

Abstract
We describe the computational design of a single-chain four-helix bundle that noncovalently self-assembles with fully synthetic non-natural porphyrin cofactors. With this strategy, both the electronic structure of the cofactor as well as its protein environment may be varied to explore and modulate the functional and photophysical properties of the assembly. Solution characterization (NMR, UV/vis) of the protein showed that it bound with high specificity to the desired cofactors, suggesting that a uniquely structured protein and well-defined site had indeed been created. This provides a genetically expressed single-chain protein scaffold that will allow highly facile, flexible, and asymmetric variations to enable selective incorporation of different cofactors, surface-immobilization and introduction of spectroscopic probes.
Introduction
The design of optically and electronically responsive molecular assemblies capable of diverse photophysical processes is becoming increasingly feasible due to progress in the study of electron transfer and charge separation in both biological systems and synthetic small molecules.1–4 Recently, studies aimed at combining the strengths of these two types of systems have been initiated through the computational design of four-helix bundles that noncovalently self-assemble with fully synthetic non-natural porphyrin cofactors. With this strategy, both the electronic structure of the cofactor as well as its protein environment may be varied to explore and modulate the functional and photophysical properties of the assembly. These systems are unlike previous de novo “maquettes” 3,5 in that these peptides were computationally designed to recognize a unique porphyrin cofactor. Thus, while maquettes bind a large number of porphyrins with low specificity, the computationally designed protein should have much higher specificity. Indeed, they bound with high specificity to their desired cofactors, suggesting that a uniquely structured protein and well-defined site had successfully been created. The initial assemblies had D2 symmetry (PATET, Figure 1b),6–8 comprising four identical helices that encapsulate two iron diphenylporphyrin molecules (FeDPP(III), Figure 1a).
Figure 1.
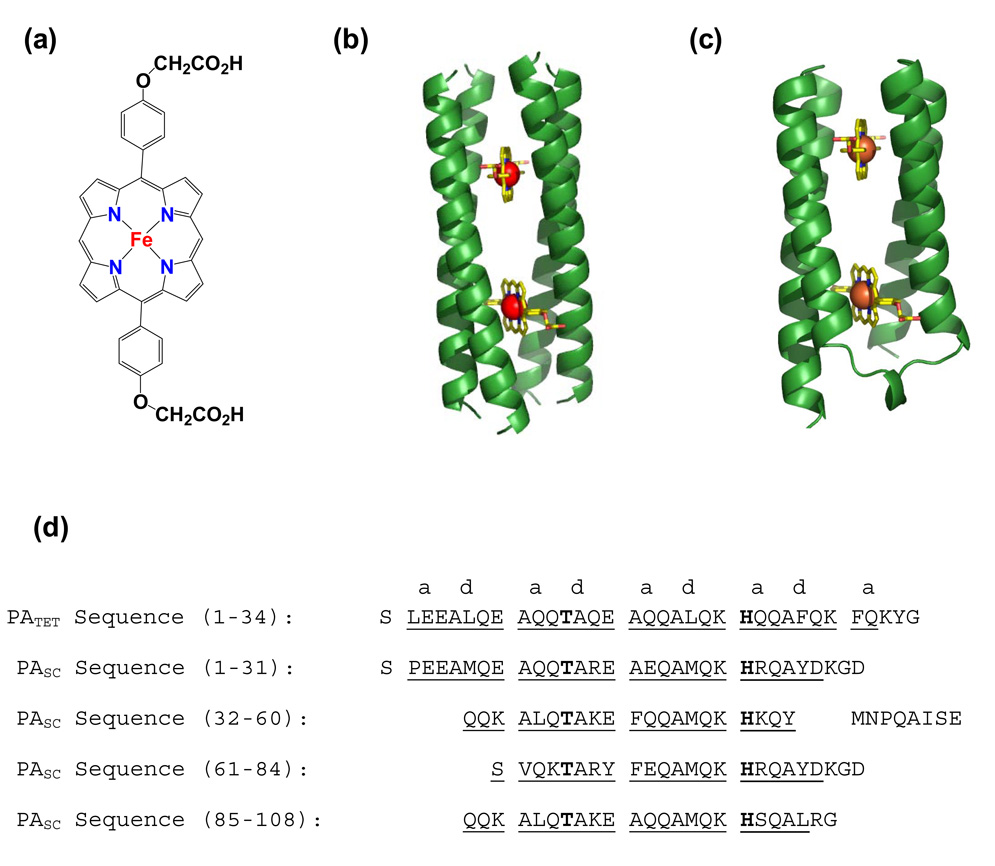
Model and sequence of Porphyrin Assemblers. (a) Iron diphenylporphyrin cofactor (b) Model of PATET 8 (c) Model of PASC (d) Sequences of PATET and PASC, which have the helical regions underlined, and the heptad positions of the residues are lettered. Keystone residues are bolded.
Porphyrin-binding peptides have been extensively studied for the past 15 years, utilizing helix-loop-helix designs with disulfide bonds, and template-assisted design strategies.1–4,9–16 Until now, however, there have been no genetically expressed single-chain proteins that allow highly facile, flexible, and asymmetric variations of the protein to allow selective incorporation of different cofactors, surface-immobilization and introduction of spectroscopic probes. Additionally, a single-chain variant would allow for the modification of Cys residues, semi synthesis,17 or genetic strategies for introducing unique cofactors and probes.18 To this end, we present a single-chain version, PASC, of the originally designed four-chain helical bundle (Figure 1c).
Despite the fact that many porphyrin-binding helical bundles have been designed and extensively characterized, there have been significant problems associated with the determination of their three-dimensional structures, in part because many have conformationally fluctuating interiors.19,20 Over the years, this problem has been alleviated somewhat through the synthesis of variants of the original “maquettes”, resulting in better dispersion in the NMR spectra of the complexes.21 Smaller peptide-porphyrin complexes with two helices per complex have also been structurally characterized.21–23 The availability of a single-chain protein, such as PASC, would greatly expedite structural determination due to the ease of incorporation of isotopic labels for heteronuclear NMR studies.
Methods
Design of PASC
Requirements for efficient cofactor binding dictated the precise protein backbone geometry. A single low-energy structure was computed with a Monte Carlo simulated annealing protocol that considered the following constraints: (i) a metal-metal distance between 17 and 19 Å, (ii) optimal His N(ε) to Fe bonding interactions, (iii) second-shell hydrogen bonds between His N(δ) and Thr O(γ), (iv) minimal steric clashes, and (v) maintenance of D2 symmetry. This process led to imidazole rings in near-perpendicular alignment as a result of the second-shell hydrogen bonds. These steps to obtain protein backbone and cofactor coordinates were repeated from Cochran et al.8 and led to the same tetrameric four-helix bundle, which was then modified. All four 32-residue helices of the original PATET bundle were shortened by four residues at each end, except for the N-terminus of one helix, which would eventually provide the N-terminus of the entire single-chain four-helix bundle. The helices were shortened so as to obtain a compact (108-residue) single-chain peptide, PASC.
With regard to stabilizing the bundle, the loops take the place of hydrophobic interactions at each end of the bundle in the previous tetrameric complex.8 The resulting bundle structure contained three shortened 24-residue helices and one 28-residue helix. The helices were connected through loops of natural protein structures taken from a protein database with reduced redundancy, PDB Select.24 A program called STITCH was developed in order to identify loops that would be most suitable to connect a given pair of unconnected helices. STITCH enumerates helix-loop-helix superpositions and rank-orders these superpositions according to root mean square differences (RMSD) between Cα atoms of the helical residues that terminate the loops and the Cα atoms of the helices. Loop structures were selected having overlaps of at least five Cα positions at each loop-helix transition. Overlapping backbone fragments were deleted. Connection points were identified by visual inspection, looking for closest overlap of loop and helix backbone atoms. Residues near the connection points were relaxed using CHARMM25 steepest decent optimization for 25 steps. Bond lengths and bond angles of the resulting backbone structure were checked using PROCHECK.26
Protein Design Steps
The identities of the four Fe-coordinating His residues and the four Thr residues to which these are hydrogen bonded were held fixed to support the six-point coordination of the Fe(III) cations: T12, H23, T38, H49, T65, H76, T91, H102. The identities of the remaining 100 out of 108 positions were determined by recursive calculations using the computational design algorithm SCADS.27,28 SCADS (which provides site-dependent amino acid probabilities) has been used previously in a number of successful protein design studies.8,24–29 All amino acid identities were allowed, except for cysteine and histidine to eliminate the possibility of oxidative degradation and unwanted disulfide bridges, and to avoid cofactor coordination at sites other than those in the positions designated above. After the first round of SCADS calculations, identities in 24 positions were fixed because they were the most probable amino acids with high probabilities of at least p>0.5, and frequently p>0.8: A9, A13, A16, M20, A26, K34, A35, A39, F42, A45, M46, K50, A66, F69, A72, M73, A79, K87, A88, A92, A95, A98, and M99. Of these, A16, M20, F42, M46, F69, M73, A95, and M99 form a complementary, asymmetric hydrophobic core between the two porphyrin macrocycles. Note that this nonsymmetrical packing illustrates a major advantage of a single-chain design as opposed to a tetramer because, as symmetry is not required, more complementary packing options become available. The hydrophobic core is shown in Figure 4.
Figure 4.
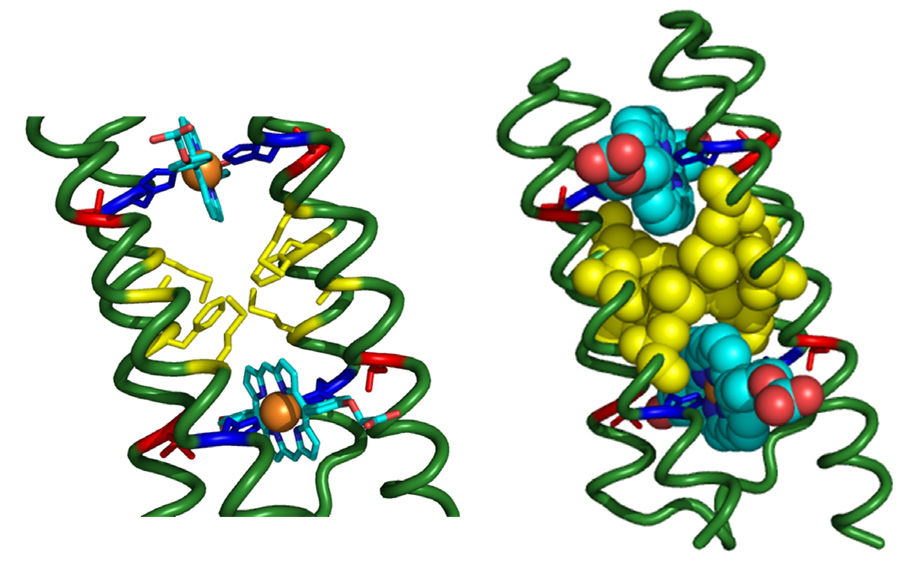
The asymmetric hydrophobic interior of PASC. Dark Blue = His, Yellow = Met and Phe, Red = Ala; blue ribbon section are the Histidines. The non-symmetric hydrophobic core of PASC consists of residues A16, M20, F42, M46, F69, M73, A95, and M99. These residues were picked by SCADS as residues dominating the probability profiles at their respective positions.
In addition to these fixed residues, for the second-round of SCADS calculation nine interior positions 6, 27, 36, 53, 58, 62, 80, 89, and 106, each facing one of the cofactors, were limited to non-charged or hydrophobic amino acid identities. After this second round of SCADS calculations, decisions about the remaining 76 amino acid identities were made based on the resulting SCADS probability profiles in conjunction with other considerations relevant to the design. Residues M6, Q11, Q18, R24, Q25, Q37, K40, Q44, Q51, Y52, I58, E60, V62, K64, Q71, R77, Q78, Q90, K93, Q100, S103, Q104, and L106 were chosen because their SCADS probabilities were the highest at their respective sites. That is, in a total of 55 out of 108 amino acid identities were fixed based on either the requirement for the hexacoordinated Fe(III) ions or on the highest SCADS probabilities. N-terminal residues S1, P2, E3, E4, and the final G108 form known helix capping motifs. Residues A5, Q7, E8, Q10, E15, Q21, K22, Q33, E41, Q47, K48, Q63, Q74, K75, Q86, E94, Q96, Q97, K101, and A105 were chosen because they were probable and because they also occupied the same positions relative to the cofactors in the earlier successful four-helix bundle design.8 In particular, interhelical salt bridges E15-K48, K22-E41, K75-E94 were also introduced to stabilize the fold. Positively charged R14, R67, and R107 (together with K50 from the first round of SCADS calculations) were chosen near the negatively charged carboxylate groups of the cofactors. D28, K29, G30, D31, D81, K82, G83, and D84 were identified based on a statistical analysis of alpha-helical hairpin turns by Lahr et al,.30,31 in which the authors analyzed the relative frequencies of amino acids in such hairpin turns and found that some amino acids are preferred. Similarly, in the larger loop, identities of the wild type residues at positions P55, A57, S59, and S61, taken from 1T06.pdb, were kept. Q32, Q43, Q56, and Q85 were chosen over more probable charged amino acids to reduce the overall number of charged amino acids in the protein. L36, M53, and L89 are hydrophobic residues in interior positions facing the cofactors, which were preferred over more probable polar amino acids. Conversely, N54 and Y68 are polar residues facing the protein exterior, which were selected over slightly more probable hydrophobic residues. Charged residues E17 and E70 were introduced to exploit potentially favorable intrahelical i, i+3 interactions with R14 and R67, respectively. Finally, Y27 and Y80 were chosen over the more probable W due to their more hydrophilic character at these partially exposed sites.
PASC/FeDPP(III) Complex Preparation
The synthetic gene for PASC was obtained and the codon sequence optimized using software from DNA2.0. The sequence was transformed into the pET28a vector from the NcoI to BamHI restriction sites (Novagen), which contains the IPTG-inducible T7 promoter. The protein was over-expressed in E.coli, harvested by centrifugation, and the cells lysed by sonication. The supernatant was heat denatured for 30 minutes at 70 °C, centrifuged, and the supernatant purified to homogeneity by reverse-phase HPLC on a preparative C4 column (Vydac). The molecular weight was confirmed by MALDI mass spectrometry. The lyophilized protein was reconstituted in 50 mM phosphate, 100 mM NaCl at pH 7.5. As previously described, the chemically synthesized FeDPP(III) cofactor was added from a stock solution in DMSO.8 Aggregation caused by this addition was minimized by briefly heating the complex to 70 °C for 15 minutes (above the Tm of the holo-protein). After heating, the samples were equilibrated at room temperature for an hour, and filtered though 0.22 µm PVDF membrane centrifuge filter (Millipore) at 13,200 rpm in an Eppendorf 5415D microcentrifuge.
UV-visible and CD Spectroscopy
To verify binding of the cofactor, complex formation was monitored at the Soret band at 410 nm. UV-visible data of the complex (as prepared above) was obtained on a Cary 300 Bio UV-Visible spectrophotometer at room temperature in a 1 cm quartz cuvette. To evaluate the backbone and stability of the protein and the complex, CD spectroscopy was carried out at 25 °C on a Jasco 810 Spectropolarimeter in 0.1 cm cuvettes. Buffer conditions for CD experiments were 10 mM phosphate buffer, pH 7.5, and the protein concentration was 25 to 50 µM. For thermal denaturation experiments, the ellipticity at 222 nm was monitored as a function of temperature. Data were collected every 2 °C with a heating rate of 1°C/min followed by a 4 minute equilibration time. Melting temperature mid-points (Tm’s) were estimated by plotting temperature (x-axis) versus Ffold (y-axis) in which the fraction folded was estimated from the observed ellipticity versus that for the folded and unfolded baselines. The apo-states of the proteins were too unstable to resolve a folded baseline, so the parameters for the holo-protein were used to approximate these values:
Determination of Aggregation State
Size Exclusion Chromatography
Gel filtration profiles were obtained using a Superdex 75 10/300GL column on an FPLC system (GE Healthcare AKTA FPLC System). To evaluate the oligomeric state, 100 µL of the complex was injected onto the column and eluted with a flow rate of 0.5mL/min with a mobile phase of 50 mM phosphate buffer and 100 mM NaCl, pH 7.5. The monomeric species eluted at 12.3 mL, yielding an apparent molecular weight of 18,500 D. MWapp was calculated from a standard curve done with mass standards blue dextran, Vo, aprotinin, 6,500 D, cytochrome C, 12,400 D, carbonic anhydrase, 29,000 D, and albumin, 66,000 D.
Analytical Ultracentrifugation
For analytical ultracentrifugation, a 300 µL sample of 150 µM PASC with 300 µM FeDPP(III) in 50 mM phosphate buffer, 100 mM NaCl at pH 7.5 was prepared as previously mentioned. After centrifuging for 10 minutes at 13,200 rpm in an Eppendorf 5415D microcentrifuge to remove insoluble material, an aliquot of 110 µL was used for sedimentation equilibrium experiments. These were performed at 25 °C using a Beckman XL-I analytical ultracentrifuge. The absorbance was monitored at 280 nm, and the sample was centrifuged at 25,000, 35,000, and 45,000 rpm (supplementary information, Figure S1). The data were analyzed using a modified global fitting routine in IGOR Pro (Wavemetric, Inc.). The data were well described by assuming a single molecular weight species. The protein maintains its calculated partial specific volume when the monomeric molecular weight is held constant. The partial specific volume of 0.7137 was calculated using SEDNTERP.23
Stoichiometry
Binding stoichiometry was determined using the monomeric species isolated through size exclusion chromatography (SEC). The monomeric PASC/FeDPP(III) complex was monitored for formation by UV-vis spectroscopy at 410 nm. By holding the protein concentration fixed, various cofactor concentrations were evaluated using UV-visible spectroscopy to monitor the formation of the Soret band at 410 nm. The stoichiometry of the SEC fractions were determined by a hemochrome assay32 for FeDPP(III) concentration, and an HPLC assay for protein concentration (Figure S2). Binding stoichiometry was determined to be 2:1, as there was no significant increase in absorbance at 410 nm with excess cofactor (Figure S3).
NMR Analysis
Multiple preparations of 100 µM 15N-labelled PASC with 200 µM FeDPP(III) were incubated at 70 °C for 15 minutes, allowed to equilibrate at room temperature, filtered through a 0.45 µm Nylon syringe filter, and 2 mL injected over a preparative Superdex 75 XK16/70 column (GE Healthcare AKTA FPLC system). The monomeric fractions were collected and concentrated using an Amicon Ultra-15 MWCO 10,000 filter (Millipore). The sample was buffer-exchanged into 50 mM phosphate buffer, 200 mM NaCl, pH 7.5 over a PD-10 column (GE Healthcare) and concentrated to 270 µL for the NMR experiment. The final sample was diluted to 300 µL with D20, and 2 µL of 150 mM 3-(Trimethylsilyl)-1-propanesulfonic acid sodium salt stock (Fluka) in water was added. The final concentrations of cofactor and protein were determined by hemochrome and HPLC assays (Figure S2).
The 1H-15N HSQC spectrum was recorded at 45 °C on a Bruker DMX-600 spectrometer equipped with a 5-mm x, y, z-shielded pulsed-field gradient triple-resonance probe. 2048 (1H) × 128 (15N) complex data points were collected for each dimension. The spectral widths used for were 8992.806 Hz for 1H and 1702.903 Hz for 15N dimensions. The data were processed by using Felix 2002 (Felix NMR, Inc.). The final matrix contains 2048 × 2048 data points along 1H and 15N dimensions.
MD Simulations
The designed cofactor-bound PASC was taken as the starting structure for the simulations. The protein was solvated within a 50 × 50 × 72 Å3 water box, and periodic boundary conditions were employed in all cell dimensions. Simulations were performed using the NAMD 2.5 package33 and the CHARMM22 all-atom force field.34 The parameratization of the six coordinate DPP-Fe( III) macrocyle and substituents was derived from CHARM22 parameters for the iron heme and from functional groups of amino acid side chains.34 A time step of 2 fs was used for bonded and nonbonded interactions, and a cutoff distance of 10 Å was maintained to calculate nonbonded interactions. Full electrostatic forces were evaluated at every other time step through application of the particle-mesh Ewald (PME) method.35 All bonds between hydrogen atoms and heavy atoms were constrained using the SHAKE algorithm with a tolerance of 10−5 Å.36
Results
Design
The structure of the 108-residue single chain protein, PASC (Porphyrin Assembler, Single Chain, Figure 1c) was designed, using the model for the corresponding homotetramer (Figure 1b, PATET) as a starting point. As described in the Methods section, interhelical loops with favorable conformations and sequences were introduced into the structure by grafting known interhelical backbone structures onto the PATET model. The precise conformation of the loops in the structure were obtained by: 1) superimposing helical hairpins from proteins of known structure onto the helices of PATET; 2) selecting hairpins with a low r.m.s.d. for the superimposed helical residues; 3) inserting the loop at a position where the chains are particularly well superimposed; and 4) selecting structures that did not interfere with cofactor binding (Figure 2).30 As described in previous work,8,28 eight “keystone” residues directly involved in cofactor ligation or second-shell interactions were predefined (four histidines and four threonines, Figure 3), while the remaining positions of the protein were specified using the computational methodology SCADS (Figure 1d). The resulting protein, PASC, exhibits complementary, asymmetric hydrophobic interactions within the interior that are difficult to realize in symmetric bundles (Figure 4). PASC was expressed from a synthetic gene (10 – 15 mg/L pure protein) in E. coli.
Figure 2.
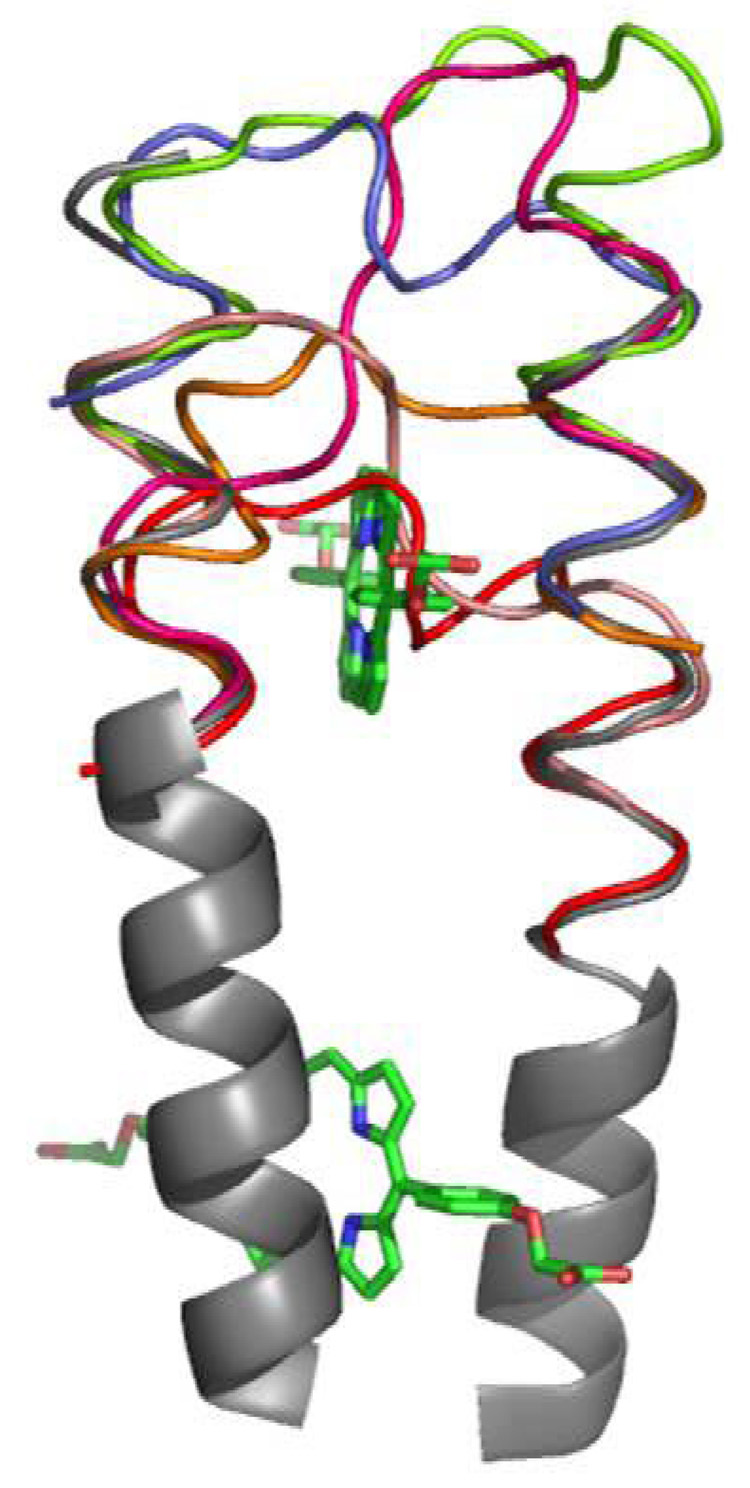
Model of loop choices for PASC. Each color corresponds to a section from the structures selected from the PDB: 2spc (light pink), lim8 (red), 1t06 (blue), 1fzn (magenta), 1g03 (orange), and lea3 (light green). The loop 1t06 (in blue) had the lowest deviation in RMSD from the PATET helices (shown in gray, they correspond to Helix 2 and 3 of PASC), and was chosen for the PASC scaffold.
Figure 3.
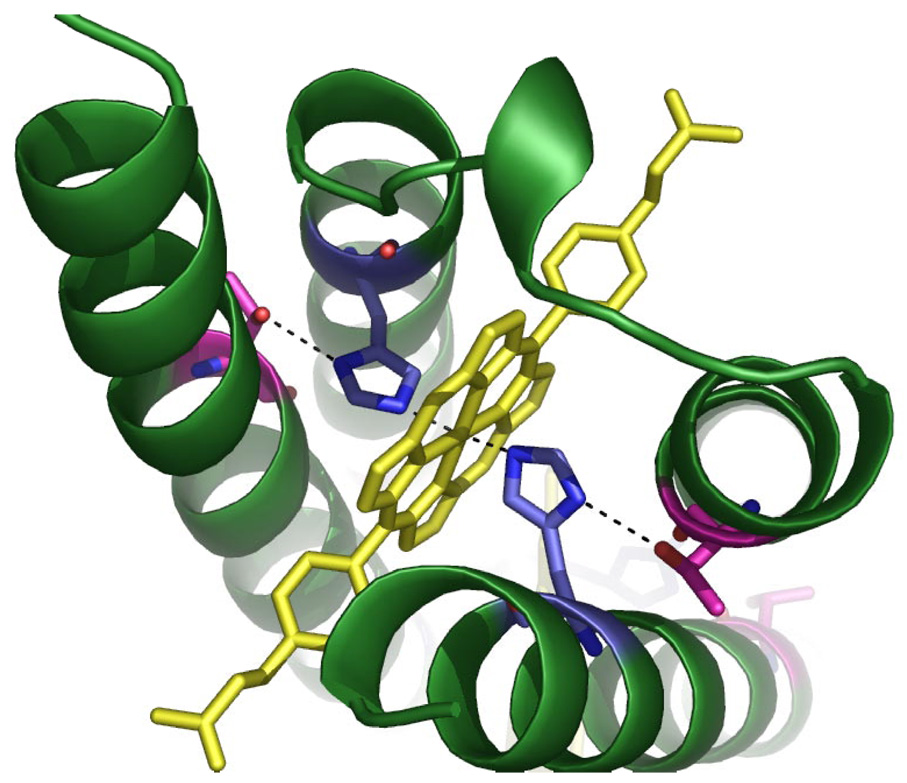
Keystone residues in PASC. Histidines are shown in blue, Threonines are shown in pink, and the porphyrin ring of FeDPP(III) is shown in yellow; hydrogen bonds are shown as dotted lines from the Fe to N(ε) of His, and from the Thr O(γ) to N(δ) of His.
UV-visible and CD spectroscopy
UV-visible spectroscopy demonstrated that PASC selectively binds FeDPP(III) in the intended stoichiometry (2:1). Addition of protein to two equivalents of FeDPP(III) resulted in a sharpening of the Soret band and a shift from 405 to 410 nm (Figure 5). Titration of PASC with FeDPP(III) indicates formation of a 2:1 cofactor to protein complex (Figure S2). Reduction of the PASC/FeDPP(III) complex with sodium dithionite resulted in a ferrous complex with spectral signatures typical of a bis-His ligated complex (λmax = 525 nm).6,37 The Soret peak for PASC is at 410 nm, and shifts to 419 nm when reduced with solid sodium dithionite. For comparison, PATET showed a Soret peak at 408 nm, which shifts to 417 nm when reduced.8 The PASC/FeDPP(III) complex gives an extinction coefficient of 177,000 M−1 cm−1, whereas the PATET complex gives an extinction coefficient of 160,000 M−1 cm−1, which are the same within the experimental error of the protein concentration determination.
Figure 5.

Cofactor binding selectivity. (a) PASC demonstrates cofactor selectivity for FeDPP(III) over a biological cofactor, (b) iron protoporphyrin IX, FePPIX(III). Cofactor was added to the protein in 50 mM phosphate buffer, 100 mM NaCl, pH 7.5 as described in the methods.
Furthermore, the visible CD spectrum showed that each cofactor was bound in a unique chiral environment in PASC.19 The UV-visible CD spectrum of the reduced species shows that both cofactors have been incorporated into the protein, with a change in the transition at 419 nm, consistent with the location of the peak of the ferrous species (Figure 6a). In order to evaluate the second binding site, the spectrum of one equivalent each of protein and cofactor was subtracted from the spectrum of one equivalent protein and two equivalents of cofactor (Figure 6b). This revealed that there is little difference in cofactor ligation at each site, because both sites produce a signal that is similar in magnitude and shape.
Figure 6.

Circular Dichroism spectroscopy in the visible-UV region. (a) The addition of sub-stoichiometric (25 µM) and stoichiometric (50 µM) amounts of FeDPP(II) to 25 µM PASC at 25 °C in 10 mM phosphate buffer at pH 7.5. (b) The spectra demonstrate equality of binding sites within the PASC scaffold.
Size exclusion chromatograph and analytical ultracentrifugation (AUC) of PASC/FeDPP(III)
The association of PASC was examined by size exclusion chromatography, using a Superdex 75 analytical column (Figure 7). In the absence of cofactor, the protein elutes in a symmetrical peak. Upon addition of one or two equivalents of FeDPP(III) per mole of PASC, the protein eluted at a smaller volume consistent with the compaction of the structure upon association with the cofactor. A second minor peak is also observed, and is indicative of aggregation in the reconstitution assay. This peak can be minimized by reconstituting at a high dilution and annealing the sample at 70 °C. Once purified, the monomeric form is stable at millimolar concentrations for several weeks at room temperature. The monomeric peak, representing the PASC /FeDPP(III) complex, was further examined by AUC. Analytical ultracentrifugation of the PASC/FeDPP(III) complex revealed that it sedimented as a single molecular species with molecular weight of 14,150 D, in excellent agreement with that for the monomeric complex, 13,932 D (Figure S1).
Figure 7.

Size Exclusion Chromatography of the PASC apo- and holo-complexes. The apo-protein eluted at a higher retention time (11 mL, black line) than the holo-protein (12.3 mL, dotted line), indicating that once the cofactor binds, the holo-protein is more compact.
Far UV-CD and NMR investigation of backbone conformation of PASC
Far UV circular dichroism spectroscopy was used to examine the helical structure, stability, and the effects of porphyrin binding in the protein as well as in PATET. The spectra of both apo-PASC and apo-PATET were nearly identical at room temperature (Figure 8a and 8b). The mean residue ellipticity for apo-PATET ([θ]222nm, 25°C) is reported to be −8,300 deg cm2 dmol−1. Apo-PASC is slightly less helical ([θ]222nm, 25°C = −7,200 deg cm2 dmol−1) due to the inclusion of the loop, and also disorder near the N- and C-terminus of PASC. This trend is also observed for the holo-scaffolds; both show an increase in helical content upon addition of the cofactor and PASC remains less helical than PATET (Table 1).
Figure 8.

Circular Dichroism spectroscopy in the near-UV region. (a) The spectra of apo-PASC (25 µM protein, shown in circles) and holo-PASC (25 µM protein + 50 µM FeDPP(III), shown in squares) at 25°C demonstrate the increase in helicity upon addition of FeDPP(III). (b) Spectra of apo-PATET (50 µM protein, shown in triangles) and holo-PATET (50 µM protein + 25 µM FeDPP(III), shown in diamonds). (c) Fraction folded for PATET (apo, holo) and PASC (apo, holo) demonstrate that the Tm’s (°C) for PASC are higher than those for PATET, indicating increased stability.
Table 1.
Summary of CD data for PATET and PASC, both apo- and holo-proteins.
| [θ222 nm] 0°C[a] | Tm[b] | Concentration [c] | |
|---|---|---|---|
| PATET (holo) [d] | −23,800 | 56 | 50 |
| PATET (apo) | −4,200 | 4 | 50 |
| PASC (holo) [e] | −14,500 | 68 | 25 |
| PASC (apo) | −3,900 | 22 | 25 |
(deg • cm2 dmol−1)
Temperature (°C)
Concentration of protein scaffold (µM)
25 µM FeDPP added to scaffold PATET
50 µM FeDPP added to scaffold PASC
However, PASC demonstrated higher stability than PATET, in both the apo- and holo-forms of the protein (Figure 8c). The addition of porphyrin resulted in a very large increase in the stability of PASC (Tm of the apo- and holo-protein were 22 °C and 68 °C, respectively, Table 1) and the steepness of the transition. Comparatively, the melting midpoints were determined to be 4 °C for apo-PATET and 56 °C for holo-PATET. This demonstrates the increased stability of the scaffold PASC, due to inclusion of inter-helical links as well as better interior packing. Given the increased stability and the fact that the scaffold demonstrated its designed behavior, it was important to establish whether or not the scaffold was a good structural candidate by evaluating the complex using NMR.
The NMR spectrum of PASC should provide valuable information concerning the degree to which the protein is uniquely folded, versus adopting a molten maquette-like conformation. Figure 9a shows that the 15N proton HSQC spectrum of PASC with FeDPP(III) at pH 7.5 is indeed well dispersed, with approximately 70 resonances observed. The amide backbone resonances are spread over 7 – 11 ppm in the proton dimension and 110 – 132 ppm in the 15N dimension, indicative of a well defined, presumably uniquely folded structure. At pH 6.0, where the exchange with solvent is slower, we expect to observe more resonances that are better resolved (Figure 9b). The approximately 95 resonances are observed at pH 6.0 and they are dispersed over 6 – 11 ppm in the proton dimension, 108 – 134 ppm in the 15N dimension, consistent with a natively folded structure.
Figure 9.

Heteronuclear Single Quantum Correlation NMR of PASC (a) The 1H-15N HSQC spectrum of 0.83 mM PASC and 1.6 mM FeDPP(III) in 50 mM phosphate buffer, 200 mM NaCl, pH 7.5, at 45 °C. (b) The 1H-15N HSQC spectrum of 1.1 mM PASC and 2.2 mM FeDPP(III) in 100 mM phosphate buffer, 150 mM NaCl, pH 6.0, at 45 °C.
Molecular Dynamics
Molecular dynamics (MD) calculations were performed to validate the retention of key design elements during an all-atom simulation of the molecular structure. If the sequence is well suited to stabilize the starting three-dimensional model, the structure should equilibrate rapidly and then show only small local deviations from its equilibrium position. However, starting structures that are either unstable, far from their energy minimum, or dynamically averaging on the nanosecond time scale might be expected to continuously drift from their starting configuration during MD simulations.38 The motions of PASC in a 50 × 50 × 72 Å3 periodic box of water were simulated for 7 ns. A plot of the RMSD of the observed structure (relative to the starting model) versus time shows the wild type protein with FeDPP(III) equilibrates within 200 ps, and then remains essentially constant over the remaining 7 ns (Figure 10a). Examination of the contributions of the individual helices to the RMSD fluctuations showed that the first and fourth helices were most variable, due to fraying of their N-terminal and C-terminal sequences (Figure S4).
Figure 10.

Molecular Dynamics simulations. Comparison of trajectories of the backbone RMSD and the metal-to-metal distance versus time for the PASC scaffold. The wild type system (PASC + FeDPP(III)) backbone RMSD is shown in (a), whereas the mutant (PASC-ALA + FeDPP(III)) backbone RMSD is in (c). The metal-to-metal distances for the wild type system (b) are shown in contrast to that for the mutant system (d).
To determine the significance of the observed structural conservation of the structure over this time scale, we introduced four mutations at positions i+4 from the His ligand. Previous work from the groups of Suslick and Benson13–16 had shown that hydrophobic residues at these positions contribute significantly to the stability of heme-peptide complexes. Thus, Y27, M53, Y80, and L06 were mutated to alanine in a protein designated PASC-ALA, and the dynamics of this protein were simulated for 7 ns. Similar to the trajectory for PASC, PASC-ALA showed a rapid equilibration in approximately 200 ps, but then its structure continued to vary over the remainder of the simulation, never reaching a stable configuration (Figure 10c). This finding is reflected not only in the plots of RMSD versus time but also from an examination of the distance between the metal ions in the two cofactors, which were essentially constant for PASC but became continuously smaller for PASC-ALA (Figure 10b and 10d). The computationally designed loops were also examined for stability, on the 7 ns timescale, and in PASC both the short as well as the long inter-helical loops were highly stable on the ns time scale (Table S1). Within the time of the simulation the phi and psi angles of the wild type residues remained in the same basins of the Ramachandran plot (Table S1).
Conclusion
Here we demonstrate that modern methods of computational protein design were successfully used to yield a fully asymmetric protein that holds considerable promise for structural characterization as well as functional studies. Specifically, this genetically expressed scaffold allows for the facile incorporation of isotopic labels such as 15N and 13C necessary for multidimensional NMR experiments. Structural studies to understand the nature of the binding are underway, and are made possible by not only the successful incorporation of the non-biological cofactor, but also by the stability of PASC in solution. The selectivity of PASC for FeDPP(III) demonstrates the specificity of binding for a given cofactor. The design and incorporation of additional cofactors with various functionalities is now possible and such designs are currently underway. In conclusion, de novo protein design is a promising and expanding approach to examine the delicate interplay between a cofactor’s environment and its functional properties.
Supplementary Material
Analytical ultracentrifugation data, FeDPP(III) quantification, hemochrome assays, and molecular dynamics simulation information. This material is available free of charge via the Internet at http://pubs.acs.org.
ACKNOWLEDGMENT
This work was supported by the DOE grant DE-FG02-04ER46156 (MJT, JGS, WFD) and the National Science Foundation grant NSEC DMR-0425780. Partial support was also provided by the NIH Grants GM54616 to W.F.D., P0148130 to J.G.S., and GM071628 to M.J.T. We also thank James Lear for interpreting the AUC data.
REFERENCES
- 1.Lombardi A, Nastri F, Pavone V. Chemical Reviews. 2001;101:3165–3189. doi: 10.1021/cr000055j. [DOI] [PubMed] [Google Scholar]
- 2.Reedy CJ, Gibney BR. Chemical Reviews. 2004;104:617–649. doi: 10.1021/cr0206115. [DOI] [PubMed] [Google Scholar]
- 3.Kennedy ML, Gibney BR. Current Opinion in Structural Biology. 2001;11:485–490. doi: 10.1016/s0959-440x(00)00237-2. [DOI] [PubMed] [Google Scholar]
- 4.Xu T, Wu SP, Miloradovic I, Therien MJ, Blasie JK. Nano Letters. 2006;6:2387–2394. doi: 10.1021/nl062091p. [DOI] [PubMed] [Google Scholar]
- 5.Discher BM, Koder RL, Moser CC, Dutton PL. Current Opinion in Chemical Biology. 2003;7:741–748. doi: 10.1016/j.cbpa.2003.10.013. [DOI] [PubMed] [Google Scholar]
- 6.Ghirlanda G, Osyczka A, Liu W, Antolovich M, Smith KP, Dutton PL, Wand AJ, DeGrado WF. Journal of the American Chemical Society. 2004;126:6. doi: 10.1021/ja039935g. [DOI] [PubMed] [Google Scholar]
- 7.North B, Summa CM, Ghirlanda G, DeGrado WF. Journal of Molecular Biology. 2001;311:1081–1090. doi: 10.1006/jmbi.2001.4900. [DOI] [PubMed] [Google Scholar]
- 8.Cochran FV, Wu S, Nanda V, Wang W, Saven JG, Therien MJ, DeGrado WF. Protein Science. 2004;13:157–157. [Google Scholar]
- 9.DeGrado WF, Summa CM, Pavone V, Nastri F, Lombardi A. Annual Review of Biochemistry. 1999;68:779–819. doi: 10.1146/annurev.biochem.68.1.779. [DOI] [PubMed] [Google Scholar]
- 10.Shifman JM, Gibney BR, Sharp RE, Dutton PL. Biochemistry. 2000;39:14813–14821. doi: 10.1021/bi000927b. [DOI] [PubMed] [Google Scholar]
- 11.Rau HK, Haehnel W. Journal of the American Chemical Society. 1998;120:468–476. [Google Scholar]
- 12.Mutter M, Tuchscherer GG, Miller C, Altmann KH, Carey RI, Wyss DF, Labhardt AM, Rivier JE. Journal of the American Chemical Society. 1992;114:1463–1470. [Google Scholar]
- 13.Liu DH, Williamson DA, Kennedy ML, Williams TD, Morton MM, Benson DR. Journal of the American Chemical Society. 1999;121:11798–11812. [Google Scholar]
- 14.Huffman DL, Rosenblatt MM, Suslick KS. Journal of the American Chemical Society. 1998;120:6183–6184. [Google Scholar]
- 15.Rosenblatt MM, Wang JY, Suslick KS. Proceedings of the National Academy of Sciences of the United States of America. 2003;100:13140–13145. doi: 10.1073/pnas.2231273100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Arnold PA, Benson DR, Brink DJ, Hendrich MP, Jas GS, Kennedy ML, Petasis DT, Wang MX. Inorganic Chemistry. 1997;36:5306–5315. [Google Scholar]
- 17.Muir TW. Annual Review of Biochemistry. 2003;72:249–289. doi: 10.1146/annurev.biochem.72.121801.161900. [DOI] [PubMed] [Google Scholar]
- 18.Wang L, Xie J, Schultz PG. Annual Review of Biophysics and Biomolecular Structure. 2006;35:225–249. doi: 10.1146/annurev.biophys.35.101105.121507. [DOI] [PubMed] [Google Scholar]
- 19.Gibney BR, Rabanal F, Reddy KS, Dutton PL. Biochemistry. 1998;37:4635–4643. doi: 10.1021/bi971856s. [DOI] [PubMed] [Google Scholar]
- 20.Betz SF, Raleigh DP, Degrado WF. Current Opinion in Structural Biology. 1993;3:601–610. [Google Scholar]
- 21.Huang SS, Koder RL, Lewis M, Wand AJ, Dutton PL. Proceedings of the National Academy of Sciences of the United States of America. 2004;101:5536–5541. doi: 10.1073/pnas.0306676101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Huang SS, Gibney BR, Stayrook SE, Dutton PL, Lewis M. Journal of Molecular Biology. 2003;326:1219–1225. doi: 10.1016/s0022-2836(02)01441-9. [DOI] [PubMed] [Google Scholar]
- 23.Laue TM, Shah BD, Ridgeway TM, Pelletier SL. Computer-aided interpretation of analytical sedimentation data for proteins. In: Harding SE, Rowe AJ, Horton JC, editors. Analytical Ultracentrifugation in Biochemistry and Polymer Science. Cambridge: Royal Society of Chemistry; 1992. pp. 90–125. [Google Scholar]
- 24.Hobohm U, Sander C. Protein Science. 1994;3:522–524. doi: 10.1002/pro.5560030317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Brooks BR, Bruccoleri RE, Olafson BD, States DJ, Swaminathan S, Karplus M. Journal of Computational Chemistry. 1983;4:187–217. [Google Scholar]
- 26.Laskowski RA, Macarthur MW, Moss DS, Thornton JM. Journal of Applied Crystallography. 1993;26:283–291. [Google Scholar]
- 27.Kono H, Saven JG. Journal of Molecular Biology. 2001;306:607–628. doi: 10.1006/jmbi.2000.4422. [DOI] [PubMed] [Google Scholar]
- 28.Calhoun JR, Kono H, Lahr S, Wang W, DeGrado WF, Saven JG. Journal of Molecular Biology. 2003;334:1101–1115. doi: 10.1016/j.jmb.2003.10.004. [DOI] [PubMed] [Google Scholar]
- 29.Nanda V, Rosenblatt MM, Osyczka A, Kono H, Getahun Z, Dutton PL, Saven JG, DeGrado WF. Journal of the American Chemical Society. 2005;127:5804–5805. doi: 10.1021/ja050553f. [DOI] [PubMed] [Google Scholar]
- 30.Lahr SJ, Engel DE, Stayrook SE, Maglio O, North B, Geremia S, Lombardi A, DeGrado WF. Journal of Molecular Biology. 2005;346:1441–1454. doi: 10.1016/j.jmb.2004.12.016. [DOI] [PubMed] [Google Scholar]
- 31.Engel DE, DeGrado WF. Journal of Molecular Biology. 2004;337:1195–1205. doi: 10.1016/j.jmb.2004.02.004. [DOI] [PubMed] [Google Scholar]
- 32.Berry EA, Trumpower BL. Analytical Biochemistry. 1987;161:1–15. doi: 10.1016/0003-2697(87)90643-9. [DOI] [PubMed] [Google Scholar]
- 33.Kale L, Skeel R, Bhandarkar M, Brunner R, Gursoy A, Krawetz N, Phillips J, Shinozaki A, Varadarajan K, Schulten K. Journal of Computational Physics. 1999;151:283–312. [Google Scholar]
- 34.MacKerell AD, Bashford D, Bellott M, Dunbrack RL, Evanseck JD, Field MJ, Fischer S, Gao J, Guo H, Ha S, Joseph-McCarthy D, Kuchnir L, Kuczera K, Lau FTK, Mattos C, Michnick S, Ngo T, Nguyen DT, Prodhom B, Reiher WE, Roux B, Schlenkrich M, Smith JC, Stote R, Straub J, Watanabe M, Wiorkiewicz-Kuczera J, Yin D, Karplus M. Journal of Physical Chemistry B. 1998;102:3586–3616. doi: 10.1021/jp973084f. [DOI] [PubMed] [Google Scholar]
- 35.Darden T, York D, Pedersen L. Journal of Chemical Physics. 1993;98:10089–10092. [Google Scholar]
- 36.Ryckaert JP, Ciccotti G, Berendsen HJC. Journal of Computational Physics. 1977;23:327–341. [Google Scholar]
- 37.Robertson DE, Farid RS, Moser CC, Urbauer JL, Mulholland SE, Pidikiti R, Lear JD, Wand AJ, Degrado WF, Dutton PL. Nature. 1994;368:425–431. doi: 10.1038/368425a0. [DOI] [PubMed] [Google Scholar]
- 38.Zou HL, Strzalka J, Xu T, Tronin A, Blasie JK. Journal of Physical Chemistry B. 2007;111:1823–1833. doi: 10.1021/jp0666378. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Analytical ultracentrifugation data, FeDPP(III) quantification, hemochrome assays, and molecular dynamics simulation information. This material is available free of charge via the Internet at http://pubs.acs.org.