (100:15:1 EtOAc/MeOH/TEA); [α]D21 −23.3 (c 1.81, MeOH); 1H NMR (600 MHz, CD3OD) δ 1.24 (t, J = 7.8 Hz, 9H), 3.13 (q, J = 7.8 Hz, 6H), 4.30 (dd, J = 6.6, 3.0 Hz, 1H), 4.34–4.42 (m, 3H), 4.70 (t, J = 5.4 Hz, 1H), 6.07 (d, J = 5.4 Hz, 1H), 6.74 (dd, J = 8.4, 1.2 Hz, 1H), 6.79 (d, J = 1.2 Hz, 1H), 7.88 (d, J = 8.4 Hz, 1H), 8.16 (s, 1H), 8.47 (s, 1H); 13C NMR (150 MHz, CD3OD) δ 8.1, 46.7, 60.3 68.5, 71.2, 74.8, 83.3, 88.1, 116.6, 118.3, 119.0, 131.5, 138.3, 139.9, 149.7, 152.7, 156.1, 161.7, 172.6; HRMS (ESI+) calcd for C17H18ClN6O8S [M+H]+ 501.0595, found 501.0592 (error 0.6 ppm).
5′-O-[N-(5-Chloro-2-hydroxybenzoyl)sulfamoyl]-2′,3′-O-isopropylideneadenosine (38c)
This was prepared from 5-chlorosalicylic acid (86 mg, 0.50 mmol) using the general procedure for CDI mediated acylation. Purification by flash chromatography (100:5:1.5 EtOAc/MeOH/TEA) afforded the title compound (33 mg, 12%): Rf 0.18 (100:5:1.5 EtOAc/MeOH/TEA); [α]D21 −96.0 (c 1.26, MeOH); 1H NMR (600 MHz, CD3OD) δ 1.33 (s, 3H), 1.56 (s, 3H), 4.30–4.33 (m, 2H), 4.53 (br s, 1H), 5.10 (d, J = 6.0 Hz, 1H), 5.35 (dd, J = 6.0, 2.4 Hz, 1H), 6.19 (d, J = 2.4 Hz, 1H), 6.75 (d, J = 8.4 Hz, 1H), 7.22 (dd, J = 8.4, 2.4 Hz, 1H), 7.84 (d, J = 2.4 Hz, 1H), 8.13 (s, 1H), 8.39 (s, 1H); 13C NMR (150 MHz, CD3OD) δ 24.2, 26.2, 60.3, 68.9, 82.1, 84.5, 84.6, 90.8, 104.7, 114.1, 118.5, 120.5, 122.7, 129.3, 132.8, 140.2, 152.7, 156.0, 159.5, 172.4; HRMS (ESI+) calcd for C20H22ClN6O8S [M+H]+ 541.0903, found 541.0914 (error 2.0 ppm).
5′-O-[N-(5-Chloro-2-hydroxybenzoyl)sulfamoyl]adenosine triethylammonium salt (14)
This was prepared from 38c (28 mg, 0.050 mmol) using the general procedure for TFA deprotection. Purification by flash chromatography (100:20:1.5 EtOAc/MeOH/TEA) afforded the title compound (15 mg, 59%): Rf 0.15 (100:20:1.5 EtOAc/MeOH/TEA); [α]D21 −23.3 (c 1.72, MeOH); 1H NMR (600 MHz, CD3OD) δ 1.17 (t, J = 7.2 Hz, 9H), 2.90 (q, J = 7.2 Hz, 6H), 4.31–4.42 (m, 4H), 4.68 (t, J = 5.4 Hz, 1H), 6.06 (d, J = 5.4 Hz, 1H), 6.76 (d, J = 8.4 Hz, 1H), 7.22 (d, J = 8.4 Hz, 1H), 7.87 (s, 1H), 8.15 (s, 1H), 8.44 (s, 1H); 13C NMR (150 MHz, CD3OD) δ 8.8, 46.3, 60.4, 68.5, 71.1, 74.8, 83.2, 88.3, 116.8, 118.0, 118.4, 120.6, 129.4, 132.8, 139.9, 152.7, 156.1, 159.5, 172.4; HRMS (ESI+) calcd for C17H18ClN6O8S [M+H]+ 501.0595, found 501.0594 (error 0.2 ppm).
5′-O-[N-(4-Fluoro-2-hydroxybenzoyl)sulfamoyl]adenosine sodium salt (15)
This was prepared from 4-fluorosalicylic acid (16 mg, 0.10 mmol) using the general procedure for CDI mediated acylation to afford 38d. The crude product was used directly for the next step.
Crude 38d prepared above was deprotected using the general procedure for TFA deprotection. Purification by flash chromatography (80:20:1 EtOAc/MeOH/TEA) followed by conversion to the sodium salt using the general procedure for ion-exchange afforded the title compound (25 mg, 48% overall yield): Rf 0.21 (8:2 EtOAc/MeOH); [α]D21 −0.05 (c 0.33, CH3OH); 1H NMR (600 MHz, CD3OD) δ 4.29–4.31 (m, 1H), 4.35 (dd, J = 10.8, 3.0 Hz, 1H), 4.38–4.40 (m, 2H), 4.69 (t, J = 5.4 Hz, 1H), 6.07 (d, J = 6.0 Hz, 1H), 6.47–6.50 (m, 2H), 7.94 (t, J = 7.8 Hz, 1H), 8.15 (s, 1H), 8.49 (s, 1H); 13C NMR (150 MHz, CD3OD) δ 69.5, 72.4, 76.0, 84.6, 89.2, 104.1 (d, 2JC-F = 23.4), 106.5 (d, 2JC-F = 21.8), 120.1, 133.44, 133.51, 141.1, 150.9, 153.8, 157.2, 164.1, 167.2 (d, JC-F = 247.8), 174.0; HRMS (ESI−) calcd for C17H16FN6O8S [M−H]− 483.0739, found 483.0758 (error 3.9 ppm).
5′-O-[N-(4-Bromo-2-hydroxybenzoyl)sulfamoyl]adenosine sodium salt (16)
This was prepared from 4-bromosalicylic acid (60 mg, 0.28 mmol) using the general procedure for CDI mediated acylation to afford 38e. The crude product was used directly for the next step.
Crude 38e prepared above was deprotected using the general procedure for TFA deprotection. Purification by flash chromatography (80:20:1 EtOAc/MeOH/TEA) followed by conversion to the sodium salt using the general procedure for ion-exchange afforded the title compound (116 mg, 74% overall yield): Rf 0.23 (8:2 EtOAc/MeOH); [α]D21 −0.13 (c 0.53, CH3OH); 1H NMR (600 MHz, CD3OD) δ 4.31–4.32 (m, 1H), 4.35–4.37 (m, 1H), 4.40–4.42 (m, 2H), 4.70 (t, J = 5.4 Hz, 1H), 6.08 (d, J = 6.0 Hz, 1H), 6.91 (dd, J = 7.8 Hz, 1.8 Hz, 1H), 6.97 (d, J = 1.8, 1H), 7.81 (d, J = 7.8 Hz, 1H), 8.17 (s, 1H), 8.50 (s, 1H); 13C NMR (150 MHz, CD3OD) δ 69.7, 72.4, 76.1, 84.6, 89.3, 119.9, 120.2, 120.9, 122.5, 127.8, 132.8, 141.1, 150.9, 153.94, 157.3, 162.9, 174.1; HRMS (ESI−) calcd for C17H16BrN6O8S [M−H]− 542.9939, found 542.9945 (error 1.1 ppm).
5′-O-[N-(2-Hydroxy-4-methylbenzoyl)sulfamoyl]adenosine sodium salt (17)
This was prepared from 4-methylsalicylic acid (76 mg, 0.50 mmol) using the general procedure for CDI mediated acylation to afford 38f. The crude product was used directly for the next step.
Crude 38f prepared above was deprotected using the general procedure for TFA deprotection. Purification by flash chromatography (80:20:1 EtOAc/MeOH/TEA) followed by conversion to the sodium salt using the general procedure for ion-exchange afforded the title compound (113 mg, 45% overall yield): Rf 0.20 (8:2 EtOAc/MeOH); [α]D21 −0.15 (c 0.56, CH3OH); 1H NMR (600 MHz, CD3OD) δ 2.27 (s, 3H), 4.31–4.32 (m, 1H), 4.34–4.37 (m, 1H), 4.39–4.42 (m, 2H), 4.71 (t, J = 5.4 Hz, 1H), 6.08 (d, J = 6.0 Hz, 1H), 6.59–6.61 (m, 2H), 7.81 (d, J = 7.8 Hz, 1H), 8.17 (s, 1H), 8.51 (s, 1H); 13C NMR (150 MHz, CD3OD) δ 20.7, 68.6, 71.5, 75.2, 83.7, 88.3, 117.1, 119.3, 119.5, 130.4, 140.2, 144.4, 150.0, 152.9, 156.4, 161.1, 174.4 (missing 1 C); HRMS (ESI−) calcd for C18H19N6O8S [M−H]− 479.0990, found 479.1009 (error 4.0 ppm).
5′-O–[N-(4-Trifluoromethyl-2-hydroxybenzoyl)sulfamoyl]adenosine sodium salt (18)
This was prepared from 4-trifluoromethylsalicylic acid (20.6 mg, 0.10 mmol) using the general procedure for CDI mediated acylation to afford 38g. The crude product was used directly for the next step.
Crude 38g prepared above was deprotected using the general procedure for TFA deprotection. Purification by flash chromatography (80:20:1 EtOAc/MeOH/TEA) followed by conversion to the sodium salt using the general procedure for ion-exchange afforded the title compound (27 mg, 48% overall yield): Rf 0.25 (8:2 EtOAc/MeOH); [α]D21 −0.04 (c 0.25, CH3OH); 1H NMR (600 MHz, CD3OD) δ 4.30–4.33 (m, 1H), 4.37–4.45 (m, 3H), 4.70 (t, J = 5.4 Hz, 1H), 6.09 (d, J = 6.0 Hz, 1H), 7.02 (d, J = 7.8 Hz, 1H), 7.04 (s, 1H), 8.08 (d, J = 7.8 Hz, 1H), 8.17 (s, 1H), 8.51 (s, 1H); 13C NMR (150 MHz, CD3OD) δ 68.8, 71.5, 75.2, 83.7, 88.3, 114.0, 114.3, 119.3, 123.0, 131.4, 136.3, 140.2, 150.0, 153.0, 156.4, 161.3, 172.5 (missing 1 aryl C and CF3); HRMS (ESI−) calcd for C18H16F3N6O8S [M−H]− 533.0707, found 533.0714 (error 1.1 ppm).
4-Benzyloxycarbonylamino-2-hydroxybenzoic acid (34h)
To a solution of 4-aminosalicylic acid (2.0 g, 13.1 mmol) in THF (40 mL) was added saturated aq NaHCO3 (40 mL) followed by benzyl chloroformate (2.04 mL, 14.4 mmol, 1.1 equiv). The reaction was stirred for 16 h at rt then concentrated to remove THF. The remaining aqueous layer was acidified with 6 N HCl to pH 3–4 then extracted with EtOAc (3× 50 mL). The combined organic layers were dried (Na2SO4) and concentrated to afford the title compound (2.30 g, 61%) as a light brown solid: 1H NMR (600 MHz, CD3OD) δ 5.17 (s, 2H), 6.91 (dd, J = 9.0, 1.8 Hz, 1H), 7.14 (d, J = 1.8 Hz, 1H), 7.29–7.39 (m, 5H), 7.72 (d, J = 9.0 Hz, 1H), 9.57 (s, 1H); 13C NMR (150 MHz, CD3OD) δ 66.6, 105.2, 107.2, 109.3, 127.9, 128.0, 128.3, 131.1, 136.6, 145.9, 154.0, 163.1, 172.0; HRMS (ESI−) calcd for C15H12NO5 [M−H]− 286.0721, found 286.0727 (error 2.1 ppm)
5′-O-[N-(4-Benzyloxycarbonylamino-2-hydroxybenzoyl)sulfamoyl]-2′,3′-O-isopropylideneadenosine (38h)
This was prepared from 34h (71 mg, 0.25 mmol) using the general procedure for CDI mediated acylation. Purification by flash chromatography (100:7 MeOH/EtOAc) afforded the title compound (75 mg, 38%) as a white solid: Rf 0.20 (100:7 MeOH/EtOAc); [α]D21 86.1 (c 1.29, CH3OH); 1H NMR (600 MHz, CD3OD) δ 1.32 (s, 3H), 1.56 (s, 3H), 4.30 (t, J = 4.2 Hz, 2H), 4.54 (br s, 1H), 5.10 (d, J = 5.4 Hz, 1H), 5.15 (s, 2H), 5.35 (br s, 1H), 6.20 (d, J = 3.0 Hz, 1H), 6.81 (d, J = 8.4 Hz, 1H), 7.28 (t, J = 7.2 Hz, 1H), 7.34 (t, J = 7.2 Hz, 2H), 7.38 (d, J = 7.2 Hz, 2H), 7.66 (s, 1H), 7.80 (d, J = 8.4 Hz, 1H), 8.13 (s, 1H), 8.40 (s, 1H); 13C NMR (150 MHz, CD3OD) δ 24.2, 26.2, 66.4, 68.7, 82.2, 84.5, 90.8, 105.6, 108.8, 114.1, 119.0, 121.4, 127.8, 127.9, 128.3, 130.9, 135.1, 136.8, 140.2, 143.9, 149.2, 152.7, 154.2, 156.1, 161.8, 173.9; HRMS (ESI+) calcd for C28H30N7O10S [M+H]+ 656.1769, found 656.1707 (error 9.4 ppm).
5′-O-[N-(4-Amino-2-hydroxybenzoyl)sulfamoyl]-2′,3′-O-isopropylideneadenosine triethylammonium salt (39)
To a solution of compound 38h (120 mg, 0.18 mmol) in MeOH (5 mL) was added 10% Pd/C (12 mg, 10% by wt) and the reaction stirred under H2 (1 atm) for 12 h. The reaction mixture was filtered through a plug of Celite. Purification by flash chromatography (100:10:1 MeOH/EtOAc/TEA) afforded the title compound (84 mg, 89%) as a white solid: Rf 0.10 (100:10:1 MeOH/EtOAc/TEA); [α]D21 −153 (c 1.88, MeOH); 1H NMR (600 MHz, CD3OD) δ 1.18 (t, J = 7.2 Hz, 9H), 1.33 (s, 3H), 1.58 (s, 3H), 2.97 (q, J = 7.2 Hz, 6H), 4.27 (d, J = 3.6 Hz, 2H), 4.55 (br s, 1H), 5.11 (d, J = 6.0 Hz, 1H), 5.37 (dd, J = 6.0, 2.4 Hz, 1H), 6.02 (d, J = 2.4 Hz, 1H), 6.11 (dd, J = 8.4, 1.8 Hz, 1H), 6.20 (d, J = 1.8 Hz, 1H), 7.64 (d, J = 8.4 Hz, 1H), 8.15 (s, 1H), 8.48 (s, 1H); 13C NMR (150 MHz, CD3OD) δ 8.6, 24.2, 26.2, 46.4, 68.6, 82.3, 84.4, 84.5, 90.8, 100.3, 106.2, 108.9, 114.1, 118.9, 131.5, 131.7, 140.2, 149.3, 152.8, 153.8, 156.1, 162.7; HRMS (ESI+) calcd for C20H24N7O8S [M+H]+ 522.1402, found 522.1385 (error 3.3 ppm).
5′-O-[N-(4-Amino-2-hydroxybenzoyl)sulfamoyl]adenosine (19)
This was prepared from 39 (50 mg, 0.090 mmol) using the general procedure for TFA deprotection. Purification by flash chromatography (100:30:3 MeOH/EtOAc/TEA) afforded the title compound (15 mg, 35%) as a white solid: Rf 0.10 (100:30:3 MeOH/EtOAc/TEA); [α]D21 −55.1 (c 3.75, MeOH); 1H NMR (600 MHz, CD3OD) δ 1.18 (t, J = 7.2 Hz, 9H), 3.17 (q, J = 7.2 Hz, 6H), 4.29–4.40 (m, 4H), 4.72 (t, J = 6.0 Hz, 1H), 6.02 (br s, 1H), 6.08 (d, J = 6.0 Hz, 1H), 6.10 (d, J = 9.0 Hz, 1H), 7.66 (d, J = 9.0 Hz, 1H), 8.16 (s, 1H), 8.50 (s, 1H); 13C NMR (150 MHz, CD3OD) δ 8.0, 46.4, 68.2, 71.3, 74.9, 83.5, 88.0, 100.3, 106.2, 109.0, 118.9, 131.7, 139.9, 149.7, 152.7, 153.8, 156.1, 162.7, 177.8; HRMS (ESI−) calcd for C17H18N7O8S [M−H]− 480.0943, found 480.0941 (error 0.4 ppm).
5′-O-[N-(6-Fluoro-2-hydroxybenzoyl)sulfamoyl]-2′,3′-O-isopropylideneadenosine triethylammonium salt (38i)
This was prepared from 6-fluorosalicylic acid (70 mg, 0.50 mmol) using the general procedure for CDI mediated acylation. Purification by flash chromatography (100:7.5:1 EtOAc/MeOH/TEA) afforded the title compound (157 mg, 60%): Rf 0.15 (100:7.5:1 EtOAc/MeOH/TEA); [α]D21 −52.8 (c 0.420, MeOH); 1H NMR (600 MHz, CD3OD) δ 1.23 (t, J = 7.2 Hz, 9H), 1.35 (s, 3H), 1.58 (s, 3H), 3.08 (q, J = 7.2 Hz, 6H), 4.31 (dd, J = 10.8, 4.2 Hz, 1H), 4.35 (dd, J = 10.8, 4.2 Hz, 1H), 4.54 (dd, J = 4.2, 1.8 Hz, 1H), 5.14 (dd, J = 3.0, 1.8 Hz, 1H), 5.37 (dd, J = 6.0, 3.0 Hz, 1H), 6.22 (d, J = 3.0 Hz, 1H), 6.44–6.50 (m, 1H), 6.58–6.62 (m, 1H), 7.17–7.22 (m, 1H), 8.15 (s, 1H), 8.42 (s, 1H); 13C NMR (150 MHz, CD3OD) δ8.2, 24.3, 26.3, 46.6, 68.9, 82.2, 84.5, 90.8, 106.1 (d, JC-F = 24 Hz), 111.9, 112.6, 114.1, 119.0, 131.8 (d, JC-F = 11.7 Hz), 132.3 (d, JC-F = 12.2 Hz), 140.2, 149.3, 152.8, 156.1, 162.0 (d, J = 131 Hz), 164.2, 170.7 (d, JC-F = 2.7 Hz); HRMS (ESI+) calcd for C20H22FN6O8S [M+H]+ 525.1198, found 525.1215 (error 3.2 ppm).
5′-O-[N-(6-Fluoro-2-hydroxybenzoyl)sulfamoyl]adenosine triethylammonium salt (20)
This was prepared from 38i (50 mg, 0.10 mmol) using the general procedure for TFA deprotection. Purification by flash chromatography (100:16:3 EtOAc/MeOH/TEA) afforded the title compound (36 mg, 76%): Rf 0.09 (100:16:3 EtOAc/MeOH/TEA); [α]D21 −47.0 (c 0.340, MeOH); 1H NMR (600 MHz, CD3OD) δ 1.14 (t, J = 7.2 Hz, 9H), 2.85 (q, J = 7.2 Hz, 6H), 4.33 (dd, J = 6.6, 3.0 Hz, 1H), 4.38–4.45 (m, 3H), 4.69 (t, J = 5.4 Hz, 1H), 6.08 (d, J = 5.4 Hz, 1H), 6.47 (dd, J = 11.4, 8.4 Hz, 1H), 6.60 (d, J = 8.4 Hz, 1H), 7.16–7.21 (m, 1H), 8.15 (s, 1H), 8.41 (s, 1H); 13C NMR (150 MHz, CD3OD) δ 8.9, 46.2, 68.6, 71.1, 74.8, 83.2, 88.3, 106.1 (d, JC-F = 24 Hz), 109.8 (d, JC-F = 9.0 Hz), 112.7 (d, JC-F = 3.3 Hz), 119.1, 132.4 (d, JC-F = 12.3 Hz), 139.9, 149.5, 152.7, 156.1, 162.0 (d, JC-F = 125 Hz), 164.1, 171.1 (d, J = 3.3 Hz); HRMS (ESI+) calcd for C17H18FN6O8S [M+H]+ 485.0891, found 485.0906 (error 3.1 ppm).
N-Hydroxysuccinimdyl-2,3-dibenzyloxybenzoate (41c)
This was prepared from 2,3-dibenzyloxybenzoic acid29 (1.00 g, 2.99 mmol, 1.0 equiv) using the general procedure for NHS ester synthesis. Purification by flash chromatography (4:1 EtOAc/hexanes) afforded the title compound (0.80 g, 62%): Rf 0.85 (EtOAc); 1H NMR (600 MHz, CDCl3) δ 4.49 (m, 4H), 5.13 (s, 4H), 7.12 (t, J = 8.4 Hz, 1H), 7.20–7.30 (m, 4H), 7.32–7.39 (m, 4H), 7.39–7.44 (m, 3H), 7.85 (dd, J = 8.4, 1.2 Hz, 1H); 13C NMR (150 MHz, CDCl3) δ 25.9, 71.6, 76.1, 120.4, 121.3, 123.9, 124.4, 127.8, 128.2, 128.4, 128.5, 128.8, 128.9, 136.5, 137.2, 150.1, 153.2, 160.9, 169.4.
5′-O-[N-(2,3-Dibenzyloxybenzoyl)sulfamoyl]-2′,3′-O-isopropylideneadenosine triethylammonium salt (42c)
This was prepared from 41c (837 mg, 1.88 mmol, 2.50 equiv) using the general procedure for NHS mediated acylation. Purification by flash chromatography (95:5:1 MeOH/EtOAc/Et3N) afforded the title compound (460 mg, 77%): Rf 0.30 (1:5 MeOH/EtOAc); [α]20D −43.5 (c 0.540, MeOH); 1H NMR (600 MHz, CD3OD) δ 1.08 (t, J = 7.2 Hz, 9H), 1.32 (s, 3H), 1.57 (s, 3H), 2.77 (q, J = 7.2 Hz, 6H), 4.24 (d, J = 3.6 Hz, 2H), 4.40–4.48 (m, 1H), 5.04–5.12 (m, 5H), 5.29 (dd, J = 5.4, 3.0 Hz, 1H), 6.19 (d, J = 3.0 Hz, 1H), 7.00–7.06 (m, 2H), 7.06–7.10 (m, 1H), 7.14–7.22 (m, 3H), 7.26–7.36 (m, 3H), 7.38–7.46 (m, 4H), 8.17 (s, 1H), 8.44 (s, 1H); 13C NMR (150 MHz, CD3OD) δ 10.2, 25.6, 27.5, 47.3, 69.8, 72.0, 76.6, 83.2, 85.6, 85.7, 91.8, 115.2, 116.1, 120.2, 121.8, 125.1, 128.6, 128.9, 129.0, 129.1, 129.5, 129.6, 137.7, 138.5, 139.4, 141.4, 146.8, 150.5, 153.5, 153.9, 157.3, 176.4; HRMS (ESI−) calcd for C34H33N6O9S [M−H]− 701.2035, found 701.2082 (error 6.7 ppm).
5′-O-[N-(2,3-Dihydroxybenzoyl)sulfamoyl]adenosine triethylammonium salt (21)
Compound 42c (0.45 g, 0.56 mmol) was treated with 10% Pd/C (90 mg, 20% by wt) at rt under H2 (1 atm) in MeOH (20 mL) for 12 h. The reaction mixture was filtered through Celite and concentrated to afford 43. The crude product was used directly for the next step.
Compound 43 prepared above was deprotected using the general procedure for TFA deprotection. Purification by flash chromatography (25:75:1 MeOH/EtOAc/Et3N) afforded the title compound (287 mg, 88%) as a white solid: mp 138–140 °C; Rf 0.2 (1:1 MeOH/EtOAc); [α]20D −23.5 (c 0.480, MeOH); 1H NMR (600 MHz, CD3OD) δ 1.24 (t, J = 7.8 Hz, 9H), 3.12 (q, J = 7.8 Hz, 6H), 4.26–4.50 (m, 4H), 4.71 (t, J = 4.8 Hz, 1H), 6.08 (d, J = 5.4 Hz, 1H), 6.61 (t, J = 7.8 Hz, 1H), 6.84 (d, J = 7.8 Hz, 1H), 7.43 (d, J = 7.8 Hz, 1H), 8.16 (s, 1H), 8.50 (s, 1H); 13C NMR (150 MHz, CD3OD) δ 9.3, 47.8, 69.6, 72.4, 76.0, 84.6, 89.2, 118.6, 119.3, 120.2, 120.9, 121.9, 141.1, 146.1, 146.8, 150.9, 153.9, 157.3, 175.0; HRMS (ESI+) calcd for C17H19N6O9S [M+H]+ 483.0929, found 483.0917 (error 2.5 ppm).
2′,3′-OO, -bis(t-Butyldimethylsilyl)-5′-O-[N-(2-pyridon-1-yl)sulfamoyl]adenosine triethylammonium salt (38j)
This was prepared from 2-hydroxynicotinic acid (83 mg, 0.60 mmol) using the general procedure for CDI mediated acylation. Purification by flash chromatography (100:10:2 MeOH/EtOAc/TEA) afforded the title compound (62 mg, 15%): Rf 0.27 (100:10:2 MeOH/EtOAc/TEA); [α]D21 −148 (c 4.20, MeOH); 1H NMR (600 MHz, CD3OD) δ −0.39 (s, 3H), −0.07 (s, 3H), 0.13 (s, 3H), 0.15 (s, 3H), 0.69 (s, 9H), 0.95 (s, 9H), 1.27 (t, J = 7.2 Hz, 9H), 3.18 (q, J = 7.2 Hz, 6H), 4.31 (br s, 1H), 4.44–4.47 (m, 3H), 4.87 (dd, J = 7.2, 4.8 Hz, 2H), 6.10 (d, J = 7.2 Hz, 1H), 6.76 (br s, 1H), 7.98 (br s, 1H), 8.18 (s, 1H), 8.34 (d, J = 6.0 Hz, 1H), 8.57 (s, 1H); 13C NMR (150 MHz, CD3OD) δ −5.1, −4.2, −4.17, −4.11, 9.4, 18.7, 19.0, 26.3, 26.5, 47.8, 69.7, 74.8, 77.5, 86.1, 88.6, 105.9, 114.1 (br), 120.3, 141.6 (br), 142.9, 148.0 (br), 151.1, 154.0, 157.5, 166.0 (br), 172.6; HRMS (ESI−) calcd for C28H44N7O8SSi2 [M−H]− 694.2516, found 694.2548 (error 4.6 ppm).
5′-O-[N-(2-Pyridon-1-yl)sulfamoyl]adenosine triethylammonium salt (23)
This was prepared from 38j (35 mg, 0.050 mmol) using the general procedure for TBAF deprotection. Purification by flash chromatography (90:5:7 MeOH/EtOAc/TEA) afforded the title compound (6.1 mg, 87%): Rf 0.03 (90:5:7 MeOH/EtOAc/TEA); [α]D21 −105 (c 0.760, MeOH); 1H NMR (600 MHz, DMSO-d6) δ 1.15 (t, J = 6.6 Hz, 9H), 2.95 (q, J = 6.6 Hz, 6H), 4.11–4.17 (m, 2H), 4.19 (dd, J = 7.8, 4.2 Hz, 1H), 4.24 (dd, J = 10.8, 4.2 Hz, 1H), 4.60 (q, J = 6.0 Hz, 1H), 5.38 (d, J = 4.2 Hz, 1H), 5.53 (d, J = 6.0 Hz, 1H), 5.92 (d, J = 6.0 Hz, 1H), 6.82 (br s, 1H), 7.31 (br s, 2H), 8.11 (s, 1H), 8.12–8.18 (br m, 1H), 8.39 (s, 1H); 13C NMR (150 MHz, DMSO-d6) δ 9.3, 45.7, 68.3, 70.7, 73.5, 82.4, 86.9, 115.3, 118.9, 139.1 (br), 139.3, 149.6, 151.4 (br), 152.7, 156.1, 165.4, 169.6 (missing one aryl carbon); HRMS (ESI−) calcd for C16H16N7O8S [M−H]− 466.0787, found 466.0767 (error 4.3 ppm).
N-Hydroxysuccinimdyl 2-fluoropyridine-3-carboxylate (41d)
This was prepared from 2-fluoronicotinic acid (300 mg, 2.1 mmol) using the general procedure for NHS ester synthesis. Purification by flash chromatograpy (1:1 EtOAc/Hexane) afforded the title compound (250 mg, 50%): Rf 0.18 (1:1 EtOAc/Hexane); 1H NMR (600 MHz, CDCl3) δ 2.91 (s, 4H), 7.38 (t, J = 6.0 Hz, 1H), 8.49–8.52 (m, 2H); 13C NMR (150 MHz, CDCl3) δ 23.3, 107.0 (d, 2JC-F = 25.1 Hz), 119.4 (d, 4JC-F = 5.0 Hz), 141.4, 151.4 (d, 3JC-F = 15.6 Hz), 155.9 (d, 3JC-F = 15.6 Hz), 159.0 (d, 1JC-F = 251 Hz), 166.4; HRMS (ESI+) calcd for C10H8FN2O4 [M+H]+ 239.0463, found 239.0469 (error 2.5 ppm).
5′-O-{N-[(2-Fluoropyridin-3-yl)carbonyl]sulfamoyl}-2′,3′-O-isopropylideneadenosine (42d)
This was prepared from 41d (100 mg, 0.40 mmol) using the general procedure for NHS ester mediated acylation. Purification by flash chromatography (1:7 MeOH/EtOAc) afforded the title compound (58 mg, 27%): Rf 0.17 (1:7 MeOH/EtOAc); [α]D21 −63.0 (c 2.50, MeOH); 1H NMR (600 MHz, CD3OD) δ 1.36 (s, 3H), 1.59 (s, 3H), 4.32 (d, J = 3.6 Hz, 2H), 4.56–4.58 (m, 1H), 5.15 (dd, J = 6.0, 2.4 Hz, 1H), 5.37 (dd, J = 6.0, 3.0 Hz, 1H), 6.22 (d, J = 3.0 Hz, 1H), 7.26–7.30 (m, 1H), 8.16 (s, 1H), 8.17–8.23 (m, 2H), 8.45 (s, 1H); 13C NMR (150 MHz, CD3OD) δ 24.3, 26.3, 68.7, 82.1, 84.5, 84.6, 90.7, 114.1, 118.9, 121.4, 140.2, 142.2, 148.2, 148.3, 149.3, 152.7, 156.1, 170.1 (d, 1JC-F = 252 Hz), 173.7; HRMS (ESI−) calcd for C19H19FN7O7S [M−H]− 508.1056, found 508.1059 (error 0.6 ppm).
5′-O-{N-[(2-Fluoropyridin-3-yl)carbonyl]sulfamoyl}adenosine triethylammonium salt (24)
This was prepared from 42d (30 mg, 0.060 mmol) using the general procedure for TFA deprotection. Purification by flash chromatography (100:20:1.5 MeOH/EtOAc/TEA) afforded the title compound (14 mg, 50%): Rf 0.17 (100:20:1.5 MeOH/EtOAc/TEA); [α]D21 −69.6 (c 2.55, MeOH); 1H NMR (600 MHz, CD3OD) δ 1.24 (t, J = 7.2 Hz, 9H), 3.13 (q, J = 7.2 Hz, 6H), 4.31–4.32 (m, 1H), 4.32–4.43 (m, 3H), 4.70 (t, J = 4.8 Hz, 1H), 6.08 (d, J = 5.4 Hz, 1H), 7.27–7.29 (m, 1H), 8.16 (s, 1H), 8.18 (d, J = 4.2 Hz, 1H), 8.24–8.27 (m, 1H), 8.50 (s, 1H); 13C NMR (150 MHz, CD3OD) δ 8.1, 46.5, 68.4, 71.2, 74.8, 83.4, 87.9, 118.9, 121.5 (d, JC-F = 4.5 Hz), 122.1 (d, JC-F = 25.7 Hz), 139.9, 142.2 (d, JC-F = 2.7 Hz), 148.3 (d, JC-F =14.0 Hz), 149.6, 152.7, 156.1, 160.6 (d, JC-F = 243 Hz), 170.1 (d, JC-F = 6.2 Hz); HRMS (ESI−) calcd for C16H15FN7O7S [M−H]− 468.0743, found 468.0740 (error 0.6 ppm);.
N-Hydroxysuccinimdyl 2-chloropyridine-3-carboxylate (41e)
This was prepared from 2-chloronicotinic acid (780 mg, 5.00 mmol) using the general procedure for NHS ester synthesis. Purification by flash chromatography (1:1 EtOAc/Hexane) afforded the title compound (660 mg, 52%): Rf = 0.21 (1:1 EtOAc/Hexane); 1H NMR (600 MHz, CDCl3) δ 2.92 (s, 4H), 7.41 (t, J = 7.8 Hz, 1H), 8.42 (dd, J = 7.8, 1.8 Hz, 1H), 8.63 (dd, J = 7.8, 1.8 Hz, 1H); 13C NMR (150 MHz, CDCl3) δ 25.9, 122.1, 122.4, 141.4, 151.7, 153.9, 159.6, 168.9; HRMS (ESI+) calcd for C10H8ClN2O4 [M+H]+ 255.0167, found 255.0163 (error 1.6 ppm).
5′-O-{N-[(2-Chloropyridin-3-yl)carbonyl]sulfamoyl}-2′,3′-O-isopropylideneadenosine triethylammonium salt (42e)
This was prepared from 41e (50 mg, 0.20 mmol) using the general procedure for NHS mediated acylation. Purification by flash chromatography (100:10:1 EtOAc/MeOH/TEA) afforded the title compound (86 mg, 82%): Rf 0.15 (100:10:1 EtOAc/MeOH/TEA); [α]D21 = −94.3 (c 0.340, MeOH); 1H NMR (600 MHz, CD3OD) δ 1.27 (t, J = 7.8 Hz, 9H), 1.36 (s, 3H), 1.59 (s, 3H), 3.16 (q, J = 7.8 Hz, 6H), 4.33–4.35 (m, 2H), 4.57 (dd, J = 5.4, 2.4 Hz, 1H), 5.20 (dd, J = 6.0, 2.4 Hz, 1H), 5.39 (dd, J = 6.0, 3.0 Hz, 1H), 6.24 (d, J = 3.0 Hz, 1H), 7.35 (dd, J = 7.8, 4.8 Hz, 1H), 7.90 (dd, J = 7.8, 1.8 Hz, 1H), 8.19 (s, 1H), 8.32 (dd, J = 4.8, 1.8 Hz, 1H), 8.45 (s, 1H); 13C NMR (150 MHz, CD3OD) δ 8.0, 24.4, 26.3, 46.7, 68.9, 82.1, 84.5, 84.6, 90.7, 114.2, 119.0, 122.7, 136.1, 138.3, 140.2, 147.3, 149.0, 149.3, 152.8, 156.1, 172.3; HRMS (ESI+) calcd for C19H21Cl N7O7S [M+H]+ 526.0906, found 526.0909 (error 0.6 ppm).
5′-O-{N-[(2-Chloropyridin-3-yl)carbonyl]sulfamoyl}adenosine triethylammonium salt (25)
This was prepared from 42e (40 mg, 0.080 mmol) using the general procedure for TFA deprotection. Purification by flash chromatography (100:20:1 EtOAc/MeOH/TEA) afforded the title compound (21 mg, 53%): Rf 0.10 (100:20:1 EtOAc/MeOH/TEA); [α]D21 −64.7 (c 0.340, MeOH); 1H NMR (600 MHz, CD3OD) δ 1.26 (t, J = 7.8 Hz, 9H), 3.15 (q, J = 7.8 Hz, 6H), 4.33 (dd, J = 6.6, 3.0 Hz, 1H), 4.39–4.46 (m, 3H), 4.73 (t, J = 6.0 Hz, 1H), 6.09 (d, J = 4.8 Hz, 1H), 7.35 (dd, J = 7.2, 4.2 Hz, 1H), 7.94 (dd, J = 7.2, 1.8 Hz, 1H), 8.19 (s, 1H), 8.31 (dd, J = 4.2, 1.8 Hz, 1H), 8.49 (s, 1H); 13C NMR (150 MHz, CD3OD) δ 8.1, 46.7, 68.5, 71.2, 74.8, 83.4, 88.1, 119.0, 122.6, 136.2, 138.3, 139.9, 147.3, 148.9, 149.7, 152.7, 156.1, 172.4; HRMS (ESI+) calcd for C16H17ClN7O7S [M+H]+ 486.0593, found 486.0598 (error 1.0 ppm).
5′-O-{N-[(Morpholino)carbonyl]sulfamoyl}adenosine sodium salt (26)
Morpholine (54 μL, 3.0 mmol, 1.0 equiv) was added dropwise to a solution of chlorosulfonyl isocyanate (54 μL, 3.0 mmol, 1.0 equiv) in CH2Cl2 (15 mL) at −20 °C. The reaction mixture was stirred at −15 °C to −20 °C for 4 h, then the solvent was evaporated in vacuo and the crude product 52 was used for the subsequent step without any further workup.
To the above intermediate 52 was added a solution of 2′,3′-O-isopropylideneadenosine (155 mg, 2 mmol, 0.66 equiv) and pyridine (48 μL, 3 mmol, 1 equiv) in MeCN (38 mL). The resulting solution was stirred at rt for 20 h. The solvent was evaporated in vacuo and purification by flash column chromatography (70:30:1 EtOAc/MeOH/TEA) afforded 2′,3′-O-isopropylidene-5′-O{N-[(morpholino)carbonyl]sulfamoyl}adenosine triethylammonium salt 54 that was approximately 50–75% pure based on 1H NMR.
Crude 54 prepared above was deprotected using the general procedure for TFA deprotection. Purification by flash column chromatography (70:30:1 EtOAc/MeOH/TEA) followed by conversion to the sodium salt using the general procedure for ion-exchange afforded the title compound (12 mg, 4% overall yield): Rf 0.29 (6:4 EtOAc/MeOH); [α]D21 −0.06 (c 0.22, CH3OH); 1H NMR (600 MHz, CD3OD) δ 3.40–3.60 (m, 4H), 3.58–3.60 (m, 4H), 4.25–4.30 (m, 3H), 4.41–4.43 (m, 1H), 4.70 (t, J = 5.4 Hz, 1H), 6.09 (d, J = 6.0 Hz, 1H), 8.19 (s, 1H), 8.54 (s, 1H); 13C NMR (150 MHz, CD3OD) δ 49.7, 68.1, 68.8, 72.5, 76.3, 85.0, 89.1, 120.2, 141.4, 151.0, 154.0, 157.4, 162.4; HRMS (ESI−) calcd for C15H20N7O8S [M−H]− 458.1099, found 458.1106 (error 1.4 ppm).
2′,3′-OO, -bis(t-Butyldimethylsilyl)-5′-O-{N-[(cyclopentyl)carbonyl]sulfamoyl}adenosine triethylammonium salt (42f)
Cyclopentanecarbonyl chloride (920 μL, 7.54 mmol) was converted to the NHS ester 41f using the general procedure B for NHS ester synthesis. The crude NHS ester was used directly for the next step.
The title compound was prepared from 41f (126 mg, 0.60 mmol) using the general procedure for NHS ester mediated acylation. Purification by flash chromatography (80:20:1 EtOAc/MeOH/TEA) afforded the title compound (270 mg, 67% overall yield): Rf 0.23 (6:4 EtOAc/MeOH); [α]D21 −64.7 (c 0.34, MeOH); 1H NMR (600 MHz, CD3OD) δ −0.37 (s, 3H), −0.01 (s, 3H), 0.16 (s, 3H); 0.17 (s, 3H), 0.71 (s, 9H), 0.98 (s, 9H), 1.23 (t, J = 7.2 Hz, 9H), 1.54–1.57 (m, 2H), 1.70–1.72 (m, 2H), 1.80–1.84 (m, 4H), 2.65 (p, J = 8.4 Hz, 1H), 3.03 (q, J = 7.2 Hz, 6H), 4.29–4.34 (m, 3H), 4.45 (d, J = 4.2 Hz, 1H), 4.84–4.86 (m, 1H, obscured by CD3OD: assigned by gCOSY as H-2′), 6.12 (d, J = 7.2 Hz, 1H), 8.18 (s, 1H), 8.57 (s, 1H); 13C NMR (600 MHz, CD3OD) δ −5.2, −4.2, −4.14 (2C), 9.8, 18.8, 19.0, 26.3, 26.5, 27.1, 31.9, 32.0 47.8, 69.1, 75.4, 77.7, 86.6, 88.3, 120.2, 141.7, 151.2, 154.0, 157.5, 186.9; MS (ESI−) calcd for C28H49N6O7SSi2 [M−H]− 669.3, found 669.3.
5′-O-{N-[(Cyclopentyl)carbonyl]sulfamoyl}adenosine sodium salt (27)
This was prepared from 42f (270 mg, 0.40 mmol) using the general procedure for TBS protection. Purification by flash chromatography (80:20:1 EtOAc/MeOH/TEA) followed by conversion to the sodium salt using the general procedure for ion-exchange afforded the title compound (96 mg, 55%): Rf 0.25 (8:2 EtOAc/MeOH); [α]D21 −0.23 (c 0.49, CH3OH); 1H NMR (600 MHz, CD3OD) δ 1.52–1.54 (m, 2H), 1.66–1.68 (m, 2H), 1.72–1.77 (m, 2H), 1.80–1.83 (m, 2H), 2.63 (p, J = 8.4 Hz, 1H), 4.24–4.31 (m, 3H), 4.38–4.39 (m, 1H), 4.69 (t, J = 6.0 Hz, 1H), 6.08 (d, J = 6.0 Hz, 1H), 8.19 (s, 1H), 8.48 (s, 1H); 13C NMR (150 MHz, CD3OD) δ 27.1, 27.1, 31.3, 31.9, 69.2, 72.5, 76.2, 84.8, 89.3, 120.3, 141.3, 151.0, 154.0, 157.4, 186.3; HRMS (ESI−) calcd for C16H21N6O7S [M−H]− 441.1197, found 441.1183 (error 3.2 ppm).
2′,3′-OO, -bis(t-Butyldimethylsilyl)-5′-O-{N-[(cyclohexyl)carbonyl]sulfamoyl}adenosine triethylammonium salt (42g)
Cyclohexanecarbonyl chloride (930 μL, 6.82 mmol) was converted to the NHS ester 41g using the general procedure B for NHS ester synthesis. The crude NHS ester was used directly for the next step.
The title compound was prepared from 41g (88 mg, 0.39 mmol) using the general procedure for NHS ester mediated acylation. Purification by flash chromatography (80:20:1 EtOAc/MeOH/TEA) afforded the title compound (230 mg, 87%); Rf 0.22 (6:4 EtOAc/MeOH); 1H NMR (600 MHz, CD3OD) δ −0.37 (s, 3H), −0.01 (s, 3H), 0.16 (s, 3H), 0.17 (s, 3H), 0.71 (s, 9H), 0.98 (s, 9H), 1.23 (t, J = 7.2 Hz, 9H), 1.28–1.33 (m, 3H), 1.43–1.46 (m, 2H), 1.65–1.66 (m, 1H), 1.75–1.76 (m, 2H), 1.86–1.88 (m, 2H), 2.16 (tt, J = 11.4, 3.0 Hz, 1H), 3.06 (q, J = 7.2 Hz, 6H), 4.30–4.33 (m, 3H), 4.45 (d, J = 4.2 Hz, 1H), 4.84–4.86 (m, 1H, obscured by CD3OD: assigned by gCOSY as H-2′), 6.11 (d, J = 7.8 Hz, 1H), 8.18 (s, 1H), 8.56 (s, 1H); 13C NMR (150 MHz, CD3OD) δ −5.3, −4.33, −4.28 (2C), 9.7, 18.6, 18.9, 26.2, 26.4, 27.0, 27.4, 31.25, 31.29, 47.6, 69.0, 74.9, 77.5, 86.4, 88.2, 120.1, 141.5, 151.1, 153.9, 157.3, 185.8; MS (ESI−) calcd for C29H51N6O7SSi2 [M−H]− 683.3, found 683.3.
5′-O-{N-[(Cyclohexyl)carbonyl]sulfamoyl}adenosine sodium salt (28)
This was prepared from 42g (230 mg, 0.33 mmol) using the general procedure for TBS protection. Purification by flash chromatography (80:20:1 EtOAc/MeOH/TEA) followed by conversion to the sodium salt using the general procedure for ion-exchange afforded the title compound (82 mg, 51%): Rf 0.22 (8:2 EtOAc/MeOH); [α]D21 −0.26 (c 0.50, CH3OH); 1H NMR (600 MHz, CD3OD) δ 1.17–1.30 (m, 3H), 1.37–1.42 (m, 2H), 1.63–1.65 (m, 1H) 1.73–1.74 (m, 2H), 1.82–1.84 (m, 2H), 2.14 (tt, J= 11.4, 3.0 Hz, 1H), 4.23–4.30 (m, 3H), 4.37–4.39 (m, 1H), 4.69 (t, J = 6.0 Hz, 1H), 6.08 (d, J = 6.0 Hz, 1H), 8.19 (s, 1H), 8.48 (s, 1H); 13C NMR (150 MHz, CD3OD) δ 27.2, 27.3, 31.27, 31.28, 69.2, 72.6, 76.2, 84.8, 89.3, 120.3, 141.3, 151.0, 153.9, 157.4, 186.4; HRMS (ESI−) calcd for C17H23N6O7S [M−H]− 455.1354, found 455.1348 (error 1.3 ppm).
2′,3′-OO, -bis(t-Butyldimethylsilyl)-5′-O-[(N-(acetyl)sulfamoyl]adenosine triethylammonium salt (42h)
Acetic anhydride (920 μL, 9.8 mmol) was converted to the NHS ester 41h using the general procedure B for NHS ester synthesis. The crude NHS ester was used directly for the next step.
The title compound was prepared from 41h (96 mg, 0.61 mmol) using the general procedure for NHS ester mediated acylation. Purification by flash chromatography (70:30:1 EtOAc/MeOH/TEA) afforded the title compound (350 mg, 93%): Rf 0.20 (85:15 EtOAc/MeOH); 1H NMR (600 MHz, CD3OD) δ −0.35 (s, 3H), −0.01 (s, 3H), 0.16 (s, 3H), 0.17 (s, 3H), 0.71 (s, 9H), 0.98 (s, 9H), 1.29 (t, J = 7.2 Hz, 9H), 1.96 (s, 3H), 3.17 (q, J = 7.2 Hz, 6H), 4.46 (d, J = 4.2 Hz, 1H), 4.30–4.35 (m, 3H), 4.82–4.84 (1H, m, obscured by CD3OD: assigned by gCOSY as H-2′), 6.12 (d, J = 7.2 Hz, 1H), 8.19 (s, 1H), 8.56 (s, 1H); 13C NMR (150 MHz, CD3OD) δ −5.2, −4.24, −4.20, −4.17, 9.5, 18.8, 19.0, 26.2, 26.3, 26.5, 47.9, 69.2, 74.9, 77.7, 86.4, 88.3, 120.2, 141.6, 151.2, 154.0, 157.5, 180.4; MS (ESI−) calcd for C24H43N6O7SSi2 [M−H]− 615.2, found 615.2.
5′-O-[N-(Acetyl)sulfamoyl]adenosine sodium salt (29)
This was prepared from 42h (350 mg, 0.57 mmol) using the general procedure for TBS protection. Purification by flash chromatography (80:20:1 EtOAc/MeOH/TEA) followed by conversion to the sodium salt using the general procedure for ion-exchange afforded the title compound (148 mg, 74%): Rf 0.26 (7:3 EtOAc/MeOH); [α]D21 −0.25 (c 0.56, CH3OH); 1H NMR (600 MHz, CD3OD) δ 1.95 (s, 3H), 4.26–4.33 (m, 3H), 4.38–4.39 (m, 1H), 4.66 (t, J = 6.0 Hz, 1H), 6.08 (d, J = 6.0 Hz, 1H), 8.19 (s, 1H), 8.49 (s, 1H); 13C NMR (150 MHz, CD3OD) δ 26.2, 69.2, 72.4, 76.2, 84.7, 89.3, 120.3, 141.2, 151.0, 154.0, 157.4, 180.8, HRMS (ESI−) calcd for C16H21N6O7S [M−H]− 387.0728, found 387.0722 (error 1.5 ppm).
5′-O-{N-[(Methoxy)carbonyl]sulfamoyl}adenosine sodium salt (30)
A solution of 36 (80 mg, 0.2 mmol, 1.0 equiv) and CDI (65 mg, 0.4 mmol, 2 equiv) in MeCN (10 mL) was stirred at rt for 2 h. Next, MeOH (3 mL) and Et3N (55 μL, 0.4 mmol, 2.0 equiv) were added and the reaction was stirred for an additional 2 h, then the reaction mixture was concentrated in vacuo. Purification by flash column chromatography (80:20:1 EtOAc/MeOH/Et3N) afforded 2′,3′-O-isopropylidene-5′-O-{N-[(methoxy)carbonyl]sulfamoyl}adenosine triethylammonium salt.
2′,3′-O-isopropylidene-5′-O-{N-[(methoxy)carbonyl]sulfamoyl}adenosine triethylammonium salt prepared above was deprotected using the general procedure for TFA deprotection. Purification by flash chromatography (70:30:1 EtOAc/MeOH/TEA) followed by conversion to the sodium salt using the general procedure for ion-exchange afforded the title compound (84 mg, 64% overall yield): Rf 0.20 (7:3 EtOAc/MeOH); [α]D21 −0.16 (c 0.58, CH3OH); 1H NMR (600 MHz, CD3OD) δ 3.56 (s, 3H), 4.23–4.31 (m, 3H), 4.36–4.38 (m, 1H), 4.65 (t, J = 5.4 Hz, 1H), 6.08 (d, J = 6.0 Hz, 1H), 8.18 (s, 1H), 8.48 (s, 1H); 13C NMR (150 MHz, CD3OD) δ 51.8, 68.2, 71.4, 75.3, 83.8, 88.3, 119.3, 140.2, 145.0, 153.0, 156.4, 161.2; HRMS (ESI−) calcd for C17H23N6O7S [M−H]− 403.0677, found 403.0690 (error 3.1 ppm).
5′-O-{N-[(Benzyl)carbonyl]sulfamoyl}adenosine sodium salt (31)
Phenylacetic acid (1.00 g, 7.3 mmol) was converted to the NHS ester 41i using the general procedure A for NHS ester synthesis. The crude NHS ester was used directly for the next step.
Compound 41i (85 mg, 0.37 mmol) prepared above was treated with 36 using the general procedure for NHS ester mediated acylation to afford crude 2′,3′-O,O -bis(t-butyldimethylsilyl)-5′-O-{N-[(benzyl)carbonyl]sulfamoyl}triethylammonium salt that was used directly for the next step.
2′,3′-OO, -bis(t-Butyldimethylsilyl)-5′-O-{N-[(benzyl)carbonyl]sulfamoyl}triethylammonium salt prepared above was deprotected using the general procedure for TBS deprotection. Purification by flash chromatography (75:25:1EtOAc/MeOH/TEA) followed by conversion to the sodium salt using the general procedure for ion-exchange afforded the title compound (110 mg, 65% overall yield: Rf 0.29 (7:3 EtOAc/MeOH); [α]D21 −0.14 (c 0.43, CH3OH); 1H NMR (600 MHz, CD3OD) δ 3.48 (s, 2H), 4.18–4.20 (m, 1H), 4.22–4.26 (m, 2H), 4.29–4.30 (m, 1H), 4.62 (t, J = 5.4 Hz, 1H), 6.06 (d, J = 6.0 Hz, 1H), 7.13 (t, J = 7.2 Hz, 1H), 7.21 (t, J = 7.8 Hz, 2H), 7.30 (d, J = 7.8 Hz, 2H), 8.19 (s, 1H), 8.45 (s, 1H); 13C NMR (150 MHz, CD3OD) δ 47.2, 69.3, 72.4, 76.2, 84.6, 89.3, 120.3, 127.3, 129.3, 130.4, 138.5, 141.2, 150.9, 154.0, 157.4, 181.1; HRMS (ESI−) calcd for C18H19N6O7S [M−H]− 463.1041, found 463.1046 (error 1.1 ppm).
5′-O–[N-(4-Azido-2,3,5,6-tetrafluorobenzoyl)sulfamoyl]adenosine triethylammonium salt (32)
N-hydroxysuccinimidyl-4-azidotetrafluorobenzoate26 (235 mg, 1.0 mmol, 1.0 equiv) was treated with 36 using the general procedure for NHS ester mediated acylation. Purification by flash column chromatography (80:20:1 EtOAc/MeOH/Et3N) afforded 5′-O–[N-(4-azido-2,3,5,6-tetrafluorobenzoyl)sulfamoyl]-2′,3′-O-isopropylideneadenosine triethylammonium salt (141 mg, 39%).
5′-O–[N-(4-azido-2,3,5,6-tetrafluorobenzoyl)sulfamoyl]-2′,3′-O-isopropylideneadenosine triethylammonium salt (105 mg, 0.2 mmol) prepared above was deprotected using the general procedure for TFA deprotection. Purification by flash column chromatography (80:20:1 EtOAc/MeOH/Et3N) afforded the title compound (62 mg, 47%): Rf 0.29 (8:2 EtOAc/MeOH); [α]D21 −0.11 (c 0.37, CH3OH); 1H NMR (600 MHz, CD3OD) δ 1.29 (t, J = 7.8 Hz, 9H), 3.21 (q, J = 7.8 Hz, 6H), 4.32–4.42 (m, 4H), 4.70 (t, J = 5.4 Hz, 1H), 6.09 (d, J = 5.4 Hz, 1H), 8.19 (s, 1H), 8.48 (s, 1H); 13C NMR (150 MHz, CD3OD) δ 9.3, 48.1, 69.9, 72.5, 76.1, 84.7, 89.3, 120.3, 141.2, 151.0, 154.0, 157.4, 166.1 (missing 3 aryl carbons and C=O); HRMS (ESI−) calcd for C17H12F4N9O7S [M−H]− 562.0522, found 562.0548 (error 4.6 ppm).
N-Hydroxysuccinimdyl N-(t-butyloxycarbonyl)-L-phenylalanine (41k)
This was prepared from N-(t-butyloxycarbonyl)-L-phenylalanine (795 mg, 3.00 mmol) using the general procedure for NHS ester synthesis. Purification by flash chromatography (2:5 EtOAc/Hexane) afforded the title compound (800 mg, 74%): Rf 0.33 (3:1 Hexane/EtOAc); [α]D21 −37.1 (c 0.730, MeOH); 1H NMR (600 MHz, CDCl3) δ 1.39 (s, 9H), 2.84 (s, 4H), 3.18 (dd, J = 13.2, 5.4 Hz, 1H), 3.30 (dd, J = 13.2, 5.4 Hz, 1H), 4.90 (t, J = 5.4 Hz, 1H), 7.25–7.33 (m, 5H); 13C NMR (150 MHz, CDCl3) δ 25.8, 28.2, 38.4, 52.8, 80.8, 127.6, 128.9, 129.9, 134.9, 154.8, 167.9, 168.7; MS (ESI+) C13H15N2O4 [M+H] calcd 263.1, found 263.1.
5′-O-{N-[N-(t-Butyloxycarbonyl)-L-phenylalanyl]sulfamoyl}-2′,3′-O-isopropylideneadenosine (42k)
This was prepared from 41k (363 mg, 1.00 mmol) using the general procedure for NHS mediated acylation. Purification by flash chromatography (100:10:1 EtOAc/MeOH/TEA) afforded the title compound (600 mg, 95%): Rf 0.16 (10:1 EtOAc/MeOH); [α]D21 − 19.1 (c 1.00, MeOH); 1H NMR (600 MHz, CD3OD) δ 1.33 (s, 9H), 1.36 (s, 3H), 1.58 (s, 3H), 2.84 (dd, J = 13.2, 9.0 Hz, 1H), 3.10 (dd, J = 13.2, 4.8 Hz, 1H), 4.16–4.23 (m, 3H), 4.47–4.51 (m, 1H), 5.06 (d, J = 6.0 Hz, 1H), 5.32 (br s, 1H), 6.21 (br s, 1H), 7.11–7.22 (m, 5H), 8.19 (s, 1H), 8.41 (s, 1H); 13C NMR (150 MHz, CD3OD) δ 25.1, 26.3, 27.5, 57.9, 80.5, 82.0, 84.4, 84.6, 85.3, 85.9, 90.7, 92.3, 114.1, 118.9, 126.2, 128.1, 129.4, 139.4, 140.3, 149.1, 149.3, 152.7, 156.0; HRMS (ESI+) calcd for C27H36N7O9S 634.2295, found 634.2309 (error 3.0 ppm).
5′-O-[N-(L-Phenylalanyl)sulfamoyl]adenosine sodium salt (33)
This was prepared from 42k (200 mg, 0.32 mmol) using the general procedure for TFA deprotection. Purification by HPLC (Microsorb™ C18 Dynamax™ preparative HPLC column, 41.4 × 250 mm, pore size 100 Å, 20–80% A in 45 min, A = 40 mM TEAB buffer, B = 70% CH3CN in H2O, flow rate = 40 mL/min, monitored at 254 nm) and lypholization of the pooled fractions followed by conversion to the sodium salt using the general procedure for ion-exchange afforded the title compound (90 mg, 58%): tR = 12.7 min; [α]D21 −7.5 (c 0.74, MeOH); 1H NMR (600 MHz, CD3OD) δ 2.87 (dd, J = 13.2, 5.4 Hz, 1H), 3.17 (dd, J = 13.2, 5.4 Hz 1H), 3.64 (t, J = 6.6 Hz, 1H), 4.22–4.29 (m, 3H), 4.36 (t, J = 4. 2 Hz, 1H), 4.62 (t, J = 4.8 Hz, 1H), 6.07 (d, J = 5.4 Hz, 1H), 7.13–7.15 (m, 1H), 7.22–7.25 (m, 4H), 8.18 (s, 1H), 8.50 (s, 1H); 13C NMR (150 MHz, CD3OD) δ 41.6, 58.5, 67.9, 70.9, 75.1, 83.2, 88.3, 119.0, 126.2, 128.2, 129.3, 138.4, 140.1, 149.6, 152.7, 156.1, 181.6 (missing 2C’s); HRMS (ESI+) calcd for C19H24N7O7S 494.1458 found 494.1489 (error 0.6 ppm).
5′-O-[N-(3-{[(Morpholino)carbonyl]amino}propanoyl)sulfamoyl]adenosine sodium salt (47)
Morpholinocarbonyl chloride (0.77 mg, 6.7 mmol) was converted to the NHS ester 44 using the general procedure B for NHS ester synthesis. The crude NHS ester was used directly for the next step.
Compound 44 (82 mg, 0.36 mmol) prepared above was treated with 37 using the general procedure for NHS mediated acylation. The crude product 46 was used directly for the next step.
Compound 46 prepared above was deprotected using the general procedure for TBS deprotection. Purification by flash chromatography (70:30:1 EtOAc/MeOH/TEA) followed by conversion to the sodium salt using the general procedure for ion-exchange afforded the title compound (107 mg, 56% overall yield): Rf 0.21 (6:4 EtOAc/MeOH); [α]D21 −0.08 (c 0.42, CH3OH); 1H NMR (600 MHz, CD3OD) δ 2.40 (t, J = 6.6 Hz, 2H), 3.29 (t, J = 4.8 Hz, 4H), 3.42 (t, J = 6.6 Hz, 2H), 3.58 (t, J = 4.8 Hz, 4H), 4.27–4.33 (m, 3H), 4.39 (t, J = 4.2 Hz, 1H), 4.66 (t, J = 5.4 Hz, 1H), 6.08 (d, J = 5.4 Hz, 1H), 8.20 (s, 1H), 8.51 (s, 1H), 13C NMR (150 MHz, CD3OD) δ 38.8, 40.3, 45.2, 67.7, 69.2, 72.4, 76.3, 84.6, 89.4, 120.3, 141.3, 150.9, 154.0, 157.4, 160.2, 181.4; HRMS (ESI−) calcd for C18H25N8O9S [M−H]− 529.1470, found 529.1489 (error 3.5 ppm).
5′-Azido-N6-butyloxylcarbonyl-5′-deoxy-2′,3′-O-isopropylideneadenosine
To a solution of 5′-azido-5′-deoxy-2′,3′-O-isopropylideneadenosine22 (146 mg, 0.44 mmol, 1.0 equiv) in THF/DMF (4:1, 10 mL) was added sodium hydride (60% dispension in mineral oil, 20 mg, 0.5 mmol, 1.15 equiv) at rt. The resulting solution was stirred 30 min then solid di-tert-butyl dicarbonate (106 mg, 0.48 mmol, 1.1 equiv) was added in one portion. The reaction was monitored by TLC until complete consumption of the amine was observed (3 h). The solution was diluted with CH2Cl2 (20 mL) and washed with saturated aq NaHCO3 (20 mL). The aqueous phase was back extracted with CH2Cl2 (2× 10 mL) and the combined organic phase was washed with H2O (20 mL), saturated aq NaCl (30 mL), dried (Na2SO4) and concentrated. Purification by flash chromatography (4:6 Hexane/EtOAc) afforded the title compound (144 mg, 60%): Rf 0.55 (1:2 Hexane/EtOAc); [α]D21 −33.1 (c 0.972, CH3OH); 1H NMR (600 MHz, CDCl3) δ 1.35 (s, 3H), 1.53 (s, 9H), 1.58 (s, 3H), 3.54 (d, J = 5.4 Hz, 2H), 4.36 (dd, J = 3.6, 5.4 Hz, 1H), 5.01 (dd, J = 6.6, 3.6 Hz, 1H), 5.42 (dd, J = 6.6, 2.4 Hz, 1H), 6.13 (d, J = 2.4 Hz, 1H), 8.09 (s, 1H), 8.74 (s, 1H); 13C NMR (150 MHz, CDCl3) δ 25.5, 27.3, 28.3, 52.4, 82.0, 82.6, 84.2, 85.8, 90.9, 115.1, 122.3, 141.8, 149.8, 150.3, 150.4, 153.2; MS (APCI+) calcd for C18H25O5N8 [M+H]+ 433.2, found 433.2.
5′-Amino-N6-t-butoxycarbonyl-5′-deoxy-2′,3′-O-isopropylideneadenosine (56)
To a solution of 5′-azido-N6-tert-butoxycarbonyl-5′-deoxy-2′,3′-O-isopropylideneadenosine (150 mg, 0.35 mmol, 1.0 equiv) in MeOH (5.0 mL) was added 10% Pd/C (10 mg, 7% by wt) in MeOH (0.5 mL) and the reaction stirred under H2 (2.5 atm) for 12 h. The reaction mixture was filtered through a plug of Celite. Purification by flash chromatography (100:17:1 EtOAc/MeOH/TEA) afforded the title compound 132 mg, 94%). 1H NMR, 13C NMR, and HRMS agreed with literature values.11
5′-Amino-5′-N-[(chromon-3-yl)carbonyl]adenosine (58)
To a solution of chromone 3-carboxylic acid (74 mg, 0.39 mmol) and CDI (63.3 mg, 0.39 mmol, 1.0 equiv) in DMF (6 mL) was stirred at 60 °C for 2 h. The solution was cooled to rt and a mixture of 56 (100 mg, 0.32 mmol, 0.8 equiv) and DBU (92 μL, 0.62 mmol, 1.5 equiv) were then added and the reaction stirred overnight at rt. The reaction mixture was concentrated in vacuo and purified by flash chromatography to afford 57. The crude product was used directly for the next step.
Crude 57 prepared above was deprotected using the general procedure for TFA deprotection. Purification by flash chromatography (60:40 EtOAc/MeOH) afforded the title compound (29 mg, 20% overall yield): Rf 0.22 (7:3 EtOAc/MeOH); [α]D21 −31.7 (c 0.11, MeOH); 1H NMR (600 MHz, DMSO) δ 3.97 (br s, 1H), 4.08 (dd, J = 5.4, 4.8 Hz, 1H), 4.16 (dd, J = 6.6, 3.6 Hz, 1H), 4.22 (br s, 1H), 4.67–4.71 (m, 1H), 5.43 (br s, 1H), 5.57 (br s, 1H), 5.92 (d, J = 6.0 Hz, 1H), 7.26–7.30 (m, 4H), 7.63 (t, J = 7.8 Hz, 1H), 7.88 (t, J = 7.8 Hz, 1H), 8.10 (br s, 1H), 8.33 (s, 1H), 8.43–8.45 (m, 1H), 10.39 (br s, 1H); 13C NMR (150 MHz, DMSO) δ 51.7, 70.8, 72.7, 82.1, 87.7, 96.0, 116.9, 119.3, 120.2, 123.9, 125.3, 134.3, 139.9, 149.3, 152.6, 154.2, 156.1, 162.6, 163.2, 179.4; MS (APCI−) calcd for C20H18N6O6 [M−H]− 437.1215, found 437.1378.
2-Benzyloxybenzamide (59)
To a solution of 2-hydroxybenzamide (2.74 g, 20 mmol, 1.0 equiv) and K2CO3 (3.30 g, 24 mmol, 1.2 equiv) in acetone (100 mL) was added benzyl bromide (2.80 mL, 24 mmol, 1.2 equiv) and the reaction heated at reflux for 16 h. After cooling to rt, the solution was filtered and the filtrate was concentrated under reduced pressure. The crude solid was recrystallized from acetone to afford the title compound (2.89 g, 62%) as a white solid: 1H NMR (CDCl3, 600 MHz) δ 5.19 (s, 2H), 5.92 (br s, 1H), 7.05 (d, J = 8.4 Hz, 1H), 7.09 (t, J = 8.4 Hz, 1H), 7.36–7.48 (m, 6H), 7.73 (br s, 1H), 8.23 (d, J = 7.8 Hz, 1H); 13C NMR (CDCl3, 150 MHz) δ 71.5, 112.9, 121.3, 121.8, 128.1, 129.0, 129.2, 132.9, 133.6, 135.8, 157.4, 167.2; HRMS (APCI+) calcd for C14H14NO2 [M+H]+ 228.1019, found 228.1042 (error 10.0 ppm).
5′-Amino-5′-N-{[(2-benzyloxybenzoyl)amino]carbonyl}-N6-t-butoxycarbonyl-5′-deoxy-2′,3′-O-isopropylideneadenosine (61)
To a stirred solution of 59 (114 mg, 0.50 mmol, 1.0 equiv) in CH2Cl2 (20 mL) oxalyl chloride (129 μL, 1.5 mmol, 3 equiv) was added dropwise at rt. The solution was then heated at 50 °C for 3 h. After cooling to rt, the reaction was concentrated in vacuo to afford a light yellow solid. Next, a solution of 56 (244 mg, 0.6 mmol, 1.2 equiv) in CH3CN (10 mL) was added and the resulting mixture was stirred 16 h at rt. The crude reaction was concentrated in vacuo and the residue was partitioned between CH2Cl2 (25 mL) and H2O (25 mL). The aqueous layer was extracted with CH2Cl2 (2×25 mL). The combined organic extracts were dried (Na2SO4) and concentrated under reduced pressure. Purification by flash chromatography (7:3 EtOAc/Hexane) afforded the title compound (290 mg, 88%) as a white solid: Rf 0.10 (6:4 EtOAc/Hexane); 1H NMR (CDCl3, 600 MHz) δ 1.35 (s, 3H), 1.54 (s, 9H), 1.59 (s, 3H), 3.64–3.71 (m, 2H), 4.41–4.44 (m, 1H), 4.99 (dd, J = 6.0, 3.6 Hz, 1H), 5.28 (s, 2H), 5.37 (dd, J = 6.0, 3.0 Hz, 1H), 6.12 (d, J = 3.0 Hz, 1H), 7.05 (d, J = 8.4 Hz, 1H), 7.10 (t, J = 7.8 Hz, 1H), 7.35–7.37 (m, 1H), 7.40–7.43 (m, 4H), 7.48 (dt, J = 8.4, 1.2 Hz, 1H), 8.05 (br s, 1H, NH), 8.12 (dd, J = 7.8, 1.2 Hz, 1H), 8.18 (s, 1H), 8.74 (s, 1H), 8.85 (t, J = 6.0 Hz, 1H, NH), 9.97 (s, 1H, NH); 13C NMR (CDCl3, 150 MHz) δ 25.7, 27.5, 28.3, 41.5, 71.8, 81.9, 82.5, 84.2, 85.3, 90.4, 113.5, 115.2, 119.9, 122.1, 122.3, 127.7, 129.1, 129.2, 132.9, 134.9, 135.0, 141.7, 149.7, 150.2, 150.7, 153.4, 153.9, 157.2, 165.9; HRMS (ESI+) calcd for C33H38N7O8 [M+H]+ 660.2776, found 660.2667 (error 16.5 ppm).
5′-Amino-5′-N-{[(2-benzyloxybenzoyl)amino]carbonyl}-5′-deoxyadenosine (62)
This was prepared from 61 (100 mg, 0.15 mmol) using the general procedure for TFA deprotection. Purification by flash chromatography (500:3 EtOAc/MeOH) afforded the title compound (55 mg, 70%): Rf 0.3 (500:3 EtOAc/MeOH); 1H NMR (CD3OD, 600 MHz) δ 3.64 (dt, J = 14.4, 4.8 Hz, 1H), 3.76 (dt, J = 14.4, 6.0 Hz, 1H), 4.18 (q, J = 4.8 Hz, 1H), 4.40 (t, J = 5.4 Hz, 1H), 4.72 (t, J = 5.4 Hz, 1H), 5.31 (s, 2H), 5.99 (d, J = 5.4 Hz, 1H), 7.09 (t, J = 7.8 Hz, 1H), 7.24 (d, J = 8.4 Hz, 1H), 7.30 (t, J = 7.2 Hz, 1H), 7.36 (t, J = 7.8 Hz, 2H), 7.46 (d, J = 7.8 Hz, 2H), 7.54 (t, J = 7.2 Hz, 1H), 7.91 (d, J = 7.8 Hz, 1H), 8.15 (s, 1H), 8.29 (s, 1H), 8.96 (br t, J = 6.0 Hz, 1H, 5′-NH); 13C NMR (CD3OD, 150 MHz) δ 40.7, 71.2, 71.4, 74.0, 83.2, 89.2, 113.8, 119.4, 120.5, 121.5, 127.6, 128.3, 128.7, 131.6, 134.6, 135.8, 140.7, 149.4, 151.8, 154.7, 155.5, 157.2, 166.6; HRMS (ESI−) calcd for C25H24N7O6 [M−H]− 518.1794, found 518.1795 (error 0.1 ppm).
5′-Amino-5′-deoxy-5′-N-{[(2-hydroxybenzoyl)amino]carbonyl}adenosine (63)
To a solution of 62 (50 mg, 0.096 mmol) in MeOH (10 mL) was added 10% Pd/C (10 mg, 20% by wt) and the reaction stirred at rt under H2 (1 atm). After 2 h, the reaction was filtered through Celite and the filtrate concentrated under reduced pressure. Purification by flash chromatography (10:2 EtOAc/MeOH) afforded the title compound (21 mg, 51%): Rf 0.12 (100:20 EtOAc/MeOH); 1H NMR (CD3OD, 600 MHz) δ 3.66 (dd, J = 14.4, 4.2 Hz, 1H), 3.80 (dd, J = 14.4 Hz, 4.8 Hz, 1H), 4.20 (q, J = 4.8 Hz, 1H), 4.42 (t, J = 4.8 Hz, 1H), 4.74 (t, J = 4.8 Hz, 1H), 6.00 (d, J = 4.8 Hz, 1H), 6.90 (ovlp t, J = 7.8 Hz, 1H), 6.91 (ovlp d, J = 7.8 Hz, 1H), 7.40 (t, J = 7.8 Hz, 1H), 7.91 (d, J = 7.8 Hz, 1H), 8.16 (s, 1H), 8.32 (s, 1H); 13C NMR (CD3OD, 150 MHz) δ 40.7, 71.2, 73.9, 83.3, 89.0, 116.7, 117.4, 119.3, 119.4, 130.9, 134.8, 140.4, 149.5, 152.7, 155.1, 156.1, 158.9, 167.5; HRMS (ESI−) calcd for C18H18N7O6 [M−H]− 428.1324, found 428.1327 (error 0.7 ppm).
Enzyme kinetic studies. ATP/PPi Exchange Assay.30
MbtA was expressed in E. coli and purified as described.15 The inhibition assays were performed as described in duplicate.15 In brief the reaction was initiated by adding 10 μL [32P]PPi with 7 nM MbtA in 90 μL reaction buffer (250 μM salicylic acid, 10 mM ATP, 1 mM PPi, 75 mM Tris-HCl, pH 7.5, 10 mM MgCl2, 2 mM DTT) at 37°C in the presence of five different concentrations of the inhibitor. The reaction was terminated by the addition of 200 μL of quenching buffer (350 mM HClO4, 100 mM PPi, 1.8 % w/v activated charcoal). The charcoal was pelleted by centrifugation and washed once with 500 μLH2O and analyzed by liquid scintillation counting as described.30 The KIapp values were calculated using either the Hill Morrison (eq 1) or Morrison (eq 2) equations as described.15, 31
| (1) |
| (2) |
M. tuberculosis H37Rv MIC Assay
All compounds MICs were experimentally determined as previously described.11 Minimum inhibitory concentrations (MICs) were determined in quadruplicate in iron-deficient GAST and GAST supplemented with 200 μM FeCl3 according to the broth microdilution method using compounds from DMSO stock solutions or with control wells treated with an equivalent amount of DMSO. Isoniazid was used as positive controls while DMSO was employed as a negative control. All measurements reported herein used an initial cell density of 104–105 cells/assay and growth monitored at 10–14 days, with the untreated and DMSO-treated control cultures reaching an OD620 0.2~0.3. Plates were incubated at 37 °C (100 μL/well) and growth was recorded by measurement of optical density at 620 nm.
Docking Studies
The homology model and docking runs were configured as described12, except that the side chains of residues 235, 240, 329, and 337 were allowed to move along with the ligand.
Supplementary Material
Table of HPLC purities of the inhibitors disclosed herein. This material is available free of charge via the Internet at http://pubs.acs.org.
Acknowledgments
This research is supported by a grant from NIH (R01AI070219) and funding from the Center for Drug Design in the Academic Health Center of the University of Minnesota to C.C.A. We thank the Minnesota Supercomputing Institute VWL lab for computer time.
ABBREVIATIONS
- CDI
carbonyl diimidazole
- DBU
1,8-diazabicyclo[5.4.0]undec-7-ene
- MDR-TB
multidrug resistant tuberculosis MIC, minimum inhibitory concentration
- NHS
N-hydroxysuccinimide
- NRPS
nonribosomal peptide synthetase
- PAS
para-aminosalicylic acid
- PDB
protein data bank
- PKS
polyketide synthase
- SAR
structure activity relationships
- TB
tuberculosis
- TBAF
tetrabutylammonium fluoride
- TBS
tert-butyldimethylsilyl
- TFA
trifluoroacetic acid
References
- 1.World Health Organization 2005 Fact sheet on tuberculosis, http://www.who.int/mediacentre/factsheets/fs104/en/print.html.
- 2.Ratledge C, Dover LG. Iron metabolism in pathogenic bacteria. Annu Rev Microbiol. 2000;54:881–941. doi: 10.1146/annurev.micro.54.1.881. [DOI] [PubMed] [Google Scholar]
- 3.Posey JE, Gherardini FC. Lack of a role for iron in the lyme disease pathogen. Science. 2000;288:1651–1653. doi: 10.1126/science.288.5471.1651. [DOI] [PubMed] [Google Scholar]
- 4.Raymond KN, Dertz EA, Kim SS. Enterobactin: an archetype for microbial iron transport. Proc Natl Acad Sci U S A. 2003;100:3584–3588. doi: 10.1073/pnas.0630018100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Crosa JH, Walsh CT. Genetics and assembly line enzymology of siderophore biosynthesis in bacteria. Microbiol Mol Biol Rev. 2002;66:223–249. doi: 10.1128/MMBR.66.2.223-249.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Vergne AF, Walz AJ, Miller MJ. Iron chelators from mycobacteria (1954–1999) and potential therapeutic applications. Nat Prod Rep. 2000;17:99–116. doi: 10.1039/a809397k. [DOI] [PubMed] [Google Scholar]
- 7.Snow GA. Mycobactins: iron-chelating growth factors from mycobacteria. Bacteriol Rev. 1970;34:99–125. doi: 10.1128/br.34.2.99-125.1970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Quadri LEN, Sello J, Keating TA, Weinreb PH, Walsh CT. Identification of a Mycobacterium tuberculosis gene cluster encoding the biosynthetic enzymes for assembly of the virulence-conferring siderophore mycobactin. Chem Biol. 1998;5:631–645. doi: 10.1016/s1074-5521(98)90291-5. [DOI] [PubMed] [Google Scholar]
- 9.Schimmel P, Tao J, Hill J. Aminoacyl t-RNA synthetases as targets for new anti-infectives. FASEB J. 1998;12:1599–1609. [PubMed] [Google Scholar]
- 10.Kim S, Lee SW, Choi EC, Choi SY. Aminoacyl-tRNA synthetases and their inhibitors as a novel family of antibiotics. Appl Microbiol Biotechnol. 2003;61:278–288. doi: 10.1007/s00253-003-1243-5. [DOI] [PubMed] [Google Scholar]
- 11.Somu RV, Boshoff H, Qiao CH, Bennett EM, Barry CE, Aldrich CC. Rationally designed nucleoside antibiotics that inhibit siderophore biosynthesis of Mycobacterium tuberculosis. J Med Chem. 2006;49:31–34. doi: 10.1021/jm051060o. [DOI] [PubMed] [Google Scholar]
- 12.Vannada J, Bennett EM, Wilson DJ, Boshoff HI, Barry CE, 3rd, Aldrich CC. Design, synthesis, and biological evaluation of beta-ketosulfonamide adenylation inhibitors as potential antitubercular agents. Org Lett. 2006;8:4707–4710. doi: 10.1021/ol0617289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Qiao C, Wilson DJ, Bennett EM, Aldrich CC. A mechanism-based aryl carrier protein/thiolation domain affinity probe. J Am Chem Soc. 2007;129:6350–6351. doi: 10.1021/ja069201e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ferreras JA, Ryu JS, Di Lello F, Tan DS, Quadri LE. Small-molecule inhibition of siderophore biosynthesis in Mycobacterium tuberculosis and Yersinia pestis. Nat Chem Biol. 2005;1:29–32. doi: 10.1038/nchembio706. [DOI] [PubMed] [Google Scholar]
- 15.Somu RV, Wilson DJ, Bennett EM, Boshoff HI, Celia L, Beck BJ, Barry CE, 3rd, Aldrich CC. Antitubercular nucleosides that inhibit siderophore biosynthesis: SAR of the glycosyl domain. J Med Chem. 2006;49:7623–7635. doi: 10.1021/jm061068d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Miethke M, Bisseret P, Beckering CL, Vignard D, Eustache J, Marahiel MA. Inhibition of aryl acid adenylation domains involved in bacterial siderophore synthesis. FEBS J. 2006;273:409–419. doi: 10.1111/j.1742-4658.2005.05077.x. [DOI] [PubMed] [Google Scholar]
- 17.Pfleger BF, Lee JY, Somu RV, Aldrich CC, Hanna PC, Sherman DH. Characterization and analysis of early enzymes for petrobactin biosynthesis in Bacillus anthracis. Biochemistry. 2007;46:4147–4157. doi: 10.1021/bi6023995. [DOI] [PubMed] [Google Scholar]
- 18.Castro-Pichel J, García-López MT, De las Heras FG. A facile synthesis of ascamycin and related analogues. Tetrahedron. 1987;43:383–389. [Google Scholar]
- 19.For complete characterization, including HRMS data see the Experimental Section. The additional -(C=O)CH2CH2- fragment was confirmed by NMR (The atoms for this fragment are numbered as: C=O = C-1, α-CH2 = C-2, β-CH2 = C-3). 1H NMR: 2.40 (t, J = 6.6 Hz, 2H, H-2) and 3.42 (t, J = 6.6 Hz, 2H, H-3); 13C NMR: 181.4 (C=O, C-1), 40.3 (C-2), 38.8 (C-3). HMBC H-2 (coupled to: C-1, C-3), H-3 (coupled to: C-1, H-2, and C=O at 160.2 from (morpholino)carbonyl fragment).
- 20.Vasilevich NI, Sachinvala ND, Maskos K, Coy DH. Selective conversion of O-succinimidyl carbamates to N-(O-carbamoyl)-succinmonoamides and ureas. Tetrahedron Lett. 2002;43:3443–3445. [Google Scholar]
- 21.May JJ, Kessler N, Marahiel MA, Stubbs MT. Crystal structure of DhbE, an archetype for aryl acid activating domains of modular nonribosomal peptide synthetases. Proc Natl Acad Sci U S A. 2002;99:12120–12125. doi: 10.1073/pnas.182156699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Liu F, Austin DJ. A general synthesis of 5′-azido-5′-deoxy-2′.3′-O-isopropylidene nucleosides. J Org Chem. 2001;66:8643–8645. doi: 10.1021/jo015800m. [DOI] [PubMed] [Google Scholar]
- 23.Yu M, Magalhães MLB, Cook PF, Blanchard JS. Bisubstrate inhibition: Theory and application to N-acetyltransferases. Biochemistry. 2006;45:14788–14794. doi: 10.1021/bi061621t. [DOI] [PubMed] [Google Scholar]
- 24.May JJ, Wendrich TM, Marahiel MA. The dhb operon of Bacillus subtilis encodes the biosynthetic template for the catecholic siderophore 2,3-dihydroxybenzoate-glycine-threonine trimeric ester bacillibactin. J Biol Chem. 2001;276:7209–7217. doi: 10.1074/jbc.M009140200. [DOI] [PubMed] [Google Scholar]
- 25.Cendrowski S, MacArthur W, Hanna P. Bacillus anthracis requires siderophore biosynthesis for Growth in macrophages and mouse virulence. Mol Microbiol. 2004;51:407–417. doi: 10.1046/j.1365-2958.2003.03861.x. [DOI] [PubMed] [Google Scholar]
- 26.Keana JFW, Cai SX. New reagents for photoaffinity-labeling - Synthesis and photolysis of functionalized perfluorophenyl azides. J Org Chem. 1990;55:3640–3647. [Google Scholar]
- 27.Trivedi OA, Arora P, Sridharan V, Tickoo R, Mohanty D, Gokhale RS. Enzymatic activation and transfer of fatty acids as acyl-adenylates in mycobacteria. Nature. 2004;428:441–445. doi: 10.1038/nature02384. [DOI] [PubMed] [Google Scholar]
- 28.Gokhale RS, Saxena P, Chopra T, Mohanty D. Versatile polyketide enzymatic machinery for the biosynthesis of complex mycobacterial lipids. Nat Prod Rep. 2007;24:267–277. doi: 10.1039/b616817p. [DOI] [PubMed] [Google Scholar]
- 29.Tse B, Kishi Y. Conformationally rigid tricyclic tripods: Synthesis and application to preparation of enterobactin analogs. J Org Chem. 1994;59:7807–7814. [Google Scholar]
- 30.Linne U, Marahiel MA. Reactions catalyzed by mature and recombinant nonribosomal peptide synthetases. Methods Enzymol. 2004;388:293–315. doi: 10.1016/S0076-6879(04)88024-8. [DOI] [PubMed] [Google Scholar]
- 31.Copeland RA. Evaluation of Enzyme Inhibitors in Drug Discovery. Wiley; Hoboken, NJ: 2005. [PubMed] [Google Scholar]
- 32.(a) Hu J, Miller MJ. Total synthesis of mycobactin S, a siderophore growth promoter of Mycobacterium smegmatis, and determination of its growth inhibitory activity against Mycobacterium tuberculosis. J Am Chem Soc. 1997;119:3462–3468. [Google Scholar]; (b) Xu Y, Miller MJ. Total synthesis of mycobactin analoguesas potent antimycobacterial agents using a minimal protecting group strategy. J Org Chem. 1998;63:4314–4322. [Google Scholar]; (c) Walz AJ, Mollmann U, Miller MJ. Synthesis and studies of catechol-containing mycobactin S and T analogs. Org Biomol Chem. 2007;5:1621–1628. doi: 10.1039/b703116e. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table of HPLC purities of the inhibitors disclosed herein. This material is available free of charge via the Internet at http://pubs.acs.org.