

Abstract
The myocyte enhancer factor-2 (MEF2) family of transcription factors play key roles in the activation of muscle structural genes. In Drosophila, MEF2 accumulates at high levels in the embryonic muscles, where it activates target genes throughout the mesoderm. Here, we identify the Transglutaminase gene (Tg; CG7356) as a direct transcriptional target of MEF2 in the cardiac musculature. Tg is expressed in cells forming the inflow tracts of the dorsal vessel, and we identify the enhancer responsible for this expression. The enhancer contains three binding sites for MEF2, and can be activated by MEF2 in tissue culture and in vivo. Moreover loss of MEF2 function, or removal of the MEF2 binding sites from the enhancer, results in loss of Tg expression. These studies identify a new MEF2 target in the cardiac musculature. Furthermore, given the relevance of transglutaminase genes to human disease, these studies provide a possible mechanism for their activation.
INTRODUCTION
Transglutaminases comprise a family of proteins whose function is to carry out protein-protein cross-linking activities. A number of vertebrate transglutaminase genes have been characterized functionally, and are known to be involved in cellular processes as diverse as blood clotting, maintenance of epithelial cell sheets, atherosclerosis, apoptosis and semen coagulation (reviewed in Griffin et al., 2002). Mutations in transglutaminase genes are known to result in a number of disorders including lamellar ichthyosis (Huber et al., 1995), peeling skin syndrome (Cassidy et al., 2005), and Factor XIII deficiency (see for example Standen and Bowen, 1993). Furthermore, transglutaminase dysregulation is associated with a number of human pathologies (Griffin et al., 2002). Clearly, understanding how the expression of transglutaminase genes is controlled has significant biomedical relevance.
Given the relatively broad expression patterns of several transglutaminase genes, the regulatory mechanisms contributing to their expression are likely to be highly variable. Indeed, a number of promoter and enhancer elements have been identified which impact transglutaminase gene expression in tissue culture, and these characterized elements respond to both ubiquitous and signal-specific factors, such as SP1 (Dubbink et al., 1999; Kida et al., 1999), AP2 (Mariniello et al., 1995), and retinoid receptors (Nagy et al., 1996). However, direct in vivo transcriptional activators of transglutaminase genes have yet to be identified.
The insect Drosophila has a single Transglutaminase gene (Transglutaminase, Tg; also called CG7356) whose predominant expression in the embryo appears to be in the cardiac tissue. The embryonic cardiac tissue comprises the dorsal vessel, a small pulsating tube which transports hemolymph from the posterior of the animal towards the head (reviewed in Cripps and Olson, 2002; Tao and Schulz, 2007). Hemolymph enters the dorsal vessel via specialized inflow tracts termed ostia, and is dispersed near the brain. Hemolymph then percolates back through the body cavity, before re-entering the cardiac tube for further circulation (Rizki, 1978).
The cardiac inflow tracts are generated from a subset of cardial cells which express the orphan nuclear receptor gene seven-up (svp). There are seven sets of Svp cells in the dorsal vessel, the most posterior three of which form ostia in the embryo and larva (Molina and Cripps, 2001; Ponzielli et al., 2002), and the remaining Svp cells form ostia in the adult stages (Monier et al., 2005; Sellin et al., 2006; Wasserthal, 2007). Since a mammalian Svp ortholog named COUPTFII is required for cardiac inflow tract formation in mice (Pereira et al., 1999), understanding gene regulatory patterns in the Svp cells in Drosophila is likely to provide important information into how cardiac inflow tracts might be genetically specified.
Recent gene expression analyses carried out by the Berkeley Drosophila Genome Project (Tomacak et al., 2002) indicated that Tg expression might occur in the Svp cells of the embryo, although in the absence of double-stained samples this point has yet to be fully demonstrated. If we could confirm the cardiac-specific expression of Tg, identifying the transcriptional regulators of this gene would provide important insight into cardiac gene regulatory mechanisms. Such findings would also impact our understanding of transglutaminase gene regulation in general.
Here, we have identified the enhancer which regulates expression of Tg, and we have determined that indeed Tg is expressed within the cardiac tissue. By analyzing the expression of both Tg and the Tg-lacZ fusion in mutant backgrounds, we show that Drosophila Transglutaminase expression is activated by the muscle transcription factor Myocyte enhancer factor-2 (MEF2), via three specific MEF2 binding sites. Removal of these binding sites from the enhancer completely eliminates enhancer activity. These studies define the first mechanism for tissue-specific transcriptional regulation of a transglutaminase gene in vivo, and stand to provide critical insight into the transcriptional regulation of transglutaminase genes.
EXPERIMENTAL PROCEDURES
In situ hybridization
To generate an antisense probe specific for Tg, the plasmid pFLC1/Tg was obtained from the Drosophila Genomics Resource Center (clone number: RE08173). This plasmid was linearized with EcoR1 and probe was synthesized using T3 RNA polymerase. Sense probe was synthesized using T7 RNA polymerase, following linearization of the plasmid with BamH1.
In situ hybridization was carried out as described by O’Neill and Bier (1994). Detection was either using a digoxigenin labeled riboprobe and alkaline phosphatase-linked secondary antibody, stained using NBT/BCIP; alternatively, fluorescent in situ hybridization was carried out using a biotinylated riboprobe and fluorescein-labeled anti-biotin. No consistent stain was observed using sense probes.
Antibody staining
Antibody detection of epitopes was carried out according to Patel (1994). Primary antibodies were anti-ß-galactosidase (Promega Corp., 1:1,000), anti-MEF2 (Lilly et al., 1995, 1:1000), anti-Tinman (Xu et al., 1998 1:500), and anti-Pericardin (Chartier et al., 2002; 1:4). For immunohistochemistry, primary antibody localization was detected using the ABC detection kit and diaminobenzidine staining reagent from Vector Laboratories. Stained embryos were cleared in glycerol and photographed using an Olympus BX51 microscope. For fluorescent detection, secondary antibodies were 488- or 568-Alexa fluor conjugates (Molecular Probes, Inc.), used at 1:2,000. Fluorescent images were captured on an MRC-600 confocal microscope using T1 and T2A filters (Biorad, Inc.). Digital or scanned images were assembled using Adobe Photoshop.
DNA methods
DNA manipulations were performed using standard techniques (Sambrook et al., 1998). Candidate Tg enhancer fragments were generated by PCR, and cloned into the pGEM-T Easy vector (Promega Corp.). DNA was then excised and cloned into the hsp-lacZ transformation vector CHAB (Thummell and Pirrotta, 1992). Primers used for each construct were as follows. For region 1: 1a enhancer fwd (5′-AGCCATAAAGCGCACTGC) and 1a enhancer rev (5′-CGATCGGTCGTCTTCTCCC); for region 2: 1b enhancer fwd (5′-GTTGGTTTCAATCGCCAA) and 1b enhancer rev (CGGTTAACGAGAGGGGCTA); for region 3: 1b-Copia fwd (5′-CATTAAAATGGGATTTTAATTGG) and 1b-Copia rev (5′-GTGAATAGTTGAACCGCCC) ; for region 4 (+12,231/+13,046): Doc-exon2+ (5′-GCATATTGCTGCAATTGG) and Doc-exon2- (5′-TAAGTATTTTCGGCAGGG). Mutagenesis was carried out according to Horton (1993).
Double-stranded oligonucleotides for DNA binding assays were generated by annealing complementary oligonucleotides corresponding to the wild-type or mutant sequences to be tested. Each 10-bp MEF2 site was flanked by 10bp of corresponding genomic sequence. Oligonucleotide pairs were designed so that, once annealed, each end of the molecule had a 5′-GG overhang. Overhangs were filled in using Klenow enzyme (New England Biolabs) and 32P-dCTP. Radioactive probes were purified in G25 spin columns (Roche), and diluted to 50,000cpm per μl. The control MEF2 binding sequence is from the Act57B gene (Kelly et al., 2002).
MEF2 protein was synthesized in vitro using rabbit reticulocyte lysate (Promega Corp.) and a Mef2 expression plasmid (Lilly et al., 1995).
DNA binding assays
Electrophoretic mobility shift assays were essentially as described by Sambrook et al. (1998). Each shift reaction contained radioactively labeled probe DNA (50,000cpm), competitor DNA (if required; 100-fold molar excess), polydI.dC (1μg), 5X binding buffer (2μl; Gossett et al., 1989), and reticulocyte lysate (3μl). Reactions took place at room temperature for 20 minutes, after which the reactions were loaded on a 5% w/v non-denaturing polyacrylamide gel and separated by electrophoresis. Gels were dried and exposed to film.
Tissue culture
Drosophila S2 cells (SL2, Drosophila Genomics Resource Center) were cultured in Schneider’s medium (Invitrogen) supplemented with 10% fetal bovine serum (Gibco), at 25°C. Reporter assays were carried out essentially as described before (Kelly Tanaka et al., 2008). Briefly, cells were plated in 24-well plates and transfected with 0.5 μg/well plasmid DNA of various compositions and 5μl/well transfection reagent Cellfectin (Invitrogen), diluted in serum-free Schneider’s medium. Plasmid DNAs that were used for assays consisted of activator plasmid pPac-Pl-Mef2, coding for wild type Mef2 (Kelly Tanaka et al., 2008) and one of the reporter plasmids (either +12,231/+13,046 or +12,702/+12,953 or +12,702/+12,952-mutant) mixed at ratio 1:9. For control, the activator plasmid was substituted with an empty vector pPac-Pl. Transfections were carried out with duplicated samples. After overnight incubation fresh full growth medium was added and cells were incubated for an additional 24 hours. At the end of incubation cells were lysed in M-Per Extraction Reagent (Pierce) and β-galactosidase activity was determined using All-in-One Mammalian β-Galactosidase Assay Kit (Pierce). β-galactosidase activity was normalized by protein concentration determined for each sample in a separate, Bradford-based protein assay (Bio-Rad). Activities of reporter constructs were calculated as folds of difference between β-galactosidase activities obtained for samples cotransfected with the activator plasmid versus empty vector. Final results represent data pooled from three independent transfections ± standard error of the mean (SEM).
Drosophila methods
Drosophila were grown at 25°C on Carpenter’s medium (Carpenter, 1950). 69B-Gal4 (Brand and Perrimon, 1993) was obtained from the Bloomington Drosophila Stock Center; UAS-Mef2 was described in Cripps et al. (2004). Mef2 mutant embryos were obtained from the stock CyO, wg-lacZ / Mef2P544, and were identified at stage 16 based upon the abnormal gut phenotype characteristic of these mutants (Ranganayakulu et al., 1995). P-element mediated germline transformation was carried out according to Rubin and Spradling (1984). Briefly, injected y w embryos were screened in the G2 generation for the rescue of the white eye color phenotype. Homozygous lines were established using standard methods. At least three transgenic lines were analyzed for each construct.
For ectopic expression of Mef2, homozygous UAS-Mef2 females were crossed to either homozygous 69B-gal4 or Tg-lacZ/+; 69B-gal4/+ heterozygotes. In the latter instance, initial stains were carried out using fluorescent detection, and immunohistochemical stains were carried out for documentation in the images.
RESULTS
Identification of Tg as a cardiac-restricted transcript
The Tg gene is located on the second chromosome at cytological location 28D3. Genome annotations have identified that it comprises two alternate promoters, for which both exons are spliced to two downstream exons, thus generating two putative polypeptide products (Grumbling et al., 2006; Figure 1A).
Figure 1. Structure and expression of Transglutaminase.

A: GBrowse image from FlyBase (Grumbling et al., 2006; url: http://flybase.bio.indiana.edu) depicting the genomic region of Tg (CG7356). Blue bars represent transcribed regions; black boxes and lines represent exons and introns of the mature mRNA; yellow boxes and lines represent coding regions. The copia and Doc transposable elements are indicated by pink lines. The extent of the riboprobe used for in situ hybridization is indicated at the bottom, as are the regions (numbered 1-4) tested for enhancer activity. B-D: Hybridization pattern of the antisense Tg riboprobe to wild-type embryos. B: At stage 14, the earliest expression was seen in the region close to the pharynx (ph). C, D: At stage 15 hybridization was detected in the Svp cells of the heart region (ht) and in the developing cardiac outflow region (cor). E: By stage 16, mature expression of Tg was observed in the Svp cells of the dorsal vessel (arrowheads), in the cardiac outflow region, and at lower levels in the non-Svp cells of the heart. All embryos are oriented with anterior to the left. B and C are sagittal views, D and E are dorsal views. Bar, 100μm.
In order to confirm the reported expression pattern of Tg, we generated a digoxigenin-labeled riboprobe from a cDNA for the gene, and hybridized this probe to Drosophila embryos. We found that the earliest specific expression of Tg was at stage 14, with hybridization detected in a location just ventral to the pharynx (Figure 1B). By stage 15, Tg transcripts were detected in a dorsal region close to the head which was identified as the precursors of the cardiac outflow region (Zikova et al., 2003), as well as at low levels in seven bilateral pairs of cells corresponding to the cardiac Svp cells (Figure 1C, D). By stage 16 (Figure 1E) strong Tg expression was observed in the cardiac outflow tract, the seven sets of Svp cells, and also at low levels in Tin-expressing cardial cells in the heart. These data confirm and expand upon the expression data previously reported.
Given that the predicted Tg gene structure comprises two alternate promoters, we also sought to determine if each promoter is used during development. RT-PCR analyses confirmed that transcripts containing each exon could be identified, and during the embryonic stage transcripts utilizing the downstream promoter predominated (data not shown). However, due to the relatively small size of the first exons, we were unable to generate exon-specific probes which were sensitive enough to detect transcripts via in situ hybridization.
Identification of the cardiac enhancer for Tg
The transcribed region of Tg spans 16kb, much of which is comprised of two transposable elements (copia and Doc). These elements are located within the large intron separating the two 5′ exons from the two 3′ exons. In order to identify sequences responsible for expression of this gene, we amplified genomic fragments and tested them for enhancer activity in transgenic animals (see Figure 1A for locations of regions tested).
Only one of the genomic regions tested showed cardiac-specific enhancer activity. When fused to a minimal promoter and lacZ reporter gene, region 4 directed lacZ expression in embryos in all of the major locations of expression of the endogenous Tg gene. This included the pharyngeal region and Svp cells at stage 15 (Figure 2A, B), and additionally the cardiac outflow region by stage 16. The timing of expression is slightly delayed compared to the endogenous gene, although that is likely a result of detecting ß-galactosidase as a reporter rather than lacZ transcripts.
Figure 2. Identification and characterization of the Tg cardiac enhancer.
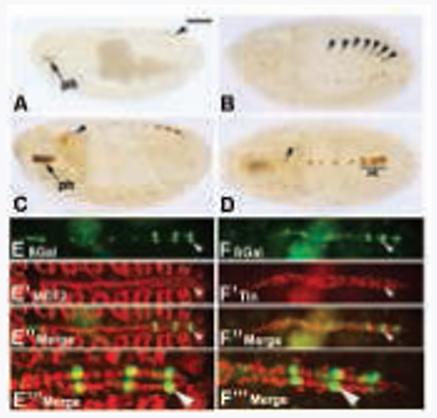
Embryos were stained for accumulation of ß-galactosidase in Tg enhancer-lacZ transgenic embryos. A-D: Diaminobenzidine-stained embryos document the time-course of enhancer-lacZ expression. A, B: Enhancer activity was detected at stage 15 in the pharyngeal region (ph) and in the Svp cells (arrowheads) of stage 15 embryos. C, D: By stage 16, there was strong pharyngeal expression, faint signal in the cardiac outflow region (arrowhead), and robust ß-galactosidase accumulation in the Svp cells. Note also that there was some reporter expression in the Tin cells of the heart (Ht), mirroring expression of the endogenous gene in the same cells. E-E”’: Accumulation of ß-galactosidase in the dorsal vessel of transgenic embryos (E, green) overlapped with that of MEF2 (E’, red). Both channels are shown in E” and at higher magnification in E”’. Arrowhead indicates a cardial cell which was positive for both ß-galactosidase and MEF2. F-F”’: Accumulation of ß-galactosidase in the dorsal vessel of transgenic embryos (F, green) did not overlap with that of Tin (F’, red). Both channels are shown in F” and at higher magnification in F”’. Arrowhead indicates a Svp cell which was positive for ß-galactosidase and negative for Tinman. All embryos are oriented with anterior to the left. A and C are sagittal views, the rest are dorsal views. Panels in E and F were generated by confocal microscopy. Bar: 100μm for A-D; 75μm for E-E” and F-F”; 30μm for E”’ and F”’.
To more effectively confirm that the enhancer (and thus the endogenous Tg) was active precisely in the cardiac cells, we carried out double-immunofluorescent staining of transgenic embryos, using antibodies for the ß-galactosidase reporter and cardiac markers. Stained embryos were analyzed by confocal microscopy.
When we stained embryos for accumulation of the muscle transcription factor myocyte enhancer factor-2 (MEF2) alongside ß-galactosidase, we observed co-localization of MEF2 with ß-galactosidase in the Tg-lacZ expressing cells of the cardiac tube. This confirmed that Tg-lacZ was expressed in cardial cells of the dorsal vessel, and not in pericardial cells nor in nearby tissues (Figure 2E-E”’). In order to determine which cardial cells expressed Tg the most strongly, we next determined if the accumulation of Tin and ß-galactosidase were overlapping. In this case, there was not overlapping accumulation of Tinman protein with the cells expressing the highest levels of Tg-lacZ. Since the muscular dorsal vessel comprises cells which either express tin, or cells which express svp and not tin, these data indicated that Tg is expressed most strongly in the Svp cells of the dorsal vessel (Figure 2F-F”’). Taken together, these studies confirm the expression of Tg in Svp cells of the dorsal vessel. This finding is important, since relatively few Svp cell-specific enhancers have been identified to date (Gajewski et al., 2000; Ryan et al., 2007; this study).
Dependence of enhancer activity upon MEF2
In order to identify candidate regulators of Tg in the embryo, we next sought to identify regulatory genes whose function was required for Tg expression. Whereas Tg was still expressed normally in homozygous svp mutants (data not shown), we found that endogenous Tg was not expressed in Mef2 mutant animals (Figure 3A,B). This result suggested that at least the cardiac component of Tg expression might depend directly upon MEF2. By contrast, Tg expression in the region below the pharynx was unaffected in Mef2 mutants (Figure 3 A, B insets). To confirm this result, we studied the cardiac expression of Tg-lacZ in mutant embryos, and found that this reporter was also inactive in a Mef2 mutant background (Figure 3 C,D).
Figure 3. Dependence of Tg expression upon MEF2.
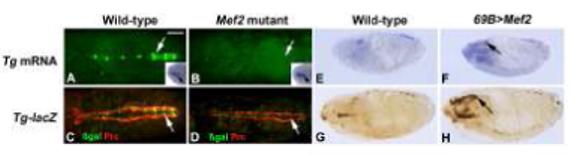
Tg transcription (A, B) and Tg-lacZ expression (C, D) were robust in the dorsal vessel of wild-type (A, C), but absent in Mef2 mutants (B, D). Note that Tg expression in the sub-pharyngeal region was not affected by the absence of MEF2 (insets in A, B). Tg transcripts in A and B were visualized either using fluorescent in situ hybridization and detected by confocal microscopy, or by immunohistochemical staining following in situ hybridization (insets). Tg-lacZ expression was detected in C and D via immunofluorescence for ß-galactosidase protein (green), and the location of the dorsal vessel was demonstrated by immunofluorescence for the pericardial cell marker Pericardin (red). Tg transcription (E, F) and Tg-lacZ expression (G, H) were also detected in either wild-type embryos (E, G) or those in which ectopic Mef2 expression had been induced (F, H). In this experiment, only mild ectopic activation of the target gene was observed in the head regions of embryos (arrows in F and H). Embryos are oriented with anterior to the left. A-D are dorsal views; E-H and insets are sagittal views. Bar, 100μm.
In order to explore the regulation of Tg by MEF2 in more detail, we next studied the expression of Tg and Tg-lacZ in the presence of ectopically-expressed Mef2. To achieve this, we utilized the Gal4-UAS expression system (Brand and Perrimon, 1993): here, we crossed the ectodermal driver 69B-gal4 to a UAS-Mef2 line, and studied the expression of Tg and Tg-lacZ in the resulting embryos. For the endogenous Tg gene, we observed a very slight pattern of ectopic gene activation in the experimental embryos (compare Figure 3E with F). There was a mild induction of Tg expression in several tissues in the head region (Figure 3F), as well as in the salivary glands (not shown). Given the broad activity of 69B-gal4 in the ectoderm, this pattern of induction seemed remarkably subdued for a putative MEF2 target gene. Furthermore, what little activation we saw was largely confined to stage 16 embryos.
To determine if this pattern of ectopic activation by MEF2 was shared between the endogenous gene and the enhancer-lacZ transgene, we carried out the same experiment in Tg-lacZ embryos. Here, we observed a similar result: there was some ectopic activation of Tg-lacZ expression in the head (Figure 3G,H), although the number of tissues showing ectopic activation was relatively small.
Whereas the strongest activation of a UAS-lacZ transgene by 69B-gal4 at stage 16 is in the head region and the salivary glands (data not shown), consistent with the most obvious ectopic activation of Tg and Tg-lacZ, our ectopic expression assays nevertheless indicated only a mild ability for MEF2 to activate Tg sequences. One possible explanation for this result is that MEF2 does not activate Tg directly and it instead functions via an intermediate regulator. An alternative hypothesis is that the effect of MEF2 upon Tg is attenuated due to there being only a few sub-optimal MEF2 binding sites within the gene.
Arrangement of MEF2 binding sites in the Tg enhancer
To distinguish between these possibilities we first sought to identify any MEF2 binding sites within the enhancer that we had isolated, by examining the sequence for instances of the MEF2 consensus 5′YTAWWWWTAR (where Y = C or T; W = A or T; R = A or G; Andres et al., 1995). Examination of the enhancer revealed that there were no consensus MEF2 binding sites, however there were five instances of a sequence comprising a single mis-match to this consensus. These sequences are labeled M-a through M-e in the schematic in Figure 4A, and their sequences are shown alongside the MEF2 consensus sequence (Andres et al., 1995) in Figure 4B. Regarding the M-a sequence which contains a C rather than a purine at the last position: while this site does not match the MEF2 consensus, we note that this sequence can bind MEF2 according to the initial studies of Andres et al (1995). Furthermore, a MEF2 site of this sequence is biologically relevant in the murine HRC gene (Anderson et al., 2004).
Figure 4. Identification of putative MEF2 sites in the Tg enhancer.
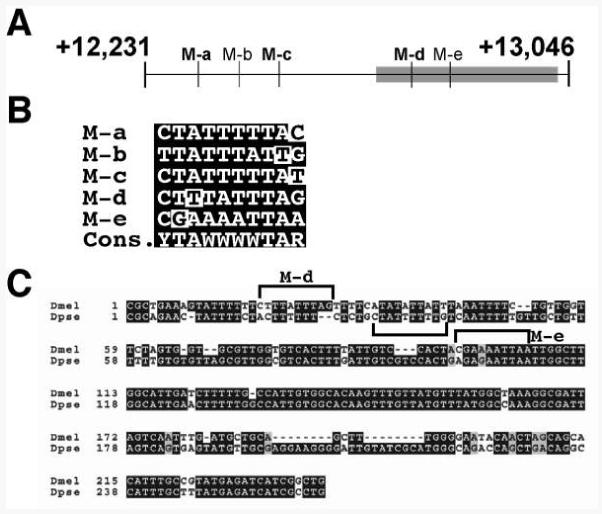
A: Schematic diagram of the enhancer, showing the locations of five putative MEF2 binding sites (labeled M-a through M-e). The region of the enhancer most conserved with D. pseudoobscura is indicated as a gray box. B: Comparison of the sequences of the five sites with the MEF2 consensus. Mismatches to consensus are indicated with white highlights. C: Conservation of Tg enhancer sequences between D. melanogaster and D. pseudoobscura. Note that, whereas the M-d sequence is not conserved, a nearby sequence in D. pseudoobscura (bracketed) contains a putative MEF2 site.
In order to further define the potential significance of these sequences, we also compared the Tg enhancer to that of the corresponding gene from the related species D. pseudoobscura. Interestingly, only the 3′ end of the enhancer showed strong sequence conservation, encompassing the M-d and M-e putative binding sites (Figure 4C). Within this region, neither putative MEF2 site was directly conserved, although a motif close in location and sequence to M-d was present. The M-e sequence was quite well conserved in D. pseudoobscura, however it contained an additional point change which rendered it less likely to represent a MEF2 binding site.
To determine if any of the putative MEF2 binding sites were capable of interacting with MEF2 protein, we carried out electrophoretic mobility shift assays. In an initial round of experiments, we mixed each probe with MEF2-containing lysate and separated the samples by electrophoresis. This revealed that three of the probe sequences (M-a, M-c, and M-d) showed binding to MEF2 protein, whereas the remaining probes (M-b and M-e) could not interact with MEF2 (data not shown).
Next, to determine if the observed interactions between the probes and MEF2 were sequence-specific, we carried out binding assays which also included competition with specific and non-specific sequences (Figure 5). For each of the double-stranded oligonucleotides tested, we observed the presence of a band corresponding to complexed MEF2-DNA; furthermore, the intensity of this band was strongly sensitive to the presence of non-radioactive competitor, but band intensity was not diminished in the presence of mutant competitor. These studies indicated that MEF2 could interact specifically with each of the candidate MEF2 binding sites M-a, M-c and M-d.
Figure 5. DNA binding assays identify three MEF2 sites within the Tg enhancer.
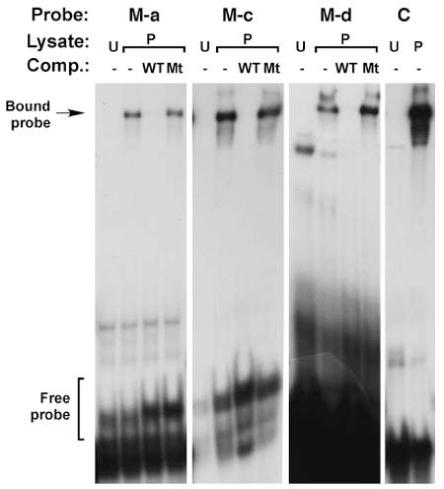
Results from electrophoretic mobility shift assays demonstrating specific interaction between the M-a, M-c, and M-d dsDNA probes and MEF2 protein. Note the absence of a band at the level of Bound probe when unprogrammed lysate (U) was added to the reaction. Inclusion of programmed lysate (P) in the absence of competitors resulted in the formation of a specific band. The intensity of this band was strongly attenuated in the presence of 100-fold excess of non-radioactive wild-type dsDNA competitor, but band intensity was not affected when unlabeled mutant dsDNA competitor is added. C refers to a control MEF2-binding probe from the Act57B gene (Kelly et al., 2002).
To further test the ability of MEF2 to interact with and to activate the Tg enhancer, we carried out co-transfection assays. Here, Drosophila Schneider Line 2 (SL2) cells were transfected with an expression plasmid containing a Mef2 cDNA, and a reporter plasmid carrying a version of the Tg enhancer. The ability of MEF2 to activate the various enhancer fragments was calculated as a fold-activation of reporter expression when compared to the absence of the activator. All the different reporter plasmids had essentially the same background expression levels in the absence of activator protein. For the reporter plasmids, we assayed three constructs: the wild-type full-length enhancer (+12,231/+13,046); a truncated enhancer which removed the two 5′ MEF2 sites but which left M-d intact (+12,702/+12,952); and the truncated enhancer in which the M-d sequence had been mutated (+12,702/+12,952mutant). The data from this experiment are summarized in Figure 6.
Figure 6. Co-transfection assays demonstrate that MEF2 is an activator of the Tg enhancer.
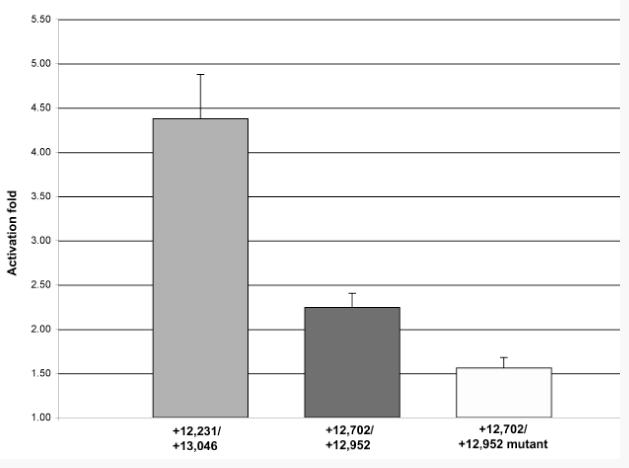
The relative levels of ß-galactosidase accumulation in SL2 cells were calculated after a Mef2 expression plasmid was co-transfected alongside Tg enhancer-lacZ reporters. The wild-type enhancer (+12,231/+13,046) was reproducibly activated by MEF2 (first bar), compared to activation by empty expression vector (set at 1.0). Truncation of this enhancer fragment to +12,702/+12,952, which removed the two 5′-most MEF2 binding sites, resulted in a strong reduction in the level of activation of the reporter by MEF2 (second bar). Finally, mutation of the remaining M-d site in this truncated enhancer further reduced activation to background levels (third bar).
We found that the activation of these enhancers followed precisely the presence and number of MEF2 binding sites that we had identified in the DNA binding assays: the full-length enhancer was activated four-fold in the presence of MEF2; the truncated enhancer was activated only two-fold in the presence of MEF2; and the truncated mutant enhancer was not efficiently activated by MEF2. These studies established that MEF2 could activate the Tg enhancer via the characterized MEF2 binding sites.
To further test the relationship between the presence of the MEF2 binding sites and the function of the Tg enhancer, we also tested the activity of the different enhancer constructs in transgenic animals in vivo. Whereas the wild-type enhancer could generate robust reporter gene expression in the Svp cells of the dorsal vessel (Figure 7A) the truncation which removed the two 5′ MEF2 sites strongly reduced reporter gene expression (Figure 7B). Further mutation of this truncated enhancer to ablate the M-d MEF2 site completely abolished reporter expression in all six transgenic lines assayed (Figure 7C). These studies thus further supported the model that MEF2 functions in vivo as an activator of the transglutaminase gene Tg.
Figure 7. In vivo activity of Tg enhancers.
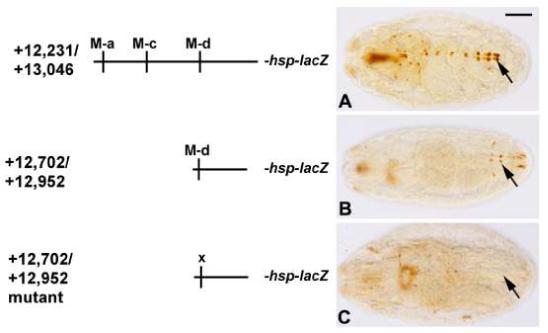
As diagrammed on the Left, the same enhancers used for co-transfection assays were tested for enhancer activity in transgenic animals in vivo. Whereas the full-length enhancer was strongly active in the cardiac cells (A), the truncated enhancer only showed very weak activity in a few Svp cells (B, and arrows in B). Mutation of the M-d MEF2 site in this truncated enhancer abolished reporter gene expression (C). Embryos are at stage 16 and oriented dorsal side uppermost and anterior to the left. Bar, 100μm.
DISCUSSION
Transglutaminase genes play a variety of roles during mammalian development and disease, and are of particular interest given their versatile functions in biology. Significant effort has been expended in identifying regulators of transglutaminase genes, and some candidate activators have been identified. Here we have taken an approach to define the Drosophila regulators of the insect transglutaminase, Tg: we have characterized the expression pattern of this gene, located the enhancer responsible for the expression pattern, and we have demonstrated that the enhancer is a direct transcriptional target of the muscle regulator MEF2. These findings are significant since they identify yet another function for MEF2, already characterized as an important transcription factor active in a variety of animal tissues (Pothoff and Olson, 2007).
At the genomic level, Tg occupies a relatively congested area of the chromosome, in which a large number of transcriptional units have been identified. There is currently no evidence that any of these transcriptional units might be functionally related. Nevertheless, the compact nature of the genomic region made it relatively simple to identify the Tg enhancer: one fragment was positive amongst only four fragments tested.
The function of Tg in the embryo has yet to be fully defined by mutant analysis. While outside the scope of this work, we note that many transglutaminase proteins are secreted from the cell, and a major transglutaminase in humans is the clotting factor XIIIa. The location of Tg expression in the cardiac inflow and outflow tracts suggests that, if it also is secreted, it would be readily dispersed within the hemolymph and might thus function in the characterized wound healing response (reviewed in Galko and Krasnow, 2004; Theopold et al., 2004). Indeed, transglutaminase activity has recently been characterized at sites of clot formation in Drosophila larvae (Karlsson et al. 2004), and systemic knock-down of Tg expression affects clot formation (Lindgren et al 2008). Nevertheless, accumulation of Tg transcripts in cells other than the cardiac tissue (such as in the vicinity of the pharynx), suggest that Tg protein might play additional roles during development.
What is the role of MEF2 in Tg activation ? Clearly MEF2 is not sufficient for Tg expression given the ectopic expression data presented above, which showed only mild ectopic activation of the endogenous gene or the cardiac enhancer. Similarly, while MEF2 can activate Tg-lacZ in tissue culture, under these conditions MEF2 is likely to be present at super-physiological levels. Moreover, MEF2 accumulation in the embryo is broad within the mesoderm, whereas Tg expression is restricted to the Svp cardial cells. On the other hand, MEF2 is essential for Tg expression, based upon the mutant analyses presented, and also based upon the enhancer deletion and mutagenesis assays. The most reasonable conclusion from all of these data is that MEF2 somehow provides a muscle or cardiac context, in which other regulatory factors must function. Perhaps MEF2 occupancy of the enhancer is necessary to provide an epigenetic signal in which other factors function to activate gene expression. Alternatively, it is possible that MEF2 provides a context for activation throughout the cardiac tube, but where expression in Tin cells is repressed. For either of these possibilities, it is likely that MEF2 functions on the Tg enhancer alongside tissue-restricted co-factors, and that these co-factors must be present to enable MEF2 to fully activate the Tg gene.
What might be the identity of the additional Tg regulators ? The Svp cells are named based upon the expression of the orphan nuclear receptor gene svp (Bodmer and Frasch, 1999; Gajewski et al., 2000), however Tg expression is normal in svp mutants. Genetically downstream of svp in the cardial cells are the T-box transcription factors Doc1-3. While Doc1-3 are expressed in Svp cells, their expression there depends upon svp function (Lo and Frasch, 2001), thus these factors seem unlikely to be candidate regulators for Tg. It is therefore possible that whatever factors regulate svp expression in the cardiac tube might also impact Tg expression. Identification of how Tg expression is restricted to the Svp cells should provide additional new insight into how this novel pattern of expression is controlled.
Acknowledgements
We are grateful to Bruce Paterson, Manfred Frasch and Danielle Gratecos for generously providing antibodies, and we thank Maryann Jaramillo for excellent technical assistance. This research was supported by HL080545 from the NIH to RMC. We acknowledge technical support from the Department of Biology’s Molecular Biology Facility, supported by NIH grant number 1P20RR18754 from the Institute Development Award (IdeA) Program of the National Center for Research Resources.
References
- Anderson JP, Dodou E, Heidt AB, De Val SJ, Jaehnig EJ, Greene SB, Olson EN, Black BL. HRC is a direct transcriptional target of MEF2 during cardiac, skeletal, and arterial smooth muscle development in vivo. Mol. Cell. Biol. 2004;24:3757–3768. doi: 10.1128/MCB.24.9.3757-3768.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andres V, Cervera M, Mahdavi V. Determination of the consensus binding site for MEF2 expressed in muscle and brain reveals tissue-specific sequence constraints. J. Biol. Chem. 1995;270:23246–9. doi: 10.1074/jbc.270.40.23246. [DOI] [PubMed] [Google Scholar]
- Bodmer R, Frasch M. Genetic determination of the Drosophila heart development. In: Rosenthal N, Harvey R, editors. Heart Development. Academic Press; New York: 1999. pp. 65–90. [Google Scholar]
- Brand AH, Perrimon N. Targeted gene expression as a means of altering cell fates and generating dominant phenotypes. Development. 1993;118:401–15. doi: 10.1242/dev.118.2.401. [DOI] [PubMed] [Google Scholar]
- Carpenter JM. A new semisynthetic food medium for Drosophila. Drosophila Information Service. 1950;24:96–97. [Google Scholar]
- Cassidy AJ, van Steensel MAM, Steijlen PM, van Geel M, van der Velden J, Morley SM, Terrinoni A, Melino G, Candi E, McLean WHI. A homozygous missense mutation in TGM5 abolishes epidermal transglutaminase 5 activity and causes acral peeling skin syndrome. Am. J. Hum. Genet. 2005;77:909–917. doi: 10.1086/497707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chartier A, Zaffran S, Astier M, Semeriva M, Gratecos D. Pericardin, a Drosophila type IV collagen-like protein is involved in the morphogenesis and maintenance of the heart epithelium during dorsal ectoderm closure. Development. 2002;129:3241–3253. doi: 10.1242/dev.129.13.3241. [DOI] [PubMed] [Google Scholar]
- Cripps RM, Olson EN. Control of cardiac development by an evolutionarily conserved transcriptional network. Dev. Biol. 2002;246:14–28. doi: 10.1006/dbio.2002.0666. [DOI] [PubMed] [Google Scholar]
- Cripps RM, Lovato TL, Olson EN. Positive autoregulation of the Myocyte enhancer factor-2 myogenic control gene during somatic muscle development in Drosophila. Dev. Biol. 2004;267:536–47. doi: 10.1016/j.ydbio.2003.12.004. [DOI] [PubMed] [Google Scholar]
- Dubbink HJ, Cleutjens KBJM, van der Korput HAGM, Trapman J, Romijn JC. An Sp1 binding site is essential for basal activity of the human prostate-specific transglutaminase gene (TGM4) promoter. Gene. 1999;240:261–267. doi: 10.1016/s0378-1119(99)00454-0. [DOI] [PubMed] [Google Scholar]
- Gajewski K, Choi CY, Kim Y, Schulz RA. Genetically distinct cardial cells within the Drosophila dorsal vessel. Genesis. 2000;28:36–43. doi: 10.1002/1526-968x(200009)28:1<36::aid-gene50>3.0.co;2-4. [DOI] [PubMed] [Google Scholar]
- Galko MJ, Krasnow MA. Cellular and genetic analysis of wound healing in Drosophila larvae. PLOS Biology. 2004;2:1114–1126. doi: 10.1371/journal.pbio.0020239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gossett LA, Kelvin DJ, Sternberg EA, Olson EN. A new myocyte-specific enhancer-binding factor that recognizes a conserved element associated with multiple muscle-specific genes. Mol. Cell. Biol. 1989;9:5022–33. doi: 10.1128/mcb.9.11.5022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffin M, Casadio R, Bergamini CM. Transglutaminases: Nature’s biological glues. Biochem J. 2002;368:377–396. doi: 10.1042/BJ20021234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grumbling G, Strelets V, The Flybase Consortium Flybase: anatomical data, images, and queries. Nucleic Acids Research. 2006;34:D484–D488. doi: 10.1093/nar/gkj068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horton RM. PCR-mediated recombination and mutagenesis. Molecular Biotechnology. 1995;3:93–99. doi: 10.1007/BF02789105. [DOI] [PubMed] [Google Scholar]
- Huber M, Rettler I, Bernasconi K, Frenk E, Lavrijsen SPM, Ponec M, Bon A, Lautenschlager S, Schorderet DF, Hohl D. Mutations of keratinocyte transglutaminase in lamellar ichthyosis. Science. 1995;267:525–528. doi: 10.1126/science.7824952. [DOI] [PubMed] [Google Scholar]
- Karlsson C, Korayem AM, Scherfer C, Dushay MS, Theopold U. Proteomic analysis of the Drosophila hemolymph clot. J. Biol. Chem. 2004;279:52033–52041. doi: 10.1074/jbc.M408220200. [DOI] [PubMed] [Google Scholar]
- Kelly KK, Meadows SM, Cripps RM. Drosophila MEF2 is a direct regulator of Actin57B transcription in cardiac, skeletal, and visceral muscle lineages. Mech. Dev. 2002;110:39–50. doi: 10.1016/s0925-4773(01)00586-x. [DOI] [PubMed] [Google Scholar]
- Kelly Tanaka KK, Bryantsev AL, Cripps RM. Myocyte enhancer factor 2 and Chorion factor 2 collaborate in activation of the myogenic program in Drosophila. Mol. Cell. Biol. 2008;28:1616–1629. doi: 10.1128/MCB.01169-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kida M, Souri M, Yamamoto M, Saito H, Ichinose A. Transcriptional regulation of cell-type specific expression of the TATA-less A subunit for human coagulation factor XIII. J. Biol. Chem. 1999;274:6138–6147. doi: 10.1074/jbc.274.10.6138. [DOI] [PubMed] [Google Scholar]
- Lilly B, Zhao B, Ranganayakulu G, Paterson BM, Schulz RA, Olson EN. Requirement of MADS domain transcription factor D-MEF2 for muscle formation in Drosophila. Science. 1995;267:688–93. doi: 10.1126/science.7839146. [DOI] [PubMed] [Google Scholar]
- Lindgren M, Riazi R, Lesch C, Wilhelmsson CW, Theopold U, Dushay MS. Fondue and transglutaminase in the Drosophila larval clot. J. Insect Physiol. 2008;54:586–592. doi: 10.1016/j.jinsphys.2007.12.008. [DOI] [PubMed] [Google Scholar]
- Lo P, Frasch M. A role for the COUP-TF-related gene seven-up in the diversification of cardioblast identities in the dorsal vessel of Drosophila. Mech. Dev. 2001;104:49–60. doi: 10.1016/s0925-4773(01)00361-6. [DOI] [PubMed] [Google Scholar]
- Mariniello L, Qin Q, Jessen BA, Rice RH. Keratinocyte transglutaminase promoter analysis. J. Biol. Chem. 1995;270:31358–31363. doi: 10.1074/jbc.270.52.31358. [DOI] [PubMed] [Google Scholar]
- Molina MR, Cripps RM. Ostia, the inflow tracts of the Drosphila heart, develop from a genetically distict subset of cardial cells. Mech. Dev. 2001;109:51–59. doi: 10.1016/s0925-4773(01)00509-3. [DOI] [PubMed] [Google Scholar]
- Monier B, Astier M, Semeriva M, Perrin L. Steroid-dependent modification of Hx function drives myocyte reprogramming in the Drosophila heart. Development. 2005;132:5283–5293. doi: 10.1242/dev.02091. [DOI] [PubMed] [Google Scholar]
- Nagy L, Saydak M, Shipley N, Lu S, Basilion JP, Yan ZH, Syka P, Chandraratna RAS, Stein JP, Heyman RA, Davies PJA. Identification and characterization of a versatile retinoid response element (retinoic acid receptor response element-retinoid X receptor response element) in the mouse tissue transglutaminase gene promoter. J. Biol. Chem. 1996;271:4355–4365. doi: 10.1074/jbc.271.8.4355. [DOI] [PubMed] [Google Scholar]
- O’Neill JW, Bier E. Double-label in situ hybridization using biotin and digoxigenin-tagged RNA probes. Biotechniques. 1994;17:870, 874–5. [PubMed] [Google Scholar]
- Patel NH. Imaging neuronal subsets and other cell types in whole-mount Drosophila embryos and larvae using antibody probes. Methods Cell. Biol. 1994;44:445–87. doi: 10.1016/s0091-679x(08)60927-9. [DOI] [PubMed] [Google Scholar]
- Pereira FA, Qiu Y, Ahou G, Tsai MJ, Tsai SY. The orphan nuclear receptor COUPTFII is required for angiogenesis and heart development. Genes and Development. 1999;13:1037–1049. doi: 10.1101/gad.13.8.1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ponzielli R, Astier M, Chartier A, Therond A, Semeriva M. Heart tube patterning in Drosophila requires integration of axial and segmental information provided by the Bithorax Complex genes and Hedgehog signaling. Development. 2002;129:4509–4521. doi: 10.1242/dev.129.19.4509. [DOI] [PubMed] [Google Scholar]
- Pothoff MJ, Olson EN. MEF2: a central regulator of diverse developmental programs. Development. 2007;134:4131–4140. doi: 10.1242/dev.008367. [DOI] [PubMed] [Google Scholar]
- Ranganayakulu G, Zhao B, Dokidis A, Molkentin JD, Olson EN, Schulz RA. A series of mutations in the D-MEF2 transcription factor reveal multiple functions in larval and adult myogenesis in Drosophila. Dev. Biol. 1995;171:169–81. doi: 10.1006/dbio.1995.1269. [DOI] [PubMed] [Google Scholar]
- Rizki TM. The circulatory system and associated cells and tissues. In: Ashburner M, Wright TRF, editors. The genetics and biology of Drosophila. 2b. Academic Press; New York: 1978. pp. 397–452. [Google Scholar]
- Rubin GM, Spradling AC. Genetic transformation of Drosophila with transposable element vectors. Science. 1982;218:348–53. doi: 10.1126/science.6289436. [DOI] [PubMed] [Google Scholar]
- Ryan KM, Hendren JD, Helander LA, Cripps RM. The NK homeodomain transcription factor Tinman is a direct activator of seven-up in the Drosophila dorsal vessel. Dev Biol. 2007;302:694–702. doi: 10.1016/j.ydbio.2006.10.025. [DOI] [PubMed] [Google Scholar]
- Sambrook J, Fritsch EF, Maniatis T. Molecular cloning: a laboratory manual. Cold Spring Harbor Laboratory; Cold Spring Harbor, N.Y.: 1998. [Google Scholar]
- Sellin J, Albrecht S, Kolsch V, Paululat A. Dynamics of heart differentiation, visualized utilizing heart enhancer elements of the Drosophila melanogaster bHLH transcription factor Hand. Gene. Exp. Patt. 2006;6:360–375. doi: 10.1016/j.modgep.2005.09.012. [DOI] [PubMed] [Google Scholar]
- Standen GR, Bowen DJ. Factor XIII A-Bristol 1: detection of a nonsense mutation (arg171-to-stop codon) in factor XIII A subunit deficiency. Brit. J. Haemat. 1993;85:769–772. doi: 10.1111/j.1365-2141.1993.tb03221.x. [DOI] [PubMed] [Google Scholar]
- Tao Y, Schulz RA. Heart development in Drosophila. Seminars Cell. Dev. Biol. 2007;18:3–15. doi: 10.1016/j.semcdb.2006.12.001. [DOI] [PubMed] [Google Scholar]
- Theopold U, Schmidt O, Soderhall K, Dushay MS. Coagulation in arthropods: defense, wound closure and healing. Trends Immunol. 2004;25:289–294. doi: 10.1016/j.it.2004.03.004. [DOI] [PubMed] [Google Scholar]
- Thummel CS, Pirrotta V. New pCaSpeR P element vectors. Drosophila Information Service. 1992;71:150. [Google Scholar]
- Tomancak P, Beaton A, Weiszmann R, Kwan E, Shu S, Lewis SE, Richards S, Ashburner M, Hartenstein V, Celniker SE, Rubin GM. Systematic determination of patterns of gene expression during Drosophila embryogenesis. Genome Biology. 2002;3:1–14. doi: 10.1186/gb-2002-3-12-research0088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wasserthal LT. Drosophila flies combine periodic heartbeat reversal with a circulation in the anterior body mediated by a newly discovered anterior pair of ostial valves and ‘venous’ channels. J. Exp. Biol. 2007;210:3707–3719. doi: 10.1242/jeb.007864. [DOI] [PubMed] [Google Scholar]
- Xu X, Yin Z, Hudson JB, Ferguson EL, Frasch M. Smad proteins act in combination with synergistic and antagonistic regulators to target Dpp responses to the Drosophila mesoderm. Genes and Development. 1998;12:2354–2370. doi: 10.1101/gad.12.15.2354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zikova M, Da Ponte J-P, Dastugue B, Jagla K. Patterning of the cardiac outflow region in Drosophila. Proc. Natl. Acad. Sci. USA. 2003;100:12189–12194. doi: 10.1073/pnas.2133156100. [DOI] [PMC free article] [PubMed] [Google Scholar]