

Abstract
Cytoplasmic dynein has been implicated in diverse mitotic functions, several involving its association with kinetochores. Much of the supporting evidence comes from inhibition of dynein regulatory factors. To obtain direct insight into kinetochore dynein function, we expressed a series of dynein tail fragments, which we find displace motor-containing dynein heavy chain (HC) from kinetochores without affecting other subunits, regulatory factors, or microtubule binding proteins. Cells with bipolar mitotic spindles progress to late prometaphase-metaphase at normal rates. However, the dynein tail, dynactin, Mad1, and BubR1 persist at the aligned kinetochores, which is consistent with a role for dynein in self-removal and spindle assembly checkpoint inactivation. Kinetochore pairs also show evidence of misorientation relative to the spindle equator and abnormal oscillatory behavior. Further, kinetochore microtubule bundles are severely destabilized at reduced temperatures. Dynein HC RNAi and injection of anti-dynein antibody in MG132-arrested metaphase cells produced similar effects. These results identify a novel function for the dynein motor in stable microtubule attachment and maintenance of kinetochore orientation during metaphase chromosome alignment.
Introduction
Cytoplasmic dynein has been implicated in spindle organization, chromosome capture and congression, spindle assembly checkpoint (SAC) protein removal, and anaphase chromosome motility (Vaisberg et al., 1993; Faulkner et al., 2000; Savoian et al., 2000; Sharp et al., 2000; Howell et al., 2001; Basto et al., 2004; Yang et al., 2007). The motor protein consists of two heavy chains (HC) and accessory intermediate chain (IC), light IC, and light chains. The C-terminal 380 kD of the HC includes the motor domain, which contains sites for ATP hydrolysis and microtubule binding (Gee et al., 1997; Kon et al., 2004; Reck-Peterson and Vale, 2004). The N-terminal region of the HC contains sites for dimerization and accessory subunit binding (Habura et al., 1999; Tynan et al., 2000). The ICs interact with the dynactin complex (Karki and Holzbaur, 1995; Vaughan and Vallee, 1995), which, in turn, interacts with the zeste white 10 (ZW10) complex to target cytoplasmic dynein to kinetochores and membranous organelles (Starr et al., 1998; Varma et al., 2006). Expression of the dynactin subunit dynamitin has been widely used as a tool to displace dynein from kinetochores (Echeverri et al., 1996; Starr et al., 1998).
Perturbation of diverse genes in the dynein pathway causes altered spindle morphology, prometaphase or metaphase delay, decreased rates of anaphase chromosome movement, a hyperactivated or hypoactivated SAC, spindle misorientation, and defective chromosome capture (Vaisberg et al., 1993; Echeverri et al., 1996; Faulkner et al., 2000; Savoian et al., 2000; Sharp et al., 2000; Howell et al., 2001; Basto et al., 2004; Stehman et al., 2007). The contribution of dynein itself to these functions has been difficult to discern. The levels of dynein at kinetochores are high during prometaphase and decline markedly when they become attached to spindle microtubules (Hoffman et al., 2001). The initial tangential “capture” of kinetochores by microtubules followed by rapid poleward movement occurs during the stage of elevated dynein levels during early prometaphase (Rieder and Alexander, 1990; Yang et al., 2007). Whether dynein participates in subsequent congression of chromosomes to the metaphase plate is uncertain. Cytoplasmic dynein has not been considered for a role in the end-on “attachment” of microtubules to kinetochores, a transition which parallels the dramatic loss of the motor protein from these sites (King et al., 2000). Loss of dynein is blocked by microtubule depolymerization (Echeverri et al., 1996) and by injection of anti-dynein antibody or recombinant dynamitin into dividing cells (Howell et al., 2001) with concomitant retention of the checkpoint protein Mad2 and metaphase arrest (Howell et al., 2001). These results together led to the proposal that dynein removes itself from kinetochores along with SAC proteins (Howell et al., 2001; Wojcik et al., 2001; Basto et al., 2004).
The current study was initiated to develop a means to address the specific contribution of dynein motor activity to kinetochore function while minimizing phenotypic contributions from perturbation of other proteins in the dynein pathway. We find that dynein tail fragments specifically displace endogenous dynein from kinetochores. Early stages in mitosis still occur, but removal of SAC proteins from kinetochores is largely abolished. Surprisingly, we find kinetochore microtubule attachment to be defective and unstable, suggesting a novel and basic role for the dynein motor in this process.
Results and discussion
Phenotypic characterization of dynein HC tail fragments
To define minimal boundaries for the dynein tail domain, we tested a series of tagged truncation and point mutant dynein HC constructs, which have been biochemically characterized previously (Gee et al., 1997; Tynan et al., 2000; this study). Overexpression of the tail fragments caused a pronounced increase in mitotic index (Fig. 1 B, n = 500), an effect which was eliminated by C-terminal truncation of the tail to aa 674 (C674) or N-terminal truncation to aa 300 (N300-C1140; Fig. 1 B). Addition of an N-terminal GFP tag had no appreciable effect on the expression or potency of the fragments, whereas a C-terminal GFP tag caused a severe reduction in expression and hence was not used in our analysis. Two point mutations, Loa (legs at odd angles; F580Y) and Cra1 (cramping 1; Y1055C), which are known to cause motor neuron degeneration in mice (Hafezparast et al., 2003), caused no apparent change in the potency of the tail constructs (unpublished data). Mitotic spindle defects were dramatically increased with the longer constructs (Fig. S1, A and C, available at http://www.jcb.org/cgi/content/full/jcb.200710106/DC1). Monopolar spindles were particularly prominent (∼42% of total mitotic figures; Fig. S1, A and D), which is consistent with a role for dynein in spindle pole separation (Vaisberg et al., 1993; Sharp et al., 2000). Approximately 30% of dividing cells showed normal bipolar spindle morphology, typically with normally focused spindle poles (Fig. S1, A and D). In interphase cells, the tail fragments also caused Golgi disruption, dispersion of mitochondria, and inhibition of minus end–directed mitochondrial movement (unpublished data).
Figure 1.

Displacement of endogenous dynein HC from kinetochores by dynein tail fragments. (A) Dynein fragments used in this study. (B) Mitotic index produced by expression of tail fragments in COS7 cells (n = 500). (C) Localization of the tail fragment myc-C1140 to prometaphase kinetochores in COS7 cells. (D and E) Displacement of endogenous dynein HC from prometaphase kinetochores in COS7 cells expressing the myc-C1140 tail fragment. (D) Kinetochore staining with an antibody recognizing endogenous dynein HC motor domain is highly reduced in cells expressing the tail. (E) Quantitation of dynein HC immunofluorescence at nonaligned prometaphase kinetochores (n = 35). P = 0.0025; two-tailed t test. (F) Kinetochore staining by antibody to endogenous dynein HC in control cells. Error bars indicate SD from mean from three independent experiments. Bar, 5 μM.
Kinetochore binding of dynein tail fragments and displacement of endogenous dynein
To test the ability of the dynein tail to associate with kinetochores, we examined the distribution of the fragments in transfected cells. Fragments C1140, C907, and C800 each clearly associated with kinetochores and centrosomes (Fig. 1 C and not depicted) in normal and nocodazole-treated cells (Fig. 2, A–E; and Fig. S2 A, available at http://www.jcb.org/cgi/content/full/jcb.200710106/DC1). Double labeling was used to test the effect of the tail fragments on the distribution of endogenous kinetochore proteins. A dynein motor domain–specific antibody showed clear staining at kinetochores of unaligned chromosomes in control cells (Fig. 1 F), but staining was severely reduced at kinetochores expressing the tail fragments in both untreated prometaphase cells (Fig. 1, D and E) and nocodazole-treated cells (not depicted). Quantitative analysis of this effect indicated an 86% decrease in dynein HC fluorescence on prometaphase kinetochores as compared with control cells (Fig. 1 E, n = 35). In contrast, the dynactin subunit p150Glued and the other dynein accessory proteins Lis1, NudE, ZW10, and cytoplasmic linker protein 170 (CLIP170) were all retained at kinetochores in most cells expressing the dynein tail (Fig. 2, A–E) at levels comparable to that of control cells (Fig. 2, A′–E′). These results suggest that the tail fragments specifically and selectively displace the active dynein HC motors from subcellular sites, whereas dynein targeting and regulatory factors are retained.
Figure 2.

Association of dynein accessory proteins with prometaphase kinetochores in dynein tail–expressing cells. COS7 cells transfected with the myc-C1140 construct were treated with nocodazole for 3 h. The presence of dynein accessory polypeptides was tested by immunofluorescence microscopy (A–C) or by use of GFP-tagged coexpressed polypeptides (D and E). Kinetochore staining of the respective proteins in nocodazole-treated control cells are shown in A′–E′. Bars, 5 μM.
Effects of dynein tail constructs on kinetochore function
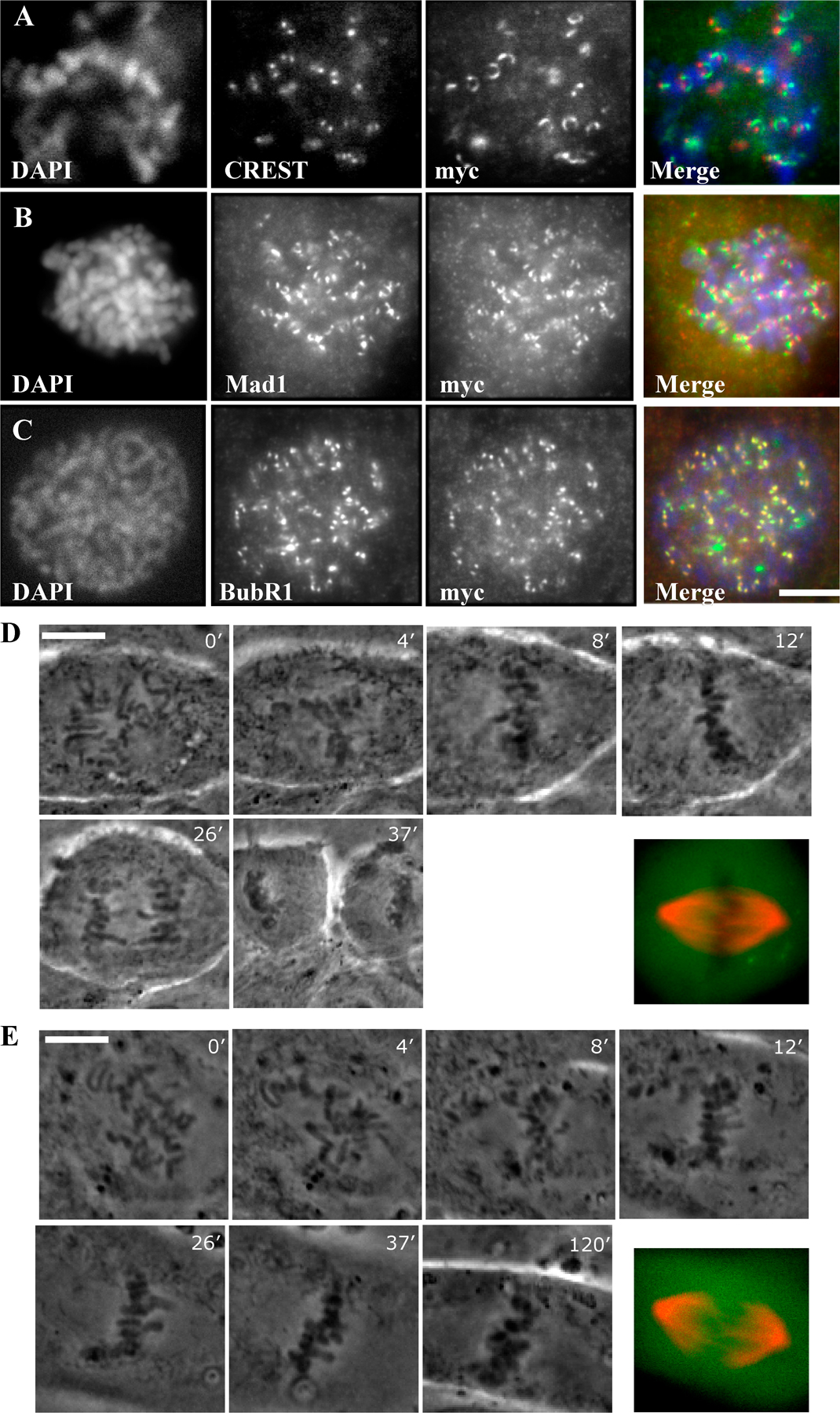
Analysis of bipolar spindles in dynein tail–expressing cells revealed ∼30% to be in early/mid prometaphase, which is similar to controls. However, close to 60% showed extensive chromosome alignment and were judged to be in late prometaphase or metaphase, which is a much higher fraction than the 16% seen for controls (Fig. 3 A). Live imaging of mitotic LLCPK1 cells overexpressing the GFP-C907 tail construct along with an mCherry-tubulin construct, beginning at early prometaphase, revealed comparable rates of chromosome alignment (mean: 8 min, n = 28 vs. mean: 7 min, n = 13 for controls). 26 of 28 cells arrested in late prometaphase-metaphase (Fig. S2, D and E), most of which eventually underwent cell death by 2–2.5 h (Videos 1 and 2, available at http://www.jcb.org/cgi/content/full/jcb.200710106/DC1). We saw no examples of bipolar spindles collapsing to produce monopolar spindles.
Figure 3.

Effects of dynein tail expression on mitotic stage and kinetochore composition during metaphase. (A) Dynein tail–expressing COS7 cells with bipolar spindles were evaluated for chromosome distribution, revealing an increase in late prometaphase cells versus untransfected controls (n = 300). Error bars indicate SD from mean from three independent experiments. (B) Example of cells used for A stained with anti-myc, anti-tubulin, and CREST (not depicted) antibodies versus DAPI (inset). Numbered boxes indicate images magnified below. (C–E) Control COS7 cell expressing a myc vector stained with anti-p150Glued, anti-Mad1, and anti-BubR1 antibodies in each case versus anti-tubulin and anti-myc (not depicted) versus DAPI (inset), showing loss of these proteins from kinetochores at metaphase. (C′–E′) COS7 cells expressing the tail construct and stained as in C–E. The numbered boxes indicate images magnified at right. Dynein tail, p150Glued, Mad1, and BubR1 are all observed to associate with both kinetochores of aligned chromatid pairs, including those kinetochores associated with microtubules. Note examples of misoriented kinetochore pairs in numbered insets (and see Fig. 4). Note that these proteins are absent at spindle poles in tail-expressing cells, in contrast to controls. (F–H and F′–H′) Analysis of Hec1, MCAK, and Kif2b immunoreactivity at aligned kinetochores. Control and tail-expressing COS7 cells were stained using anti-myc (insets) and anti-Hec1, anti-MCAK, and anti-Kif2b antibodies, respectively versus DAPI (n = 150). Mean kinetochore spacing in Hec1-stained cells was decreased relative to controls (see text), which is consistent with loss of tension at kinetochores. MCAK and Kif2b localize to prometaphase kinetochores (G and H) and were normally redistributed to spindle poles in dynein-inhibited cells (G′ and H′). Bars, 5 μM.
In normal mitotic cells, dynein is prominent at unattached kinetochores and greatly reduced at attached kinetochores (King et al., 2000; Hoffman et al., 2001). In marked contrast, the dynein tail could still be detected at each kinetochore of aligned sister chromatid pairs in 38% of expressing cells (n = 50; Fig. 3 B, insets and numbered boxes). The dynactin subunit p150Glued was also found at tail-positive kinetochores (Fig. 3, C and C′, insets and numbered boxes). These observations together support a role for dynein in self-removal. Other proteins were also retained at tail-positive kinetochores, although the microtubule disassembly factors Kif2b and MCAK were not enriched relative to controls (Fig. 3, G, G′, H, and H′; and Table I). In tail-induced monopolar spindles, the tail fragments, as well as endogenous p150Glued, were observed on each member of kinetochore pairs (Fig. S3 B, insets and numbered boxes, available at http://www.jcb.org/cgi/content/full/jcb.200710106/DC1). This pattern contrasts with that in untransfected monastrol-treated cells, in which we observe p150Glued only at the outward-facing kinetochores (Fig. S3 A, insets and numbered boxes). We note that chromosomes also tended to be located more peripherally within the tail-induced (Fig. S3 C) versus the monastrol-induced (Fig. S3 D) monopolar spindles.
Table I. Composition of dynein tail–decorated kinetochores.
| Protein | (−) Nocodazole | (+) Nocodazole |
|---|---|---|
| Dynein motor | − | − |
| p150Glued | ++ | + |
| CLIP170 | + | + |
| CLASP1 | ++ | + |
| LIS1 | ND | + |
| NudE | ND | + |
| ZW10 | ND | + |
| Hec1 | + | + |
| Aurora B | + | + |
| Kif2b | − | + |
| MCAK | −a | + |
A summary of results on the localization of kinetochore proteins in the presence of dynein tail. (−) Nocodazole data refer only to aligned kinetochores.
Despite reported kinetochore localization, we observe most MCAK at spindle poles in control and tail-expressing late prometaphase/metaphase cells.
The accumulation in late prometaphase/metaphase of the dynein tail-expressing cells prompted us to investigate their SAC status. BubR1 and Mad1 were each prominent at nocodazole-treated kinetochores (Fig. S2, B and C), indicating that recruitment of SAC proteins to kinetochores was normal. In control transfected cells, SAC proteins normally depart kinetochores as chromosomes become attached to microtubules and are largely depleted from aligned kinetochores (Fig. 3, D and E). In striking contrast, Mad1 and BubR1 were strongly associated with many of the aligned kinetochores in tail-expressing cells (Fig. 3, D′ and E′). We also observed that Mad1 and BubR1 immunostaining clearly persisted at paired kinetochores in tail-induced monopolar spindles (Fig. S3, E and F).
Proper end-on microtubule attachment to kinetochores is dependent on the Hec1–Ndc80 complex (Cheeseman et al., 2006; DeLuca et al., 2006). One indicator of defective attachment is a loss in kinetochore microtubule stability. Recent studies have found that inhibition of NudE/NudEL or of ZW10 (Stehman et al., 2007; Yang et al., 2007) have moderate effects on microtubule stability, but whether these effects involve dynein or other factors that these proteins recruit, such as dynactin, CLIP-170, and LIS1, is unknown. To test whether loss of the dynein motor from kinetochores affects microtubule stability, we briefly chilled dynein tail–expressing cells to 4°C. We observed a marked reduction in cold-resistant kinetochore microtubule bundles in comparison to controls using either LLCPK1 (Fig. 4, A–C) or COS7 cells (not depicted), although kinetochore staining by anti-Hec1 antibody persisted (Fig. 3, F and F′).
Figure 4.

Role of dynein in kinetochore microtubule stability and kinetochore orientation. LLCPK1 cells transfected with the dynein tail fragment were incubated at 4°C for 10 min and then examined by confocal microscopy. (A) Dynein tail–expressing cell stained with anti-tubulin, DAPI, and anti-myc (inset) showing few remaining kinetochore microtubule bundles. (B) A control cell stained as in A showing numerous cold-stable kinetochore microtubule bundles. (C) Fraction of cells exhibiting decrease in the number of cold-stable microtubule bundles in tail-expressing versus control untransfected cells after cold treatment (n = 90). P = 0.012; two-tailed t test. (D–I) Analysis of kinetochore orientation in tail-expressing LLCPK1 cells. (D–F and D′–F′) Cells in late prometaphase/metaphase (D, D′, E, and E′, pretreated with MG132 as indicated) or prometaphase (F and F′) transfected with the tail were stained with anti-tubulin, CREST, and anti-myc antibodies (D′–F′) as indicated versus DAPI (not depicted). (G and H) Control LLCPK1 cells expressing GFP. The numbered boxes indicate images magnified at right. Insets show GFP staining. (I) Fraction of aligned kinetochore pairs exhibiting different attachment patterns. P = 0.0025; two-tailed t test. Error bars indicate SD from mean from three independent experiments. Bars, 5 μM.
Another indicator of defects in microtubule attachment is interkinetochore spacing. We determined a mean separation of 1.21 ± 0.22 μm for kinetochore pairs of control aligned COS7 cell chromosomes (n = 61) using Hec1 as a kinetochore marker. This value was reduced to 0.89 ± 0.17 μm (n = 68) for aligned chromosomes in dynein tail–expressing cells (n = 68), representing a 69% decrease in tension (Fig. 3, F and F′). The reduced spacing was close to that for nonaligned kinetochore pairs in these cells (0.748 ± 0.13 μm; n = 61), untransfected controls (0.745 ± 0.13 μm; n = 41), and nocodazole-treated control cells (0.746 ± 0.09 μm; n = 58).
We also noted a striking misorientation of many aligned kinetochore pairs in the dynein tail–expressing cells relative to the major spindle axis (Fig. 4, D and D′, insets and numbered boxes) as compared with control cells (Fig. 4, E and E′, insets and numbered boxes). Inspection revealed fewer biattached kinetochore pairs relative to controls (Fig. 4, G and H, insets and numbered boxes) but a substantial increase in unattached or monoattached ones, with the latter occurring predominantly at the distal end of the kinetochore-microtubule bundles (Fig. 4 I). Lateral attachments were commonly observed during prometaphase in both control GFP-expressing and tail-expressing cells (Fig. 4, F and F′). Related results were obtained using Hec1 as a kinetochore marker, which was also found to be clearly retained at the misoriented kinetochores (Fig. 3, F and F′).
RNAi of dynein HC expression in COS7 cells revealed a similar reduction in spindle microtubule stability (Fig. S3, G and G′). We also injected the function-blocking anti–dynein IC antibody into cells arrested at metaphase by treatment with the proteasome inhibitor MG132. Kinetochore microtubule bundles in control antibody-injected cells were stable to cold treatment as expected (Fig. 5, A and B). In contrast, by 1 h after anti-dynein antibody injection, few cold-resistant kinetochore microtubule bundles remained (Fig. 5, A′ and B′). 72% of the dynein antibody-injected cells showed partial to complete loss of stable kinetochore microtubules in comparison with 10% in control antibody-injected cells (n = 25). Apparent misorientation of kinetochores was observed even at 37°C, although by this stage distortion of the kinetochore microtubules in many injected cells complicated a detailed analysis (Fig. 5 A′). The latter effects may relate to those observed in dynein tail–expressing cells and could result from improper kinetochore-microtubule attachment.
Figure 5.

Behavior of kinetochores and kinetochore microtubules. (A, A′, B, and B′) LLCPK1 cells were treated with MG132 for 1 h, injected with antibody, fixed, and stained 1 h later with or without prior cold treatment using anti-tubulin, CREST, and anti–mouse IgG (insets) antibodies versus DAPI (not depicted). Kinetochore microtubules were unaffected by anti-dynein antibody in cells retained at 37°C (A and A′) but were severely destabilized in these cells after cold treatment (B, B′) as compared with the control antibody. (C and D) HeLa cells stably expressing YFP–CENP-A, a cotransfection marker mCherryC1 with (D) or without (C) the myc-C1140 dynein tail construct, were incubated with MG132 for 45 min and imaged at one frame per 15 s for 15 min (C′ and D′) Time-lapse images of individual kinetochore pairs (boxed) followed for 3 min (C′) from a control cell and (D′) a tail-expressing cell. The maximal amplitude of the oscillation of each pair is indicated at the bottom. Arrows indicate the midline of metaphase plate. (E) Maximum amplitude of kinetochore pair oscillations from control and dynein tail–expressing cells from four and six dividing cells, respectively. (F and G) COS7 cells expressing GFP-Hec1 were injected either with a control mouse IgG (F, n = 6 cells) or dynein IC (G, n = 12 cells) antibody 30 min after MG132 treatment. Live images of kinetochores were acquired every 15 s for 6–9 min starting 30 min after injection. (F′ and G′) Time-lapse images of kinetochore pairs (F and G, boxes). (H) Analysis of kinetochore behavior in dynein IC antibody-injected versus control antibody-injected cells (n = 6 cells, 55 kinetochore pairs for each condition). (I) Models for dynein role in kinetochore microtubule attachment, orientation, and alignment. Model I: Dynein removes disassembly factor (blue oval) from kinetochore, a model not supported by the data (see text). Model II: (a) Dynein-mediated tension reorients kinetochores; (b) dynein brings microtubule ends toward core microtubule attachment sites; and (c) dynein-mediated tension persists during metaphase as indicated by role in kinetochore oscillations. Bars, 5 μM.
Finally, we analyzed the effects of dynein inhibition on kinetochore behavior by live imaging in HeLa cells stably expressing YFP–centromeric protein (CENP) A, transfected with the myc-C1140 tail construct and an mCherry cotransfection marker and treated with MG132 for 1 h (Videos 3 and 4, available at http://www.jcb.org/cgi/content/full/jcb.200710106/DC1). The most dramatic effect was an increase in the magnitude of kinetochore oscillations along the major spindle axis, resulting in a broadening of the metaphase plate (Fig. 5, C, C′, D, D′, and E). Misorientation of kinetochore pairs could also be observed, an effect which occurred over a period of a few minutes (Fig. 5 D′, boxed). We also injected the anti–dynein IC antibody into COS7 cells transiently expressing Hec1-GFP as a kinetochore marker and arrested in metaphase by treatment with MG132 (Videos 5 and 6). The normal oscillatory behavior of kinetochores became even more strikingly irregular, with some kinetochore pairs drifting away from the metaphase plate and often becoming misoriented (Fig. 5, F, F′, G, G′, and H). As observed by fixed imaging (Fig. 4, D–I), kinetochore pairs became progressively misaligned with time (unpublished data).
Roles of kinetochore dynein
Collectively, our data support a role for kinetochore dynein in its own removal and that of other kinetochore proteins upon microtubule attachment. More surprisingly, replacement of dynein with a truncated version affected stable end-on attachment itself. Also noteworthy was the substantial congression of chromosomes to the metaphase plate within the range of times for normal cells. The latter result is consistent with a role for other motors, such as CENP-E, (Kapoor et al., 2006), or for microtubule assembly-disassembly dynamics in this process. The instability of kinetochore microtubules in dynein tail–expressing cells is unexpected. Dynein is well-established to interact with microtubules laterally in general (Paschal and Vallee, 1987) and in the early stages of mitosis (Rieder and Alexander, 1990; Emanuele and Stukenberg, 2007; Vorozhko et al., 2008), but the possibility of a microtubule plus-end interaction has not been extensively explored. We envision two principal models for how dynein might function at microtubule ends and at these sites regulate kinetochore microtubule stability. First, dynein might remove microtubule plus end destabilization factors (Fig. 5 I, model I; and Table I). However, we observe no apparent increase in Kif2b or MCAK at metaphase-aligned kinetochores of dynein tail–expressing cells. We do, however, observe retention of the microtubule stabilization factors p150Glued and CLASP1 (Fig. 3 B and Table I).
Alternatively, we believe that dynein-mediated tension at microtubule plus ends may have several important effects on kinetochore behavior (Fig. 5 I, model II, a–c). First, we propose that dynein interacts sequentially with the sides and then the plus ends of kinetochore microtubules (Fig. 5 I, model II, a). Dynein could participate in this largely unexplored phenomenon by producing tension at both members of paired kinetochores (Fig. 5 I, model II, a) or even at individual kinetochores. We note that an analogous shift in the mode of interaction between dynein and microtubules is already suggested by observations of cytoplasmic microtubule behavior in the yeast Saccharomyces cerevisiae. As the mitotic nucleus enters the bud, microtubules emanating from the leading pole body interact with the lateral cell cortex in a manner akin to gliding in in vitro assays (Adames and Cooper, 2000). Ultimately, however, these microtubules achieve an apparent dynein-dependent end-on interaction with the bud cortex (Carminati and Stearns, 1997). Thus, these states may be comparable to those we propose to be involved in the interaction between kinetochores and microtubules in higher eukaryotic cells. It is additionally possible that dynein-generated tension serves to draw microtubules into close contact with the core microtubule attachment sites within the kinetochore (Fig. 5 H, II, b).
Finally, our evidence identifies an as yet unreported reversibility in kinetochore-microtubule attachment. This conclusion is based on injection of anti-dynein antibody, which interfered with stable microtubule attachment even in cells arrested in metaphase by MG132 (Fig. 5, B and B′). Whether dynein is required to maintain attachments or, instead, to repair spontaneous reversals of microtubule–kinetochore interactions, remains to be explored. In addition, live imaging revealed marked effects on oscillatory movements of kinetochore pairs and progressive loss in their proper orientation and alignment. These results provide strong evidence for persistent dynein activity at kinetochores (Fig 5 I, model IIc) despite the sharp reduction in dynein levels upon microtubule attachment (King et al., 2000). Our results also implicate cytoplasmic dynein along with Kif18A in regulating metaphase chromosome oscillations (Stumpff et al., 2008).
Materials and methods
cDNA constructs
N-terminal myc-tagged C1140, C907, C800, C674, and C260 dynein HC cDNAs were characterized previously (Gee et al., 1997; Tynan et al., 2000). GFP, myc, and HA tags were added using pEGFP-C1, pCMV-myc, and a pkHA3-derived plasmid, respectively. The Loa and Cra1 mutations were inserted using the QuikChange II site-directed mutagenesis kit (Stratagene). Cytoplasmic dynein HC RNAi was performed using the pRNAT vector (GenScript) characterized previously (Tsai et al., 2007). ZW10-GFP, GFP-Kif2b, GFP-Hec1, and mCherry-tubulin constructs were gifts from G. Chan (University of Alberta, Edmonton, Canada), D. Compton (Dartmouth Medical School, Hanover, NH), E. Salmon (University of North Carolina, Chapel Hill, NC), and R. Tsien (University of California, San Diego, La Jolla, CA), respectively.
Antibodies
The dynein HC motor domain-specific polyclonal, anti-myc polyclonal, and rat anti-tubulin monoclonal antibodies have been described previously (Tynan et al., 2000; Mikami et al., 2002; Stehman et al., 2007). Monoclonal antibodies included anti-dynein IC (IC 74.1; Millipore), anti-p150Glued and anti-BubR1 (BD Biosciences), anti–α-tubulin (Sigma-Aldrich), anti-Hec1 (Abcam), anti-GFP (Invitrogen), and anti-HA (Covance). CREST autoimmune antiserum, anti-LIS1, anti-MAD1, anti-CLIP170 monoclonals, and anti-MCAK and anti-CLASP1 polyclonals were generously provided by B. Brinkley (Baylor College of Medicine, Houston, TX), O. Reiner (The Weizman Institute of Science, Rehovot, Israel), A. Mussachio (European Institute of Oncology, Milan, Italy), H. Goodson (University of Notre Dame, Notre Dame, IN), C. Walczak (Indiana University, Bloomington, IN), and A. Akhmanova (Erasmus Medical Center, Rotterdam, Netherlands), respectively.
Cell culture
COS7, HeLa cells stably expressing YFP—CENP-A (gift from D. Cleveland, University of California, San Diego, La Jolla, CA), and LLCPK1 cells were grown at 37°C in DME with 3% (LLCPKI) or 10% FBS plus 100 U/ml penicillin and 100 mg/ml streptomycin. Cells were plated on 18-mm coverslips to 70–80% confluency, transfected using Effectene (QIAGEN), and examined after 36–48 h. Drug treatments included 10 μM nocodazole for 3 h, 100 μM monastrol for 4 h, and 10 μM MG132 for 1–2 h. Cold treatment was for 10 min in ice cold PBS.
Fixed and live imaging
Immunofluorescence microscopy.
Cells were rinsed in PHEM buffer (120 mM Pipes, 50 mM Hepes, 20 mM EGTA, and 4 mM magnesium acetate, pH 6.9) and typically fixed for 6 min at −20°C in methanol. For anti-LIS1 and CLIP170 staining, cells were extracted with 0.05% Triton X-100 at RT for 1 min and fixed with 4% PFA at RT for 20 min and −20°C methanol for 4 min. 0.1% Triton X-100 preextraction was used to monitor kinetochore-microtubule attachments. For MCAK staining, cells were fixed in 2% PFA. For the analysis of dynein displacement from kinetochores, cells were incubated sequentially with rabbit anti-dynein HC, Cy3 anti–rabbit, rabbit anti-myc, and Cy2 anti–rabbit antibodies. All antibody incubations were at 37°C for 1 h in PBS plus 0.05% BSA. DAPI staining (0.1 μg/ml) was performed for 10 min, and cells were mounted using Prolong Antifade (Invitrogen). Images were acquired with a microscope (DMIRBE; Leica) equipped with a camera (ORCA 100; Hamamatsu Photonics) and MetaMorph software (MDS Analytical Technologies). Confocal microscopy was performed using a multiphoton (Carl Zeiss, Inc.) or 510 META LSM (Carl Zeiss, Inc.) using 0.3–0.6-μm Z-series steps. Images were cropped and processed using Photoshop 7.0 (Adobe). For quantitation of the effects of dynein inhibition (dynein tail expression and antibody injection) on the stability of kinetochore-microtubule bundles, we defined a bundle as thick linear arrays of tubulin-positive structures >2–3 μm in length.
Antibody microinjection.
LLCPK1 cells were treated with MG132 for 1 h and then injected with 5 μg/ml of purified 74.1 monoclonal anti-dynein antibody (Millipore) or control Mouse IgG antibody using an Eppendorf FemtoJet system. Cells were examined 30–60 min after injection as required.
Live-cell imaging.
LLCPK1 cells were cotransfected with GFP-C907 or GFP- and mCherry-tubulin constructs. After 1 d, phase contrast images were acquired every 1 min for up to 2.5 h using MetaMorph software and a microscope (DMIRBE) equipped with an incubation chamber for temperature/CO2 control. The rate of chromosome alignment was measured based on the time required for the chromosomes to align at the metaphase plate starting from early prometaphase. For live imaging of mitotic kinetochores, COS7 cells transiently expressing Hec1-GFP were injected with a control mouse IgG or dynein IC74 antibody 30 min after the proteasome inhibitor MG132 treatment, and live-imaging was performed 30 min later. HeLa cells stably expressing YFP–CENP-A were treated with MG132 for 45 min. Fluorescent images of Hec1-GFP or YFP–CENP-A were acquired every 15 s for 6–15 min as required. Images were visualized using PLAN FLUOTAR 40× 1 NA, PLAN APO 63× 1.32 NA, or PLAN APO 100× 1.4 NA oil-immersion objective lenses as required.
Online supplemental material
Fig. S1 is a summary of spindle defects produced by dynein tail expression. Fig. S2 shows Mad1 and BubR1 localization to tail-decorated prometaphase kinetochores and a montage of stills from Videos 1 and 2. Fig. S3 is the analysis of monopolar spindles in tail-expressing cells. Videos 1 and 2 are live imaging of mitosis in control and tail-expressing cells. Videos 3, 4, 5, and 6 are live imaging of kinetochores in tail-expressing and dynein antibody-injected cells.
Supplementary Material
Acknowledgments
We thank Y. Mao and T. Maresca for helpful advice, Y. Chen for cDNA cloning, and Drs. B. Brinkley, O. Reiner, A. Mussachio, H. Goodson, A. Akhmanova, C. Walczak, D. Cleveland, E. Salmon, and D. Compton for generous help with reagents.
This research was supported by National Institutes of Health (GM47434 to R.B. Vallee).
Abbreviations used in this paper: CENP, centromeric protein; CLIP170, cytoplasmic linker protein 170; HC, heavy chain; IC, intermediate chain; SAC, spindle assembly checkpoint; ZW10, zeste white 10.
References
- Adames, N.R., and J.A. Cooper. 2000. Microtubule interactions with the cell cortex causing nuclear movements in Saccharomyces cerevisiae. J. Cell Biol. 149:863–874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basto, R., F. Scaerou, S. Mische, E. Wojcik, C. Lefebvre, R. Gomes, T. Hays, and R. Karess. 2004. In vivo dynamics of the rough deal checkpoint protein during Drosophila mitosis. Curr. Biol. 14:56–61. [DOI] [PubMed] [Google Scholar]
- Carminati, J.L., and T. Stearns. 1997. Microtubules orient the mitotic spindle in yeast through dynein-dependent interactions with the cell cortex. J. Cell Biol. 138:629–641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheeseman, I.M., J.S. Chappie, E.M. Wilson-Kubalek, and A. Desai. 2006. The conserved KMN network constitutes the core microtubule-binding site of the kinetochore. Cell. 127:983–997. [DOI] [PubMed] [Google Scholar]
- DeLuca, J.G., W.E. Gall, C. Ciferri, D. Cimini, A. Musacchio, and E.D. Salmon. 2006. Kinetochore microtubule dynamics and attachment stability are regulated by Hec1. Cell. 127:969–982. [DOI] [PubMed] [Google Scholar]
- Echeverri, C.J., B.M. Paschal, K.T. Vaughan, and R.B. Vallee. 1996. Molecular characterization of the 50-kD subunit of dynactin reveals function for the complex in chromosome alignment and spindle organization during mitosis. J. Cell Biol. 132:617–633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emanuele, M.J., and P.T. Stukenberg. 2007. Xenopus Cep57 is a novel kinetochore component involved in microtubule attachment. Cell. 130:893–905. [DOI] [PubMed] [Google Scholar]
- Faulkner, N.E., D.L. Dujardin, C.Y. Tai, K.T. Vaughan, C.B. O'Connell, Y. Wang, and R.B. Vallee. 2000. A role for the lissencephaly gene LIS1 in mitosis and cytoplasmic dynein function. Nat. Cell Biol. 2:784–791. [DOI] [PubMed] [Google Scholar]
- Gee, M.A., J.E. Heuser, and R.B. Vallee. 1997. An extended microtubule-binding structure within the dynein motor domain. Nature. 390:636–639. [DOI] [PubMed] [Google Scholar]
- Habura, A., I. Tikhonenko, R.L. Chisholm, and M.P. Koonce. 1999. Interaction mapping of a dynein heavy chain. Identification of dimerization and intermediate-chain binding domains. J. Biol. Chem. 274:15447–15453. [DOI] [PubMed] [Google Scholar]
- Hafezparast, M., R. Klocke, C. Ruhrberg, A. Marquardt, A. Ahmad-Annuar, S. Bowen, G. Lalli, A.S. Witherden, H. Hummerich, S. Nicholson, et al. 2003. Mutations in dynein link motor neuron degeneration to defects in retrograde transport. Science. 300:808–812. [DOI] [PubMed] [Google Scholar]
- Hoffman, D.B., C.G. Pearson, T.J. Yen, B.J. Howell, and E.D. Salmon. 2001. Microtubule-dependent changes in assembly of microtubule motor proteins and mitotic spindle checkpoint proteins at PtK1 kinetochores. Mol. Biol. Cell. 12:1995–2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howell, B.J., B.F. McEwen, J.C. Canman, D.B. Hoffman, E.M. Farrar, C.L. Rieder, and E.D. Salmon. 2001. Cytoplasmic dynein/dynactin drives kinetochore protein transport to the spindle poles and has a role in mitotic spindle checkpoint inactivation. J. Cell Biol. 155:1159–1172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapoor, T.M., M.A. Lampson, P. Hergert, L. Cameron, D. Cimini, E.D. Salmon, B.F. McEwen, and A. Khodjakov. 2006. Chromosomes can congress to the metaphase plate before biorientation. Science. 311:388–391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karki, S., and E.L. Holzbaur. 1995. Affinity chromatography demonstrates a direct binding between cytoplasmic dynein and the dynactin complex. J. Biol. Chem. 270:28806–28811. [DOI] [PubMed] [Google Scholar]
- King, J.M., T.S. Hays, and R.B. Nicklas. 2000. Dynein is a transient kinetochore component whose binding is regulated by microtubule attachment, not tension. J. Cell Biol. 151:739–748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kon, T., M. Nishiura, R. Ohkura, Y.Y. Toyoshima, and K. Sutoh. 2004. Distinct functions of nucleotide-binding/hydrolysis sites in the four AAA modules of cytoplasmic dynein. Biochemistry. 43:11266–11274. [DOI] [PubMed] [Google Scholar]
- Mikami, A., S.H. Tynan, T. Hama, K. Luby-Phelps, T. Saito, J.E. Crandall, J.C. Besharse, and R.B. Vallee. 2002. Molecular structure of cytoplasmic dynein 2 and its distribution in neuronal and ciliated cells. J. Cell Sci. 115:4801–4808. [DOI] [PubMed] [Google Scholar]
- Paschal, B.M., and R.B. Vallee. 1987. Retrograde transport by the microtubule associated protein MAP 1C. Nature. 330:181–183. [DOI] [PubMed] [Google Scholar]
- Reck-Peterson, S.L., and R.D. Vale. 2004. Molecular dissection of the roles of nucleotide binding and hydrolysis in dynein's AAA domains in Saccharomyces cerevisiae. Proc. Natl. Acad. Sci. USA. 101:1491–1495. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Rieder, C.L., and S.P. Alexander. 1990. Kinetochores are transported poleward along a single astral microtubule during chromosome attachment to the spindle in newt lung cells. J. Cell Biol. 110:81–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savoian, M.S., M.L. Goldberg, and C.L. Rieder. 2000. The rate of poleward chromosome motion is attenuated in Drosophila zw10 and rod mutants. Nat. Cell Biol. 2:948–952. [DOI] [PubMed] [Google Scholar]
- Sharp, D.J., G.C. Rogers, and J.M. Scholey. 2000. Cytoplasmic dynein is required for poleward chromosome movement during mitosis in Drosophila embryos. Nat. Cell Biol. 2:922–930. [DOI] [PubMed] [Google Scholar]
- Starr, D.A., B.C. Williams, T.S. Hays, and M.L. Goldberg. 1998. ZW10 helps recruit dynactin and dynein to the kinetochore. J. Cell Biol. 142:763–774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stehman, S.A., Y. Chen, R.J. McKenney, and R.B. Vallee. 2007. NudE and NudEL are required for mitotic progression and are involved in dynein recruitment to kinetochores. J. Cell Biol. 178:583–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stumpff, J., G. von Dassow, M. Wagenbach, C. Asbury, and L. Wordeman. 2008. The kinesin-8 motor Kif18A suppresses kinetochore movements to control mitotic chromosome alignment. Dev. Cell. 14:252–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai, J.W., K.H. Bremner, and R.B. Vallee. 2007. Dual subcellular roles for LIS1 and dynein in radial neuronal migration in live brain tissue. Nat. Neurosci. 10:970–979. [DOI] [PubMed] [Google Scholar]
- Tynan, S.H., M.A. Gee, and R.B. Vallee. 2000. Distinct but overlapping sites within the cytoplasmic dynein heavy chain for dimerization and for intermediate chain and light intermediate chain binding. J. Biol. Chem. 275:32769–32774. [DOI] [PubMed] [Google Scholar]
- Vaisberg, E.A., M.P. Koonce, and J.R. McIntosh. 1993. Cytoplasmic dynein plays a role in mammalian mitotic spindle formation. J. Cell Biol. 123:849–858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varma, D., D.L. Dujardin, S.A. Stehman, and R.B. Vallee. 2006. Role of the kinetochore/cell cycle checkpoint protein ZW10 in interphase cytoplasmic dynein function. J. Cell Biol. 172:655–662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaughan, K.T., and R.B. Vallee. 1995. Cytoplasmic dynein binds dynactin through a direct interaction between the intermediate chains and p150Glued. J. Cell Biol. 131:1507–1516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vorozhko, V.V., M.J. Emanuele, M.J. Kallio, P.T. Stukenberg, and G.J. Gorbsky. 2008. Multiple mechanisms of chromosome movement in vertebrate cells mediated through the Ndc80 complex and dynein/dynactin. Chromosoma. 117:169–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wojcik, E., R. Basto, M. Serr, F. Scaerou, R. Karess, and T. Hays. 2001. Kinetochore dynein: its dynamics and role in the transport of the Rough deal checkpoint protein. Nat. Cell Biol. 3:1001–1007. [DOI] [PubMed] [Google Scholar]
- Yang, Z., U.S. Tulu, P. Wadsworth, and C.L. Rieder. 2007. Kinetochore dynein is required for chromosome motion and congression independent of the spindle checkpoint. Curr. Biol. 17:973–980. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}