

Abstract
Effective targeting of drugs to cells requires that the drug reach the target cell and interact specifically with it. In this study we synthesized a biomacromolecular, multivalent construct intended to target glioblastoma tumors. The construct was created by linking three dodecapeptides, reported to bind the α6β1 integrin, with poly(ethylene glycol) linkers. The construct is intended to be delivered locally, and it demonstrates a more homogenous and more rapid perfusion profile in comparison with quantum dots. The binding specificity of the construct was investigated using glioblastoma cells and normal human astrocyte cells. The results reveal qualitative differences in binding between glioma and normal human astrocyte cells, with a moderate increase in binding avidity due to multivalency (0.79 μM for the trivalent construct versus 4.28 μM for the dodecapeptide). Overall, biomacromolecular constructs appear to be a promising approach for targeting with high biocompatibility, good perfusion abilities, and specificity.
Introduction
Drug targeting promises to improve treatment of cellular pathologies such as cancer by allowing earlier detection, determining the position of the tumor(s), and increasing local concentration of therapeutics at the tumor sites.1 One method for accomplishing this is to conjugate an imaging or therapeutic modality to a ligand or antibody for a cell surface receptor over-expressed by the target cells.2, 3 The conjugation can take many forms including linkage of the targeting molecule via a short polymeric linker,4 or by conjugating to nanoscale materials such as liposomes,5 nanoshells,6 or quantum dots.7
Recent interest in nanoparticles such as nanoshells and quantum dots has led to several concerns for these technologies. First, it is likely that much of the targeting achieved by these nanoparticles is due to their size and not receptor-ligand binding. Experiments have shown enhanced accumulation of poly(ethylene glycol) (PEG) coated nanoparticles at tumor sites.8 Likewise these particles accumulate in the liver and spleen in even greater concentrations than they do at the tumor site.9 Alone this might not present a major problem, but most nanoparticles studied to date are not rapidly degraded which will likely lead to phagocytosis by macrophages and, in the case of quantum dots, this leads to concerns of toxicity from the metals in their cores.10
Polymeric constructs have the potential to avoid both of these concerns, but they have not been developed or characterized sufficiently to realize this potential. Drug-antibody conjugates are the most developed targeting constructs; however, antibodies are large – almost as large as nanoparticles. Caplan and Rosca demonstrated that increased specificity due to multivalency can only be reached at ligand concentrations less than the affinity of the receptor ligand interactions.11 Antibodies bind very tightly to their antigens (KA ~ 1011), thus specificity at nM or μM concentration would be no greater than the ratio of receptor overexpression.12 Investigations using peptide ligands have also been shown to mediate binding of targeting constructs to cell surface receptors,13 but to date these constructs have not been shown to target pathological cells in greater concentration than they do non-pathological cells. Additionally, it has not been demonstrated that these smaller constructs exhibit more rapid diffusion and/or perfusion in tissue.14
In this study we synthesized and characterized the function of a biomacromolecular, multivalent targeting construct. A dodecapeptide ligand, reported to bind specifically to the α6β1-integrin,15 was linked via PEG to create a construct consisting of three identical ligands. The avidity of this construct was determined relative to the unlinked dodecapeptide ligand. Cellular specificity is assayed by exposing a glioblastoma cell line and a normal human astrocyte cell line to fluorescently-labeled multivalent constructs. Finally the concentration profile of these biomacromolecular constructs was compared to quantum dots in a perfusion assay. These characterizations tested the hypothesis that a relatively small biomacromolecular construct will achieve specific targeting while enhancing its ability to perfuse/diffuse through tissue analogs.
Experimental Details
Synthesis of constructs
Peptides were synthesized using TentaGel R-NH2 resin (Rapp Polymere, Germany), and all Fmoc protected amino acids were purchased from Novabiochem (San Diego, CA). All additional chemicals were purchased from Sigma-Aldrich (St. Louis, MO). The synthesis was accomplished using standard solid phase synthesis described in detail by Handl et al.13 After completion of each dodecapeptidetide (TWYKIAFQRNRK), three molecules of PEG were added manually via the following protocol. The N-terminus of the previous peptide segment was de-protected and the resin was incubated with 2 molar equivalence of Fmoc-NH-(PEG)2-COOH (20 atoms, Nα-Fmoc-19-amino-5-oxo-3,10,13,16-tetraoxa-6-azanonadecan-1-oic acid, Novabiochem) and 2-(1H-9-Azobenzotriazole-1-yl)-1,1,3,3-tetramethylaminium Hexafluorophosphate (HATU) in a 3:1 mixture of Dichloromethane: N, N,-Dimethylformamide. The reaction was allowed to proceed overnight. This procedure was repeated two additional times to create a linker containing three molecules of PEG. After this manual addition, the second peptide sequence was synthesized using the automatic synthesizer as above. The trivalent construct consists of three peptide sequences and two linkers (each containing three PEG molecules). After synthesis was completed, the protecting groups were cleaved, and the final peptide construct was precipitated using standard techniques.4 The molecular masses of the constructs were checked using MALDI-TOF, and the products were purified using reverse phase chromatography as described in detail by Handl et al.13 Constructs for imaging were synthesized in the same manner; however, at the end of the sequence a biotin molecule was added. The Europium binding assays were in competition with Europium-labeled dodecapeptide which was synthesized by adding a chelator molecule, Diethylenetriaminepentaacetic Acid (DTPA), for Europium to a dodecapeptide as described in detail by Handl et al.13
Europium binding assays
Normal human astrocytes (NHA) and glioma cells (SF767) were generously supplied by Michael Berens (Translational Genomic Institute, Phoenix, Arizona). Cells were maintained under standard conditions (37 °C and 5% CO2) and were grown in Dulbecco’s modified Eagle medium (DMEM) supplemented with 10% bovine growth serum (BGS). The day prior to the experiment, cells were passaged. On the day of the experiments, the cells were harvested using PBS/EDTA (7mM EDTA) and then incubated in DMEM supplemented with 3% bovine serum albumin. 50,000 cells were added to each well in a round bottom 96-well plate (Sarstead). The cells were incubated with 50 μL of binding media (DMEM, 1mM 1,10-phenanthroline, 200mg/L bacitracin, 0.5mg/L leupeptin, and 3% BSA) and 50 μL of un-labeled construct in binding media. After 30 minutes incubation at 37 °C and 5% CO2, 50 μL of Eu-dodecapeptide in binding media was added and incubated with the cells for an additional 30 minutes. Following incubation, cells were washed three times with wash buffer (50mM Tris-HCl, 30mM NaCl, and 0.5% BSA). Enhancement solution (Perkin Elmer) was added (100 μL/well) and incubated at 37 °C and 5% CO2, for approximately 1 hour. Cells were pelleted at 700 rpm for 10 minutes, and the supernatant was transferred to a 96-well black, clear-bottom plate and fluorescence was read on Wallac Victor3 V instrument using the standard Time Resolved Fluorescence (TRF) protocol (emission 340 nm, excitation 615 nm). Each data point is the average of quadruplicates and the error bars represent standard error of the mean.
The affinities (KD) of constructs were calculated by fitting a scalar factor to theoretical binding, calculated using a well established cell surface receptor binding model,16 and then comparing it to the binding assay data: saturation assay for the Eu-dodecapeptide and competitive assay for the dodecapeptide and trivalent constructs.
Imaging of binding
Visual investigation of binding was performed on the same cells using the biotinylated constructs. Prior to the assay the cells were labeled with CellTracker blue (SF767) or green (NHA) (Molecular Probes, Invitrogen). Cells were incubated with biotinylated constructs (trivalent) for 30 minutes, at 37 °C and 5% CO2. Binding was visualized using streptavidin conjugated to Texas Red fluorophore (Product #21624, Pierce) using the manufacturer recommended protocol. Random fields (2 per well) were chosen and imaged. Contrast levels for each image were adjusted in Adobe Photoshop using the auto levels feature to allow visualization of all cells. An area corresponding to a cell was selected and the pixel intensity was calculated using the histogram tool (mean pixel intensity). This measurement was performed for each cell type, and a t-test was performed to determine if the mean pixel intensity was significantly different between the two cell types.
Perfusion
Perfusion studies were performed in 0.6 % agarose gels molded in 24 well plates. Either FITC-conjugated construct (FITC-trivalent) or quantum dots (Qtracker® 655, Molecular Probes) were injected (22 gauge blunt end needle) into the gel at a rate of 3μL/min using a syringe pump (Harvard Instruments) for two hours. The perfusion distance was measured in Adobe Photoshop (version 5.0.2) using concentric circles generated by choosing pixels of similar intensity by setting a low tolerance level (10) for the tool. Three measurements were performed in each set of concentric circles, North, East, South, and the background fluorescence was measured at the edge of the well. The pixel intensities were normalized against the intensity at the site of injection where the concentration should equal that of the injectate. The error bars depict standard error of the mean for the measured distance.
Toxicity
The level of toxicity of the construct was assessed using the LIVE/DEAD Viability/Cytotoxicity Kit (Molecular Probes). Cells were plated in a 96-well plate at a density of5000 cell/well (4 replicates) and incubated in the presence of construct (concentration varied from 30 μM to 30 nM) for five days. Cells were labeled using the cytotoxicity kit according to the manufacturer protocol, and two images of randomly selected fields were collected per well. The contrast of the image was adjusted in Adobe Photoshop to allow visualization of all cells. The number of dead cells was counted visually, and the number of live cells was calculated by dividing the number of green pixels by the average number of pixels per cell. Error bars depict standard error of the mean.
Results
Construct synthesis
Figure 2 illustrates the typical mass spectroscopy results for the trivalent construct. The group of peaks at 5966, 6285, 6603, and 6922 Da represents a mixture of trivalent constructs linked by one, two or three PEG molecules per linker, for a total of three, four, five or six molecules per construct. The peaks centered around 3200 Da are the corresponding doubly charged products. Products containing a total of four, five and six PEG molecules are the most abundant. Isolation of trivalent constructs containing linkers of different lengths was difficult due to similarity in degree of hydrophilicity, therefore all of these compounds eluted in the same chromatography fraction.
Figure 2.
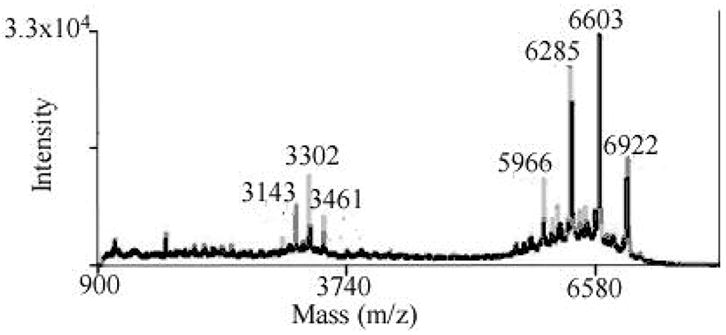
MALDI-TOF results of the trivalent construct. The major group of peaks represents the mass of constructs containing three (5966 Da), four (6284 Da), five (6603 Da) and six (6921Da) molecules of PEG. The group of peaks around 3200 Da represents the corresponding mass of doubly charged molecules.
Binding activity
The activity of the constructs was investigated through binding assays. Since the constructs were created using peptide sequences demonstrated to bind to integrins,15, 18 binding assays were performed using non-adherent cells to ensure that all integrins were available for binding. Cells were harvested using a PBS/EDTA mixture to prevent the cleavage of the integrins, and then incubated with the ligand. The assay is competitive, in which the binding of the Eu-dodecapeptide is competed off by either the dodecapeptide or the trivalent construct. The results in Figure 3 illustrate the percent binding of the labeled competitor to glioma cells in the presence of each construct. It was expected that the trivalent construct would exhibit stronger avidity in comparison to the dodecapeptide due to cooperativity of multiple ligands. We found the value of the affinity for the trivalent construct and the dodecapeptide by minimizing the square of the error between theoretical and actual binding to cells.16 Replicates were obtained by measuring additional binding vs. concentration curves and fitting EC50 values for each curve (DDP, n=8; Eu-DDP, n=4; Trivalent, n=8). The trivalent construct was best fit by an affinity (KD) value of 0.79μM in comparison with the weaker affinity of the dodecapeptide of approximately 4.28 μM. The two values are statistically significant (p<0.05) based on t-test.
Figure 3.
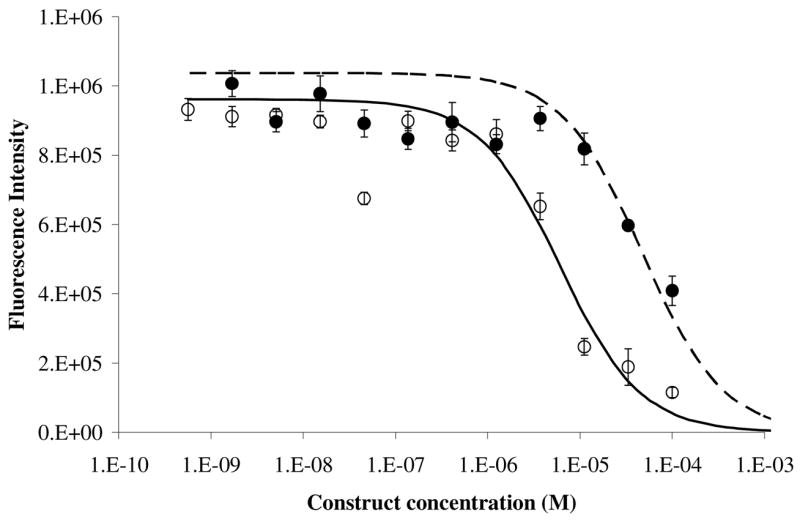
Competitive binding of dodecapeptide (solid circle) and trivalent construct (open circles) to glioblastoma cells. Trivalent construct exhibits an avidity of approximately 0.79 μM while the dodecapeptide has an affinity of approximately 4.28 μM. Continuous lines depict theoretical binding fit for the affinity constant, dashed (dodecapeptide) and solid (trivalent). Error bars depict standard error of the mean.
Imaging of binding
To visualize binding of the constructs to the cells, we examined the binding of biotin-Trivalent construct with cells. Prior to the experiment, the two cell types, astrocytes and glioblastoma cells were labeled with green and blue cell tracker respectively, and the construct was detected using streptavidin conjugated to Texas Red fluorophore. Figure 4 shows a field of cells for which green/blue (4A) and red (4B) fluorescence were imaged. Figure (4B) shows that glioma cells, denoted by filled arrow heads, are consistently outlined by the trivalent constructs; whereas, normal human astrocytes, denoted by open arrow heads, are not. Although total fluorescence per cell shows no statistical significance (average intensity per pixel), there is a qualitative difference in the binding to the two cell types. The glioma cells consistently display peripheral staining, outlining the cells, most likely indicative of binding to integrins, while some but not all of the normal astrocytes exhibit punctuate staining throughout the cell. This is possibly indicative of non-specific staining or internalization of construct. Because the punctuate staining is particularly intense, no statistical difference in intensity/pixel is observed due to large variance in the astrocyte data (data not shown).
Figure 4.
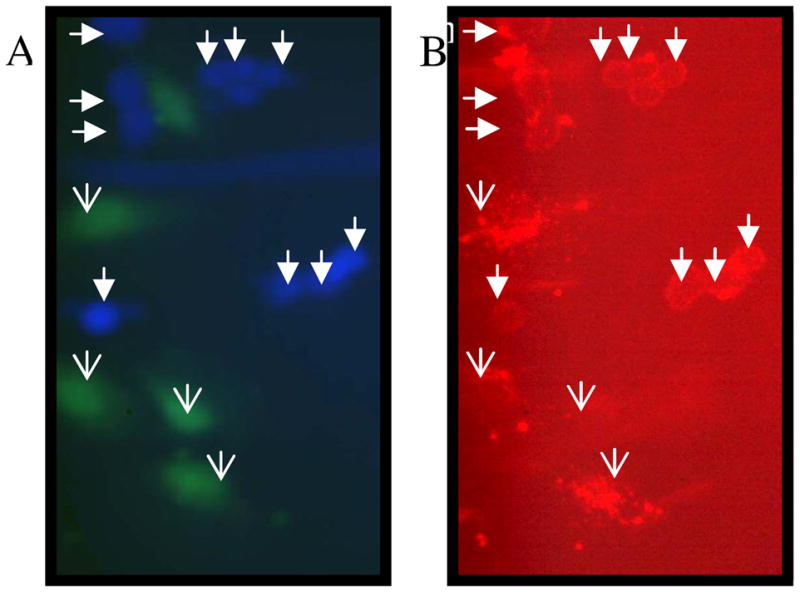
Perfusion studies
The constructs investigated in this study are directed toward glioblastoma cells, and it is possible to envision future application to specific targeting by direct injection to the tumor site via intracranial cannula (i.e. Ommaya reservoir or intrathecal cannula).19 To simulate such conditions we injected the constructs into 0.6 % agarose gels, a typical brain tissue mimetic.20 The constructs were delivered using a flow rate of 3 μL/min, a practical injection rate for intracranial delivery, for 2 hours.21 The time of infusion was determined experimentally as the optimal elapsed time for visualization. The multivalent constructs demonstrate good perfusion qualities, a circular pattern of perfusion with an approximate radius of 8.2 mm versus the quantum dots with an area of approximately 5.2 mm at a normalized concentration of 35% of the initial injection concentration. Statistical analyses revealed significant difference (p<0.05) in the distance perfused at percent concentration of 63, 44, 35, and 29%. To characterize this difference we fit the data to a well established mass transport model of flow through porous media by fitting the diffusion coefficient (D) and the retardation coefficient (f), which characterize the diffusion and the hindrance of convective transport through porous media.22 We found that the profile of the multivalent construct was best fit by D = 6×10−6 cm2/sec and f = 1, (a value greater than one would provide a better fit; however f = 1 represents no retardation), while the profile of the quantum dots was best fit by D = 1×10−6 cm2/sec and f = 0.25.
Discussion
One of the reasons to synthesize biomacromolecular constructs is to provide constructs that are eliminated from the body after injection. Here we synthesized, biodegradable multivalent constructs of low molecular weight (<8000 Da). Since molecules that are smaller than 40 kDa can pass through the kidney, these constructs can likely be eliminated without the need to be degraded.23 Additionally the peptide segments of these molecules will likely undergo hydrolysis further decreasing the size of any remaining constructs thus allowing their elimination. Achieving this goal, however, raises the concern that intravenous injection of such low molecular weight constructs could lead to their clearance from the blood before achieving efficacious targeting.23 This concern can be avoided by using localized injection near the site suspected to contain tumors.
For the purposes of this study, we created constructs that are targeted to glioblastoma tumors in the brain for which local delivery is preferred due to the presence of the blood-brain barrier. Injection of therapeutic molecules directly into brain tissue has been investigated and used clinically;21, 24 however, this technique presupposes knowledge of the tumor site. Combining this technique with targeting constructs allows the clinician to inject in a region suspected to contain tumor sites, and the construct will theoretically establish concentration gradients within the tissue based on receptor-ligand binding.25 Constructs that do not bind to cells will eventually diffuse/perfuse away from the injection site and/or be taken up into the blood stream from which they will be rapidly cleared. Thus, rapid clearance of these constructs is likely to enhance contrast between the tumor site and normal tissue while at the same time minimizing accumulation of drug in the liver and spleen.
The ability of the constructs to perfuse through tissue will determine the size of the region that the clinician can image or treat with one injection of the constructs. Here we compare the perfusion of biomacromolecular constructs with quantum dots in a tissue analog. The data show that biomacromolecular constructs perfuse more readily and more homogeneously than do quantum dots. This is further indication that quantum dots injected intravenously are likely limited to delivery to tissues in which the vasculature has been permeabilized, most likely by growth factors, near well-established tumors.26 Direct injection into the tissue will allow imaging and treatment of nascent tumors that have not permeabilized their vasculature. In addition to these considerations, the data indicate that polymeric constructs injected locally can perfuse a region of approximately 3 cm diameter in two hours. Extrapolation based on best-fit retardation and diffusion coefficients suggests that these constructs could image or treat a region of approximately 7.5 cm in diameter after 12 hour injection duration. Although several of these injections at various positions in the brain may need to be performed concurrently to image or treat the entire brain, it is likely that this would be clinically viable if the increase in patient prognosis is substantial.
It has been demonstrated that linker length and flexibility play a critical role in effective binding of multimeric constructs. A study performed by Arnold et al. demonstrated that a separation of 25–73 nm leads to integrin clustering, i.e. binding of multiple integrins.27 In our constructs each NH2-(PEG)2-COOH molecule is approximately 10 nm, in the elongated form; thus, constructs with three PEG molecules per linker (30nm in elongated form) are likely to be able to bind multiple integrins.
The mechanisms of binding, increased avidity, and effects of linkers on multivalent constructs have been reviewed extensively.13, 28, 29 In this study we observe that the trivalent construct exhibits slightly stronger avidity than does the dodecapeptide, 0.79μM vs. 4.28μM. The Eu-dodecapeptide appears to have a stronger affinity in comparison with the dodecapeptide (0.62μM); however, the KD for the labeled dodecapeptide was determined using a saturation assay which might result in estimation of stronger KD due to readings above the linear range of the TRF at high concentrations (above 10μM). However, the difference in binding affinity might also be due to the addition of the DTPA molecule, as it has been reported in a study by Benyhe et al. to slightly enhance affinity.17 Although there is a difference in the affinity of the two constructs, dodecapeptide with or without DTPA-Eu, the increase in affinity due to multivalency is determined using competitive assay which is identical to the method used to obtain affinity of the unlabeled dodecapeptide, so the comparison between the two unlabeled constructs (DDP and trivalent) is performed using identical procedures.
Although the trivalent construct did exhibit slightly stronger avidity than the dodecapeptide, 0.79μM vs. 4.28μM, there is not as great an increase as would be expected from cooperativity. This is most likely due to sub-optimal linker properties. Krishnamurthy et al. have demonstrated that linkers longer than necessary are able to bind cooperatively but that they lose additional entropy in order to do so.29 Handl and co-workers have shown that the avidity of multivalent constructs can be greatly increased by optimization of the length and rigidity of the linkers.4 The results here could be greatly improved by such optimization. In addition to the properties of the linker, the placement of peptides within the construct most likely affects individual ligand-receptor affinity. For the trivalent construct, affinity of the peptides at the N-terminus, C-terminus, and middle of the construct may differ significantly. It would be possible to test this by making constructs in which one dodecapeptide is functional (TWYKIAFQRNRK) while the other two dodecapeptide sequences are scrambled and then repeating the competitive binding assay to quantify affinity for each of the three possible arrangements. Since cooperativity is not possible in a construct with only one functional peptide, such a study would help distinguish suboptimal avidity due to peptide placement within the construct versus penalties to cooperativity due to sub-optimal linker properties.
The α6β1-integrin was chosen because it has been shown to be over-expressed by glioma cells30 and also because a ligand for this receptor has been identified. Nakahara et al., demonstrated that the dodecameric peptide binds specifically to the α6β1-integrin but also acts on the integrin signaling of invasion by stimulating invadopodial activity. This could present a limitation to this particular choice of ligand if these constructs are not utilized either as a therapeutic or for imaging immediately followed by treatment. More importantly, this work is intended to demonstrate proof-of-principle for receptor targeting. Thus, if this issue is found to be problematic, identification of alternate ligands which do not signal for undesired cell behavior would be warranted to achieve specific targeting while avoiding this issue.
Finally, construct binding to α6β1-integrin receptors results in qualitative differences in binding between glioblastoma cells (SF767s) and normal human astrocytes (NHAs). Mathematical modeling work by two of us (Caplan and Rosca) suggests that multivalency can potentially increase the contrast between cell types beyond that achievable by monovalent drug-ligand constructs.11 According to that theoretical work, monovalent constructs will establish a ratio of binding equal to the ratio of receptor numbers on the target/non-target cells. Increasing the affinity of this single bond will increase the number of monomers bound to each cell type, but the relative numbers bound to each cell type should not change. Achieving cooperativity through multiple weak interactions, however, takes advantage of the fact that cells with a greater number of receptors are more likely to form multiple bonds with each construct. Therefore, multivalent constructs have potential to achieve specificity much greater than the ratio of receptor over-expression. Although the specificity achieved in this study is not quantified sufficiently to draw conclusions of this sort, it is promising that the trivalent construct studied shows qualitative differences between the glioblastoma (peripheral) and the normal human astrocyte (punctuate) cell lines suggesting that multivalent constructs may be bound in greater numbers to cell surface receptors on the glioblastoma cells.
Conclusion
Biomacromolecular, multivalent constructs bind with greater avidity than a dodecapeptide to glioblastoma cells. Visualization demonstrates qualitative differences in binding between glioblastoma (peripheral) and normal human astrocytes (punctuate) when fluorescently-labeled constructs are added to in vitro cell culture. The relatively small size of these constructs enhances their ability (vis-à-vis quantum dots) to perfuse and diffuse through tissue analogs. Overall, the biomacromolecular targeting constructs show promise as a biocompatible method for tumor targeting via direct injection into tissue.
Figure 1.
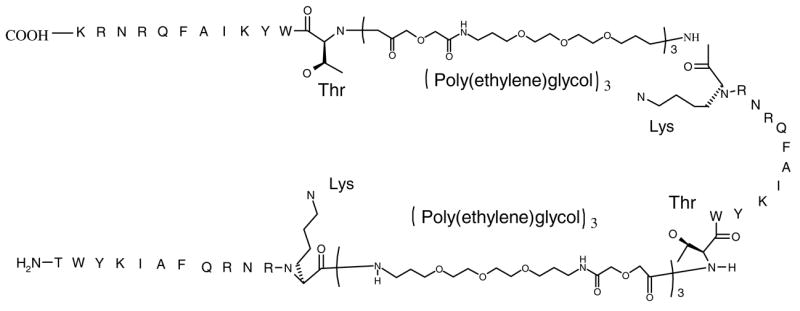
Structure of the trivalent construct. The amine end of the dodecapeptide sequence (the amine end of the Threonine) attaches to the carboxylic end of the PEG linker. The amino acid residues present at the end of the peptide sequence (Lys and Thr) are expanded for full visualization of peptide bond formation.
Figure 5.

Live/dead analysis. (A) Open symbols represent toxicity results of the dodecapeptide, triangles of the normal astrocytes, and diamonds of the glioblastoma cells. Solid symbols represent results of the toxicity of the trivalent construct, circles of the normal astrocyte cells and squares of the glioma cells. Statistical difference in toxicity levels are marked, (*) for the trivalent (p<0.01) and (#) for dodecapeptide (p<0.01). Note the compressed scale of the y-axis. Error bars denote standard error of the mean (n=4). (B) Representative picture of the live/dead assay on normal astrocyte cells. (C) Picture illustrating live/dead assay on glioma cells. Dead cells appear at the edge of the cell clusters leading to large error bars when the imaging field is selected randomly.
Figure 6.

(A) Perfusion profiles of quantum dots (diamond, n=2), dodecapeptide construct (square, n=2), and trivalent construct (circle, n=3). Continuous lines depict model results fit for the diffusion and retardation coefficients, dashed (QD) and solid (trivalent). Perfusion patterns of quantum dots (B) and trivalent construct (C) after two hours of infusion. Scale = 1mm.
Table 1.
Structure and activity of the constructs
| Construct Structure | Construct Name | EC50 ± SE (μM) |
|---|---|---|
|
|
PEG | Not applicable |
| NH2-TWYKIAFQRNRK-COOH | Dodecapeptide (DDP) | 4.28 ± 0.58 |
| DTPA(Eu)-TWYKIAFQRNRK-COOH | Eu-Dodecapeptide | 0.62 ± 0.02 |
| FITC-TWYKIAFQRNRK-COOH | FITC-Dodecapeptide | Not determined |
| Biotin-TWYKIAFQRNRK-COOH | Biotin-Dodecapeptide | Not determined |
| NH2-DDP-(PEG)3-DDP-(PEG)3-DDP-COOH | Trivalent | 0.79 ± 0.13 |
| Biotin-NH-DDP-(PEG)3-DDP-(PEG)3-DDP-COOH | Biotin-Trivalent | Not determined |
| FITC-NH-DDP-(PEG)3-DDP-(PEG)3-DDP-COOH | FITC-Trivalent | Not determined |
Acknowledgments
The authors thank our funding sources: NIH (R21 NS051310, K22 DE014386), Arizona Biomedical Research Commission Grant 0606 and our collaborators Michael Berens (Translational Genomic Institute), and Heather Maynard (UCLA). Recognition is given to Dr. Michael Berens, Dr. Dominique Hoelzinger, and Dr. Tim Demuth for assistance with obtaining and validating cell lines. We thank Dr. Heather Handl and Dr. Liping Xu for assisting with the binding assay. We also thank Dr. Dan Brune and John Lopez for their generous assistance and advice with the construct synthesis.
References
- 1.Balthasar S, Michaelis K, Dinauer N, von Briesen H, Kreuter J, Langer K. Preparation and characterisation of antibody modified gelatin nanoparticles as drug carrier system for uptake in lymphocytes. Biomaterials. 2005;26(15):2723–32. doi: 10.1016/j.biomaterials.2004.07.047. [DOI] [PubMed] [Google Scholar]
- 2.Shahied LS, Tang Y, Alpaugh RK, Somer R, Greenspon D, Weiner LM. Bispecific minibodies targeting HER2/neu and CD16 exhibit improved tumor lysis when placed in a divalent tumor antigen binding format. J Biol Chem. 2004;279(52):53907–14. doi: 10.1074/jbc.M407888200. [DOI] [PubMed] [Google Scholar]
- 3.Handl HL, Vagner J, Han H, Mash E, Hruby VJ, Gillies RJ. Hitting multiple targets with multimeric ligands. Expert Opin Ther Targets. 2004;8(6):565–86. doi: 10.1517/14728222.8.6.565. [DOI] [PubMed] [Google Scholar]
- 4.Vagner J, Handl HL, Gillies RJ, Hruby VJ. Novel targeting strategy based on multimeric ligands for drug delivery and molecular imaging: homooligomers of alpha-MSH. Bioorg Med Chem Lett. 2004;14(1):211–5. doi: 10.1016/j.bmcl.2003.09.079. [DOI] [PubMed] [Google Scholar]
- 5.Madhankumar AB, Slagle-Webb B, Mintz A, Sheehan JM, Connor JR. Interleukin-13 receptor-targeted nanovesicles are a potential therapy for glioblastoma multiforme. Mol Cancer Ther. 2006;5(12):3162–9. doi: 10.1158/1535-7163.MCT-06-0480. [DOI] [PubMed] [Google Scholar]
- 6.Gobin AM, O’Neal DP, Watkins DM, Halas NJ, Drezek RA, West JL. Near infrared laser-tissue welding using nanoshells as an exogenous absorber. Lasers Surg Med. 2005;37(2):123–9. doi: 10.1002/lsm.20206. [DOI] [PubMed] [Google Scholar]
- 7.Gao X, Cui Y, Levenson RM, Chung LW, Nie S. In vivo cancer targeting and imaging with semiconductor quantum dots. Nat Biotechnol. 2004;22(8):969–76. doi: 10.1038/nbt994. [DOI] [PubMed] [Google Scholar]
- 8.O’Neal DP, Hirsch LR, Halas NJ, Payne JD, West JL. Photo-thermal tumor ablation in mice using near infrared-absorbing nanoparticles. Cancer Lett. 2004;209(2):171–6. doi: 10.1016/j.canlet.2004.02.004. [DOI] [PubMed] [Google Scholar]
- 9.Michalet X, Pinaud FF, Bentolila LA, Tsay JM, Doose S, Li JJ, Sundaresan G, Wu AM, Gambhir SS, Weiss S. Quantum dots for live cells, in vivo imaging, and diagnostics. Science. 2005;307(5709):538–44. doi: 10.1126/science.1104274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jackson H, Muhammad O, Daneshvar H, Nelms J, Popescu A, Vogelbaum MA, Bruchez M, Toms SA. Quantum dots are phagocytized by macrophages and colocalize with experimental gliomas. Neurosurgery. 2007;60(3):524–9; . doi: 10.1227/01.NEU.0000255334.95532.DD. discussion 529–30. [DOI] [PubMed] [Google Scholar]
- 11.Caplan MR, Rosca EV. Targeting drugs to combinations of receptors: a modeling analysis of potential specificity. Ann Biomed Eng. 2005;33(8):1113–24. doi: 10.1007/s10439-005-5779-1. [DOI] [PubMed] [Google Scholar]
- 12.Alberts B, Johnson A, Lewis J, Raff M, Roberts K, Walter P. Molecular biology of the cell. 4. Garland Science, Taylor & Francis Group; New York: 2002. p. 1381. [Google Scholar]
- 13.Handl HL, Vagner J, Yamamura HI, Hruby VJ, Gillies RJ. Lanthanide-based time-resolved fluorescence of in cyto ligand-receptor interactions. Anal Biochem. 2004;330(2):242–50. doi: 10.1016/j.ab.2004.04.012. [DOI] [PubMed] [Google Scholar]
- 14.Wang Y, Yuan F. Delivery of viral vectors to tumor cells: extracellular transport, systemic distribution, and strategies for improvement. Ann Biomed Eng. 2006;34(1):114–27. doi: 10.1007/s10439-005-9007-2. [DOI] [PubMed] [Google Scholar]
- 15.Nakahara H, Nomizu M, Akiyama SK, Yamada Y, Yeh Y, Chen WT. A mechanism for regulation of melanoma invasion. Ligation of alpha6beta1 integrin by laminin G peptides. J Biol Chem. 1996;271(44):27221–4. doi: 10.1074/jbc.271.44.27221. [DOI] [PubMed] [Google Scholar]
- 16.Lauffenburger DA, Linderman JJ. Receptors Models for binding, traffciking and signaling. Oxford University Press; New York: 1993. p. 365. [Google Scholar]
- 17.Ligeti M, Gunduz O, Magyar A, Kato E, Ronai AZ, Vita C, Varga I, Hudecz F, Toth G, Borsodi A, Benyhe S. Synthesis and biological studies of nociceptin derivatives containing the DTPA chelating group for further labeling with therapeutic radionuclides. Peptides. 2005;26(7):1159–66. doi: 10.1016/j.peptides.2005.01.018. [DOI] [PubMed] [Google Scholar]
- 18.Nomizu M, Kuratomi Y, Malinda KM, Song SY, Miyoshi K, Otaka A, Powell SK, Hoffman MP, Kleinman HK, Yamada Y. Cell binding sequences in mouse laminin alpha1 chain. J Biol Chem. 1998;273(49):32491–9. doi: 10.1074/jbc.273.49.32491. [DOI] [PubMed] [Google Scholar]
- 19.Oshiro S, Tsugu H, Komatsu F, Ohnishi H, Ueno Y, Sakamoto S, Fukushima T, Soma G. Evaluation of intratumoral administration of tumor necrosis factor-alpha in patients with malignant glioma. Anticancer Res. 2006;26(6A):4027–32. [PubMed] [Google Scholar]
- 20.Chen ZJ, Gillies GT, Broaddus WC, Prabhu SS, Fillmore H, Mitchell RM, Corwin FD, Fatouros PP. A realistic brain tissue phantom for intraparenchymal infusion studies. J Neurosurg. 2004;101(2):314–22. doi: 10.3171/jns.2004.101.2.0314. [DOI] [PubMed] [Google Scholar]
- 21.Chen ZJ, Broaddus WC, Viswanathan RR, Raghavan R, Gillies GT. Intraparenchymal drug delivery via positive-pressure infusion: experimental and modeling studies of poroelasticity in brain phantom gels. IEEE Trans Biomed Eng. 2002;49(2):85–96. doi: 10.1109/10.979348. [DOI] [PubMed] [Google Scholar]
- 22.Truskey GA, Yuan F, Katz DF. Transport Phenomena in Biological Systems. Pearson Prentice Hall; Upper Saddle River, New Jersey: 2004. p. 793. [Google Scholar]
- 23.DeNardo SJ, Yao Z, Lam KS, Song A, Burke PA, Mirick GR, Lamborn KR, O’Donnell RT, DeNardo GL. Effect of molecular size of pegylated peptide on the pharmacokinetics and tumor targeting in lymphoma-bearing mice. Clin Cancer Res. 2003;9(10 Pt 2):3854S–64S. [PubMed] [Google Scholar]
- 24.Shimamura T, Husain SR, Puri RK. The IL-4 and IL-13 pseudomonas exotoxins: new hope for brain tumor therapy. Neurosurg Focus. 2006;20(4):E11. doi: 10.3171/foc.2006.20.4.6. [DOI] [PubMed] [Google Scholar]
- 25.Stukel JM, Heys JJ, Caplan MR. Optimizing Delivery of Multivalent Targeting Constructs for Detection of Secondary Tumors. doi: 10.1007/s10439-008-9498-8. submitted. [DOI] [PubMed] [Google Scholar]
- 26.Folkman J. Tumor angiogenesis: therapeutic implications. N Engl J Med. 1971;285(21):1182–6. doi: 10.1056/NEJM197111182852108. [DOI] [PubMed] [Google Scholar]
- 27.Arnold M, Cavalcanti-Adam EA, Glass R, Blummel J, Eck W, Kantlehner M, Kessler H, Spatz JP. Activation of integrin function by nanopatterned adhesive interfaces. Chemphyschem. 2004;5(3):383–8. doi: 10.1002/cphc.200301014. [DOI] [PubMed] [Google Scholar]
- 28.Kiessling LL, Gestwicki JE, Strong LE. Synthetic multivalent ligands in the exploration of cell-surface interactions. Curr Opin Chem Biol. 2000;4(6):696–703. doi: 10.1016/s1367-5931(00)00153-8. [DOI] [PubMed] [Google Scholar]
- 29.Krishnamurthy VM, Semetey V, Bracher PJ, Shen N, Whitesides GM. Dependence of effective molarity on linker length for an intramolecular protein-ligand system. J Am Chem Soc. 2007;129(5):1312–20. doi: 10.1021/ja066780e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gingras MC, Roussel E, Bruner JM, Branch CD, Moser RP. Comparison of cell adhesion molecule expression between glioblastoma multiforme and autologous normal brain tissue. Journal of Neuroimmunology. 1995;57:143–53. doi: 10.1016/0165-5728(94)00178-q. [DOI] [PubMed] [Google Scholar]