

Abstract
SacIp dysfunction results in bypass of the requirement for phosphatidylinositol transfer protein (Sec14p) function in yeast Golgi processes. This effect is accompanied by alterations in inositol phospholipid metabolism and inositol auxotrophy. Elucidation of how sac1 mutants effect “bypass Sec14p” will provide insights into Sec14p function in vivo. We now report that, in addition to a dramatic accumulation of phosphatidylinositol-4-phosphate, sac1 mutants also exhibit a specific acceleration of phosphatidylcholine biosynthesis via the CDP-choline pathway. This phosphatidylcholine metabolic phenotype is sensitive to the two physiological challenges that abolish bypass Sec14p in sac1 strains; i.e. phospholipase D inactivation and expression of bacterial diacylglycerol (DAG) kinase. Moreover, we demonstrate that accumulation of phosphatidylinositol-4-phosphate in sac1 mutants is insufficient to effect bypass Sec14p. These data support a model in which phospholipase D activity contributes to generation of DAG that, in turn, effects bypass Sec14p. A significant fate for this DAG is consumption by the CDP-choline pathway. Finally, we determine that CDP-choline pathway activity contributes to the inositol auxotrophy of sac1 strains in a novel manner that does not involve obvious defects in transcriptional expression of the INO1 gene.
INTRODUCTION
The yeast Sec14p is essential for glycoprotein transport from the Golgi complex and for cell viability (Novick et al., 1980; Bankaitis et al., 1989, 1990; Franzusoff and Schekman, 1989). In the absence of Sec14p function, vesicles fail to bud from the Golgi, and secretory proteins accumulate within this organelle. The role of Sec14p in stimulating Golgi secretory function appears to be a direct one, because a pool of Sec14p exists in a specific association with yeast Golgi membranes in vivo (Cleves et al., 1991b).
Mutations in any one of at least seven genes effect an efficient bypass of the essential Sec14p requirement for Golgi function and cell viability (Cleves et al., 1991a; Alb et al., 1996; Fang et al., 1996; Kearns et al., 1998). Three of these “bypass Sec14p” loci define structural enzymes of the CDP-choline pathway for phosphatidylcholine (PtdCho) biosynthesis (Cleves et al., 1991b; McGee et al., 1994), one of the two pathways for PtdCho biosynthesis in yeast (Figure 1). Both genetic and biochemical evidence suggests that the PtdCho-bound form of Sec14p downregulates the CDP-choline pathway by effecting an inhibition of CCTase, the rate-determining enzyme of the CDP-choline pathway (McGee et al., 1994; Skinner et al., 1995). These data exemplify how a fundamentally antagonistic relationship can exist between household phospholipid biosynthesis and essential cellular processes.
Figure 1.

CDP-choline and PtdEtn methylation pathways for PtdCho biosynthesis. Cho, choline; Cho-P, choline phosphate; CDP-Cho, CDP-choline; PtdMME, phosphatidylmonomethylethanolamine; PtdDME, phosphatidyldimethylethanolamine; SAM, S-adenosylmethionine; SAHC, S-adenosylhomocysteine.
Yeast sac1 strains also exhibit bypass Sec14p phenotypes, and these strains are characterized by unselected inositol auxotrophies (Cleves et al., 1989; Whitters et al., 1993; Kearns et al., 1997). Although the molecular basis of the inositol auxotrophy of sac1 strains has not been characterized, considerable data have been obtained regarding the role of Sac1p in regulating inositol phospholipid metabolism and Golgi secretory function. Sac1p is an integral membrane protein of the Golgi and endoplasmic reticulum, and Sac1p defects result in a bypass Sec14p phenotype, an inositol auxotrophy, a cold sensitivity for growth, allele-specific suppression of yeast actin mutations, and dramatic alterations in inositol phospholipid metabolism (Cleves et al., 1989; Novick et al., 1989; Whitters et al., 1993; Kearns et al., 1997). We proposed that Sac1p dysfunction effects its bypass Sec14p phenotypes by resulting in an expansion of a Golgi diacylglycerol (DAG) pool that is required for secretory vesicle formation (Kearns et al., 1997). Because the CDP-choline pathway is a potent DAG consumer (Figure 1), we further proposed that the toxic effect of CDP-choline pathway activity on Golgi membrane secretory function is related to depletion of this Golgi DAG pool when Sec14p is absent (McGee et al., 1994; Kearns et al., 1997).
In this report, we describe the pleiotropic changes in lipid metabolism that occur in sac1 strains. Three conclusions are derived. First, sac1 yeast strains exhibit a dramatic elevation in metabolic flux through the CDP-choline pathway for PtdCho biosynthesis. Genetic and biochemical data suggest that these increased rates of PtdCho biosynthesis are driven by elevated amounts of DAG produced in sac1 strains, and that this DAG production is phospholipase D (PLD) dependent. The hyperactivity of the CDP-choline pathway (an index of increased DAG availability) correlates with all conditions under which sac1 mutations are known to support bypass Sec14p. By contrast, the massive accumulation of inositol phospholipid that is a signature of sac1 strains is by itself insufficient to effect bypass Sec14p. Second, we find that CDP-choline pathway activity in sac1 strains contributes to the Ino− phenotype of these mutants. Finally, we demonstrate that the Ino− phenotype of sac1 strains is not the result of inability to induce INO1 expression under conditions of inositol depletion. These results provide the first example of which we are aware regarding an inositol auxotrophy in yeast that occurs via mechanisms independent of INO1 transcriptional defects.
MATERIALS AND METHODS
Yeast Strains
The yeast strains used in this study included: CTY182 (MATa ura3-52, lys2–801, Δhis3-200), CTY1-1A (CTY182 sec14-1ts), CTY100 (CTY1-1A sac1-26), CTY243 (CTY1-1A sac1Δ-356::HIS3), CTY165 (MATa ura3-52, ade2-101, Δhis3-200, sac1-22), CTY244 (CTY182, sac1Δ-356::HIS3), CTY1079 (CTY1-1A spo14Δ::HIS3), CTY1127 (CTY100 spo14Δ::URA3), CTY1129 (CTY124 spo14Δ::HIS3), CTY1098 (CTY159 spo14Δ::URA3), and CTY1099 (CTY160 spo14Δ::URA3).
Media and Genetic Techniques
YPD and defined yeast minimal medium either containing (I+) or lacking (I−) inositol have been described (Sherman et al., 1983). Plasmids used in this study have also been described (Kagiwada et al., 1996; Kearns et al., 1997). [14C]Choline chloride, [methyl-14C]methionine, and [32P]orthophosphate were purchased from Amersham (Arlington Heights, IL). myo-Inositol and other media ingredients were purchased from Sigma (St. Louis, MO). Standard yeast genetic methods and procedures for transformation have been described (Ito et al., 1983; Rothstein, 1983).
[14C]Choline and [14C]Methyl-Methionine Labeling of Yeast Lipids
Yeast strains were grown to midlogarithmic growth phase (3 ml; OD600 = 0.8–1.0) in defined minimal medium containing 0.1 mM inositol and 1 mM choline and presented with [14C]choline chloride (1 μCi/ml) for 20 min at 25°C with shaking (McGee et al., 1994). For [14C]methyl-methionine labelings, strains were grown and labeled as previously described (McGee et al., 1994). Incorporation of label was terminated by the addition of trichloroacetic acid (TCA) to 5%; the cells were subsequently incubated in 5% TCA for 20 min on ice and washed once in 5% TCA, and lipids were extracted by the method of Atkinson (1984). Briefly, after washing, yeast cells were pelleted by low-speed centrifugation in a clinical centrifuge, and the pellets were resuspended in 1 ml of polar extraction solvent (McGee et al., 1994) for 20 min at 65°C. Lipids were recovered by the addition of 5 ml of CHCl3:CH3OH:butylated hydroxytoluene (2:1:0.0005%) and 0.5 ml of H2O, followed by vigorous vortexing for 0.5 min. The mixed organic solutions were centrifuged in a clinical centrifuge for 5 min to separate the organic and aqueous phases. The organic phase was removed and dried under a gentle stream of N2 gas, followed by resuspension of the dried lipids in 60 ml of CHCl3:CH3OH:butylated hydroxytoluene for resolution by one-dimensional paper chromatography using Whatman (Maidstone, United Kingdom) SG81 paper treated as described by Steiner and Lester (1972) and the solvent system CHCl3:CH3OH:NH4OH (22:5.7:1). Radiolabeled PtdCho was visualized and quantitated using the PhosphorImager 425 instrument (Molecular Dynamics, Sunnyvale, CA).
For normalization of [14C]choline incorporation into PtdCho, identical cultures were labeled with [32P]orthophosphate (10 μCi/ml) for the same time as [14C]choline chloride-labeled cultures. After TCA precipitation and washing, 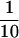 of the culture volume was removed, immobilized on 0.5-mm glass fiber filters, and washed with 50 vol of 50 mM phosphate buffer, pH 7.0. The immobilized cells and filters were dried and placed in scintillation vials for counting. [32P]Orthophosphate incorporation values were used to normalize loading of lipid samples from [14C]choline-labeled cells. This was done to compensate for the fact that strains that efficiently incorporate [14C]choline into PtdCho invariably incorporate more total [14C]choline label into cells.
of the culture volume was removed, immobilized on 0.5-mm glass fiber filters, and washed with 50 vol of 50 mM phosphate buffer, pH 7.0. The immobilized cells and filters were dried and placed in scintillation vials for counting. [32P]Orthophosphate incorporation values were used to normalize loading of lipid samples from [14C]choline-labeled cells. This was done to compensate for the fact that strains that efficiently incorporate [14C]choline into PtdCho invariably incorporate more total [14C]choline label into cells.
Determination of Bulk Phospholipid Content by Radiolabeling with [32P]Orthophosphate
Yeast strains were grown to midlogarithmic growth phase (3 ml; OD600 = 0.8–1.0) in defined minimal medium containing 0.1 mM inositol and 1 mM choline and presented with [32P]orthophosphate (10 μCi/ml) for 20 min at 25°C with shaking. One-tenth of the culture was removed after TCA precipitation to assess incorporation of label. Lipids were extracted as for [14C]choline chloride-labeled cultures, and equal amounts of incorporated counts were resolved by two-dimensional paper chromatography using Whatman SG81 paper. The first-dimension solvent was CHCl3:CH3OH:NH4OH:H2O (22:9:1:0.26), and the second-dimension solvent was CHCl3:CH3OH:CH3COOH:H2O (8:1:1.25:0.25). Radiolabeled phospholipid species were visualized and quantified by phosphorimaging.
Phosphatidylinositol-4-Phosphate (PtdIns-4-P) Measurements
Cells grown in medium containing 0.1 mM inositol and 1 mM choline were pulse radiolabeled with [3H]inositol (8 μCi/ml) for 45 min at 25°C and shaking. Otherwise, these experiments were carried out exactly as the phospholipid labelings with the following modifications: after TCA addition the cells were washed with 4 ml of 50 mM phosphate buffer, pH 7.0, followed by the addition of 1 ml CHCl3:CH3OH:1N HCl (1:2:0.8) and glass beads to one-half volume. Cultures were vortexed for 20 s every 5 min for 1 h with incubation on ice between rounds of vortexing. Two milliliters of CHCl3, 2 ml of CH3OH, 2 ml of 0.1N HCl, and 0.1 ml of 1 M NaCl were added, followed by brief vortexing and incubation at 4°C for 60 min. The organic layer (bottom) was extracted, dried under nitrogen gas, and resuspended in 40 μl of CHCl3:CH3OH (2:1). For two-dimensional paper chromatography the first solvent system was CHCl3:CH3OH:NH4OH:H2O (12: 5:0.36:1), and the second-dimension solvent was CHCl3:CH3OH:CH3COOH:H2O (15:3.3:2:1). Lipids were visualized by autoradiography.
Assignment of the accumulated inositol phospholipid in sac1 mutants as PtdIns-4-P was as follows. CTY182 (wild-type) and CTY244 (Δsac1) cells were radiolabeled to steady state with [3H]myo-inositol in medium supplemented with inositol (50 μM), and bulk cellular lipids were extracted and deacylated by base hydrolysis as described by Stolz et al. (1998). In parallel experiments, sac1-22 mutants were pulse radiolabeled with [3H]myo-inositol (8 μCi/ml) for 45 min at 25°C in inositol (100 μM)-supplemented medium. An internal [32P]PtdIns-4-P standard was generated by preparing [32P]PtdIns-4-P from radiolabeled COS cells (York and Majerus, 1994) and including this material in the deacylation mixture with lipids recovered from the Δsac1 mutant. The deacylated products were equilibrated to 10 mM ammonium phosphate, pH 3.5, applied to a 4.6 × 125-mm Partisphere SAX-10 column (Whatman), and eluted with a linear gradient of 10–340 mM ammonium phosphate over 15 min, 340–1.02 M ammonium phosphate over 7.5 min, and isocratic 1.02 M ammonium phosphate for 5 min. Deglyceration of glycerophosphoinositols and glycerophosphoinositol phosphates was performed as described (Lips et al., 1989). Enzymatic digests of deglycerated samples were performed using recombinant inositol polyphosphate 1-phosphatase (York et al., 1994). Some 3.6 × 105 cpm of deglycerated sample from sac1 mutants was incubated with mock control or with 0.4 μg of 1-phosphatase in 170 mM HEPES, pH 7.5, 1 mM EGTA, 100 mM KCl, and 3 mM MgCl2 at 37°C for 30 min in a total volume of 50 μl. Products were analyzed by HPLC as above.
Northern Analyses
Total RNA was extracted from cells, resolved by electrophoresis in formaldehyde–agarose gels, and transferred to a Biodyne A membrane (Pall, East Hills, NY) as described (Hosaka et al., 1992). Specific probes for INO1, OPI3, and PSS1 were generated from appropriate gene fragments labeled with [α-32P]dCTP using the Megaprime DNA random-priming system marketed by Amersham.
Assessment of the Effects of Inositol Starvation on Cell Growth and Viability
Yeast strains were grown to midlogarithmic growth phase in minimal defined medium containing 1 mM inositol. At time “zero” the cells were washed twice with double distilled H2O, washed once with minimal defined medium lacking inositol (I−), and resuspended in 3 ml of the same I− medium at a cell density of 1 × 106 cells/ml. At appropriate times after shift, 50 μl of culture volume were removed and serially diluted for plating onto solid complex medium (YPD plates; Sherman et al., 1983). After 3 d of growth at 25°C, colony-forming units were counted to assess viable cell numbers.
RESULTS
sac1 Yeast Strains Accumulate PtdIns-4-P
Physiological abnormalities associated with Sac1p defects in yeast include 1) bypass Sec14p, 2) cold sensitivity for growth, 3) accumulation of an inositol phospholipid, and 4) inositol auxotrophy (Cleves et al., 1989; Kearns et al., 1997). With regard to alterations in inositol phospholipid metabolism, this accumulated inositol phospholipid is detected in lipid extracts prepared from sac1 mutants labeled with either [32P]orthophosphate (Figure 2A) or [3H]inositol (see below). This spot 1 phospholipid was originally identified by us to be the inositol sphingolipid M(IP)2C on the basis of its Rf, our ability to label this species either with [3H]inositol or [32P]orthophosphate, and by staining with orcinol (Kearns et al., 1997). That our assignment of the spot 1 phospholipid was incorrect became clear when 1) we confirmed the finding of Stock et al. (1999) that disruption of IPT1 [the gene for M(IP)2C synthase] failed to influence the bypass Sec14p phenotype of sac1 mutants, and 2) we observed that the spot 1 phospholipid accumulated in ipt1 derivatives of sac1 strains (our unpublished results). These data indicated that spot 1 phospholipid was a phosphoinositide. Several lines of evidence now indicate this spot 1 lipid species to represent PtdIns-4-P (Figure 2B).
Figure 2.

Phospholipid profiles of wild-type and sac1 mutant yeast strains. (A) Yeast strains were pulse radiolabeled for 20 min with [32P]orthophosphate at 26°C in inositol- and choline-supplemented medium. Radiolabeled phospholipids were extracted using an acidic extraction solvent suitable for recovery of phosphoinositides (see MATERIALS AND METHODS) and resolved by two-dimensional paper chromatography. Phospholipid species are numbered. Relevant phospholipids are identified as follows: 1, PtdIns-4-P; 4, PtdIns; 5, PtdSer; 6, PtdCho; 7, PtdEtn; 8, PtdOH. The spot 1 lipid also labels with [3H]inositol, and its assignment as PtdIns-4-P is justified below. The sac1-22 strain exhibited a dramatic elevation in both PtdIns-4-P and PtdCho and a significant reduction in PtdSer, relative to wild-type yeast. These aberrant levels of PtdCho, PtdIns-4-P, and PtdSer were typical of Δsac1 strains as well. Phospholipids extracted from the equivalent number of wild-type and sac1-22 cells were chromatographically resolved in this experiment. (B) Deacylation of [3H]inositol-labeled lipids from sac1 strains reveals a dramatic increase in a soluble species that coelutes with the glycerophosphoinositol 4-P standard (i.e. the deacylated product of PtdIns-4-P). Total cellular lipids prepared from equal cell equivalents of [3H]inositol-labeled wild-type (CTY182) or isogenic Δsac1 strain (CTY244) were deacylated, and resulting glycerophosphoinositols were analyzed by HPLC. The radioactive profile from the region corresponding to glycerophosphoinositol monophosphates is shown for CTY182 (dotted line, lower panel) and CTY244 (solid line, upper panel). The elution position of glycerophosphoinositol-4-P was identified by an internal 32P standard included in HPLC analysis of deacylated fractions prepared from CTY244 (open circles, upper panel). In other experiments, radiolabeled glycerophosphoinositol, glycerophosphoinositol 4-P, and glycerophosphoinositol 4,5-P2 were used as standards to further verify assignments. (C) Inositol polyphosphate 1-phosphatase digestion of soluble head groups derived by deglyceration of deacylated inositol lipids prepared from the Δsac1 strain CTY244. Reactants of control (upper panel) or inositol polyphosphate-1-phosphatase-treated (lower panel) deglycerated samples were separated by anion-exchange HPLC. Peaks corresponding to Ins-1-P, Ins-4-P, Ins-1,3-P2, and Ins-1,4-P2 eluted at 17, 18, 33, and 35 min, respectively.
The assignment of the spot 1 lipid as PtdIns-4-P rests on several criteria. First, we found that a vps34 null allele, which blocks formation of PtdIns-3-P and PtdIns-3,5-P2 in yeast (Gary et al., 1998), did not reduce the amount of spot 1 phospholipid accumulated in sac1 mutants (our unpublished results). More direct evidence was obtained from experiments in which bulk [3H]inositol phospholipids were extracted from wild-type and sac1 strains and deacylated, and the deacylated products were resolved by HPLC (see MATERIALS AND METHODS). As shown in Figure 2B, sac1 mutants exhibited a modest (2- to 2.5-fold) steady-state increase in PtdIns-3-P levels relative to SAC1 strains (Figure 2B). PtdIns-4,5-P2 levels were not increased (our unpublished results). By far the most dramatic alteration in the glyceroinositolphosphate profiles between SAC1 and Δsac1 strains was observed in a 3H-labeled species that comigrated with the glycerophosphoinositol-4-P standard. This species reproducibly accumulated in Δsac1 strains to levels that were some six- to eightfold increased over the levels of glycerophosphoinositol-4-P recovered in deacylated lipid fractions prepared from wild-type yeast. Similar results were obtained from analysis of deacylated phospholipid fractions prepared from sac1-22 mutants radiolabeled with [3H]inositol (our unpublished results). These cumulative data provided suggestive, but not conclusive, evidence that sac1 strains accumulate PtdIns-4-P.
To solidify the assignment of PtdIns-4-P for the spot 1 lipid, [3H]inositol-labeled lipids were extracted from sac1 strains radiolabeled to steady state and deacylated. The deacylated fraction was deglycerated, and the released inositide head groups were analyzed by HPLC. As shown in Figure 2C (upper panel), the expected head group species were recovered as judged by their coelution with appropriate inositol phosphate standards. Consistent with the analyses of deacylated inositol glycerolipids from sac1 strains, the accumulated species that coeluted with the inositol-1,4-bisphosphate (Ins-1,4-P2) standard was present at levels approximately sixfold in excess of the head group derived from PtdIns-3-P. To probe the orientation of the phosphates on the deglycerated head groups, we assessed susceptibility of the soluble head group species to the action of inositol polyphosphate-1-phosphatase, an enzyme that exhibits substrate selectivity for Ins-1,4-P2 (York et al., 1994). A product exhibiting the properties of Ins-4-P was generated in this enzymatic reaction (Figure 2C, lower panel), and this species was formed at the expense of the material that coeluted with the Ins-1,4-P2 standard (Figure 2C, compare upper and lower panels). These data demonstrate that the spot 1 phospholipid that accumulates in sac1 mutants represents PtdIns-4-P.
Which PtdIns 4-kinase is responsible for generating the accumulated PtdIn-4-P? [3H]Inositol pulse-labeling experiments indicated that prechallenge of sac1 strains with high concentrations of wormannin (1 mM; i.e., >1000 times the minimal growth-inhibitory concentration for sac1 mutants) had little effect on PtdIns-4-P accumulation in these strains (our unpublished results). Because wortmannin targets the Stt4p PtdIns 4-kinase, but not the Pik1p PtdIns 4-kinase, in yeast (Cutler et al., 1997), we think it likely that the PtdIns-4-P that accumulates in sac1 strains is primarily generated via Pik1p.
sac1 Yeast Strains Experience Elevated Flux through the CDP-Choline Pathway for PtdCho Biosynthesis
Examination of the data presented in Figure 2A revealed several additional abnormalities in sac1 strains in addition to accumulation of PtdIns-4-P. First, incorporation of 32P radiolabel into PtdCho appeared to be greatly enhanced in sac1-22 strains relative to its SAC1 partner. Second, sac1-22 strains exhibited reduced levels of phosphatidylserine (PtdSer). The sac1-22 allele is unique in that it is the only sac1 mutation that does not evoke inositol auxotrophy. Nevertheless, the bypass Sec14p phenotype associated with this allele, and the other manifest alterations in inositol phospholipid metabolism associated with it are inositol dependent (Kearns et al., 1997).
To more closely examine the relationship between Sac1p function and phospholipid metabolism, we quantitated the rates of phospholipid biosynthesis in sac1-22, sac1-26, and Δsac1 strains and a SAC1 partner strain. Yeast were pulse radiolabeled in inositol-containing medium for 20 min with [32P]orthophosphate at 25°C, phospholipids were extracted by methods optimal for recovery of the major yeast phospholipids, and individual phospholipid species were resolved by paper chromatography and quantitated (see MATERIALS AND METHODS). The results are shown in Figure 3A. The data demonstrate that sac1 mutants indeed exhibited dramatically increased rates of PtdCho biosynthesis. Quantitation of the phospholipid profiles revealed that the sac1-22, sac1-26 and Δsac1 mutants all incorporated nearly 70% more [32P]orthophosphate into PtdCho during the 20-min pulse than did the SAC1 strain (Figure 3A). This difference could not be attributed to generally altered rates of phospholipid synthesis in sac1 strains, because sac1 and wild-type strains exhibited very similar rates of incorporation of 32P into phospholipid (see legend to Figure 3A). Because SAC1 and sac1 strains incorporated very similar amounts of [32P] into phospholipid per OD600 cells during the labeling period, these data indicate that sac1 mutants produced some 70% more PtdCho on a per cell basis than did wild-type strains during the labeling period (Figure 3A). Elevated PtdCho production was discerned only in pulse-radiolabeling experiments, however. No significant difference in the steady-state PtdCho content of SAC1 and mutant sac1 strains was recorded (our unpublished results). Decreased amounts of radiolabel in the PtdIns fraction were also recovered from these sac1 mutant strains. Finally, these pleiotropic sac1-associated effects on phospholipid metabolism were recorded irrespective of whether these strains harbored SEC14 or sec14ts alleles. At this point, we do not know what contribution reduced PtdSer levels make to the various sac1 phenotypes.
Figure 3.

Phosphatidylcholine synthesis in wild-type and sac1 mutant yeast strains. (A) Strains with the indicated genotypes (at bottom) were grown to midlogarithmic growth phase in medium containing inositol (0.1 mM) and choline (1 mM). Cell cultures were then pulse radiolabled with [32P]orthophosphate (10 μCi/ml) for 30 min at 25°C. Phospholipids were extracted by the method of Atkinson (1984) and resolved by two-dimensional chromatography, and radiolabeled species were quantified by phosphorimaging (see MATERIALS AND METHODS). Biosynthetic rates were deduced from the relative proportion of label recovered in each individual phospholipid species (expressed as percentage of total phospholipid). Rates of total phospholipid synthesis in these strains were estimated by measuring incorporation of 32P into chloroform-soluble counts (11,300–12,600 cpm/OD600 for sac1 and sac1, Δspo14 mutants to 10,000–11,000 cpm/OD600 for wild-type strains). Error bars are indicated. Phospholipid species are presented in the following order: PtdIns (black bars), PtdCho (cross-hatched bars), PtdSer (hatched bars), PtdEtn (stippled bars), and phosphatidic acid (open bars). Strains used were CTY1-1A (sec14-1ts, SAC1, SPO14), CTY165 (sec14-1ts, sac1-22, SPO14), and CTY1261 (sec14-1ts, sac1-22, Δspo14). These data represent an average of three independent experiments, and all strains were analyzed in parallel for each trial. In a typical quantitation, PtdCho accounted for 5.0 × 105 and 2.7 × 105 phosphorimager units in sac1 and SAC1 strains, respectively, when 1 × 106 phosphorimager units of total phospholipid were analyzed from each strain. (B) Activities of the CDP-choline pathway and PtdEtn methylation pathway were evaluated in vivo by pulse radiolabeling of SAC1 and sac1-22 strain pairs (CTY1-1A and CTY165, respectively). The indicated strains were grown to midlogarithmic growth phase in choline-free medium containing inositol. Cell cultures were split, and one aliquot was pulse radiolabeled with [14C]choline chloride (1 μCi/ml) or [14C]methyl-methionine (1 μCi/ml), respectively, for 20 min at 26°C with shaking. A parallel aliquot was labeled with [32P]orthophosphate for 20 min at 26°C. Phospholipids from equal cell equivalents (determined by 32P incorporation into phospholipid in the parallel cultures) were subsequently extracted, resolved, and quantified as described in MATERIALS AND METHODS. These data represent an average of three independent experiments. For quantitative comparison of a representative experiment, wild-type and sac1 strains incorporated 3.8 × 104 and 1.3 × 105 cpm [14C]choline chloride into PtdCho per OD600 cells, respectively. (C) Strains with the indicatedgenotypes were grown to midlogarithmic growth phase in choline-free medium containing inositol. Cell cultures were split, and one aliquot was pulse radiolabeled with [14C]choline chloride (1 μCi/ml) or [32P]orthophosphate (10 μCi/ml) for 20 min at 26°C. Phospholipids were extracted, resolved, and quantified (see MATERIALS AND METHODS). Again, samples were normalized for cell equivalents based on incorporation of [32P]orthophosphate into the parallel cultures as described in B. These data represent an average of three independent experiments. Strains CTY182 (SAC1) and CTY244 (Δsac1) were transformed with the test YEp(DGK) plasmid and the vector-only control YEp(URA3) plasmid, respectively. The YEp(DGK) plasmid was represented by the pCTY85 construct that drives constitutive expression of DGK in yeast (Kearns et al., 1997).
Yeast produce PtdCho either via the CDP-choline pathway or the phosphatidylethanolamine (PtdEtn) methylation pathway (Figure 1). We determined whether the increased PtdCho biosynthetic rates observed in sac1 mutants resulted from increased activity of one or both of these pathways. To specifically monitor activity of the CDP-choline pathway, we subjected sac1-22 and SAC1 strain pairs to a [14C]choline pulse-radiolabeling regimen and measured the incorporation of radiolabel into PtdCho (see MATERIALS AND METHODS). The data indicate that sac1-22 mutants incorporated 2.8 ± 0.6-fold more [14C]choline into PtdCho per OD600 of cells than did SAC1 strains, and this effect was typical for sac1 mutants (Figure 3B). We interpret these data to indicate that the sac1 mutants sustain a rate of metabolic flux through the CDP-choline pathway that is two- to threefold greater than that exhibited by wild-type yeast.
Two independent lines of evidence further support this finding. First, [32P]orthophosphate pulse-radiolabeling experiments demonstrated that omission of choline from the medium (i.e., a condition that reduces CDP-choline pathway activity) diminished the rate of 32P radiolabel incorporation into PtdCho. Under those conditions, sac1 mutants exhibited only a 1.3-fold greater rate of 32P incorporation into PtdCho relative to wild type. Second, genetic disruption of the CDP-choline pathway in Δsac1 strains reduced flux through the CDP-choline pathway some 30-fold (as measured by [14C]choline incorporation into PtdCho in a 20-min pulse). The resultant basal level of CDP-choline pathway activity was similar to that measured for an isogenic SAC1, cki1-284::HIS3 strain (our unpublished results). These findings excluded the formal possibility that a normally cryptic choline- and DAG-utilizing pathway for PtdCho synthesis was activated in sac1 mutants.
To determine whether the PtdEtn methylation pathway for PtdCho biosynthesis was also stimulated in sac1 mutants, a [methyl-14C]methionine pulse-radiolabeling strategy was used to measure the activity of this pathway (see Figure 1). No significant differences in [methyl-14C]methionine incorporation into PtdCho were recorded between sac1 and SAC1 strains, because the sac1 mutant exhibited 1.2 ± 0.4 the wild-type rate of PtdEtn methylation pathway activity (Figure 3B).
DAG Kinase (DGK) Expression Reduces CDP-Choline Pathway Activity in sac1 Strains
The CDP-choline pathway directly consumes DAG in the process of PtdCho synthesis, whereas the methylation pathway does not (Figure 1). Pulse-radiolabeling analyses described above demonstrated that sac1 strains produce DAG at rates some two- to threefold greater than those recorded for isogenic SAC1 strains, suggesting that CDP-choline pathway hyperactivity in sac1 strains might be supported by increased DAG availability. To investigate the issue in more detail, Escherichia coli DGK, an enzyme that converts DAG to phosphatidic acid (PtdOH), was expressed in Δsac1 strains. We then assessed the effect of metabolic shunting of DAG to PtdOH on CDP-choline pathway activity in Δsac1 strains.
[14C]Choline radiolabeling experiments demonstrated that DGK expression effected a reproducible 30% decrease in CDP-choline pathway activity in sac1 strains (Figure 3C). Although the effect was relatively modest, and the rate of CDP-choline pathway activity in DGK-expressing Δsac1 strains still exceeded those of wild-type strains, this reduction in CDP-choline pathway was physiologically significant, because it strongly influenced sac1-associated inositol auxotrophy (see below).
Elevated CDP-Choline Pathway Activity in Δsac1 Strains Is PLD Dependent
Full manifestation of all presently known bypass Sec14p mechanisms, including those associated with sac1 mutations, requires the contribution of a functional SPO14 gene, which encodes the sole PIP2-activated PLD in yeast (Xie et al., 1998). In addition, the bypass Sec14p phenotype of sac1 strains is abolished by DGK expression (Kearns et al., 1997). We have proposed that PLD may play a role in DAG generation by providing a pool of PtdOH that serves as a substrate for PtdOH phosphohydrolases. A prediction of this model is that PLD is required to sustain the elevated CDP-choline pathway activity of sac1 strains because it ultimately generates excess DAG in these mutants.
To test this prediction, a [32P]orthophosphate pulse-radiolabeling regimen (which predominantly monitors PtdCho synthesis via the CDP-choline pathway; McGee et al., 1994) was used. In these experiments, excess choline (1 mM) was included in the growth medium to ensure that any influence of PLD on CDP-choline pathway activity was independent of PLD-mediated generation of choline itself. Introduction of a Δspo14 allele into the sac1-26 strain evoked a significant reduction in CDP-choline pathway activity (Figure 3A). In these experiments, PtdCho represented 29.5 ± 1.9 and 49.0 ± 1.9% of the radiolabeled phospholipid extracted from SAC1 and sac1 strains, respectively. The rate of PtdCho synthesis in the sac1-26, Δspo14 mutant was not significantly different from that of wild type, because PtdCho comprised 34.5 ± 3.4% of the radiolabled phospholipid recovered from that double mutant strain (Figure 3A). Because all three strains incorporated very similar amounts of 32P into phospholipid per OD600 cells during the labeling period (see Figure 3A legend), the relative percentages of label incorporated into PtdCho directly reflected quantitative differences in rates of PtdCho synthesis. The demonstration that sac1-26, Δspo14 double mutant strains exhibit wild-type rates of flux through the CDP-choline pathway, when coupled with the sensitivity of sac1-mediated CDP-choline pathway hyperactivity to DGK expression (Figure 3C), suggests that PLD activity supplies the DAG that drives accelerated metabolic flux through this pathway.
To determine whether PLD activity was solely responsible for the increased DAG production observed in sac1 mutants (Kearns et al., 1997), we subjected isogenic PLD-proficient (SPO14) and deficient (Δspo14) sac1-22 strain pairs to a 20-min [14C]acetate pulse at 25°C. Lipids were extracted, and DAG was resolved by TLC (Buttke and Pyle, 1982). We took care to expeditiously extract and resolve the DAG to limit the unavoidable isomerization of 1,2-DAG to 1,3-DAG. Isogenic SAC1, SPO14 strains were used as wild-type controls, and both 1,2-DAG and 1,3-DAG species were identified and quantitated to yield total DAG values. In accord with the data of Kearns et al. (1997), sac1, SPO14 strains exhibited a 70% increase in DAG production relative to wild-type control (22 ± 2 and 13 ± 1% of total extractable lipid, respectively; n = 3). This effect was observed in media with inositol concentrations of ≥300 μM, as used by Kearns et al. (1997), but not in media with inositol concentration of <100 μM. Rates of [14C]acetate incorporation into lipid in these experiments were equivalent for SAC1 strains and sac1 mutants (420 ± 60 and 418 ± 18 cpm · OD600−1 · min−1, respectively). Genetic inactivation of PLD had no effect on the elevated DAG production scored for sac1-22 strains as assayed by this regimen (21 ± 1%; n = 3).
We also used steady-state [14C]acetate labeling experiments to assess DAG pools at equilibrium in SAC1 strains, sac1-22 mutants, and sac1-22, Δspo14 double mutants. Because sac1 mutants exhibit elevated activity of at least one DAG consuming pathway (i.e., the CDP-choline pathway), we expected that sac1 mutants would not exhibit elevated DAG levels at steady state. As predicted, no steady-state differences in DAG pools were detected in these strains (13 ± 1, 12 ± 1, and 12 ± 2% of total chloroform-soluble counts, respectively; n = 3). These data are in accord with our previous determinations in which we measured DAG by radiolabeling cells to steady state with [14C]acetate and chasing for 2 h. In those experiments, there also were no significant differences in steady-state DAG pools among SAC1 strains, sac1 mutants, and sac1, Δspo14 double mutants (Xie et al., 1998). These cumulative data suggest that sac1 strains produce elevated DAG by at least two distinct pathways: one that is PLD dependent and results in a DAG pool that is robustly scavenged by CDP-choline pathway activity, and a second pathway that is revealed in [14C]acetate pulse-radiolabeling experiments, when cells are grown in media containing higher concentrations of inositol.
Accumulation of PtdIns-4-P in PLD-deficient sac1 Strains
The bypass Sec14p phenotype of sac1 strains correlates closely with the dramatic accumulation of an inositol phospholipid (Kearns et al., 1997), which is now identified as PtdIns-4-P (Figure 2A). Perhaps elevated PtdIns-4-P defines the biochemical basis for bypass Sec14p in sac1 strains. Yet, the sac1 bypass Sec14p phenotype is sensitive to both DGK expression and genetic inactivation of PLD (Kearns et al., 1997; Xie et al., 1998). We therefore tested whether either of the conditions that abrogate sac1-mediated bypass Sec14p also influenced PtdIns-4-P accumulation in sac1 strains.
Both [3H]inositol- and [32P]orthophosphate-labeling regimens were used to compare the magnitude of PtdIns-4-P accumulation in SAC1 and sac1-22 strain pairs that were either proficient or deficient in PLD activity. As shown in Figure 4A, [3H]inositol pulse-radiolabeling experiments indicated that sac1-22 strains exhibited a dramatic (more than sixfold) accumulation of PtdIns-4-P relative to SAC1 strains when cultured in inositol-containing medium. Inspection of profile obtained for the isogenic Δspo14 partner indicated that PLD inactivation had no significant effect on PtdIns-4-P accumulation in these mutants (Figure 4A). The results obtained from parallel [32P]orthophosphate-labeling experiments provided quantitative results that confirmed the conclusion derived from the [3H]inositol-labeling experiments (Figure 4B). By the lipid extraction method used, 3.3 ± 0.1% of the 32P radiolabel incorporated into phospholipid was recovered in the PtdIns-4-P fraction of SAC1 strains. By contrast, 17.8 ± 3.2 and 14.7 ± 2.5% of the 32P incorporated into phospholipid extracted from the sac1-22 and the sac1-22, Δspo14 double mutant strain was recovered in PtdIns-4-P, respectively (Figure 4B). DGK expression, which is similar to Δspo14 in that it too abrogates sac1-mediated bypass Sec14p (Kearns et al., 1997), also had no effect on PtdIns-4-P accumulation, as measured by [32P]orthophosphate pulse radiolabeling (our unpublished results). Although sac1-22 strains were used in these experiments, the same results were obtained with sac1 null strains as well (our unpublished results). We conclude that PtdIns-4-P accumulation is insufficient to support bypass Sec14p.
Figure 4.


Effects of PLD deficiency on PtdIns-4-P accumulation and bypass Sec14p. (A) Strains CTY1-1A (SAC1, SPO14), CTY165 (sac1-22, SPO14), and CTY (sac1-22, Δspo14) were grown in medium containing inositol (0.1 mM) and pulse radiolabeled with [3H]inositol (5 μCi/ml) for 1 h at 25°C and shaking. Radiolabeled inositol phospholipids were subsequently extracted and resolved by two-dimensional paper chromatography. Radiolabeled species were visualized by autoradiography. The positions of PtdIns and PtdIns-4-P are identified. These data are from a representative experiment. (B) The conditions of the experiment were exactly as in A with the exception that cells were radiolabeled with [32P]orthophosphate (10 μCi/ml). Radiolabeled species were subsequently extracted under acidic conditions optimal for recovery of phosphoinositides, resolved by two-dimensional chromatography, visualized, and quantitated by phosphorimaging. The data represent the average of three independent experiments. The bulk incorporation of label into phsopholipid for these strains during the pulse was essentially equivalent (see Figure 3A), and the relative magnitudes of PtdIns-4-P were directly related to quantities of PtdIns-4-P per cell equivalent in these strains. (C) A set of isogenic sec14-1ts, Δspo14 strains carrying the indicated bypass Sec14p mutations were streaked for single colonies on YPD medium and incubated for 72 h at 33.5°C. The corresponding sac1-26, pct1-2, kes1-1, and BSD1-124 derivatives were represented by strains CTY1127, CTY1096, CTY1098, and CTY1129, respectively. The SEC14, Δspo14 derivative (CTY1092) served as a positive growth control, whereas the sec14-1ts, Δspo14 strain (CTY1079) represented the negative control. In the Δspo14 genetic background, neither sac1, kes1 nor BSD1-124 alleles imparted any detectable phenotypic suppression of sec14 growth defects. By contrast, inactivation of the CDP-choline pathway via the pct1-2 mutation (and cki1; our unpublished results) retained a clear PLD-independent ability to suppress sec14ts growth defects under these conditions.
sac1 Mutants Are Entirely Dependent on PLD Activity for Growth under Sec14p-deficient Conditions, Whereas pct1 Mutants Are Not
PLD is an essential component of the mechanism(s) by which all known classes of bypass Sec14p alleles exert their suppressor phenotypes (Sreenivas et al., 1998; Xie et al., 1998). What remains unclear is whether PLD is the ultimate executor of these bypass Sec14p phenotypes or whether it is only one of several contributory factors. To distinguish between these possibilities, PLD-deficient sec14-1ts strains carrying individual bypass Sec14p mutations were analyzed for their abilities to grow under various conditions. Although a SEC14, Δspo14 double mutant strain grew well at 33.5°C, the isogenic sec14-1ts derivative was unable to grow at this temperature (Figure 4C). Indeed, the sec14-1ts, Δspo14 double mutant was incapable of growth at temperatures above 31.5°C. Individual sac1, kes1, or BSD1-124 mutations failed to suppress sec14-1ts growth defects at 33.5°C (Figure 4C) or any other restrictive temperature. Thus, the mechanisms for bypass Sec14p in these strains operated through PLD because no PLD-independent component for suppression could be discerned.
Different results were obtained for sec14-1ts, Δspo14 mutants carrying mutations (pct1) that inactivate the CDP-choline pathway. These triple mutant strains grew well at 33.5°C, clearly indicating a PLD-independent component to suppression of sec14 defects by this mechanism (Figure 4C). Analysis of sec14-1ts, Δspo14, cki1 triple mutants yielded the same results (our unpublished results). We conclude that PLD is a major (but not the sole) contributor to the bypass Sec14p condition elicited by CDP-choline pathway defects. Interestingly, steady-state [32P]orthophosphate radiolabeling experiments conducted at 26°C, followed by a 2-h chase at 33.5°C, revealed that sec14-1ts, Δspo14, pct1 strains exhibited similarly reduced bulk PtdOH levels to sec14-1ts, Δspo14, sac1 strains. These reductions in bulk PtdOH were 10-fold relative to isogenic SPO14 strains (2.1 ± 0.4 vs. 0.2 ± 0.0% of total phospholipid for SPO14 and Δspo14 derivatives of the pct1 strain, respectively; n = 3). Yet the pct1 strains grew well at 33.5°C, whereas the sac1 partners did not. Thus, reduced PtdOH levels can be uncoupled from the mechanisms by which CDP-choline pathway mutations effect bypass Sec14p.
Constitutive Transcription of INO1 Fails to Relieve sac1 Inositol Auxotrophy
Sac1p is a multifunctional protein that regulates ATP import into the lumen of intracellular organelles in a manner that can be uncoupled from its role in regulating inositol phospholipid metabolism (Mayinger et al., 1996; Kearns et al., 1997; Köchendorfer et al., 1999). Because Δsac1 mutations evoke an unfolded protein response in yeast (Kōchendörfer et al., 1999), and this response inhibits transcription of INO1, we analyzed whether the Ino− phenotype of sac1 strains was related to defects in INO1 expression. The INO1 gene product catalyzes the conversion of glucose-6-phosphate to inositol-1-phosphate, the committed step for inositol biosynthesis in yeast (Carman and Henry, 1989). INO1 transcription is also strongly repressed by inositol and choline, and efficient induction of INO1 expression upon shift of yeast from inositol-replete to inositol-deficient conditions is required for cellular viability in inositol-free growth media (Culbertson et al., 1976a,b).
To address whether defects in INO1 expression contributed to the Ino− phenotype of sac1 mutants, we used two independent approaches. First, the INO1 gene was placed under constitutive transcriptional control of the yeast SEC14 promoter. This PSEC14::INO1 expression cassette was introduced into ino1-13 and Δsac1 strains in the context of a high-copy yeast plasmid YEp(PSEC14::INO1). Introduction of the YEp(PSEC14::INO1) plasmid restored inositol prototrophy to ino1-13 strains, thereby providing a demonstration that YEp(PSEC14::INO1) sustained sufficient expression of INO1 to correct the inositol auxotrophy of an ino1 mutant. Strikingly, YEp(PSEC14::INO1) failed to complement the Ino− phenotype of Δsac1 strains (Figure 5A).
Figure 5.

The inositol auxotrophy of Δsac1 strains is not the consequence of defects in INO1 expression. (A) The yeast strains with the indicated genotypes were streaked for isolation on uracil-deficient medium in the presence (+Inositol) or absence (−Inositol) of inositol and incubated at 26°C for 72 h. The YEp(PSEC14:: INO1) plasmid, which drives constitutive expression of INO1 from the SEC14 promoter, was incapable of rescuing inositol prototrophy in the Δsac1 strain CTY244. That this plasmid effectively drove INO1 expression is amply demonstrated by its ability to rescue growth of the ino1-13 mutant (CTY) on inositol-free medium. (B) Wild-type (CTY182) and Δsac1 (CTY244) strains were grown in YPD medium to midlogarithmic growth phase, harvested, and washed. The cultures were split and either shifted to minimal medium containing both inositol and choline (+Ino) or to inositol- and choline-free medium (−Ino). After 4 h of incubation at 30°C with shaking, cells were harvested, and total RNA was recovered and subjected to Northern analysis using specific probes for INO1 and OPI3 mRNA that were generated by random priming. Each lane was loaded with 10 μg of RNA. The results clearly demonstrate that both INO1 and OPI3 expression was induced in cells shifted to inositol- and choline-free medium. To confirm proper normalization of RNA load, these same RNA samples were also probed for messages (e.g., PSS1) that are present under both the Ino− and Ino+ conditions. As expected, PSS1 mRNA was detected in all lanes (our unpublished results).
Second, we used Northern analyses to assess the efficiency of induction of INO1 transcription upon shift of SAC1 and Δsac1 yeast strains from inositol-containing medium to inositol-free medium. INO1 mRNA was essentially undetectable in either wild-type or Δsac1 cells shifted from YPD medium to minimal medium supplemented with inositol and choline (Figure 5B). Yet, both SAC1 and Δsac1 strains induced robust transcription of INO1 by 4 h after shift from YPD to inositol- and choline-free minimal medium. Similarly, OPI3 transcription, which is regulated in the same manner as is that of INO1 (Carman and Henry, 1989), was also induced to high levels upon shift of Δsac1 strains from YPD to inositol-free minimal medium (Figure 5B). These data demonstrated that the Ino− phenotype of sac1 strains is not a simple consequence of defects in INO1 expression.
DGK Expression and CDP-Choline Pathway Defects Suppress Inositol Auxotrophy
We noted that DGK expression strongly influenced the inositol requirement of sac1 strains. Normally, the Ino− trait of sac1 strains is a tight phenotype because these mutants fail to produce colonies when streaked for isolation on inositol-free media (Figure 6A), even when such cultures are seeded with a heavy inoculum and incubated for many days. Expression of bacterial DGK in these strains suppressed the inositol auxotrophy of even Δsac1 mutants to the extent that individual colonies could readily be observed within 72 h of incubation of cells on inositol-free agar plates (Figure 6A). This effect was specific in the sense that DGK expression influences only one other sac1 phenotype; i.e., it abolishes sac1-mediated bypass Sec14p (Kearns et al., 1997). DGK expression did not diminish the cold sensitivity for growth (cs) phenotype characteristic of sac1 strains (our unpublished results).
Figure 6.

DGK expression suppresses the inositol auxotrophy associated with Δsac1 strains. (A) The indicated yeast strains were streaked for isolation on solid uracil-deficient minimal medium that either contained inositol (+Inositol) or was inositol free (−Inositol). Plates were incubated at 26°C for 72 h. Wild-type and Δsac1 strains were CTY182 and CTY244, respectively. Episomal DGK expression plasmids and cognate vector-only controls are designated YEpDGK and YEpURA3, respectively. (B) Mutations that block PtdCho biosynthesis by the CDP-choline pathway suppress the inositol auxotrophy of Δsac1 strains. Yeast strains with the indicated genotypes were streaked for isolation on solid minimal defined medium that was either supplemented with inositol (+Inositol) or was inositol-free (−Inositol). Plates were incubated at 26°C for 72 h and scored for growth. Wild-type and Δsac1 strains were CTY182 and CTY244, respectively. The Δcki1 and Δopi3 derivatives were generated by transformation of yeast cells with the appropriate disruption constructs and confimed by genomic Southern analysis. In these experiments, heavy inocula of the Δsac1 and Δsac1, Δopi3 strains were streaked onto the −Inositol plates to illustrate the tightness of the inositol auxotrophy.
The DGK expression data suggested the possibility that the Ino− phenotype of sac1 mutants may result from accelerated CDP-choline pathway activity. A prediction of this model was that genetic inactivation of this specific PtdCho biosynthetic pathway will also restore an Ino+ phenotype to Δsac1 strains. To test this prediction, mutations that disrupt structural genes encoding CDP-choline pathway enzymes were introduced into Δsac1 mutants, and the abilities of the resultant double mutants to grow on inositol-free media were assessed. The data show that introduction of the cki1-284::HIS3 mutation (a disruption of the structural gene encoding choline kinase, the first enzyme of the CDP-choline pathway; see Figure 1), fully restored the ability of Δsac1 mutants to grow in inositol-free medium (Figure 6B). Phenotypic suppression of the sac1-associated inositol auxotrophy by the cki1-284::HIS3 allele was recapitulated when pct1::URA3 disruption alleles (which inactivate the structural gene encoding the second enzyme of the CDP-choline pathway; Figure 1) were introduced into Δsac1 strains (our unpublished results). Significantly, an opi3::URA3 allele (which inactivates the PtdEtn pathway for PtdCho biosynthesis; Figure 1) failed to suppress the Δsac1-associated Ino− phenotype (Figure 6B). The potency with which CDP-choline pathway defects suppress theΔsac1-associated Ino− phenotype was evident in shift experiments in which Δsac1, cki1-284::HIS3 double mutants displayed wild-type growth rates upon shift from inositol-replete to inositol-free liquid media (Figure 7). In the course of these experiments, we observed a distinction between the behavior of sac1 and ino1 mutants under conditions of inositol deprivation. Both ino1-13 and Δsac1 strains retained viability up to 4 h after shift to inositol-free medium. After that time, however, the ino1-13 strain experienced a rapid loss of cell viability that resulted in a 1000-fold decrease in viable cell number by 24 h after shift (Figure 7). This is characteristic of yeast mutants unable to synthesize inositol de novo (Culbertson et al., 1976a,b; Carman and Henry, 1989). By comparison, Δsac1 mutants mutants suffered a more modest 10-fold reduction in viable cell number after 24 h of inositol starvation.
Figure 7.

Effects of inositol starvation on cell viability. Wild-type (CTY182; closed circles), Δsac1 (CTY244; closed squares), Δsac1, Δcki1 (CTY; stippled cicles), and ino1-13 (CTY; open squares) strains were grown in minimal defined medium supplemented with inositol, harvested, washed, and shifted to inositol-free medium at time zero. Samples of each culture were subsequently removed at the indicated times after shift and serially diluted in YPD medium, and the serial dilutions were immediately plated onto YPD agar plates. Individual colonies were scored after 3 d of growth at 26°C to assess viable cell counts. Data are from a representative experiment.
DISCUSSION
A dissection of Sec14p function in yeast has been driven by the analysis of suppressor mutations that endow cells with the ability to execute Golgi function, and retain viability, in the absence of Sec14p (Cleves et al., 1991a; Kearns et al., 1998). The logic on which suppressor genetics is founded dictates that such bypass Sec14p mutations exert their effects by restoring a biochemical condition that normally falls under the purview of Sec14p function. From these analyses, we have proposed that Sec14p functions to maintain a Golgi DAG pool that is critical for Golgi secretory function (McGee et al., 1994; Kearns et al., 1997, 1998). Specifically, we proposed that PtdCho-bound Sec14p down-regulates DAG consumption via the CDP-choline pathway (Skinner et al., 1995; Alb et al., 1996; Kearns et al., 1998), whereas PtdIns-bound Sec14p promotes DAG generation by regulating inositol phospholipid metabolism (Fang et al., 1998; Kearns et al., 1997, 1998). In this manner, Sec14p serves as a phospholipid sensor whose phospholipid-bound states independently, but convergently, function to maintain Golgi DAG (Figure 8).
Figure 8.

Bypass Sec14p mechanisms. The relevant lipid metabolic pathways and proteins that determine the activity of these pathways are shown. The key lipids (i.e., PtdIns-4-P, PtdCho, and DAG) are indicated in bold. Key proteins and their corresponding sites of action are indicated in the black boxes. We propose two principal mechanisms for bypass Sec14p. First, we suggest that Kes1p, SacIp, and Bsd1p are involved in a PLD regulatory pathway that, in yeast mutants carrying the corresponding bypass Sec14p alleles, results in PLD activation and subsequent production of DAG. This particular pathway recapitulates the physiological function for the PtdIns-bound form of Sec14p (Sec14p∼PI) that we have proposed effects DAG production via regulation of inositol phospholipid metabolism (Kearns et al., 1997). Loss of SacIp function results in PtdIns-4-P accumulation that, in turn, either activates Bsd1p (identified by dominant bypass Sec14p alleles; Cleves et al., 1991b; Fang et al., 1996) or inactivates Kes1p (identified by recessive bypass Sec14p alleles; Fang et al., 1996). The consequence is activation of PLD, which results in DAG production through the metabolism of the PtdOH generated by PLD-mediated hydrolysis of PtdCho. We propose that the excess DAG generated via this route is consumed by the CDP-choline pathway for PtdCho biosynthesis, thereby resulting in the observed activation of this pathway biosynthetic pathway in sac1 mutants. Second, we suggest that dysfunction of any one of the enzymes of the CDP-choline pathway results in the observed recessive bypass Sec14p phenotypes (Cleves et al., 1991b), because inactivation of the CDP-choline pathway recapitulates the function of PtdCho-bound Sec14p (Sec14p∼PC) (Skinner et al., 1995). Although bypass Sec14p via this route also requires PLD activity, there is nonetheless a measurable PLD-independent component to suppression of Sec14p defects by CDP-choline pathway defects.
One of several important lines of evidence supporting the DAG model was our observation that the ability of sac1 strains to effect bypass Sec14p correlated with what we interpreted as overproduction of an inositol phospholipid that we identified as the most highly modified yeast sphingolipid, M(IP)2C, a lipid whose synthesis produces Golgi DAG (Kearns et al., 1997). In those experiments, the failure of Kearns et al. (1997) to use deacylation as an initial means for fractionating the deacylatable glycerophospholipids from sphingolipids contributed significantly to the misidentification. More rigorous analyses now indicate that the major inositol phospholipid that accumulates in sac1 strains is PtdIns-4-P (Figure 2). A biochemical basis for elevations in PtdIns-4-P is suggested by the demonstration that the Sac1p domains of other inositide phosphatases themselves represent novel phosphoinositide phosphatase modules (Guo et al., 1999). Thus, sac1 strains likely accumulate PtdIns-4-P because the primary mechanism for its degradation to PtdIns is inactivated.
Based on the collective data reported herein, we revise our interpretation of the mechanism for bypass Sec14p in sac1 strains to take into account the various new data (Figure 8). We maintain that increased DAG production represents the key physiological event that allows Sec14p-independent growth and secretion in these strains, as previously proposed (Kearns et al., 1997). The evidence now suggests that the pathway for this DAG production in sac1 mutants involves PLD activity. We also report the unanticipated discovery that the physiological basis for sac1-associated inositol auxotrophy is related to aberrant lipid metabolism in these strains and not to defects in transcriptional induction of INO1. The evidence that speaks to these various points is as follows.
First, we demonstrate that a biochemical signature of sac1 mutants is a dramatic acceleration in the rate of metabolic flux through the CDP-choline pathway for PtdCho biosynthesis (Figure 3). This effect is observed for all sac1 alleles, including Δsac1, and it is observed in sac1-22 strains only when these strains are grown in the presence of inositol. The significance of the latter point is that sac1-22 strains, although exceptional from the standpoint that these are not inositol auxotrophs, are only able to exhibit bypass Sec14p phenotypes when grown in inositol-containing medium (Kearns et al., 1997). The signature alterations in inositol phospholipid metabolism characteristic of sac1 strains are also recorded in sac1-22 strains but, again, only when these mutants are provided with inositol in the growth medium (Kearns et al., 1997; see above).
Second, our demonstration that accelerated rates of CDP-choline pathway activity correlate with bypass Sec14p in sac1 mutants is consistent with the DAG production model shown in Figure 8. Because sac1 strains exhibited wild-type levels of bulk DAG at steady state, an expected consequence of excess DAG production in sac1 strains would be a compensatory increase in the activity of a DAG degrading–consuming pathway. We conclude that the CDP-choline pathway represents a major metabolic sink for excess DAG in sac1 strains. Our finding that CDP-choline pathway hyperactivity in sac1 strains is sensitive to the metabolic conversion of DAG to PtdOH effected by DGK expression also supports this concept (Figure 3C). That this excess DAG is ultimately produced from PtdOH, a phospholipid that is a direct product of PLD action, is suggested by the demonstration that PLD inactivation (even when exogenous choline is supplied in vast excess) reduces CDP-choline pathway activity in sac1 strains to essentially wild-type levels (Figure 3A).
The stimulation of the CDP-choline pathway recorded for sac1 strains presents an intriguing paradox. The essence of the paradox is that, although hyperactivated CDP-choline pathway activity correlates with Sac1p-mediated bypass Sec14p, genetic inactivation of the CDP-choline pathway constitutes a recognized mechanism for bypass Sec14p (Cleves et al., 1991b). The DAG model illustrated in Figure 8 reconciles this contradiction in a simple manner. It posits that, although increased DAG production effects bypass Sec14p, the resultant consumption of DAG manifests itself in elevated CDP-choline pathway activity. The stimulation of the CDP-choline pathway in sac1 mutants raises the possibility that DAG availability helps set the baseline rate of metabolic flux through this pathway in yeast. Such a DAG effect could potentially be mediated by DAG stimulating the activity of CCTase, the rate-determining enzyme of the CDP-choline pathway.
Third, our results lend insight into the role of PtdIns-4-P accumulation in the sac1-mediated mechanism for bypass Sec14p. Because PLD-insufficient sac1 strains still accumulate high levels of this phosphoinositide (Figure 4, A and B), yet are incompetent for bypass Sec14p (Xie et al., 1998), we conclude that increased PtdIns-4-P is at best a contributing factor to bypass Sec14p. Indeed, the finding that PLD deficiency abolishes the bypass Sec14p phenotype of sac1 strains in the face of PtdIns-4-P accumulation suggests that excess PtdIns-4-P may contribute to bypass Sec14p by effecting a downstream activation of PLD (Figure 8). For example, PtdIns-4-P could modulate PLD activity indirectly by influencing the action of another protein whose function is to regulate PLD. The yeast oxysterol-binding protein homologue Kes1p and the BSD1 gene product represent candidate PLD regulators (Figure 8), because the bypass Sec14p growth phenotypes of kes1 and BSD1-124 mutants are also completely abolished by PLD insufficiency (Figure 4C). Kes1p is a particularly attractive candidate because it binds PtdIns-4-P, and Kes1p overproduction phenocopies PLD inactivation (our unpublished data). Interestingly, CDP-choline pathway mutations exhibit a significant PLD-independent component of their ability to suppress sec14 defects (Figure 4C). We attribute this PLD-independent component to reflect reduced rates of DAG consumption (Figure 8).
Fourth, we report insights into the mechanism that underlies the inositol auxotrophy of sac1 strains. We demonstrate that CDP-choline pathway activity contributes to this inositol requirement. Intereference with the activity of this PtdCho biosynthetic pathway at any one of several points restores the ability of Δsac1 strains to grow in the absence of exogenous inositol (Figure 6B) at wild-type rates (Figure 7). Because sac1-associated inositol auxotrophy does not result in obvious defects in transcriptional regulation of INO1 (Figure 5, A and B), nor is it accompanied by the classical “inositol-less death” of ino1 mutants (Figure 7), we conclude that Sac1p deficiency results in an inability of cells to thrive on endogenously produced inositol. In that regard, the accumulation of PtdIns-4-P in sac1 strains indicates disruption of a substantial metabolic flux from PtdIns-4-P to PtdIns. This block in PtdIns production may contribute to the unusual inositol auxotrophy of sac1 strains in a manner that does not solely operate through CDP-choline pathway hyperactivation.
Finally, we emphasize the pleiotropic nature of the phospholipid metabolic alterations that accompany Sac1p dysfunction. Some of these are involved in mediating bypass Sec14p (i.e., accumulation of PtdIns-4-P and subsequent increases in PLD-mediated DAG production). We propose others to represent indirect correlates of the bypass Sec14p condition (e.g., accelerated CDP-choline pathway activity and reduced PtdSer levels). A continuing challenge in these analyses is the recognition of which alterations in phospholipid metabolism in bypass Sec14p mutants most directly mediate Sec14p-independent growth and Golgi secretory function in yeast.
ACKNOWLEDGMENTS
These studies were supported by National Institutes of Health grant GM-44530 to V.A.B. M.P.R. and B.G.K. were each supported by a National Science Foundation predoctoral neurobiology training grant (NSF-BIR-9256853). S.G. and J.D.Y. were supported by a Burroughs Wellcome Fund Career Award in the Biomedical Sciences and by National Institutes of Health grant R01-HL-55672 to J.D.Y. The impetus for reassessing M(IP)2C levels in sac1 mutants was stimulated by experiments performed by the laboratory of Jon Takemoto (Utah State University, Logan, UT). We are grateful to Takemoto and colleagues for helpful discussions and the ipt1 disruption construct.
REFERENCES
- Aitken JF, van Heusden GPH, Temkin M, Dowhan W. The gene encoding the phosphatidylinositol transfer protein is essential for cell growth. J Biol Chem. 1990;265:4711–4717. [PubMed] [Google Scholar]
- Alb JG, Jr, Kearns MA, Bankaitis VA. Phospholipid metabolism and membrane dynamics. Curr Opin Cell Biol. 1996;8:534–541. doi: 10.1016/s0955-0674(96)80032-9. [DOI] [PubMed] [Google Scholar]
- Atkinson KD. Saccharomyces cerevisiae recessive suppressor that circumvents phosphatidylserine deficiency. Genetics. 1984;108:533–543. doi: 10.1093/genetics/108.3.533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bankaitis VA, Aitken JR, Cleves AE, Dowhan W. An essential role for a phospholipid transfer protein in yeast Golgi function. Nature. 1990;347:561–562. doi: 10.1038/347561a0. [DOI] [PubMed] [Google Scholar]
- Bankaitis VA, Malehorn DE, Emr SD, Greene R. The Saccharomyces cerevisiae SEC14 gene encodes a cytosolic factor that is required for transport of secretory proteins from the yeast Golgi complex. J Cell Biol. 1989;108:1271–1281. doi: 10.1083/jcb.108.4.1271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buttke TM, Pyle AL. Effects of unsaturated fatty acid deprivation on neutral lipid synthesis in Saccharomyces cerevisiae. J Bacteriol. 1982;152:747–756. doi: 10.1128/jb.152.2.747-756.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carman GA, Henry SA. Phospholipid biosynthesis in yeast. Annu Rev Biochem. 1989;58:635–667. doi: 10.1146/annurev.bi.58.070189.003223. [DOI] [PubMed] [Google Scholar]
- Cleves AE, McGee TP, Bankaitis VA. Phospholipid transfer proteins: a biological debut. Trends Cell Biol. 1991a;1:30–34. doi: 10.1016/0962-8924(91)90067-j. [DOI] [PubMed] [Google Scholar]
- Cleves AE, McGee TP, Whitters EA, Champion KM, Aitken JR, Dowhan W, Goebl M, Bankaitis VA. Mutations in the CDP-choline pathway for phospholipid biosynthesis bypass the requirement for an essential phospholipid transfer protein. Cell. 1991b;64:789–800. doi: 10.1016/0092-8674(91)90508-v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cleves AE, Novick PJ, Bankaitis VA. Mutations in the SAC1 gene suppress defects in yeast Golgi and yeast actin function. J Cell Biol. 1989;109:2939–2950. doi: 10.1083/jcb.109.6.2939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Culbertson M, Donahue TF, Henry S. Control of inositol biosynthesis in Saccharomyces cerevisiae: properties of a repressible enzyme system in extracts of wild-type (Ino+) cells. J Bacteriol. 1976a;126:232–242. doi: 10.1128/jb.126.1.232-242.1976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Culbertson M, Donahue TF, Henry S. Selection of spontaneous mutants by inositol starvation in yeast. J Bacteriol. 1976b;126:243–250. doi: 10.1007/BF00269415. [DOI] [PubMed] [Google Scholar]
- Cutler NS, Heitman J, Cardenas ME. STT4 is an essential phosphatidylinositol 4-kinase that is a target of wortmannin in Saccharomyces cerevisiae. J Biol Chem. 1997;272:27671–27677. doi: 10.1074/jbc.272.44.27671. [DOI] [PubMed] [Google Scholar]
- Fang M, Kearns BG, Gedvilaite A, Kagiwada S, Kearns M, Fung MKY, Bankaitis VA. Kes1p shares homology with human oxysterol binding ptrotein and participates in a novel regulatory pathway for yeast Golgi-derived transport vesicle biogenesis. EMBO J. 1996;15:6447–6459. [PMC free article] [PubMed] [Google Scholar]
- Fang M, Rivas MP, Bankaitis VA. The contribution of lipids and lipid metabolism to cellular functions of the Golgi complex. Biochim Biophys Acta. 1998;1404:85–100. doi: 10.1016/s0167-4889(98)00049-4. [DOI] [PubMed] [Google Scholar]
- Franzusoff A, Schekman R. Functional compartments of the Golgi complex are defined by the sec7 mutation. EMBO J. 1989;8:2695–2702. doi: 10.1002/j.1460-2075.1989.tb08410.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gary JD, Wurmser AE, Bonangelino CJ, Weisman LS, Emr SD. Fab1p is essential for PtdIns(3)P 5-kinase activity and the maintenance of vacuolar size and membrane homeostasis. J Cell Biol. 1998;143:65–79. doi: 10.1083/jcb.143.1.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo S, Stolz LE, Lemrow S, York JD. SAC1-like domains of yeast SAC1, INP52 and INP53, and human synaptojanin encode polyphosphoinositide phosphatases. J Biol Chem. 1999;274:12990–12995. doi: 10.1074/jbc.274.19.12990. [DOI] [PubMed] [Google Scholar]
- Hosaka K, Nikawa J, Kodaki T, Yamashita S. A dominant mutation that alters the regulation of INO1 expression in Saccharomyces cerevisiae. J Biochem. 1992;116:1317–1321. doi: 10.1093/oxfordjournals.jbchem.a123761. [DOI] [PubMed] [Google Scholar]
- Ito H, Fukuda Y, Murata K, Kimura A. Transformation of intact yeast cells treated with alkali cations. J Bacteriol. 1983;153:163–168. doi: 10.1128/jb.153.1.163-168.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kagiwada S, Kearns BG, McGee TP, Fang M, Hosaka K, Bankaitis VA. The yeast BSD2–1 mutation influences both the requirement for phosphatidylinositol transfer protein function and derepression of phospholipid biosynthetic gene expression in yeast. Genetics. 1996;143:685–697. doi: 10.1093/genetics/143.2.685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kearns BG, Alb JG, Jr, Bankaitis VA. Phosphatidylinositol transfer proteins: the long and winding road to physiological function. Trends Cell Biol. 1998;8:276–282. doi: 10.1016/s0962-8924(98)01281-1. [DOI] [PubMed] [Google Scholar]
- Kearns BG, McGee TP, Mayinger P, Gedvilaite A, Phillips SE, Kagiwada S, Bankaitis VA. An essential role for diacylglycerol in protein transport from the yeast Golgi complex. Nature. 1997;387:101–105. doi: 10.1038/387101a0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kochendörfer K-U, Then A, Kearns BG, Bankaitis VA, Mayinger P. The role for SacIp in microsomal ATP transport is distinct from its function in inositol phospholipid metabolism. EMBO J. 1999;18:1506–1515. doi: 10.1093/emboj/18.6.1506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lips DL, Majerus PW, Gorga FR, Young AT, Benjamin TL. Phosphatidylinositol 3-phosphate is present in normal and transformed fibroblasts and is resistant to hydrolysis by bovine brain phospholipase C II. J Biol Chem. 1989;264:8759–8763. [PubMed] [Google Scholar]
- Mayinger P, Bankaitis VA, Meyer DI. SacIp mediates the adenosine triphosphate transport into yeast endoplasmic reticulum that is required for protein translocation. J Cell Biol. 1996;131:1377–1386. doi: 10.1083/jcb.131.6.1377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGee TP, Skinner HB, Whitters EA, Henry SA, Bankaitis VA. A phosphatidylinositol transfer protein controls the phosphatidylcholine content of yeast Golgi membranes. J Cell Biol. 1994;124:273–287. doi: 10.1083/jcb.124.3.273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novick P, Field C, Schekman R. Identification of 23 complementation groups required for posttranslational events in the yeast secretory pathway. Cell. 1980;21:205–215. doi: 10.1016/0092-8674(80)90128-2. [DOI] [PubMed] [Google Scholar]
- Novick P, Osmond BC, Botstein DD. Suppressors of yeast actin mutations. Genetics. 1989;121:659–674. doi: 10.1093/genetics/121.4.659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothstein RJ. One step gene disruption in yeast. Methods Enzymol. 1983;101:202–211. doi: 10.1016/0076-6879(83)01015-0. [DOI] [PubMed] [Google Scholar]
- Salama SR, Cleves AE, Malehorn DE, Whitters EA, Bankaitis VA. Cloning and characterization of Kluyveromyces lactis SEC14, a gene whose product stimulates Golgi secretory function in Saccharomyces cerevisiae. J Bacteriol. 1990;172:4510–4521. doi: 10.1128/jb.172.8.4510-4521.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherman F, Fink GR, Hicks JB. Methods in Yeast Genetics. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory; 1983. pp. 1–113. [Google Scholar]
- Skinner HB, McGee TP, McMaster CR, Fry MR, Bell RM, Bankaitis VA. Phosphatidylinositol transfer protein stimulates yeast Golgi function by inhibiting choline-phosphate cytidylyltransferase activity. Proc Natl Acad Sci USA. 1995;92:112–116. doi: 10.1073/pnas.92.1.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sreenivas A, Patton-Vogt JL, Bruno V, Griac P, Henry SA. A role for phospholipase D in growth, secretion, and regulation of membrane lipid synthesis in yeast. J Biol Chem. 1998;273:16635–16638. doi: 10.1074/jbc.273.27.16635. [DOI] [PubMed] [Google Scholar]
- Steiner MR, Lester RL. In vitro studies of phospholipid biosynthesis in Saccharomyces cerevisiae. Biochim Biophys Acta. 1972;260:222–243. doi: 10.1016/0005-2760(72)90035-5. [DOI] [PubMed] [Google Scholar]
- Stock SD, Hama H, DeWald DB, Takemoto JY. SEC14-dependent secretion in Saccharomyces cerevisiae: nondependence on sphingolipid-coupled diacyglycerol synthesis. J Biol Chem. 1999;274:12979–12983. doi: 10.1074/jbc.274.19.12979. [DOI] [PubMed] [Google Scholar]
- Stolz LE, Kuo WJ, Longchamps J, Sekhon MK, York JD. Inp51, a yeast inositol polyphosphate 5-phosphatase required for phosphatidylinositol 4,5-bisphosphate homeostasis and whose absence confers a cold-resistant phenotype. J Biol Chem. 1998;273:11852–11861. doi: 10.1074/jbc.273.19.11852. [DOI] [PubMed] [Google Scholar]
- Whitters EA, Cleves AE, McGee TP, Skinner HB, Bankaitis VA. SAC1p is an integral membrane protein that influences the cellular requirement for phospholipid transfer protein function and inositol in yeast. J Cell Biol. 1993;122:79–94. doi: 10.1083/jcb.122.1.79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie Z, Fang M, Rivas MP, Faulkner A, Sternweis PC, Engebrecht J, Bankaitis VA. Phospholipase D activity is required for suppression of yeast phosphatidylinositol transfer protein defects. Proc Natl Acad Sci USA. 1998;95:12346–12351. doi: 10.1073/pnas.95.21.12346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- York JD, Chen Z, Ponder JW, Chauhan AK, Mathews FS, Majerus PW. Crystallization and initial crystallographic characterization of recombinant inositol polyphosphate 1-phosphatase produced in Spodoptera frugiperda cells. J Mol Biol. 1994;236:584–589. doi: 10.1006/jmbi.1994.1167. [DOI] [PubMed] [Google Scholar]
- York JD, Majerus PW. Nuclear phosphatidylinositols decrease during S-phase of the cell cycle in HeLa cells. J Biol Chem. 1994;269:7847–7850. [PubMed] [Google Scholar]