

Abstract
Shiga toxin (Stx)-producing Escherichia coli (STEC) is a major cause of sporadic cases of disease as well as serious outbreaks worldwide. The spectrum of illnesses includes mild nonbloody diarrhea, hemorrhagic colitis, and hemolytic-uremic syndrome. STEC produces one or more Stxs, which are subdivided into two major classes, Stx1 and Stx2. The ingestion of contaminated food or water, person-to-person spread, and contact with animals are the major transmission modes. The infective dose of STEC may be less than 100 organisms. Effective prevention of infection is dependent on rapid detection of the causative bacterial pathogen. In the present study, we examined 295 stool specimens for the presence of Stx-producing E. coli by three different methods: an Stx enzyme-linked immunosorbent assay, a conventional PCR assay, and a LightCycler PCR (LC-PCR) assay protocol recently developed by our laboratory at the Institute of Medical Microbiology at Hannover Medical School. Our intent was to compare these three methods and to examine the utility of the STEC LC-PCR protocol in a clinical laboratory. The addition of a control DNA to each sample to clearly discriminate inhibited specimens from negative ones enhanced the accuracy of the LC-PCR protocol. From our results, it can be concluded that LC-PCR is a very useful tool for the rapid and safe detection of STEC in clinical samples.
Shiga toxin (Stx)-producing Escherichia coli (STEC) is an emerging pathogen responsible for sporadic cases of infection as well as serious outbreaks worldwide. The potent cytotoxins produced by STEC are divided into two major classes, Stx1 and Stx2 (32; S. M. Scotland, H. R. Smith, and B. Rowe, Letter, Lancet 2:885-886). STEC causes a spectrum of human diseases ranging from mild nonbloody diarrhea through hemorrhagic colitis to the extraintestinal manifestation hemolytic-uremic syndrome (HUS) (17, 18). HUS is characterized by hemolytic anemia, thrombocytopenia, and acute renal failure (29). It is the major reason for an acute loss of kidney function in childhood (26, 31). Stx2e is an Stx2 variant produced mainly by E. coli isolates associated with edema disease in pigs (21), although HUS caused by an STEC strain expressing Stx2e in humans has been reported (9). The infectious dose of STEC may be very low (22); as a result, the organisms can be transmitted efficiently not only through contaminated foods but also from person to person (27). Rapid detection of the causative pathogen therefore is an important contribution to effective prevention of infection.
The identification of STEC by culture methods is a challenging task, as pathogenic strains are similar to nonpathogenic strains of E. coli (24). Currently, the best accepted techniques for diagnosis involve the identification of Stxs, either through protein detection by a cytotoxicity assay or an enzyme-linked immunosorbent assay (ELISA) or at the genomic level by PCR (22, 24). Further, STEC-specific plating media are available. Sorbitol-MacConkey (SMAC) agar and other chromogenic culturing methods are widely used (3, 4, 20); special blood agar plates allow for detection of the enterohemolytic phenotype of STEC strains (5). Cytotoxicity assays are the most sensitive methods for detecting active Stxs (24), but they are cumbersome to perform and therefore are not often established for routine microbiological diagnosis. Stx-specific ELISAs are acceptable alternatives, with several tests being commercially available. However, compared to cytotoxicity assays, they are less sensitive (11) and are also known to generate some false-positive results (1, 6). The disadvantage of plating media is that STEC may be substantially underrepresented in stool specimens from patients or asymptomatic carriers; this possibility hampers the reliable detection of these pathogens by bacterial culturing (15). Furthermore, SMAC agar only works for STEC strains which are sorbitol negative, and an enterohemolytic phenotype can also be observed with other members of the family Enterobacteriaceae.
The detection of stx genes by PCR offers certain advantages over the above-described diagnostic tools. It is exquisitely sensitive and highly specific. Therefore, even fewer than 10 Stx-producing E. coli organisms per ml of culture against a background of more than 109 other organisms per ml can be reliably detected in stool specimens (23). The serotype of the respective strain does not influence the PCR diagnosis of STEC, as detection is solely based on the presence or absence of stx genes. With new developments in PCR technology, such as using fluorescence for the identification and verification of PCR products (12, 33), the molecular identification of emerging enteropathogens becomes an economical alternative to the previously used methods.
In this study, we simultaneously examined 295 stool specimens from 271 diarrheic patients and patients with suspected STEC infections for the presence of STEC by three different methods: an Stx-specific ELISA (11), a conventional PCR (8, 30), and a recently developed LightCycler PCR (LC-PCR) (2). Our intent was to compare these three methods and to examine the utility of the STEC LC-PCR in a clinical laboratory. Further, we used an internal control DNA to identify inhibited samples.
MATERIALS AND METHODS
Collection of samples and preparation of template DNA for PCR.
A total of 295 stool specimens were collected from 271 patients with diarrhea over the periods from 1 September 2000 to 31 January 2001, 1 April 2001 to 31 January 2002, and 28 April 2003 to 8 May 2003. A small swab or 250 μl of liquid stool was incubated in EHEC toxin medium (Heipha, Heidelberg, Germany) in a horizontal shaker overnight at 37°C. Two loops (30 μl) of this broth culture were then plated directly on both blood agar and SMAC agar plates (Oxoid, Wesel, Germany) and again incubated overnight at 37°C. All colonies were suspended in 1 ml of 0.9% NaCl. The suspension was diluted 1:20 in PCR-grade H2O (AppliChem, Darmstadt, Germany); 10 μl was taken from this mixture for PCR with either a conventional block cycler or the LightCycler instrument.
Stx-specific ELISA.
An Stx-specific ELISA was performed according to the manufacturer's instructions. Briefly, the RIDASCREEN Verotoxin ELISA (R-Biopharm, Darmstadt, Germany) detects the presence of Stx1 and Stx2 by using immobilized mouse monoclonal antibodies against these two toxin molecules. For ELISA testing, 100 μl of a liquid culture from EHEC toxin medium was used. Absorbances were measured at 450 nm by using a spectrophotometer. The threshold was calculated according to the manufacturer's instructions, and positive and negative controls were run with each test.
Cytotoxicity testing.
Vero cells were grown as monolayers in 100 μl of Dulbecco's minimal essential medium (Biochrom, Berlin, Germany) supplemented with l-glutamine, antibiotics (penicillin and streptomycin), and 5% fetal bovine serum on 96-well plates (Greiner Bio-One, Frickenhausen, Germany). For the Stx-specific ELISA, supernatants from EHEC toxin medium were filter sterilized (Minisart; Sartorius, Göttingen, Germany) and diluted 1:5 and 1:25 in tissue culture medium. From each dilution step, 100 μl was added to the Vero cell culture, leading to final dilutions of 1:10 and 1:50, respectively. Assays were run in duplicate with purified Stx2 as a positive control. Cells were incubated for 24 to 48 h at 37°C in a 5% CO2 atmosphere. Cytopathic effects, characterized by lysis, vacuolization, and rounding of the cells, were measured photometrically after staining of the adherent monolayer. For staining, cells were fixed with 2% (vol/vol) formalin in phosphate-buffered saline for 15 min and then stained with a crystal violet solution (5% [vol/vol] ethanol, 2% [vol/vol] formalin, 0.13% [wt/vol] crystal violet; all chemicals were from Applichem) for 15 min at room temperature. The dye was redissolved with 100 μl of 70% ethanol, and then spectrophotometric absorbances were measured at 540 nm and compared to those for the positive and negative controls.
Conventional PCR.
Conventional PCR was performed with 50-μl assay mixtures and stxB1- and stxA2-specific primers KS7-KS8 and LP43-LP44 (all synthesized by MWG-Biotech, Ebersberg, Germany) as described by Schmidt et al. (30) and Cebula et al. (8). A culture isolate of STEC strain EDL933 (25) served as a positive control. DNA amplicons were subjected to submarine gel electrophoresis with 2% agarose gels, visualized by ethidium bromide staining, and compared to the respective PCR products from the stx1- and stx2-positive control strain.
LC-PCR.
Detection of stx genes with the LightCycler instrument was performed with a single capillary tube by using melting point analysis for the detection of the stx1, stx2, and stx2e genes as described recently (2). Primers and probes were synthesized by TIB MOLBIOL (Berlin, Germany). All DNA oligonucleotides used for conventional PCR as well as LC-PCR are listed in Table 1. As in conventional PCR, genomic DNA from STEC strain EDL933 was used as a positive control. To identify inhibited samples, we added to each capillary tube a plasmid control DNA which was amplified and detected together with the target sequences by means of the stxA2-specific primers and fluorescent hybridization probes. The construction and validation of this inhibition plasmid control DNA are described elsewhere (T. Bellin, A. Meuser, A. Karow, M. Hartmann, A. Matussek, S. Kafert, and F. Gunzer, submitted for publication). Briefly, a 917-bp fragment of the stxA2 gene was amplified by PCR with primers GK1a (GCT CTA GAA TGA AGT GTA TAT TAT TT) and AG1 (ACG CTG CAG CTG TAT TAC), bearing recognition sites for restriction endonucleases XbaI and PstI. The DNA amplicon was cloned into pUC19 linearized with XbaI and PstI. With a QuikChange site-directed mutagenesis kit from Stratagene (Amsterdam, The Netherlands), three point mutations were introduced into the probe binding region of the cloned stxA2 sequence by PCR-guided mutagenesis, thereby lowering the melting point of the control amplicon to approximately 58°C. The LightCycler signal generated by the plasmid was readily detected in samples that did not contain inhibitors.
TABLE 1.
Nucleotide sequences of primers and probes used in this studya
| Primer or probe and target | Name | Sequence (5′ → 3′) | Function | Positions | Tm (°C) | Refer- ence |
|---|---|---|---|---|---|---|
| Primers | ||||||
| stx1 | KS7 | CCC GGA TCC ATG AAA AAA ACA TTA TTA ATA GC | Sense | 1120-1142 | 54.0 | 30 |
| KS8 | CCC GAA TCC AGC TAT TCT GAG TCA ACG | Antisense | 1401-1384 | 52.0 | 30 | |
| StxA1 598 | AGT CGT ACG GGG ATG CAG ATA AAT | Sense | 598-621 | 56.9 | 2 | |
| StxA1 1015 | CCG GAC ACA TAG AAG GAA ACT CAT | Antisense | 1015-992 | 55.3 | 2 | |
| stx2 | LP43 | ATC CTA TTC CCG GGA GTT TAC G | Sense | 295-316 | 57.0 | 8 |
| LP44 | GCG TCA TCG TAT ACA CAG GAG C | Antisense | 881-860 | 57.0 | 8 | |
| StxA2 679 | TTC CGG AAT GCA AAT CAG TC | Sense | 679-698 | 52.5 | 2 | |
| StxA2 942 | CGA TAC TCC GGA AGC ACA TTG | Antisense | 942-922 | 54.6 | 2 | |
| Probes | ||||||
| stx1 | StxA1 FL 724 | CTG TCA CAG TAA CAA ACC GTA ACA TCG CTC-X | FL probe | 724-695 | 65.5 | 2 |
| StxA1 LC 693 R7 | LC-TGC CAC AGA CTG CGT CAG TGA GGT-ph | Red 705 probe | 693-670 | 67.5 | 2 | |
| stx2 | StxA2 FL 769 | CAG AGC AGT TCT GCG TTT TGT CAC TGT CA-X | FL probe | 769-797 | 65.0 | 2 |
| StxA2 LC 799 R6 | LC-AGC AGA AGC CTT ACG CTT CAG GC-ph | Red 640 probe | 799-821 | 63.3 | 2 |
Sequence M19473 (14) was used as a reference for nucleotide positions of stx1-specific primers and probes, and sequence X07865 (13) was used as a reference for all stx2-specific oligonucleotides. Tm, melting temperature. X, 3′ fluoresceine; LC, 5′ LightCycler dye; ph, 3′ phosphate. FL, fluoresceine Red 640, LightCycler dye Red 640; Red 705, LightCycler dye Red 705.
Isolation of STEC from stool specimens.
First, we tried to isolate STEC strains from positive stool specimens on EHEC agar (Heipha), a solid plating medium consisting of enterohemolysin blood agar on one half and chromogenic BCM agar on the other half of a petri dish. If these efforts were unsuccessful, colony hybridization was performed. From the plate suspension used for PCR analysis, serial 10-fold dilutions were made by using phosphate-buffered saline, and 100 μl of the 10−5, 10−6, and 10−7 dilution steps was spread on SMAC agar plates. The contents of plates with 200 to 300 well-separated bacterial colonies were replicated on polyamide round filters (Sartolon; Sartorius, Göttingen, Germany). Membrane-bound bacterial colonies were lysed, and genomic DNA was released through incubation of the filters in DNA I solution (0.5 M NaOH, 1.5 M NaCl) for 15 min, followed by a 5-min neutralization step with DNA II solution (1.0 M Tris-HCl [pH 7.5], 1.5 M NaCl) and a 20-min incubation step with 2× SSC (1× SSC is 0.15 M NaCl plus 0.015 M sodium citrate [pH 7.4]). The filters were then washed in 2× SSC- 0.1% sodium dodecyl sulfate for 2 h at 65°C in a hybridization oven. The wash buffer was changed after 1 h, and the filters were scrubbed thoroughly with a cotton towel to remove any residues from bacterial colonies, which might give false-positive signals. The hybridization membranes were then incubated at 42°C with digoxigenin-labeled PCR products amplified with LightCycler primers StxA1 598 and StxA1 1015 or StxA2 679 and StxA2 942 (2) by using Dig Easy Hyb solutions (Roche Diagnostics, Mannheim, Germany) and a digoxigenin DNA labeling and detection kit (Roche) according to the instructions provided by the manufacturer. Probe-positive colonies were visualized by chemoluminescence on X-ray film with CSPD (Applied Biosystems, Langen, Germany) as a substrate. With the aid of a light box, the corresponding colonies could then be isolated from the original plates.
Phenotypic methods.
STEC isolates were serotyped according to the method of Bockemühl et al. (7) by using antisera for E. coli somatic (O) antigens O1 to O170 and flagellar (H) antigens H1 to H56.
Statistical analysis.
Differences between groups were assessed by use of an alternative chi-square test, called McNemar's chi-square test (19), which is based on the numbers of discordant pairs r and s, as follows:
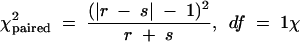 |
McNemar's chi-square test values of >3.841 were considered to be statistically significant. The use of McNemar's chi-square test is valid provided that the total number of discordant pairs is at least 10. In our studies, there were sometimes fewer than 10 discordant pairs; therefore, beyond that number, we calculated exact binomial probabilities. A P value of <0.05 was considered statistically significant. Both tests showed the same results for all comparisons; therefore, we consider McNemar's chi-square test to be robust.
RESULTS
Comparison of ELISA, conventional PCR, and LC-PCR.
The STEC LC-PCR detected one or more stx genes in 51 (17.2%) of the 295 stool specimens. The PCR profile and ELISA signal of the samples as well as the serotype and stx genotype of the isolated bacterial strains are listed in Table 2. Compared to conventional PCR, LC-PCR detected stx genes in five additional samples (Table 3). Four samples were positive for the stx1 gene, and one sample produced signals for the stx1 and stx2e genes (Table 2). None of them was found positive by the ELISA. Although the advantage of LC-PCR over conventional PCR in detecting STEC was not statistically significant (P = 0.063; χ2 = 3.2), the trend was that LC-PCR could identify such organisms in clinical samples more often. The stx genes discovered were identical in 44 of the 46 samples found positive by both PCR methods. Conventional PCR failed to detect stx1 two times in STEC strains that were found positive for both stx1 and stx2 by LC-PCR (Table 2, sample no. 38 and 40). Comparing both PCR methods to the Stx-specific ELISA revealed that significantly more STEC-positive stool specimens were identified by the molecular techniques (Table 3). Twenty additional samples harboring STEC were detected by LC-PCR (P < 0.001; χ2 = 18.05), and the immunoassay still failed 15 times when it was compared to conventional PCR (P < 0.001; χ2 = 13.07). Further, the Stx-specific ELISA produced eight false-positive results. In total, we were able to isolate STEC strains of various serotypes from 40 (78.4%) of the 51 PCR-positive stool specimens.
TABLE 2.
Results from conventional PCR, Stx-specific ELISA, and LC-PCR as well as serotypes of STEC strains isolated from 51 stool samples testing positive for stx genes with LC-PCRa
| Sample | Result of:
|
Serotype | ||||||
|---|---|---|---|---|---|---|---|---|
| No. | Name | Conventional PCR
|
ELISA | LC-PCR
|
||||
| stx1 | stx2 | stx1 | stx2 | stx2e | ||||
| 1 | D12 2001 | + | − | + | + | − | − | O55:H7 |
| 2 | D20 2001 | + | − | − | + | − | − | O91:H14 |
| 3 | D20 2002 | + | − | + | + | − | − | O91:H− |
| 4 | D20 2001 II | + | − | − | + | − | − | O91:H14 |
| 5 | D24 2001 | − | + | − | − | + | − | ND |
| 6 | D25 2002 | − | + | + | − | + | − | ND |
| 7 | D25 2004 | − | − | − | + | − | − | ND |
| 8 | D27 2004 | + | − | + | + | − | − | O145:H− |
| 9 | E21 2002 | − | + | − | − | − | + | ND |
| 10 | E21 2003 | − | + | − | − | − | + | ND |
| 11 | E22 2003 | + | − | + | + | − | − | O142:H33 |
| 12 | E22 2004 | + | − | + | + | − | − | O91:H− |
| 13 | E22 2003 II | + | − | + | + | − | − | O142:H33 |
| 14 | E25 5020 | + | − | + | + | − | − | O91:H− |
| 15 | E25 5021 | − | − | − | + | − | + | ND |
| 16 | E28 5009 | − | − | − | + | − | − | ND |
| 17 | E29 2004 | + | − | + | + | − | − | O142:H33 |
| 18 | eh 156 | + | + | + | + | − | + | O128:H2 |
| 19 | eh 179 | + | + | − | + | − | + | O113:H17 |
| 20 | eh 181 | − | + | − | − | + | − | O2:H− |
| 21 | eh 241 | + | + | − | + | − | + | O128:H2 |
| 22 | eh 507 | + | − | − | + | − | − | O103:H2 |
| 23 | eh 1478 | + | + | + | + | + | − | O157:H− |
| 24 | eh 1493 | − | + | + | − | + | − | O157:H7 |
| 25 | eh 1625 | − | + | + | − | + | − | O157:H7 |
| 26 | eh 1829 | + | − | + | + | − | − | O91:H14 |
| 27 | eh 1935 | + | − | + | + | − | − | O145:HNT |
| 28 | eh 134 II | + | − | − | + | − | − | O26:H1 |
| 29 | eh 1413 I | + | − | + | + | − | − | O111:H− |
| 30 | eh 1493 II | − | + | + | − | + | − | O111:H− |
| 31 | eh 1776 I | − | + | + | − | + | − | O157:H7 |
| 32 | eh 1870 I | + | − | − | + | − | − | ND |
| 33 | eh 1933 II | + | − | + | + | − | − | O145:HNT |
| 34 | eh 1962 II | + | − | + | + | − | − | O91:H14 |
| 35 | eh 2 | + | − | − | + | − | − | O103:H2 |
| 36 | eh 2086 II | + | − | + | + | − | − | O26:H11 |
| 37 | F01 2002 | + | − | + | + | − | − | O55:H7 |
| 38 | 1K28 2001 | − | + | − | + | + | − | O157:H7 |
| 39 | 1H20 2002 | − | + | + | − | + | − | ONT:H− |
| 40 | 1I27 2001 | − | + | + | + | + | − | O145:H− |
| 41 | 1J04 2001 | + | − | − | + | − | − | ONT:H− |
| 42 | 1J18 5018 | + | − | − | + | − | − | O91:H− |
| 43 | 1J20 2003 | − | − | − | + | − | − | ND |
| 44 | 1K06 2001 | − | − | − | + | − | − | ND |
| 45 | 2A21 2004 | + | − | + | + | − | − | O91:H− |
| 46 | 1K20 2005 | + | + | + | + | + | − | O157:H− |
| 47 | 204943 2002 | − | + | + | − | + | − | O157:H7 |
| 48 | 199414 2002 | − | + | + | − | + | − | ND |
| 49 | 287887 2003 | + | − | + | + | − | − | O91:H− |
| 50 | 288597 2003 | − | + | + | − | + | − | O157:H− |
| 51 | 288991 2003 | − | + | + | − | + | − | O157:H− |
+, positive; −, negative; ND, not detected. PCR-only-positive results were obtained for samples 5, 7, 9, 10, 15, 16, 32, 43, and 44.
TABLE 3.
Checkerboard for comparison of positive and negative results of Stx-specific ELISA, LC-PCR, and conventional PCR for all 295 stool specimens
| Result | No. of specimens with the following result:
|
|||
|---|---|---|---|---|
| LC-PCR + | LC-PCR − | PCR + | PCR − | |
| ELISA + | 31 | 8 | 31 | 8 |
| ELISA − | 20 | 236 | 15 | 241 |
| PCR + | 46 | 0 | ||
| PCR − | 5 | 244 | ||
Serotypes of STEC strains isolated from stool specimens.
Overall, isolated strains belonged to 11 different serogroups (Table 2). They contained the following O antigens (number of strains): O2 (1), O26 (2), O55 (2), O91 (10), O103 (2), O111 (2), O113 (1), O128 (2), O142 (3), O145 (4), and O157 (9). Two strains were not typeable. Five of the nine O157 STEC strains possessed H7 flagella; the four remaining ones were nonmotile (H−).
stx genes of STEC strains isolated from stool specimens.
Twenty-nine stool specimens were positive for the stx1 gene, and 14 had the stx2 and stx2e genes. Eight stool specimens contained STEC with two toxin genes, appearing either as stx1 and stx2 (n = 4) or as stx1 and stx2e (n = 4) genotypes. With respect to the isolated strains, 24 of them harbored stx1 genes, 9 were positive for stx2 genes, and 7 contained both stx1 and stx2 genes (n = 4) or both stx1 and stx2e genes (n = 3). No bacteria could be isolated from five stx2-positive stool specimens, three stx1-positive stool specimens, two stx2e-positive stool specimens, and one stool specimen harboring both stx1 and stx2e genes. Among these were five specimens found positive for stx genes solely by LC-PCR. ELISA was negative for 12 stx1-positive samples, 2 stx2-positive samples, 2 stx2e-positive samples, 1 stx1- and stx2-positive sample, and 3 stx1- and stx2e-positive samples. Isolation of bacteria from nine of these ELISA-negative stool specimens was not successful.
Cytotoxic activity of samples found positive only by PCR.
None of the nine stool specimens found positive for stx genes by one or both PCR methods (Table 2) exhibited cytotoxic activity toward Vero cells. Although we screened 2,000 to 3,000 individual colonies from each of the respective bacterial suspensions for the presence of stx genes by using colony hybridization, no STEC could be isolated.
DISCUSSION
Due to the fact that the identification of STEC by culture methods is difficult, more efficient techniques for the diagnosis of these emerging pathogens must be developed. In this study, we have shown that the detection of STEC by molecular methods is significantly more effective than detection by a licensed commercially available immunoassay. The ELISA that we used failed to detect 15 and 20 STEC-positive stool specimens identified by conventional PCR and LC-PCR, respectively. Furthermore, it delivered eight false-positive results. These findings clearly demonstrate that the Stx-specific ELISA used here has limitations for the detection of STEC in stool specimens. An additional restraint on the use of Stx detection assays is that not all E. coli strains which harbor stx genes produce the encoded protein under laboratory conditions (16).
Conventional PCR failed to identify five STEC-positive stool specimens detected by LC-PCR. Although this difference does not reach statistical significance, we assume that our choice of primers and the optimized PCR protocol account for the improved STEC detection. LC-PCR also has the advantage of rapid cycling combined with fluorescence-based identification and verification of PCR products. Further, the stx1 and stx2 genes are detected in a single reaction capillary tube, reducing the effort and time needed for diagnosis. The fact that, despite our endeavors, no STEC could be isolated from 9 of the 20 ELISA-negative stool specimens is most likely due to very low numbers of organisms in these specimens. The lack of cytotoxic activity toward Vero cells can be explained in this way also. These observations indicate the superior sensitivity of molecular techniques for the detection of STEC. Many of the traditional culture methods used for identifying STEC do not reveal the presence of non-O157 E. coli (3; H. Karch, Editorial, Eur. J. Clin. Microbiol. Infect. Dis. 15:276-280), and the detection of such organisms is beyond the scope of most routine laboratories. The use of molecular techniques is the best way to address this problem, and our study demonstrates the usefulness of LC-PCR for this purpose.
A complete analysis of 32 probes with a LightCycler instrument, including time for preparation of samples, takes about 180 min. Conventional PCR takes almost 2.5 times as long (440 min) and does not include the verification of positive PCR products by hybridization. The LC-PCR assay delivers very specific results quickly; therefore, this application is especially attractive for work involving a large number of samples, such as stool diagnostic procedures in clinical microbiology or food safety surveillance. Furthermore, melting point analysis allows discrimination between stx2 and stx2e, the gene encoding the pig edema disease toxin (21), a feature potentially interesting for epidemiological studies and risk assessment of stx-positive samples from asymptomatic patients. Friedrich et al. showed recently that infection with STEC possessing stx2e genes bears only a minimal risk of severe complications, such as hemorrhagic colitis and HUS (10).
When PCR is used to analyze clinical samples, inhibitory specimens must be safely identified to avoid false-negative results (28). Therefore, an internal standard was added to each test sample. A positive signal from this target demonstrates successful amplification and thereby validates a negative result for the primary target. Our approach is generally useful for all real-time PCR applications where melting curve analysis can be performed and certainly increases the security of any PCR-based diagnostic method. Bacterial strains were isolated from 40 out of 51 LC-PCR-positive samples and serotyped. The fact that we found an STEC content of 17.2% in the stool specimens investigated can be explained by the choice of specimens, which were taken from patients with diarrhea as well as from individuals with suspected STEC infections.
In conclusion, we have shown that LC-PCR is a very useful tool for identifying STEC in clinical samples. Compared to the ELISA used in this study, LC-PCR was significantly more effective in detecting pathogens. In addition, the costly and time-consuming hybridization of DNA amplicons for verification after conventional PCR becomes redundant. However, in addition to the rapid detection of STEC, culturing the organisms is desirable to assess their antibiotic resistance profiles and, from a public health perspective, to perform subtyping beyond stx gene detection for surveillance and outbreak investigations. Better methods are therefore still needed to isolate STEC from PCR-positive stool specimens.
Acknowledgments
We thank M. Burchardt, K. Schneider, and A. Hendricks for excellent technical assistance and K. Drexler and S. Kafert for helpful discussions. We also thank D. Bitter-Suermann for continuous support of our work and Christofer Samuelsson for assistance in preparing the manuscript.
This study was financially supported by Cytonet GmbH & Co. Hannover Management KG.
Matthias Pulz and Andreas Matussek contributed equally to this work.
REFERENCES
- 1.Ball, H. J., D. Finlay, A. Zafar, and T. Wilson. 1996. The detection of verocytotoxins in bacterial cultures from human diarrhoeal samples with monoclonal antibody-based ELISAs. J. Med. Microbiol. 44:273-276. [DOI] [PubMed] [Google Scholar]
- 2.Bellin, T., M. Pulz, A. Matussek, H. G. Hempen, and F. Gunzer. 2001. Rapid detection of enterohemorrhagic Escherichia coli by real-time PCR with fluorescent hybridization probes. J. Clin. Microbiol. 39:370-374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bettelheim, K. A. 1998. Reliability of CHROMagar O157 for the detection of enterohaemorrhagic Escherichia coli (EHEC) O157 but not EHEC belonging to other serogroups. J. Appl. Microbiol. 85:425-428. [DOI] [PubMed] [Google Scholar]
- 4.Bettelheim, K. A. 1998. Studies of Escherichia coli cultured on Rainbow Agar O157 with particular reference to enterohaemorrhagic Escherichia coli (EHEC). Microbiol. Immunol. 42:265-269. [DOI] [PubMed] [Google Scholar]
- 5.Beutin, L., S. Aleksic, S. Zimmermann, and K. Gleier. 1994. Virulence factors and phenotypical traits of verotoxigenic strains of Escherichia coli isolated from human patients in Germany. Med. Microbiol. Immunol. (Berlin) 183:13-21. [DOI] [PubMed] [Google Scholar]
- 6.Beutin, L., S. Zimmermann, and K. Gleier. 1996. Pseudomonas aeruginosa can cause false-positive identification of verotoxin (Shiga-like toxin) production by a commercial enzyme immune assay system for the detection of Shiga-like toxins (SLTs). Infection 24:267-268. [DOI] [PubMed] [Google Scholar]
- 7.Bockemühl, J., S. Aleksic, and H. Karch. 1992. Serological and biochemical properties of Shiga-like toxin (verocytotoxin)-producing strains of Escherichia coli, other than O-group 157, from patients in Germany. Zentbl. Bakteriol. 276:189-195. [DOI] [PubMed] [Google Scholar]
- 8.Cebula, T. A., W. L. Payne, and P. Feng. 1995. Simultaneous identification of strains of Escherichia coli serotype O157:H7 and their Shiga-like toxin type by mismatch amplification mutation assay-multiplex PCR. J. Clin. Microbiol. 33:248-250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Franke, S., D. Harmsen, A. Caprioli, D. Pierard, L. H. Wieler, and H. Karch. 1995. Clonal relatedness of Shiga-like toxin-producing Escherichia coli O101 strains of human and porcine origin. J. Clin. Microbiol. 33:3174-3178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Friedrich, A. W., M. Bielaszewska, W. L. Zhang, M. Pulz, T. Kuczius, A. Ammon, and H. Karch. 2002. Escherichia coli harboring Shiga toxin 2 gene variants: frequency and association with clinical symptoms. J. Infect. Dis. 185:74-84. [DOI] [PubMed] [Google Scholar]
- 11.Gerritzen, A. 1998. Comparison of two enzyme immuno assays and verocell cytotoxicity for detection of verotoxins in human feces. J. Lab. Med. 22:704-712. [Google Scholar]
- 12.Heid, C. A., J. Stevens, K. J. Livak, and P. M. Williams. 1996. Real time quantitative PCR. Genome Res. 6:986-994. [DOI] [PubMed] [Google Scholar]
- 13.Jackson, M. P., R. J. Neill, A. D. O'Brien, R. K. Holmes, and J. W. Newland. 1987. Nucleotide sequence analysis and comparison of the structural genes for Shiga-like toxin I and Shiga-like toxin II encoded by bacteriophages from Escherichia coli 933. FEMS Microbiol. Lett. 44:109-114. [DOI] [PubMed] [Google Scholar]
- 14.Jackson, M. P., J. W. Newland, R. K. Holmes, and A. D. O'Brien. 1987. Nucleotide sequence analysis of the structural genes for Shiga-like toxin I encoded by bacteriophage 933J from Escherichia coli. Microb. Pathog. 2:147-153. [DOI] [PubMed] [Google Scholar]
- 15.Karch, H., C. Janetzki-Mittmann, S. Aleksic, and M. Datz. 1996. Isolation of enterohemorrhagic Escherichia coli O157 strains from patients with hemolytic-uremic syndrome by using immunomagnetic separation, DNA-based methods, and direct culture. J. Clin. Microbiol. 34:516-519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Karch, H., T. Meyer, H. Rüssmann, and J. Heesemann. 1992. Frequent loss of Shiga-like toxin genes in clinical isolates of Escherichia coli upon subcultivation. Infect. Immun. 60:3464-3467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Karmali, M. A., M. Petric, C. Lim, P. C. Fleming, G. S. Arbus, and H. Lior. 1985. The association between idiopathic hemolytic uremic syndrome and infection by verotoxin-producing Escherichia coli. J. Infect. Dis. 151:775-782. [DOI] [PubMed] [Google Scholar]
- 18.Karmali, M. A., B. T. Steele, M. Petric, and C. Lim. 1983. Sporadic cases of haemolytic-uraemic syndrome associated with faecal cytotoxin and cytotoxin-producing Escherichia coli in stools. Lancet 1:619-620. [DOI] [PubMed] [Google Scholar]
- 19.Kirkwood, B. R. (ed.). 1988. Essentials of medical statistics, p. 94-105. Blackwell Science, Oxford, England.
- 20.March, S. B., and S. Ratnam. 1986. Sorbitol-MacConkey medium for detection of Escherichia coli O157:H7 associated with hemorrhagic colitis. J. Clin. Microbiol. 23:869-872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Marques, L. R., J. S. Peiris, S. J. Cryz, and A. D. O'Brien. 1987. Escherichia coli strains isolated from pigs with edema disease produce a variant of Shiga-like toxin II. FEMS Microbiol. Lett. 44:33-38. [Google Scholar]
- 22.Nataro, J. P., and J. B. Kaper. 1998. Diarrheagenic Escherichia coli. Clin. Microbiol. Rev. 11:142-201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Paton, A. W., J. C. Paton, P. N. Goldwater, and P. A. Manning. 1993. Direct detection of Escherichia coli Shiga-like toxin genes in primary fecal cultures by polymerase chain reaction. J. Clin. Microbiol. 31:3063-3067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Paton, J. C., and A. W. Paton. 1998. Pathogenesis and diagnosis of Shiga toxin-producing Escherichia coli infections. Clin. Microbiol. Rev. 11:450-479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Perna, N. T., G. Plunkett, III, V. Burland, B. Mau, J. D. Glasner, D. J. Rose, G. F. Mayhew, P. S. Evans, J. Gregor, H. A. Kirkpatrick, G. Posfai, J. Hackett, S. Klink, A. Boutin, Y. Shao, L. Miller, E. J. Grotbeck, N. W. Davis, A. Lim, E. T. Dimalanta, K. D. Potamousis, J. Apodaca, T. S. Anantharaman, J. Lin, G. Yen, D. C. Schwartz, R. A. Welch, and F. R. Blattner. 2001. Genome sequence of enterohaemorrhagic Escherichia coli O157:H7. Nature 409:529-533. [DOI] [PubMed] [Google Scholar]
- 26.Ray, P. E., and X. H. Liu. 2001. Pathogenesis of Shiga toxin-induced hemolytic uremic syndrome. Pediatr. Nephrol. 16:823-839. [DOI] [PubMed] [Google Scholar]
- 27.Reida, P., M. Wolff, H. W. Pohls, W. Kuhlmann, A. Lehmacher, S. Aleksic, H. Karch, and J. Bockemühl. 1994. An outbreak due to enterohaemorrhagic Escherichia coli O157:H7 in a children day care centre characterized by person-to-person transmission and environmental contamination. Zentbl. Bakteriol. 281:534-543. [DOI] [PubMed] [Google Scholar]
- 28.Rosenstraus, M., Z. Wang, S. Y. Chang, D. DeBonville, and J. P. Spadoro. 1998. An internal control for routine diagnostic PCR: design, properties, and effect on clinical performance. J. Clin. Microbiol. 36:191-197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ruggenenti, P., M. Noris, and G. Remuzzi. 2001. Thrombotic microangiopathy, hemolytic uremic syndrome, and thrombotic thrombocytopenic purpura. Kidney Int. 60:831-846. [DOI] [PubMed] [Google Scholar]
- 30.Schmidt, H., M. Montag, J. Bockemühl, J. Heesemann, and H. Karch. 1993. Shiga-like toxin II-related cytotoxins in Citrobacter freundii strains from humans and beef samples. Infect. Immun. 61:534-543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Smith, H. R., and S. M. Scotland. 1988. Vero cytotoxin-producing strains of Escherichia coli. J. Med. Microbiol. 26:77-85. [DOI] [PubMed] [Google Scholar]
- 32.Strockbine, N. A., L. R. Marques, J. W. Newland, H. W. Smith, R. K. Holmes, and A. D. O'Brien. 1986. Two toxin-converting phages from Escherichia coli O157:H7 strain 933 encode antigenically distinct toxins with similar biologic activities. Infect. Immun. 53:135-140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wittwer, C. T., K. M. Ririe, R. V. Andrew, D. A. David, R. A. Gundry, and U. J. Balis. 1997. The LightCycler: a microvolume multisample fluorimeter with rapid temperature control. BioTechniques 22:176-181. [DOI] [PubMed] [Google Scholar]