

Abstract
Individuals affected with the neurofibromatosis 2 (NF2) cancer predisposition syndrome develop specific ocular lesions. To determine whether these lesions result from altered NF2 gene expression, microdissection and PCR were used to investigate 40 ocular lesions from seven eyes of four NF2 patients for LOH, with markers that flank the NF2 gene on chromosome 22q. NF2 protein (merlin) expression was also evaluated in these lesions, using immunohistochemistry. Retinal hamartoma was observed in all seven eyes, including one with combined pigment epithelial and retinal hamartoma (CPERH). Retinal tufts were present in four eyes (three patients), retinal dysplasia in two eyes (two patients), optic nerve neurofibroma in one eye, iris naevoid hyperplasia in two eyes (two patients) and pseudophakia in all eyes. Markers were informative in three patients (six eyes from three unrelated families). One patient was non-informative due to prolonged decalcification. All retinal and optic nerve, but not iris lesions, demonstrated consistent LOH for the NF2 gene. Merlin was not expressed in the retina, optic nerve, or iris lesions. These results suggest that inactivation of the NF2 gene is associated with the formation of a variety of retinal and optic nerve lesions in NF2 patients.
Keywords: retina, hamartoma, loss of heterozygosity, merlin, neurofibromatosis 2, optic nerve glioma, retinal dysplasia, Lisch nodule
Introduction
The neurofibromatoses consist of at least two distinct autosomal dominant disorders caused by mutations in different genes [1,2]. Neurofibromatosis 1 (NF1) was first described by von Recklinghausen in 1882 [3] and is the most common form, with a birth incidence of about 1 in 3000 [1]. It has also been called ‘von Recklinghausen disease’ or peripheral neurofibromatosis. The earliest description of neurofibromatosis 2 (NF2), formerly called central or bilateral acoustic neurofibromatosis, was by Wishart in 1822 [4]. Since the 1987 National Institutes of Health (NIH) Consensus Development Conference on Neurofibromatosis [5], the designations of NF1 and NF2 have gained widespread recognition and the genes of each disorder have been cloned. NF1 is caused by mutations that inactivate the NF1 gene on chromosome 17q, while the NF2 gene resides on chromosome 22q [2]. Both genes are tumour suppressor genes.
NF2 is much rarer than NF1, with a birth incidence of about 1 in 40 000 [6], and the morbidity and mortality associated with it are greater, due to the intracranial and intraspinal tumours [1,7]. The major feature of NF2 is the presence of bilateral vestibular schwannomas (acoustic neuromas). Schwannomas of other cranial, spinal and peripheral nerves, as well as intracranial and spinal meningiomas, are also frequent findings. In addition, café au lait spots and cutaneous schwannomas may be present. Previously documented ocular manifestations of NF2 include loss of vision due to optic nerve meningioma, neurotrophic keratopathy, juvenile posterior lens opacities [8,9], juvenile cortical cataracts, intraocular neurilemmoma [10], epiretinal membrane [11-13], retinal hamartoma [14,15], combined pigment epithelial and retinal hamartoma (CPERH) [16-18], optic nerve glioma [19,20] and iris Lisch nodules [21]. Juvenile posterior lens opacities and juvenile cortical cataracts are included in the diagnostic criteria for NF2 [22].
Ocular abnormalities associated with NF2 have been documented in other phacomatoses such as NF1 and tuberous sclerosis [7,23,24]. Although they may present similar clinical and histological pictures (phenotype), involve genetic mutation and/or deletion of various tumour suppressor genes and result in loss of normal protein expression at the phacomatous lesions, the genotypes of each phacomatosis are completely different and the genes of each disease locate in different chromosomes [25-27]. Tuberous sclerosis has two genes that are identified on chromosomes 16q (TSC 2) and 9q (TSC 1).
The identification of the NF2 gene in 1993 ushered in a new era of NF2 research [28-30]. Since then, germline NF2 mutations have been found in large numbers of NF2 patients, and genotype–phenotype correlations have been identified. Nonsense and frame-shift mutations are generally associated with severe disease, splice-site mutations are usually associated with variable phenotypes, and missense mutations and large deletions are frequently seen with mild disease. Schwannomas and meningiomas from NF2 patients and the same types of sporadic tumours have shown frequent loss of heterozygosity (LOH) for polymorphic DNA markers close to the NF2 gene [31,32]. In the LOH studies, the tumours lose the wild type NF2 allele and retain the inherited or somatically mutated NF2 allele, which likely produces little or no functioning NF2 protein. The NF2 protein, called merlin or schwannomin [33,34] shares sequence homology with the ERM (ezrin, radixin, and moesin) family of cytoskeleton membrane-linking proteins.
Because NF2 is not a common disease and the eyes are often difficult to obtain, there are only a few reported isolated patients with routine ocular pathology. Thus far, no molecular pathology of the eyes with NF2-associated lesions has been reported in the literature. In this study, we report the results of LOH analysis for polymorphic markers flanking the NF2 gene in 40 ocular lesions from four NF2 patients, from three unrelated families. We also studied NF2 protein expression in all abnormal ocular lesions from these four patients.
Materials and methods
Patients
The patients had been enrolled in an IRB-approved protocol at the Clinical Center, NIH. Clinical evaluation included physical and neurological examinations, a complete eye examination, including slit lamp and dilated fundoscopic evaluation of the lens and fundus, audiology, auditory brain stem-evoked responses and magnetic resonance imaging of the brain and entire spine. Peripheral blood was obtained for molecular studies. Complete study details have been reported previously [35,36]; the report by Parry et al. includes clinical and molecular details of the four patients whose eyes are the subjects of this report [36]. All four patients met standard diagnostic criteria for NF2 [22]. The patients were from three unrelated families. Patients 1 and 3 were the proband and his sib, respectively, from family G17703 in Parry et al. [36], and patient 2 was the father of patient G17900 [36]. These patients had the same exon 11 nonsense mutation. Patient 4 was the proband of family G17690 [36]; he had an exon 2 nonsense mutation. Patients 1, 3, and 4 died at ages 42, 47, and 16, respectively, from complications of central nervous system tumours. Patient 2 died at age 70 from kidney failure unrelated to NF2.
Specimens
Two eyes from patient 4 were obtained at the time of autopsy at the NIH. Two eyes from each of patients 2 and 3 and one eye from patient 1 were received from outside institutions. All four patients had a variety of clinically detected ocular findings. These included posterior capsular cataracts, retinal hamartomas including a CPERH, epiretinal membranes and iris Lisch nodules, all of which were reported previously [9,18]. The seven eyes were fixed in 10% formalin, and processed and sectioned through the pupillary optic nerve plane according to their clinical presentations. The entire material was paraffin-embedded and at least 10 serial sections were cut through each lesion from each block. All slides were thoroughly reviewed to identify various ocular lesions that were suitable for subsequent microdissection and genetic studies.
Immunohistochemistry
After deparaffinization, the sections were immunostained using the avidin–biotin–peroxidase complex technique. The primary antibodies were polyclonal rabbit antibodies against glial fibrillary acidic protein (GFAP), neuron-specific enolase (NSE) and S-100, and a monoclonal mouse antibody against HMB-45 (Dako Corporation, Santa Barbara, CA), or control rabbit IgG for polyclonal antibodies (GFAP, NSE, and S-100) and control mouse IgG for HMB-45. In addition, polyclonal rabbit antibody against merlin (WA30; YAEHRGRARDEAEMEYLKC; residues 192–209 plus a carboxyl terminal cystein residue) was also applied as a primary antibody after the deparaffinized section had been treated in 0.4% pepsin in 0.01 n HCl for 30 min at 37 °C as previously reported [37]. The secondary antibodies were biotin-conjugated goat anti-rabbit IgG or horse anti-mouse IgG (Vector Laboratories, Burlingame, CA). The substrate was avidin–biotin–peroxidase complex, and the chromogen was diaminobenzidine. A deparaffinized section from a normal eye was used as a positive control for immunostaining against merlin antibody.
Microdissection
Unstained 5 μm sections on glass slides were deparaffinized with xylene, rinsed in ethanol from 100% to 80%, briefly stained with haematoxylin and eosin, and briefly rinsed in 10% glycerol in tromethamine hydrochloride–EDTA buffer. Microdissection was performed under direct light microscopic visualization using a 30-gauge needle as previously described [38]. A total of 40 separate ocular lesions were microdissected from the seven eyes. From each eye, at least two areas of ‘normal’ tissues (retina, ciliary body, iris, or cornea) were selected and analysed as normal control tissue.
DNA extraction
Procured cells were immediately resuspended in 5–10 μl buffer containing tromethamine hydrochloride, pH 8.0; 10 mm ethylenediamine tetraacetic acid, pH 8.0; 1% polyoxyethylene 20 sorbitan monolaurate; and 0.5 mg/ml proteinase K, and were incubated at 37 °C overnight. The mixture was boiled for 10 min to inactivate the proteinase K, and 2 μl of this solution was used for polymerase chain reaction (PCR) amplification of the DNA.
Primers and PCR conditions
All samples were examined for LOH at the NF2 gene locus using markers D22S683, TOP1P2, and CYP2D on chromosome 22q. Markers D22S683 and CYP2D are 6.3 and 12.4 Mbs telomeric to the NF2 gene respectively, and marker TOP1P2 (M55630) is 4.8 Mbs centromeric to the NF2 gene. The three flanking markers, as compared to the intragenic NF2 markers, are more satisfactory for the analysis of minuscule microdissected material; they were therefore used in this study. Each PCR sample contained 2 μl of template DNA as described above, 10 pmol of each primer, 20 nmol each of dATP, dCTP, dGTP, and dTTP, 15 mM MgCl2, 0.1 U Taq DNA polymerase, 0.05 μl 32P-dCTP (6000 Ci/mmol), and 1 μl of 10 × buffer in a total volume of 10 μl. PCR was performed with 35 cycles: denaturing at 94 °C for 1 min, annealing at 55 °C for 1 min and extending at 72 °C for 1 min. The final extension was continued for 10 min.
Loss of heterozygosity (LOH) analysis
Labelled amplified DNA was mixed with an equal volume of formamide loading dye (95% formamide, 20 mm EDTA, 0.05% bromophenol blue, and 0.05% xylene cyanol). Samples were then denatured for 5 min at 95%, loaded onto a gel consisting of 6% polyacrylamide (acrylamide : bisacrylamide 49 : 1), and electrophoresed at 1800 V for 90 min. After electrophoresis, the gels were transferred to 3 mm Whatman paper and dried. Autoradiography was performed with Kodak X-OMAT film (Eastman Kodak, Rochester, NY).
A patient was considered informative for a polymorphic marker if normal tissue DNA showed two different alleles (heterozygosity). The criterion for LOH was complete or near complete absence of one allele in the DNA from any ocular lesion compared to normal DNA, as defined by direct visualization.
Results
Histopathology
Ocular lesions were found in all seven eyes (Table 1). Cataracts were evident by pseudophakia (presence of intraocular lenses) in seven eyes, iris naevoid hyperplasia at the anterior surface in two eyes of two patients, and retinal lesions in all eyes. The specific retinal lesions included epiretinal membranes and retinal hamartomas in seven eyes (Figure 1A–D) with additional CPERH in one eye (Figure 1E) [18], retinal tufts in four eyes of three patients (Figure 2A), retinal dysplasia in two eyes of two patients (Figure 2B), and optic glioma in one eye (Figure 2C). Retinal hamartomas were defined as hamartomatous growths of glial, vascular, and/or melanocytic derivation in the retina. They appeared as flat, discoid masses, or bulged forward prominently into the vitreous chamber. In a few, there were ganglion cells and neurons. Coloboma was also identified in two eyes. A Fuchs adenoma was seen in the ciliary body of one eye.
Table 1.
Ocular pathology of the four NF2 patients
| Iris: Naevoid hyperplasia |
Lens: Cataracts |
Retina |
Optic nerve: Neurofibroma |
Eye: Coloboma |
||||
|---|---|---|---|---|---|---|---|---|
| Case* | PRM† | Hamartoma | Tuft | Dysplasia | ||||
| 1 (1) | + | Pseudophakia | + | + | − | − | − | − |
| 2 (2) | − | Pseudophakia | + | +(CPERH) | + | + | − | + |
| 3 (2) | − | Pseudophakia | + | + | + | − | − | − |
| 4 (2) | + | Pseudophakia | + | + | + | + | + | + |
Cases 1, 2, and 3 had an identical exon 11 nonsense mutation; case 4 had a nonsense mutation of exon 2. cases 1 and 3 were from the same family. (x) = number of eyes in the study.
PRM, pre-retinal membrane; CPERH, combined pigment epithelial and retinal hamartoma.
Figure 1.
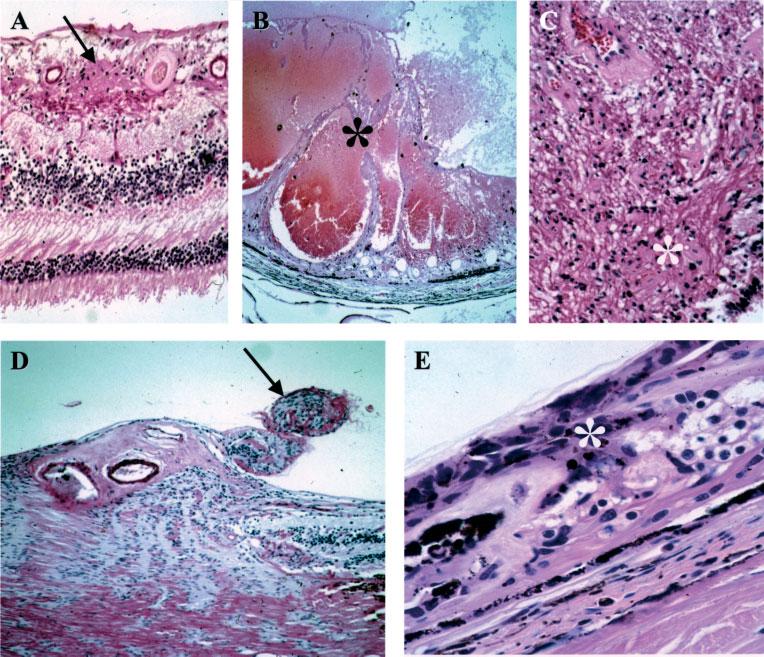
Retinal hamartomas. (A) A small vascular hamartoma (arrow) located in the inner retina. (B) A large vascular hamartoma (asterisk) infiltrated most of the retinal layers. (C) A glial hamartoma in the retina (asterisk). (D) Two small hamartomas (arrow) located at the optic nerve head. (E) CPERH (asterisk) (haematoxylin and eosin; original magnification: A, ×200; B, ×50; C, ×200; D, ×100; E, ×400)
Figure 2.
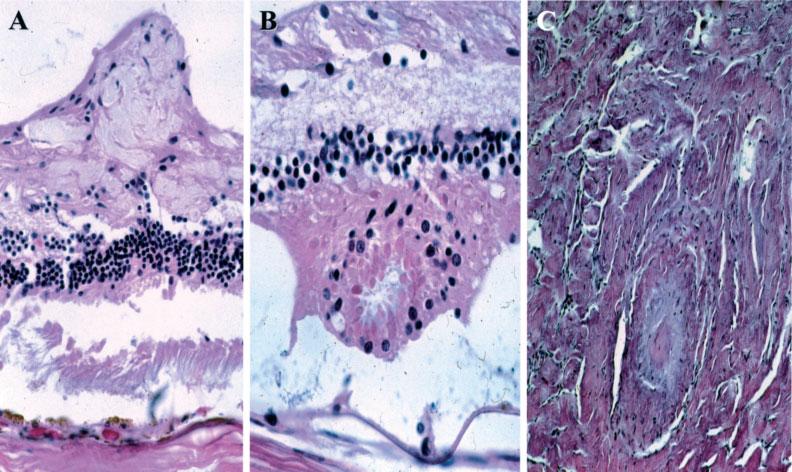
Other NF2-associated lesions. (A) Retinal tuft arising from the nerve fibre layer. (B) Retinal dysplasia showing a rosette-like configuration. (C) Neurofibroma in the optic nerve (haematoxylin and eosin; original magnification, A, ×200; B, ×400; C, ×200)
Immunohistochemistry
All retinal hamartomas consisted of cells positive for GFAP, NSE, and/or S-100. In general, the glial elements (GFAP) were located in the periphery of each lesion and neural components (NSE) appeared throughout each lesion. Retinal tufts were mostly positive for GFAP or composed mainly of glial tissue. Positive staining for melanin component (HMB-45) was found only in the CPERH.
Using the polyclonal antibody (WA30) against merlin [24], normal adult eye showed strong positive merlin staining in the lens, iris and ciliary body epithelial cells and the optic nerve and weak staining of the internal limiting membrane and the rods and cones, but not of other retinal layers where there are no myelinated nerve fibres. The data were similar to the report by Rouleau and associates [39]. All NF2-associated retinal hamartomas and the optic nerve glioma were negative for merlin by immunohistochemistry. Loss of merlin expression was also noted in the residue lens epithelial cells of the pseudophakic eyes. In addition, there was a marked decrease of anti-merlin immunoreactivity in the optic nerve surrounding the NF2 glioma lesion (Figure 3).
Figure 3.
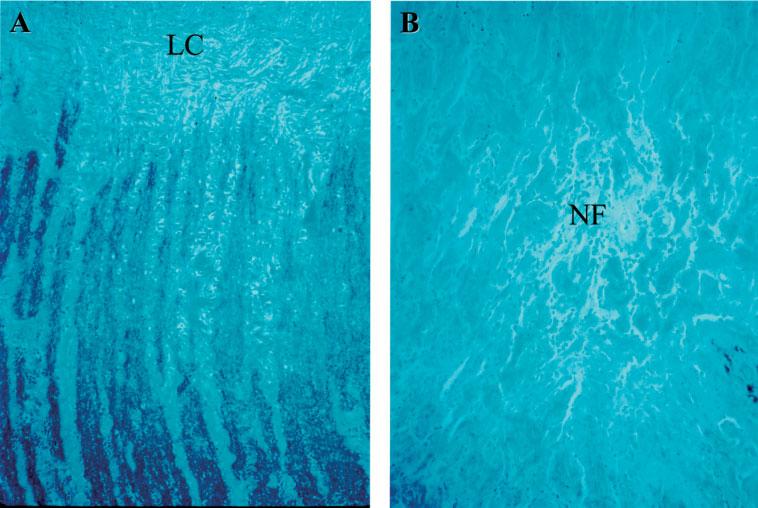
Expression of merlin in optic nerve. (A) Positive, black colour merlin staining of the optic nerve in an NF2 patient without an optic nerve lesion, showing no difference compared to the normal optic nerve control, where there are Schwann cells and myelinated nerve fibres; note, as expected, absence of staining beyond the level of the lamina cribrosa (LC). (B) Negative merlin staining of the neurofibroma (NF) and positive staining of the optic nerve (left lower corner) of case 4 (immunohistochemical staining against anti-merlin antibody, original magnification, ×100)
LOH on chromosomes 22q
Microdissected cells from a total of 40 separate ocular lesions were examined for loss of the three chromosome 22q markers. Thirty-five lesions in three patients (patients 2–4) were informative (Table 2). The eye of patient 1 had bone formation in the posterior choroid and retina and the specimen had undergone prolonged decalcification for routine histological processing; thus the eye was non-informative. LOH of D22S683, TOP1P2, and CYP2D was found in all examined retinal solid lesions as well as the optic nerve glioma (Figure 4A and B). Only one of the two patients with iris naevoid lesions was informative (patient 4); the iris naevoid lesions in that patient did not demonstrate LOH for NF2 (D22S683 and TOP1P2) markers. In addition, a Fuchs adenoma at the ciliary body did not show LOH for the three markers on chromosome 22q. These findings indicate inactivation of the NF2 gene in all types of retinal and optic nerve lesions in the eyes of the NF2 patients included in this study (Table 2).
Table 2.
LOH of NF2 gene in the 40 ocular lesions
| Iris: Naevoid hyperplasia |
Ciliary body: Fuchs adenoma |
Retina |
Optic nerve: Neurofibroma |
||||
|---|---|---|---|---|---|---|---|
| Case | PRM | Hamartoma | Tuft | Dysplasia | |||
| 1 | NI (3*) | NI (2) | |||||
| 2 | + (1) | + (9) | + (2) | + (2) | |||
| 3 | + (1) | + (7) | + (1) | ||||
| 4 | − (2) | − (1) | + (1) | + (4) | + (1) | + (1) | + (2) |
PRM, pre-retinal membrane; NI, non-informative.
(x) = number of lesions of each type.
+, LOH; −, no LOH.
Figure 4.
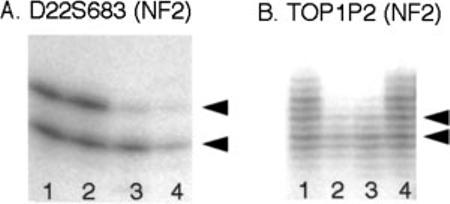
Autoradiography showing LOH at the NF2 gene locus in ocular lesions. Representative retinal hamartomas showing LOH for both markers D22S683 (A) and TOP1P2 (B). Arrowheads point to the position of the respective alleles. ‘Duplication bands’ are seen associated with the dinucleotide repeat marker TOP1P2 (B). (A: lanes 1 and 2, normal retina; lane 3, retinal hamartoma; lane 4, optic nerve neurofibroma. B: lanes 1 and 4, normal retina; lanes 2 and 3, two different retinal hamartomas)
Discussion
We have provided histopathological documentation of the occurrence of retinal hamartomas, CPERH, epiretinal membranes, retinal tufts, and optic nerve glioma in four NF2 patients whose other clinical manifestations of NF2 have also been reported [36]. We and others have reported these ocular lesions in association with NF2 [11,12,14,15,17-20,35,40-43]. Retinal hamartoma is the most commonly found lesion in the retina of NF2 patients. Unlike the hamartoma in tuberous sclerosis, composed of mainly glial tissue, NF2-associated retinal hamartoma usually contains multiple tissues of glial, vascular and even melanocytic origin [23]. In addition, we also observed the occurrence of retinal dysplasia in two eyes from two patients. This abnormality showing retinal dysmorphogenesis has not been documented in the eyes of patients with NF2. Retinal dysplasia is a malformation of the developing retina that leads to a rosette-like configuration described by ophthalmic pathologists and can only be demonstrated in pathological sections.
Our observation of such a large number and variety of abnormal ocular lesions in the seven studied eyes may reflect the fact that all four patients had germline nonsense mutations; this group of mutations is generally associated with severe clinical manifestations [36]. We found somewhat different arrays of ocular lesions in the three patients with germline mutation of the same exon (patients 1–3) and similar phenotypes in two patients with mutations in different exons (patient 2 vs. patient 4).
LOH in various hamartomas has been detected in other phacomatous lesions including NF1, tuberous sclerosis, familial adenomatous polyposis coli and Peutz–Jeghers syndrome [25,26,44]. LOH of NF2 gene is detected in schwannoma [45,46]. We have detected consistent LOH for markers flanking the NF2 gene in all the retina and optic nerve, but not iris and ciliary body, lesions in the three informative patients. In addition, we have shown that no detectable merlin protein was present in any of the hamartomatous lesions and the residual lens epithelial cells in those pseudophakic eyes that had an inserted intraocular lens after cataract extraction.
Merlin expression is observed in cells migrating from the ventricular zone to the cortical plate during mid-embryonic development of the mouse neocortex and in a majority neural crest derived cells [47]. This developmental pattern suggests that merlin may play a role in neuronal maturation and cellular migration in the developing central nervous system. Loss of merlin, through NF2 mutation and allele loss, could result in a developmental defect involving migration of the neuroblastic cells in the retina, resulting in retinal dysplasia.
In summary, we found that retinal dysplasia is an additional ocular lesion associated with NF2. All of the various types of retinal and optic nerve lesions that were present in the eyes of three informative NF2 patients demonstrated LOH for three chromosome 22q markers flanking the NF2 gene, and by inference, loss of the normal allele of that gene resulting in loss of merlin expression. These results are consistent with the NF2 gene acting as a tumour suppressor and indicate that inactivation of this gene is important in the pathogenesis of retinal and optic nerve lesions associated with NF2.
References
- 1.Pollack IF, Mulvihill JJ. Neurofibromatosis 1 and 2. Brain Pathol. 1997;7:823–836. doi: 10.1111/j.1750-3639.1997.tb01067.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zwarthoff EC. Neurofibromatosis and associated tumour suppressor genes. Pathol Res Pract. 1996;192:647–657. doi: 10.1016/S0344-0338(96)80086-0. [DOI] [PubMed] [Google Scholar]
- 3.von Recklinghausen FD. Ueber die multiplen Fibrome der Haut und ihre Beziehung zu den multiplen Neuromen. Hirschwald; Berlin: 1882. [PubMed] [Google Scholar]
- 4.Wishart J. Cases of tumors in the skull, dura mater and brain. Edingb Med Surg J. 1822;18:393–397. [PMC free article] [PubMed] [Google Scholar]
- 5.Conference NIoHCD Neurofibromatosis. Conference Statement. Arch Neurol. 1988;45:575–578. [PubMed] [Google Scholar]
- 6.Evans DG, Huson SM, Donnai D, et al. A genetic study of type 2 neurofibromatosis in the United Kingdom. II. Guidelines for genetic counselling. J Med Genet. 1992;29:847–852. doi: 10.1136/jmg.29.12.847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ragge NK. Clinical and genetic patterns of neurofibromatosis 1 and 2. Br J Ophthalmol. 1993;77:662–672. doi: 10.1136/bjo.77.10.662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pearson-Webb MA, Kaiser-Kupfer MI, Eldridge R. Eye findings in bilateral acoustic (central) neurofibromatosis: association with presenile lens opacities and cataracts but absence of Lisch nodules. N Engl J Med. 1986;315:1553–1554. doi: 10.1056/NEJM198612113152419. [DOI] [PubMed] [Google Scholar]
- 9.Kaiser-Kupfer MI, Freidlin V, Datiles MB, et al. The association of posterior capsular lens opacities with bilateral acoustic neuromas in patients with neurofibromatosis type 2. Arch Ophthalmol. 1989;107:541–544. doi: 10.1001/archopht.1989.01070010555030. [DOI] [PubMed] [Google Scholar]
- 10.Freedman SF, Elner VM, Donev I, Gunta R, Albert DM. Intraocular neurilemmoma arising from the posterior ciliary nerve in neurofibromatosis: pathologic findings. Ophthalmology. 1988;95:1559–1564. doi: 10.1016/s0161-6420(88)32983-0. [DOI] [PubMed] [Google Scholar]
- 11.Kaye LD, Rothner AD, Beauchamp GR, Meyers SM, Estes ML. Ocular findings associated with neurofibromatosis type II. Ophthalmology. 1992;99:1424–1429. doi: 10.1016/s0161-6420(92)31789-0. [DOI] [PubMed] [Google Scholar]
- 12.Meyers SM, Gutman FA, Kaye LD, Rothner AD. Retinal changes associated with neurofibromatosis 2. Trans Am Ophthalmol Soc. 1995;93:245–252. doi: 10.1016/s0002-9394(14)70558-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lueder GT, Doll JT. Pseudopapilledema in neurofibromatosis type 2. Am J Ophthalmol. 2000;129:405–407. doi: 10.1016/s0002-9394(99)00382-7. [DOI] [PubMed] [Google Scholar]
- 14.Landau K, Dossetor FM, Hoyt WF, Muci-Mendoza R. Retinal hamartoma in neurofibromatosis 2. Arch Ophthalmol. 1990;108:328–329. doi: 10.1001/archopht.1990.01070050026011. [DOI] [PubMed] [Google Scholar]
- 15.Good WV, Erodsky MC, Edwards MS, Hoyt WF. Bilateral retinal hamartomas in neurofibromatosis type 2. Br J Ophthalmol. 1991;75:190. doi: 10.1136/bjo.75.3.190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Font RL, Moura RA, Shetlar DJ, Martinez JA, McPherson AR. Combined hamartoma of sensory retina and retinal pigment epithelium. Retina. 1989;9:302–311. doi: 10.1097/00006982-198909040-00011. [DOI] [PubMed] [Google Scholar]
- 17.Sivalingam A, Augsburger J, Perilongo G, Zimmerman R, Barabas G. Combined hamartoma of the retina and retinal pigment epithelium in a patient with neurofibromatosis type 2. J Pediatr Ophthalmol Strabismus. 1991;28:320–322. doi: 10.3928/0191-3913-19911101-08. [DOI] [PubMed] [Google Scholar]
- 18.Bouzas EA, Parry DM, Eldridge R, Kaiser-Kupfer MI. Familial occurrence of combined pigment epithelial and retinal hamartomas associated with neurofibromatosis 2. Retina. 1992;12:103–107. doi: 10.1097/00006982-199212020-00005. [DOI] [PubMed] [Google Scholar]
- 19.Dossetor FM, Landau K, Hoyt WF. Optic disk glioma in neurofibromatosis type 2. Am J Ophthalmol. 1989;108:602–603. doi: 10.1016/0002-9394(89)90445-5. [DOI] [PubMed] [Google Scholar]
- 20.Rettele GA, Brodsky MC, Merin LM, Teo C, Glasier CM. Blindness, deafness, quadriparesis, and a retinal malformation: the ravages of neurofibromatosis 2. Surv Ophthalmol. 1996;41:135–141. doi: 10.1016/s0039-6257(96)80003-8. [DOI] [PubMed] [Google Scholar]
- 21.Garretto NS, Ameriso S, Molina HA, et al. Type 2 neurofibromatosis with Lisch nodules. Neurofibromatosis. 1989;2:315–321. [PubMed] [Google Scholar]
- 22.Gutmann DH, Aylsworth A, Carey JC, et al. The diagnostic evaluation and multidisciplinary management of neurofibromatosis 1 and neurofibromatosis 2. JAMA. 1997;278:51–57. [PubMed] [Google Scholar]
- 23.Font RL, Ferry AP. The phakomatoses. Int Ophthalmol Clin. 1972;12:1–50. [PubMed] [Google Scholar]
- 24.Kerrison JB. Neuro-ophthalmology of the phacomatoses. Curr Opin Ophthalmol. 2000;11:413–420. doi: 10.1097/00055735-200012000-00006. [DOI] [PubMed] [Google Scholar]
- 25.Rasmussen SA, Friedman JM. NF1 gene and neurofibromatosis 1. Am J Epidemiol. 2000;151:33–40. doi: 10.1093/oxfordjournals.aje.a010118. [DOI] [PubMed] [Google Scholar]
- 26.Niida Y, Stemmer-Rachamimov AO, Logrip M, et al. Survey of somatic mutations in tuberous sclerosis complex (tsc) hamartomas suggests different genetic mechanisms for pathogenesis of tsc lesions. Am J Hum Genet. 2001;69:493–503. doi: 10.1086/321972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Crino PB, Henske EP. New developments in the neurobiology of the tuberous sclerosis complex. Neurology. 1999;53:1384–1390. doi: 10.1212/wnl.53.7.1384. [DOI] [PubMed] [Google Scholar]
- 28.Rouleau GA, Wertelecki W, Haines JL, et al. Genetic linkage of bilateral acoustic neurofibromatosis to a DNA marker on chromosome 22. Nature. 1987;329:246–248. doi: 10.1038/329246a0. [DOI] [PubMed] [Google Scholar]
- 29.Rouleau GA, Merel P, Lutchman M, et al. Alteration in a new gene encoding a putative membrane-organizing protein causes neuro-fibromatosis type 2. Nature. 1993;363:515–521. doi: 10.1038/363515a0. [DOI] [PubMed] [Google Scholar]
- 30.Trofatter JA, MacCollin MM, Rutter JL, et al. A novel moesin-, ezrin-, radixin-like gene is a candidate for the neurofibromatosis 2 tumor suppressor. Cell. 1993;72:791–800. doi: 10.1016/0092-8674(93)90406-g. [DOI] [PubMed] [Google Scholar]
- 31.Seizinger BR, Martuza RL, Gusella JF. Loss of genes on chromosome 22 in tumorigenesis of human acoustic neuroma. Nature. 1986;322:644–647. doi: 10.1038/322644a0. [DOI] [PubMed] [Google Scholar]
- 32.Lutchman M, Rouleau GA. Neurofibromatosis type 2: a new mechanism of tumor suppression. Trends Neurosci. 1996;19:373–377. doi: 10.1016/S0166-2236(96)10044-8. [DOI] [PubMed] [Google Scholar]
- 33.Gutmann DH. Molecular insights into neurofibromatosis 2. Neurobiol Dis. 1997;3:247–261. doi: 10.1006/nbdi.1997.0128. [DOI] [PubMed] [Google Scholar]
- 34.Gusella JF, Ramesh V, MacCollin M, Jacoby LB. Merlin: the neurofibromatosis 2 tumor suppressor. Biochim Biophys Acta. 1999;1423:M29–36. doi: 10.1016/s0304-419x(99)00005-0. [DOI] [PubMed] [Google Scholar]
- 35.Parry DM, Eldridge R, Kaiser-Kupfer MI, Bouzas EA, Pikus A, Patronas N. Neurofibromatosis 2 (NF2): clinical characteristics of 63 affected individuals and clinical evidence for heterogeneity. Am J Med Genet. 1994;52:450–461. doi: 10.1002/ajmg.1320520411. [DOI] [PubMed] [Google Scholar]
- 36.Parry DM, MacCollin MM, Kaiser-Kupfer MI, et al. Germ-line mutations in the neurofibromatosis 2 gene: correlations with disease severity and retinal abnormalities. Am J Hum Genet. 1996;59:529–539. [PMC free article] [PubMed] [Google Scholar]
- 37.Perry A, Cai DX, Scheithauer BW, et al. Merlin, DAL-1, and progesterone receptor expression in clinicopathologic subsets of meningioma: a correlative immunohistochemical study of 175 cases. J Neuropathol Exp Neurol. 2000;59:872–879. doi: 10.1093/jnen/59.10.872. [DOI] [PubMed] [Google Scholar]
- 38.Chan CC, Vortmeyer AO, Chew EY, et al. VHL gene deletion and enhanced VEGF gene expression detected in the stromal cells of retinal angioma. Arch Ophthalmol. 1999;117:625–630. doi: 10.1001/archopht.117.5.625. [DOI] [PubMed] [Google Scholar]
- 39.Claudio JO, Lutchman M, Rouleau GA. Widespread but cell type-specific expression of the mouse neurofibromatosis type 2 gene. Neuroreport. 1995;6:1942–1946. doi: 10.1097/00001756-199510020-00028. [DOI] [PubMed] [Google Scholar]
- 40.Landau K, Yasargil GM. Ocular fundus in neurofibromatosis type 2. Br J Ophthalmol. 1993;77:646–649. doi: 10.1136/bjo.77.10.646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Brodsky MC, Landau K, Wilson RS, Boltshauser E. Morning glory disc anomaly in neurofibromatosis type 2. Arch Ophthalmol. 1999;117:839–41. [PubMed] [Google Scholar]
- 42.Ragge NK, Baser ME, Klein J, et al. Ocular abnormalities in neurofibromatosis 2. Am J Ophthalmol. 1995;120:634–641. doi: 10.1016/s0002-9394(14)72210-x. [DOI] [PubMed] [Google Scholar]
- 43.Ragge NK, Baser ME, Riccardi VM, Falk RE. The ocular presentation of neurofibromatosis 2. Eye. 1997;11:12–18. doi: 10.1038/eye.1997.3. [DOI] [PubMed] [Google Scholar]
- 44.Oberhuber G, Stolte M. Gastric polyps: an update of their pathology and biological significance. Virchows Arch. 2000;437:581–590. doi: 10.1007/s004280000330. [DOI] [PubMed] [Google Scholar]
- 45.Harada T, Irving RM, Xuereb JH, et al. Molecular genetic investigation of the neurofibromatosis type 2 tumor suppressor gene in sporadic meningioma. J Neurosurg. 1996;84:847–851. doi: 10.3171/jns.1996.84.5.0847. [DOI] [PubMed] [Google Scholar]
- 46.Twist EC, Ruttledge MH, Rousseau M, et al. The neurofibromatosis type 2 gene is inactivated in schwannomas. Hum Mol Genet. 1994;3:147–151. doi: 10.1093/hmg/3.1.147. [DOI] [PubMed] [Google Scholar]
- 47.Huynh DP, Tran TM, Nechiporuk T, Pulst SM. Expression of neurofibromatosis 2 transcript and gene product during mouse fetal development. Cell Growth Differ. 1996;7:1551–1561. [PubMed] [Google Scholar]