

Abstract
The synthesis of five new meso-tetraarylporphyrins having pyridine, pyrimidine or nitrile groups extending tetragonally via alkynyl linkages from the para positions is described. The radial extension is nearly double that of common porphyrins such as tetra-p-pyridylporphyrin. Practical quantities can be produced by Pd-coupling protocols when traditional methods fail. Applications of these extended porphyrins in the area of porous metal-organic frameworks are anticipated.
Keywords: Porphyrins, heterocycles, Sonagashira reaction, palladium cross-coupling, Rothemund synthesis, metal-organic frameworks
Introduction
An active area of contemporary chemistry is the assembly of new materials using porphyrin building blocks. Particularly intriguing are the possibilities with porous metal-organic frameworks (MOFs), sometimes called “artificial zeolites”. In seminal work in the early 1990s, Robson combined the tetragonal shape propagation properties of tetraarylporphyrins with metal-directed coordinate bond formation to produce porous framework solids whose voids were filled with exchangeable solvent molecules.1 In the last decade or so, a variety of infinite framework, lamellar, and ribbon structures have been discovered using tetraarylporphyrins that contain peripheral groups capable of forming metal-ligand coordinate bonds in a divergent manner1–13 and a number of reviews on this topic are now available.14–18 The control of pore size and the maintenance of structural integrity after solvent removal from the pores are two of the critical issues that must be addressed before practical applications can result.19–22 More robust structures can be produced using bridging ligands with metalloporphyrins5 or fullerenes as supramolecular pillars11 but typically at the expense of pore size.
For the most part, work in this area has been restricted to a few easily synthesized or commercially available tetraarylporphyrins, such as 5,10,15,20-tetrakis(4-cyanophenyl)porphyrin or 5,10,15,20-tetrakis(4-pyridyl)porphyrin:
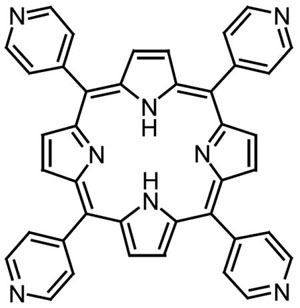
With the possibility of metal-organic frameworks with larger pore sizes in mind, we have synthesized analogues of these popular porphyrins having radial extensions of N-donor groups designed to bind metal ions.

Alkynyl linkages from the para-positions of the meso-aryl groups maintain a rigid propagation of tetragonal shape. A related meta-alkynyl derivative has recently been used to prepare a bis-porphyrin box.23
The most commonly employed method for making a symmetrical tetraarylporphyrin continues to be the Rothemund synthesis from a benzaldehyde24 using Adler and Longo’s modification.25 Many porphyrins are also amenable to Lindsey conditions.26 However, there are significant problems when benzaldehydes bearing heterocyclic moieties are used. Alder-Longo conditions typically give low yields and Lindsey conditions can fail all together. This has been attributed to the poor solubility of intermediates in the acidified solvent.27 In this paper we explore these limitations with alkynyl-linked benzaldehydes containing basic groups, and then improve the methodology by employing palladium cross-coupling reactions on a pre-formed tetra(p-bromophenyl)porphyrin.
Palladium cross-coupling methodologies are playing an increasingly important role in porphyrin synthesis.28–31 Excellent protocols have been developed for Suzuki,32 Stille,33 Negishi,34 and Sonagashira conditions involving porphyrin coupling partners. The simplicity of the experimental procedure, the wide availability of acetylenes, advances in the catalyst formulation,35 and the efficiency of coupling all contribute to the popularity of the Sonagashira reaction in porphyrin chemistry. It is these factors that prompted us to explore its utility in the current study.
Results and Discussion
Preparation of alkynyl benzaldehydes
The benzaldehydes 6, 7,36 8,37 9 and 10 were each produced in good yield (78–88%) by the Sonagashira reaction of 4-ethynylbenzaldehyde with the appropriate bromide (Scheme 1). Compound 6 was also produced from the reverse coupling partners in equally good yield (80%) (Scheme 2).
Scheme 1.

Scheme 2.

Compounds 6–9 were isolated as colored solids which, when necessary, could be purified by sublimation to colorless compounds, albeit with some product loss due to decomposition. Compound 10 could not be sublimed but could be recrystallized to analytically purity, despite the retention of a golden color.
Preparations of porphyrins under Alder-Longo conditions
Compounds 6–8 were reacted with pyrrole under Adler Longo conditions for twenty minutes (Scheme 3). From these reactions the corresponding porphyrins 1–3 were isolated in better-than-expected yields of ca. 11% after chromatographic purification. We found it beneficial to use aldehydes purified by sublimation in these reactions because then the porphyrin purification process was facilitated.
Scheme 3.
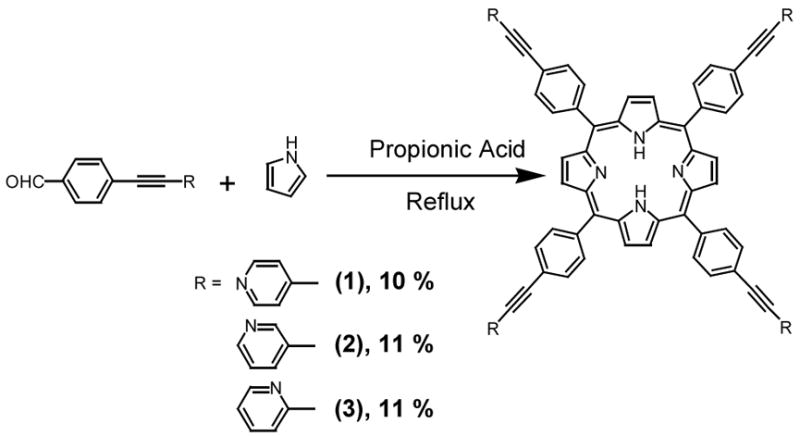
The reactions of 9 and 10 resulted in the precipitation of much solid during the porphyrin-forming reactions, presumably from decomposition. Porphyrins could be isolated after chromatography of each reaction mixture but they always contained a stubborn impurity, resistant to separation by chromatography or recrystallization. Due to the low yield of these reactions (ca. 2%) and the problem of purification, this method was abandoned for 4 and 5.
Preparation of porphyrins using Lindsey conditions
Reactions of benzaldehydes 9 and 10 were attempted using Lindsey conditions. Upon addition of BF3.OEt2 to the reaction mixture of 9 and pyrrole in chloroform, an immediate precipitate developed and no porphyrin was formed. We attribute this to complexation of BF3 with the heterocyclic base, like that encountered with other unprotected pyrimidines.38 In a separate reaction with a Brønsted acid in place of the Lewis acid, the addition of trifluoroacetic acid to the reaction mixture did not lead to the formation of any porphyrin at room temperature, even after six hours. In contrast, the addition of BF3.OEt2 to a mixture of 10 and pyrrole in chloroform led smoothly to the formation of porphyrin 5 after DDQ oxidation (Scheme 4). A pure product was isolated in 23% yield after chromatography and crystallization, providing a quite satisfactory preparation of this new, extended porphyrin.
Scheme 4.
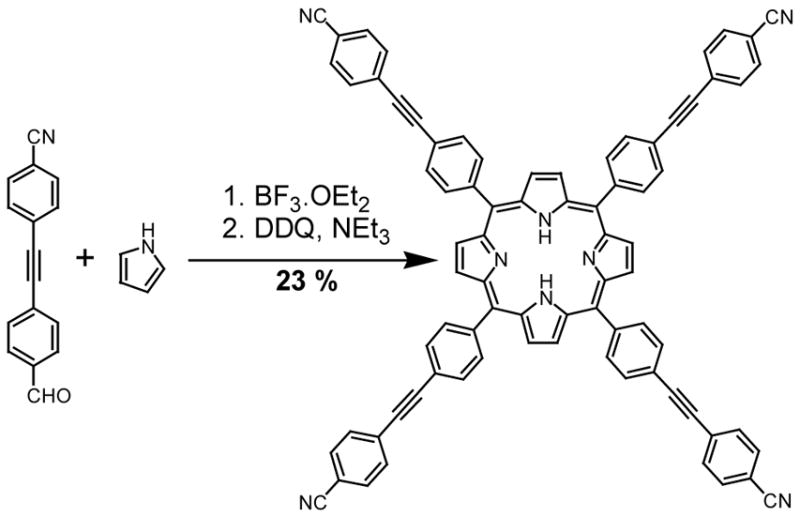
Having identified the limits of porphyrin-forming reactions using Adler-Longo and Lindsey conditions, we turned our attention towards the use of cross-coupling protocols with a pre-formed porphyrin. This takes advantage of a higher-yielding porphyrin synthesis but demands high efficiency in the subsequent four-fold coupling reaction. Successful four-fold couplings have been reported39 but not with N-heterocycles.
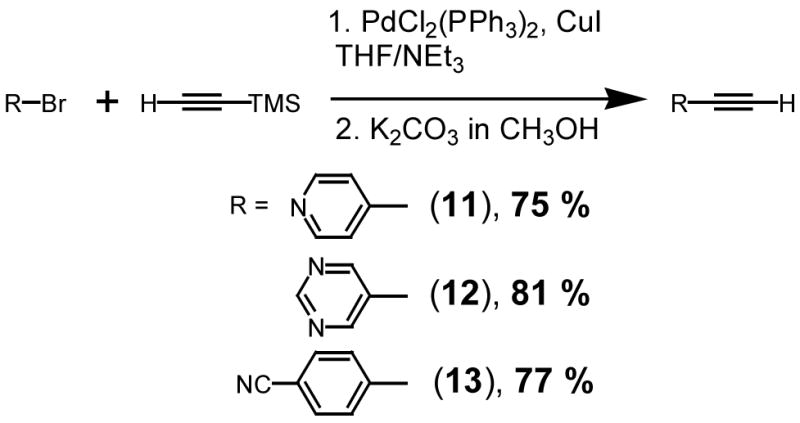
Preparation of the alkynes
Following with the excellent procedure detailed by Thorand and Krause,40 4-bromopyridine, 5-bromopyrimidine and 4-bromobenzonitrile were coupled with trimethylsilylacetylene in THF/triethylamine (Scheme 5). Standard methods were used for deprotection of the silylethers41 and compounds 11,42 12, and 1343,44 were isolated as colorless solids in good yields (75–81%).
Scheme 5.
Preparation of porphyrins by cross-coupling with alkynes
Our strategy was to use the easily-prepared 5,10,15,20-tetrakis(4-bromophenyl)porphyrin and couple it to the alkyne in the final step. This makes best use of the alkyne and also reduces the amount of Pd catalyst needed compared to the earlier procedures (Scheme 1). To the best of our knowledge, there is only one example of four-fold coupling of an alkyne to 5,10,15,20-tetrakis(4-bromophenyl)porphyrin. Chan and co-workers39 used trimethylsilylacetylene and PdCl2(PPh3)2 in triethylamine to produce the four-fold coupled product in 78% yield. These conditions, however, gave no coupled product with our alkynes. We conducted a survey of conditions using three commonly employed solvents and two common catalysts (see Supporting Information for details) and found success when using conditions similar to those reported for the synthesis of tetra-vinylated porphyrins via the Heck reaction,45 namely a Pd(0) reagent in DMF solvent at elevated temperature. Our procedure bears similarity to the recently reported Heck (Cu-free Sonogashira) alkynylation reaction of porphyrins46 and to a four-fold coupling reaction using a meso-tetrakis(ethynyl)porphyrin.47
Each of the alkynes 11–13 was reacted with 5,10,15,20-tetrakis(4-bromophenyl)porphyrin under the conditions outlined in Scheme 6 and the four-fold alkynylated compounds 1, 4 and 5 were isolated in ca. 70% yields.
Scheme 6.
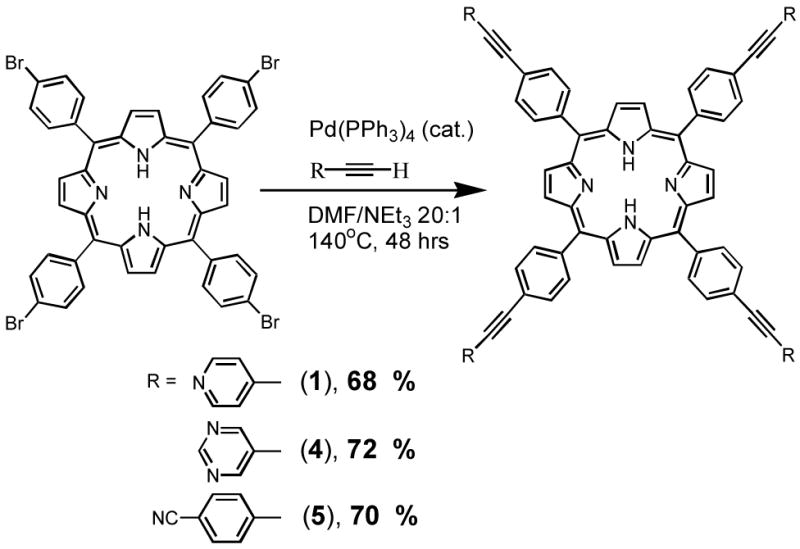
Stoichiometry was important because excess alkyne led to lower yields and MALDI mass spectrometry indicated higher than four-fold levels of alkyne incorporation. Excess alkyne incorporation has been seen in the Sonagashira reaction of other porphyrin substrates.48 Reaction time was also important because, given enough time, the alkynyl linkages become hydrogenated to vinyl linkages.
Conclusions
We have shown that acceptable yields of extended pyridine-containing porphyrins 1–3 can be obtained under Adler-Longo conditions. Lindsey conditions with BF3 catalysis are incompatible with N-heterocyclic groups but less basic cyano substituents are tolerated, allowing a good synthesis of porphyrin 5 to proceed. This finding is useful because subsequent derivatization chemistry of the cyano groups is possible.
More flexible, however, is the palladium-catalyzed route to extended porphyrins. This method did not display the functional group incompatibility problems of Adler-Longo or Lindsey conditions, allowing extended porphyrins to be synthesized on a gram scale with operational simplicity. The yield on the last step is particularly good, making efficient use of the materials and the palladium catalyst. The potential of tetraarylporphyrins with extended meso-substituents can now be explored in studies of metal-organic frameworks.
Experimental Section
General Procedure for aldehyde synthesis by Pd-catalyzed coupling
A flask is charged with 4-ethynylbenzaldehyde (1 equiv), the requisite arylbromide (1 equiv), PdCl2(PPh3)2 (1–2 mol%), and dry Et3N. The mixture is stirred magnetically for 10 min with Ar bubbled through the solution before CuI (2–4 mol%) was added and a reflux condenser attached and the mixture heated to reflux with stirring under Ar for 2 h. Following removal of Et3N by rotary evaporation, CHCl3 was added and the solution was filtered, washed with 15% aq. K2CO3, H2O, brine, dried (MgSO4 or Na2SO4) and the CHCl3 removed. Compounds were treated by passing through a plug of silica gel or by the addition of EtOH and sonication/trituration before sublimation (< 0.2 mm, heat gun), whereupon colourless solids were obtained.
4-(4-pyridinyl)benzaldehyde (6)
Following the general procedure with 4-ethynylbenzaldehyde (3.40 g, 26.1 mmol), 4-bromopyridine hydrochloride (5.26 g, 27.1 mmol), PdCl2(PPh3)2 (0.38 g, 2 mol%), Et3N (100 mL) and CuI (0.20 g, 4 mol%). Silica gel treatment and sublimation gave pure 6 (4.34 g, 80%). Compound 6 was also produced in the same manner from 4-ethynylpyridine (1.51 g, 14.6 mmol), 4-bromobenzaldehyde (2.47 g, 13.4 mmol), PdCl2(PPh3)2 (88 mg, 1 mol%) and CuI (69 mg, 2.5 mol%) in Et3N (50 mL) (2.22 g, 80 %). 1H NMR (300 MHz, CDCl3) δ: 7.38, (d 2H [JHH 4.6 Hz]); 7.69, (d 2H [JHH 8.2 Hz]); 7.88, (d 2H [JHH 6.7 Hz]); 8.62, (d 2H [JHH 4.6 Hz]); 10.02, (s 1H). 13C NMR (75 MHz, CDCl3) δ: 90.75, 93.20, 126.16, 128.82, 130.25, 131.24, 133.05, 136.73, 150.56, 191.88. m/z Calcd C14H9N1O1 207.06933; Found 207.06841. Anal.: Found: C, 81.38; H, 4.65; N, 6.61: Calcd for C14H9N1O1: C, 81.14; H, 4.38; N, 6.76.
4-(3-pyridinyl)benzaldehyde (7)
Following the general procedure from 4-ethynylbenzaldehyde (1.14 g, 8.8 mmol), 3-bromopyridine (1.45 g, 9.2 mmol), PdCl2(PPh3)2 (120 mg, 2 mol%) and CuI (70 mg, 4 mol%) in Et3N (35 mL). EtOH (ca. 1 mL) was added to the solid and the mixture sonicated/triturated before filtering off a yellow-orange powder (1.42 g, 78%). Purification by sublimation gives a colorless solid. 1H NMR (300 MHz, CDCl3) δ: 7.29, (t 1H [JHH 4.1 Hz]); 7.67, (d 2H [JHH 8.2 Hz]); 7.81, (d 1H [JHH 8.2 Hz]); 7.86, (d 2H [JHH 8.2 Hz]); 8.57, (d 1H [JHH 5.1 Hz]); 8.77, (s 1H); 10.00, (s 1H). 13C NMR (75 MHz, CDCl3) δ: 89.68, 91.52, 119.65, 123.07, 128.61, 129.55, 132.15, 135.75, 138.53, 149.12, 152.29, 191.24. m/z Calcd C14H9N1O1 207.06841; Found 207.06795. Anal.: Found: C, 81.16; H, 4.48; N, 6.51: Calcd for C14H9N1O1: C, 81.14; H, 4.38; N, 6.76.
4-(2-pyridinyl)benzaldehyde (8)
Following the general procedure from 4-ethynylbenzaldehyde (1.16 g, 8.9 mmol), 2-bromopyridine (1.43 g, 9.1 mmol), PdCl2(PPh3)2 (120 mg, 2 mol%) and CuI (70 mg, 4 mol%) in Et3N (35 mL). EtOH (ca. 1 mL) was added to the solid and the mixture sonicated/triturated before filtering off a tan powder (1.58 g, 85%). Purification by sublimation gives a colorless solid. 1H NMR (300 MHz, CDCl3) δ: 7.29 (t 1H [JHH 5.1 Hz]); 7.56, (d 1H [JHH 8.2 Hz]); 7.71, (t 1H [JHH 7.7 Hz]); 7.74, (d 2H [JHH 8.2 Hz]; 7.87, d 2H [JHH 6.2 Hz]; 8.64, d 1H [JHH 4.6 Hz); 10.02, (s 1H). 13C NMR (75 MHz, CDCl3) δ: 87.93, 91.98, 123.32, 127.42, 128.38, 129.51, 132.51, 135.89, 136.32, 142.67, 150.17, 191.31. m/z Calcd C14H9N1O1 207.06841; Found 207.06879. Anal.: Found: C, 81.10; H, 4.47; N, 6.77: Calcd for C14H9N1O1: C, 81.14; H, 4.38; N, 6.76.
4-(5-Pyrimidinyl)benzaldehyde (9)
Following the general procedure from 4-ethynylbenzaldehyde (3.14 g, 24.1 mmol), 5-bromopyrimidine (3.92 g, 24.7 mmol), PdCl2(PPh3)2 (0.20 g, 1.2 mol%) and CuI (0.12 g, 2.6 mol%) in Et3N (100 mL). Silica gel treatment and sublimation gave pure 9 (4.40 g, 88%). 1H NMR (300 MHz, CDCl3) δ: 7.71, (d 2H [JHH 6.7 Hz]); 7.90, (d 2H [JHH 8.2 Hz]); 8.88, (s 2H); 9.18, (s 1H); 10.04, (s 1H). 13C NMR (75 MHz, CDCl3) δ: 86.50, 95.70, 119.89, 128.44, 130.30, 132.98, 136.87, 157.86, 159.42, 191.85. m/z Calcd C13H8N2O1 207.05584; Found 207.05579. Anal.: Found: C, 75.26.; H, 3.70; N, 13.15: Calcd for C13H8N2O1: C, 74.94; H, 3.87; N, 13.45.
4-(4-cyanophenyl)benzaldehyde (10)
Following the general procedure from 4-ethynylbenzaldehyde (2.76 g, 21.2 mmol), 4-bromobenzonitrile (3.86 g, 21.2 mmol), PdCl2(PPh3)2 (0.30 g, 2 mol%), PPh3 (0.11 g, 2 mol%) and CuI (0.16 g, 4 mol%) in Et3N (85 mL). EtOH (ca. 1–2 mL) was added and the mixture sonicated/triturated before filtering off a solid that was crystallized by layering hexane (4–5 volumes) onto a solution of the title compound dissolved in the minimum of hot CHCl3 (4.05 g, 83%). 1H NMR (300 MHz, CDCl3) δ: 7.65, (m 4H); 7.69, (d 2H [JHH 8.2 Hz]); 7.90, (d 2H [JHH 8.7 Hz]); 10.04, (s 1H). 13C NMR (75 MHz, CDCl3) δ: 91.15, 92.39, 112.17, 118.25, 127.32, 128.29, 129.57, 132.08, 132.21, 132.28, 135.96, 191.20. m/z Calcd C16H9N1O1 231.06841; Found 231.06878. Anal.: Found: C, 83.05; H, 3.67; N, 5.95: Calcd for C16H9N1O1: C, 83.10; H, 3.92; N, 6.06.
General Procedure for porphyrins by Rothemund synthesis
Aldehydes 1–3 (1 equiv) and pyrrole (1 equiv) were placed in a flask and propionic acid added and the mixture refluxed for 20 min. In the case of easily identified crystalline product filtration was used, alternatively the solvent was removed under reduced pressure and the residue taken up in CHCl3 and loaded onto an Al2O3 column and eluted with CHCl3. In all cases chromatography on silica gel eluting with CHCl3–alcohol mixtures was used and crystalline products were obtained by layering MeOH onto concentrated CHCl3 solutions.
5,10,15,20-tetrakis(4-(4-pyridinylethynyl)phenyl)porphyrin (1)
Following the general procedure from 6 (1.00 g, 4.8 mmol), pyrrole (0.32 g, 4.8 mmol) and propionic acid (20 mL). The crystallized solid was collected on a frit and washed with hot water (3 × 10 mL) and then ethanol (3 × 5 mL) and chromatographed on silica gel (2 × 20 cm, CHCl3–MeOH, 95–5) for purification and recrystallized giving beautiful, shiny purple needles (120 mg, 10%). 1H NMR (300 MHz, CDCl3) δ: −2.78, (s(br) 2NH); 7.54, (d 8H [JHH 6.1 Hz]); 7.98, (d 8H [JHH 8.6 Hz]); 8.25 (d 8H [JHH 8.0 Hz]); 8.70, (d 8H [JHH 5.5 Hz]); 8.89, (s 8H). UV-vis: λnm (log ε) 424 (5.43), 519 (4.09), 555 (3.91), 592 (3.59), 649 (3.53). MALDI mass spectrum: Calcd m/z C72H43N8 1019.3605; Found 1019.3602. Anal.: Found: C, 84.64; H, 4.13; N, 10.99: Calcd for C72H42N8: C, 84.85; H, 4.15; N, 11.00.
5,10,15,20-tetrakis(4-(3-pyridinylethynyl)phenyl)porphyrin (2)
Following the general procedure from 7 (1.00 g, 4.8 mmol), pyrrole (0.32 g, 4.8 mmol) and propionic acid (20 mL). After the Al2O3 (8 × 10 cm, CHCl3) and silica gel chromatography (4 × 30 cm, CHCl3–EtOH, 97.5–2.5) crystallization gave shiny purple needles (131 mg, 11 %). 1H NMR (300 MHz, CDCl3) δ: −2.76 (s(br) 2NH); 7.37 (m 4H [JHH 8.2]); 7.96 (m 12H); 8.26 (d 8H [JHH 6.2 Hz]); 8.64 (d 4H [JHH 4.9 Hz]); 8.90 (s 8H); 8.94 (s 4H). UV-vis: λnm (log ε) 425 (5.76), 519 (4.36), 555 (4.19), 592 (3.87), 649 (3.83). MALDI mass spectrum: Calcd m/z C72H43N8 1019.3605; Found 1019.3627. Anal.: Found: C, 85.32; H, 4.33; N, 10.33: Calcd for C72H42N8: C, 84.85; H, 4.15; N, 11.00.
5,10,15,20-tetrakis(4-(2-pyridinylethynyl)phenyl)porphyrin (3)
Following the general procedure from 7 (1.00 g, 4.8 mmol), pyrrole (0.32 g, 4.8 mmol) and propionic acid (20 mL). After the Al2O3 (8 × 10 cm, CHCl3) and silica gel chromatography (4 × 30 cm, CHCl3–EtOH, 97–3) crystallization gave shiny purple needles (126 mg, 10.5 %). 1H NMR (300 MHz, CDCl3) δ: −2.79, (s(br) 2NH); 7.33, (t 4H [JHH 6.0 Hz]); 7.69, (d 4H [JHH 7.7 Hz]); 7.78, (t 4H [JHH 7.5 Hz]); 8.02, (d 8H [JHH 7.9 Hz]); 8.24, (d 8H [JHH 7.9 Hz]; 8.72, (d 4H [JHH 4.6 Hz]); 8.90, (s 8H). UV-vis: λnm (log ε) 424 (5.79), 518 (4.37), 554 (4.21), 593 (3.87), 648 (3.84). MALDI mass spectrum: Calcd m/z C72H43N8 1019.3605; Found 1019.3578. Anal.: Found: C, 82.07; H, 3.68; N, 9.00: Calcd for C72H42N8: C, 84.85; H, 4.15; N, 11.00.
5,10,15,20-tetrakis(4-(4-cyanophenylethynyl)phenyl)porphyrin (5)
To a two necked flask charged with 10 (0.50 g, 2.16 mmol) and a stir bar was attached a reflux condenser fitted with an Ar gas supply and a suba-seal. After three evacuation-refill cycles, dry, oxygen-free, CHCl3 (200 mL) and pyrrole (0.15 mL, 2.16 mmol) were admitted through the seal. After stirring a few minutes BF3.OEt2 (0.27 mL, 2.16 mmol) was added via syringe and the mixture stirred for 1 h at r.t. DDQ (0.45 g 1.98 mmol) and Et3N (1.5 mL) were then added and the mixture refluxed 1 h. After removing ca. ¾of the solvent by rotary evaporation the reaction mixture was loaded directly onto an Al2O3 column (8 × 15 cm, CHCl3) and a single fraction collected. The volume was reduced by rotary evaporation until near saturation and the solution loaded onto silica gel (4 × 30 cm, CHCl3) and the fraction following a small head band was collected and crystallized by layering methanol onto a concentrated CHCl3 solution to gave a purple solid (140 mg, 23 %). 1H NMR (300 MHz, CDCl3) δ: −2.78, (s(br) 2NH); 7.74, (m 16H); 7.96, (d 8H [JHH 7.9 Hz]); 7.25, (d 8H [JHH 7.9 Hz]); 8.88, (s 8H). UV-vis: λnm (log ε) 425 (5.62), 519 (4.23), 555 (4.07), 592 (3.74), 648 (3.68). MALDI mass spectrum: Calcd m/z C80H43N8 1115.3605; Found 1115.3527. Anal.: Found: C, 86.18; H, 3.91; N, 10.32: Calcd for C80H42N8: C, 86.15; H, 3.80; N, 10.05.
General Procedure for synthesis of terminal alkynes by Sonagashira reaction
A modified Schlenk flask was charged with requisite arylbromide (1 equiv), PPh3 (2.5 mol%), PdCl2(PPh3)2 (3–5 mol%) and a stir bar and evacuated-refilled three times with Ar. THF (to give ca. 0.5 M soln), Et3N (1.5 equiv [2.25 equiv in the case of 11]) and trimethylsilylacetylene (1.5 equiv) were added by syringe and the mixture stirred for 15 min under Ar before CuI (2–2.5 mol%) was added and the teflon screw valve closed and the reaction stirred at r.t. After reaction THF was removed under reduced pressure and hexane added and the mixture filtered over Celite. The filtrate was washed with H2O and the hexane removed. The residual solid was dissolved in MeOH (to give ca. 0.5 M soln) and K2CO3 (0.1 g per 10 mmol) added and the mixture stirred at r.t. for 1.5 h. The reactions were worked up by removal of some MeOH by rotary evaporation, dilution with H2O, extraction with Et2O (4x), drying (MgSO4 or Na2SO4) and removal of solvent. The residues were purified by chromatography on silica gel or sublimation (< 0.5 mm, heat gun) to give a colorless solids.
4-Ethynylpyridine (11)
Following the general procedure from 4-bromopyridine hydrochloride (1.80 g, 9.25 mmol), PPh3 (40 mg, 2.5 mol%), PdCl2(PPh3)2 (250 mg, 3 mol%), Et3N (3.0 mL, 21.5 mmol), trimethylsilylacetylene (1.10 g, 11.2 mmol) and CuI (37 mg, 1.7 mol%) in THF (20 mL) for 8 h gave a dark solid after work up. MeOH/K2CO3 treatment and subsequent work up gave a dark residue that was purified by passing through a plug of silica gel (CH2Cl2) and then sublimation (0.72 g, 75 %). NMR data were consistent with those reported.40
5-Ethynylpyrimidine (12)
Following the general procedure from 5-bromopyrimidine (5.10 g, 32.1 mmol), Ph3P (0.21 g, 0.80 mmol), PdCl2(PPh3)2 (1.10 g, 5 mol%), Et3N (4.86 g, 48.1 mmol), trimethylsilylacetylene (4.72 g, 48.1 mmol) and CuI (0.12 g, 2 mol%) in THF (60 mL) for 16 h gave a golden solid following work up. The residue after MeOH/K2CO3 treatment and work up was purified by sublimation (2.70 g, 81%). 1H NMR (300 MHz, CDCl3) δ: 3.40, (s 1H); 8.80, (s 2H); 9.15, (s 1H). 13C NMR (75 MHz, CDCl3) δ: 84.40, 119.89, 157.23, 159.29. m/z Calcd C6H4N2 104.03694; Found 104.03745. Anal.: Found: C, 68.89; H, 3.63; N, 27.00: Calcd for C6H4N2: C, 69.22; H, 3.87; N, 26.91.
4-Ethynylbenzonitrile (13)
Following the general procedure from 4-bromobenzonitrile (4.97 g, 27.3 mmol), PPh3 (0.17 g, 2.5 mol %), PdCl2(PPh3)2 (0.96 g, 5 mol%), Et3N (4.23 g, 41.8 mmol), trimethylsilylacetylene (4.05 g, 41.2 mmol) and CuI (0.17 g, 2.5 mol%) in THF (60 mL) for 16 h at r.t. gave a yellow solid after work up. The residue after MeOH/K2CO3 treatment and work up was purified by column chromatography on silica gel eluting with CH2Cl2–hexanes (1–3, Rf 0.2) (2.67 g, 77 %). 1H NMR (300 MHz, CDCl3) δ: 3.30, (s 1H); 7.61, (d 2H); 7.56, (d 2H). 13C NMR (75 MHz, CDCl3) δ: 81.49, 81.84, 112.36, 118.19, 126.99, 131.99, 132.65. m/z Calcd C9H5N1 127.04220; Found 127.04172. Anal.: Found: C, 85.01; H, 3.89; N, 11.09: Calcd for C9H5N1: C, 85.02; H, 3.96; N, 11.02.
General Procedure for porphyrin synthesis by Pd-catalyzed coupling
A modified Schlenk tube was charged with 5,10,15,20-tetrakis(4-bromophenyl)porphyrin (ca. 100mg, 0.11 mmol), the desired alkyne (4.5–5 equiv.), Pd(PPh3)4 (10 mol%), dry Et3N (1 mL), dry DMF (20 mL) and a magnetic stir bar. The mixture was degassed by three freeze-pump-thaw cycles before being back-filled with Ar and closing the teflon screw valve. The contents of the tube were stirred and heated at 140 °C for 48 h. After cooling the solvents were removed under reduced pressure to dryness and the purple solid was taken up in CHCl3 (ca. 30 mL) and sonicated before filtering through Celite. After removal of solvent the residue was redissolved in the minimum of hot CHCl3 and methanol was then layered onto the solution the crystals that formed were collected by filtration and washed with methanol and hexane.
5,10,15,20-tetrakis(4-(4-pyridinylethynyl)phenyl)porphyrin (1)
5,10,15,20-tetrakis(4-bromophenyl)porphyrin (108.3 mg, 0.116 mmol), 4-ethynylpyridine (60.7 mg, 0.589 mmol), Pd(PPh3)4 (11.3 mg, 10 mol%). Yield (80.2 mg, 68%).
5,10,15,20-tetrakis(4-(5-pyrimidinylethynyl)phenyl)porphyrin (4)
5,10,15,20-tetrakis(4-bromophenyl)porphyrin (108.7 mg, 0.117 mmol), 5-ethynylpyrimidine (56.8 mg, 0.546 mmol), Pd(PPh3)4 (13.5 mg, 10 mol%). Yield (85.6 mg, 72 %). 1H NMR (300 MHz, CDCl3) δ: −2.78, (s(br) 2NH); 7.98, (d 8H [JHH 7.9 Hz]); 8.27, (d 8H [JHH 7.9 Hz]); 8.89, (s 8H); 9.02, (s 8H); 9.24, (s 4H). UV-vis: λnm (log ε) 425 (5.62), 519 (4.23), 555 (4.07), 592 (3.74), 648 (3.68). Anal.: Found: C, 79.01; H, 4.11; N, 15.93: Calcd for C68H38N12.0.5H2O: C, 79.13; H, 3.81; N, 16.28.
5,10,15,20-tetrakis(4-(4-cyanophenylethynyl)phenyl)porphyrin (5)
5,10,15,20-tetrakis(4-bromophenyl)porphyrin (101.7 mg, 0.109 mmol), 4-ethynylbenzonitrile (64.2 mg, 0.505 mmol), Pd(PPh3)4 (12.9 mg, 10 mol%). Yield (84.1 mg, 70%).
Procedure for large scale porphyrin synthesis by Pd-catalyzed coupling
5,10,15,20-tetrakis(4-(5-pyrimidinylethynyl)phenyl)porphyrin (5)
In a single necked round bottomed flask was placed reagent grade DMF (200 mL) and reagent grade triethylamine (20 mL). Ar was bubbled through the magnetically stirred solution with a fritted bubbler for 1 h before 5,10,15,20-tetrakis(4-bromophenyl)porphyrin (1.03 g, 1.11 mmol), 5-ethynylpyrimidine (0.53 g, 5.10 mmol), and Pd(PPh3)4 (121 mg, 10 mol%) were added under flow of Ar and a reflux condenser attached. The contents were heated at 130 °C with stirring for 60 h under Ar. The same general procedure as above was followed for the isolation and purification (0.77 g, 59 %).
Supplementary Material
Further experimental procedures, 1H NMR spectra for compounds 1–13, 13C NMR spectra for compounds 6–10, 12, 13, mass spectra for compounds 1–9,11–13, UV-visible spectra for compounds 1-5. This material is available free of charge via the Internet at http://pubs.acs.org.
Acknowledgments
We thank Professor Peter D. W. Boyd for his valued input to this work. It was supported by NIH grant GM 23851.
References
- 1.Abrahams BF, Hoskins BF, Michail DM, Robson R. Nature. 1994;369:727–729. [Google Scholar]
- 2.Drain CM, Lehn JM. J Chem Soc, Chem Commun. 1994:2313–2315. [Google Scholar]
- 3.Drain CM, Nifiatus F, Vasenko A, Batteas JD. Angew Chem Int Ed Engl. 1998;37:2344–2347. doi: 10.1002/(SICI)1521-3773(19980918)37:17<2344::AID-ANIE2344>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- 4.Kumar RK, Balasubramanian S, Goldberg I. Inorg Chem. 1998;37:541–552. doi: 10.1021/ic971259u. [DOI] [PubMed] [Google Scholar]
- 5.Goldberg I. Chem Europ J. 2000;6:3863–3870. doi: 10.1002/1521-3765(20001103)6:21<3863::aid-chem3863>3.3.co;2-m. [DOI] [PubMed] [Google Scholar]
- 6.Pan L, Noll BC, Wang X. Chem Commun. 1999:157–158. [Google Scholar]
- 7.Sharma CVK, Broker GA, Huddleston JG, Baldwin JW, Metzger RM, Rogers RD. J Am Chem Soc. 1999;121:1137–1144. [Google Scholar]
- 8.Lin KJ. Angew Chem Int Ed Engl. 1999;38:2730–2732. [PubMed] [Google Scholar]
- 9.Hagrman D, Hagrman PJ, Zubieta J. Angew Chem Int Ed. 1999;38:3165–3168. doi: 10.1002/(sici)1521-3773(19991102)38:21<3165::aid-anie3165>3.0.co;2-o. [DOI] [PubMed] [Google Scholar]
- 10.Kosal ME, Suslick KS. J Solid State Chem. 2000;152:87–98. [Google Scholar]
- 11.Sun D, Tham FS, Reed CA, Boyd PDW. Proc Natl Acad Sci. 2002;99:5088–5092. doi: 10.1073/pnas.072602399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Carlucci L, Ciani G, Proserpio DM, Porta F. Angew Chem Int Ed. 2003;42:317–322. doi: 10.1002/anie.200390106. [DOI] [PubMed] [Google Scholar]
- 13.Ohmura T, Usuki A, Fukumori K, Ohta T, Ito M, Tatsumi K. Inorg Chem. 2006;45:7988–7990. doi: 10.1021/ic060358h. [DOI] [PubMed] [Google Scholar]
- 14.Smithenry DW, Suslick KS. J Porph Phthalocyanines. 2004;8:182–190. [Google Scholar]
- 15.Drain CM, Goldberg I, Sylvain I, Falber A. Topics in Current Chemistry. 2005;245:55–88. [Google Scholar]
- 16.Goldberg I. Chem Comm. 2005;10:1243–1254. doi: 10.1039/b416425c. [DOI] [PubMed] [Google Scholar]
- 17.Boyd PWD, Reed CA. Acc Chem Res. 2005;38:235–242. doi: 10.1021/ar040168f. [DOI] [PubMed] [Google Scholar]
- 18.Suslick KS, Bhyrappa P, Chou JH, Kosal ME, Nakagaki S, Smithenry DW, Wilson SR. Acc Chem Res. 2005;38:283–291. doi: 10.1021/ar040173j. [DOI] [PubMed] [Google Scholar]
- 19.Eddaoudi M, Moler DB, Li HL, Chen BL, Reineke TM, O’Keeffe M, Yaghi OM. Acc Chem Res. 2001;34:319–330. doi: 10.1021/ar000034b. [DOI] [PubMed] [Google Scholar]
- 20.Kitagawa S, Kitaura R, Noro S. Angew Chem Int Ed. 2004;43:2334–2375. doi: 10.1002/anie.200300610. [DOI] [PubMed] [Google Scholar]
- 21.Kitagawa S, Noro S, Nakamura T. Chem Comm. 2006:701–707. doi: 10.1039/b511728c. [DOI] [PubMed] [Google Scholar]
- 22.Mueller U, Schubert M, Teich F, Puetter H, Scheirle-Arndt K, Pastre J. J Mater Chem. 2006;16:626–636. [Google Scholar]
- 23.Ikeda A, Ayabe M, Shinkai S, Sakamoto S, Yamaguchi K. Org Lett. 2000;2:3707–3710. doi: 10.1021/ol006620m. [DOI] [PubMed] [Google Scholar]
- 24.Rothemund P. J Am Chem Soc. 1935;57:2010–2011. [Google Scholar]
- 25.Adler AD, Longo FR, Finarelli JD. J Org Chem. 1967;32:476. [Google Scholar]
- 26.Lindsey JS, Schreiman IC, Hsu HC, Kearney PC, Marguerettaz AM. J Org Chem. 1987;52:827–836. [Google Scholar]
- 27.Gryko D, Lindsey JS. J Org Chem. 2000;65:2249–2252. doi: 10.1021/jo9918100. [DOI] [PubMed] [Google Scholar]
- 28.Sharman WM, Van Lier JE. J Porph Phthalocyanines. 2000;4:441–453. [Google Scholar]
- 29.Setsune JI. J Porph Phthalocyanines. 2000;8:93–102. [Google Scholar]
- 30.Wagner RW, Ciringh Y, Clausen C, Lindsey JS. Chem Mat. 1999;11:2974–2983. [Google Scholar]
- 31.Loewe RS, Tomizaki K-y, Youngblood WJ, Bo Z, Lindsey JS. J Mater Chem. 2002;12:3438–3451. [Google Scholar]
- 32.Shi B, Boyle RW. J Chem Soc, Perkin Trans 1. 2002:1397–1400. [Google Scholar]
- 33.Shanmugathasan S, Johnson CK, Edwards C, Matthews EK, Dolphin D, Boyle RW. J Porph Phthalocyanines. 2000;4:228–232. [Google Scholar]
- 34.DiMagno SG, Lin VSY, Therien MJ. J Org Chem. 1993;58:5983–5993. [Google Scholar]
- 35.Wagner RW, Johnson TE, Li F, Lindsey JS. J Org Chem. 1995;60:5266–5273. [Google Scholar]
- 36.Sorensen US, Pombo-Villar E. Tetrahedron. 2005;61:2697–2703. [Google Scholar]
- 37.Gryko DT, Piechota KE. J Porph Phthalocyanines. 2002;6:81–97. [Google Scholar]
- 38.Motmans F, Ceulemans E, Smeets S, Dehaen W. Tetrahedron Lett. 1999;40:7545–7548. [Google Scholar]
- 39.Chan CS, Tse AKS, Chan KS. J Org Chem. 1994;59:6084–6089. [Google Scholar]
- 40.Thorand S, Krause N. J Org Chem. 1998;63:8551–8553. [Google Scholar]
- 41.Austin WB, Bilow N, Kelleghan WJ, Lau KSY. J Org Chem. 1981;46:2280–2286. [Google Scholar]
- 42.Holmes BT, Pennington WT, Hanks TW. Synth Commun. 2003;33:2447–2461. [Google Scholar]
- 43.Flatt AK, Yao Y, Maya F, Tour JM. J Org Chem. 2004;69:1752–1755. doi: 10.1021/jo035821b. [DOI] [PubMed] [Google Scholar]
- 44.McIlroy SP, Clo E, Nikolajsen L, Frederiksen PK, Nielsen CB, Mikkelsen KV, Gothelf KV, Ogilby PR. J Org Chem. 2005;70:1134–1146. doi: 10.1021/jo0482099. [DOI] [PubMed] [Google Scholar]
- 45.Pereira MM, Muller G, Ordinas JI, Azenha ME, Arnaut LG. J Chem Soc, Perkin Trans. 2;2002:1583–1588. [Google Scholar]
- 46.Ljungdahl T, Pettersson K, Albinsson B, Maartensson J. J Org Chem. 2006;71:1677–1687. doi: 10.1021/jo052423v. [DOI] [PubMed] [Google Scholar]
- 47.Yen WN, Lo SS, Kuo MC, Mai CL, Lee GH, Peng SM, Yeh CY. Org Lett. 2006;8:4239–4242. doi: 10.1021/ol061478w. [DOI] [PubMed] [Google Scholar]
- 48.Chen YJ, Lee GH, Peng SM, Yeh CY. Tet Lett. 2005;46:1541–1544. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Further experimental procedures, 1H NMR spectra for compounds 1–13, 13C NMR spectra for compounds 6–10, 12, 13, mass spectra for compounds 1–9,11–13, UV-visible spectra for compounds 1-5. This material is available free of charge via the Internet at http://pubs.acs.org.