

Abstract
Objective
The capacity of neutrophils to eradicate bacterial infections is dependent on normal development and the activation of functional responses, which include chemotaxis and the generation of oxygen radicals during the respiratory burst. A unique feature of the neutrophil is its highly lobulated nucleus, which is thought to facilitate chemotaxis but may also play a role in other critical neutrophil functions. Nuclear lobulation is dependent on the expression of the inner nuclear envelope protein, the lamin B receptor (LBR), mutations of which cause hypolobulated neutrophil nuclei in human Pelger-Huët anomaly (PHA) and the "ichthyosis" (ic) phenotype in mice. In this study we have investigated roles for LBR in mediating neutrophil development and the activation of multiple neutrophil functions, including chemotaxis and the respiratory burst.
Materials and Methods
A progenitor EML cell line was generated from an ic/ic mouse, and derived cells that lacked LBR expression were induced to mature neutrophils and then examined for abnormal morphology and functional responses.
Results
Neutrophils derived from EML-ic/ic cells exhibited nuclear hypolobulation identical to that observed in ichthyosis mice. The ic/ic neutrophils also displayed abnormal chemotaxis, supporting the notion that nuclear segmentation augments neutrophil extravasation. Furthermore, promyelocytic forms of ic/ic cells displayed decreased proliferative responses and produced a deficient respiratory burst upon terminal maturation.
Conclusions
Our studies of promyelocytes that lack LBR expression have identified roles for LBR in regulating not only the morphologic maturation of the neutrophil nucleus but also proliferative and functional responses that are critical to innate immunity.
Key words: Lamin B receptor, neutrophil, chemotaxis, respiratory burst, proliferation
Introduction
The capacity of the innate immune response to eradicate bacterial infections is dependent in part on the ability of neutrophils to migrate toward infected tissues and to rapidly escape the capillary network during chemotaxis. Neutrophil chemotaxis is mediated by the activation of G-protein coupled receptors, which initiate intracellular signaling cascades that result in directional F-actin polymerization (reviewed in [1,2]). While this process provides efficient neutrophil migration, neutrophils in circulation are presented with the formidable task of navigating through capillary endothelium and within infected tissues. A prominent morphologic feature of the developing neutrophil is its unique nucleus: as the neutrophil matures, chromatin condenses into heterochromatin, some of which forms thin chromatin bridges that connect separate nuclear lobes [3,4]. The lobulated nucleus is believed to provide increased fluidity to the neutrophil that may facilitate chemotaxis through the tight confines of interendothelial and interstitial spaces [4]. If this view is correct, then loss of nuclear lobulation would be expected to adversely affect neutrophil mobility and function. Support for this notion has been provided by functional studies of neutrophils with Pelger-Huët anomaly (PHA), a disorder that causes hyposegmentation of the neutrophil nucleus [5–7]. However, some reports have suggested that neutrophils with PHA function normally, and PHA in humans has classically been viewed as a benign disorder with no apparent effects on innate immune responses [8–10]. All of these studies were performed with neutrophils heterozygous for the PHA mutation, which is not surprising since homozygous forms of PHA are most commonly fatal due to associated skeletal abnormalities [11–14]. Interestingly, recent studies have revealed that mutations in the lamin B receptor (LBR) gene cause PHA, and that critical functions of LBR in mediating nuclear structural integrity and/or cholesterol metabolism may contribute to the lethal phenotype of homozygous PHA [15,16].
LBR is an inner nuclear envelope protein that binds B-type lamins and heterochromatin, and these associations are thought to help orchestrate the dynamic structural changes that occur to the nucleus during every cell cycle [17]. Evidence that LBR may play a role in neutrophil nuclear segmentation was first demonstrated in human myeloblastic HL-60 cells, in which LBR expression is increased during neutrophil differentiation, while the expression of certain lamins is downregulated [18,19]. More recent studies have demonstrated that familial PHA in humans is caused by heterozygous mutations of LBR, and that the ichthyosis (ic) mutation in mice is caused by homozygous Lbr gene mutations that completely abrogate LBR protein expression in spleen and lymphatic tissues [15,20]. Importantly, peripheral blood neutrophils from ic/ic mice displayed either bilobed nuclei typical of PHA or ovoid nuclei with reorganized heterochromatin. These nuclear abnormalities were not exclusive to the neutrophil lineage; lymphocytes in the blood and lymphatic organs showed clumping of chromatin at nuclear edges and eosinophils contained band nuclei. All of the ic/ic homozyogotes exhibited sparse hair and decreased body size. Homozygotes occasionally displayed hydrocephalus and syndactyly affecting one or more paws [20]. Homozygous mutations of LBR were also identified in a spontaneously aborted fetus with Greenberg/HEM dysplasia; peripheral blood neutrophils from the fetus’ mother displayed PHA [16]. However, possible effects of deficient LBR expression on chemotaxis or other neutrophil functions were not analyzed in any of these studies.
In an effort to study the function of neutrophils that lack LBR expression, we have generated a progenitor EML (for Erythroid, Myeloid and Lymphoid potential) cell line from bone marrow of a homozygous ichthyosis mutant mouse (C57BL/6J-Lbric-J/Lbric-J, abbreviated as ic/ic). As a control, EML cells were also generated from a normal littermate that was later identified to be heterozygous for ic. The homozygous EML-ic/ic cells contain the genetic lesion at the Lbr locus identified in the C57BL/6J-Lbric-J/Lbric-J mouse strain [20], and display significantly reduced Lbr mRNA as compared to control cells. Importantly, differentiation of EML-derived PROmyelocyte (EPRO)-ic/ic cells gave rise to neutrophils with severe nuclear hypolobulation identical to that identified in neutrophils from ic/ic mice. We have previously shown that differentiated, wild-type EPRO cells exhibit the full spectrum of normal neutrophil functional responses, including efficient chemotaxis [21]. By comparison, the hypolobulated ic/ic neutrophils exhibited deficient migration through membranes with 3.0 µm pores in response to two chemoattractants, suggesting that lobulation of the nucleus facilitates neutrophil chemotaxis. The mutant neutrophils also generated a defective oxidative burst but showed essentially normal phagocytosis. We demonstrate that the defective respiratory burst is likely due in part to a loss of gp91phox expression, suggesting that deficient LBR expression may also affect transcriptional regulation of this critical oxidase. In the article following this study, the Lbr mutation in ic/ic cells is shown to affect the expression of multiple proteins integral to the neutrophil nucleus (including LBR), and also alter the distribution of heterochromatin markers [22]. Together, our data provide novel evidence that LBR expression is essential not only for neutrophil nuclear lobulation but also for normal neutrophil functional responses and neutrophil-specific gene expression.
Materials and Methods
Derivation of EML/EPRO-+/ic and −ic/ic cell lines
EML-like cells were generated using whole bone marrow harvested from a 40 day old female C57BL/6J-Lbric-J/Lbric-J (ic/ic) mouse and a normal littermate essentially as described [23]. To express a dominant negative form of retinoic acid receptor α (RARα), cells were spinfected for 2 hours with supernatants derived from a stable GP+E86 producer line infected with the LRARα403SN vector (a kind gift of Archibald Perkins, Yale University, New Haven) plus polybrene (4 µg/ml; Sigma, St. Louis , MO) [24]. Following a two week exposure to G418 (0.4 mg/mL, Invitrogen, Carlsbad, CA), EML-like cells emerged after approximately 1 month; thereafter, cells were routinely maintained in Iscove’s Modified Dulbecco Medium (IMDM, Invitrogen) supplemented with 20% horse serum (HS) and 15% BHK/MKL-conditioned medium (as a source of SCF). EPRO-+/ic and −ic/ic cells were generated by inducing EML cells with all-trans retinoic acid (ATRA, 10 µM, Sigma) plus Interleukin-3 (25 ng/mL, Peprotech, Rocky Hill, NJ) for 3 days, washing cells in PBS and then selecting for promyelocytes in IMDM supplemented with 20% HS plus GM-CSF (10 ng/mL, Peprotech). For terminal differentiation, EPRO cells were induced with ATRA for 3–5 days. All cells were maintained at 37°C in 5% CO2, and growth medium was supplemented with 5 U/mL penicillin, 5 µg/mL streptomycin sulfate and 0.25 µg/mL amphotericin B.
Amplification of the Lbr gene locus
To isolate genomic DNA, cells (1 × 107) were washed in 1X PBS, resuspended in RSB (10 mM Tris pH 7.5, 100 mM NaCl, 10 mM EDTA, 0.5% SDS) supplemented with Proteinase K (200 µg/mL) and then incubated overnight at 55°C. The mixture was phenol/chloroform extracted, DNA was precipitated using standard procedures, and then PCR was performed using previously reported primers and conditions in a Px2 Thermal Cycler (Thermo Electron Corp., Milford, MA) [20]. The resulting products were digested with BglI and then electrophoresed on a 2% agarose gel stained with ethidium bromide (EtBr). The bands were visualized and digitally photographed with a Genius BioImaging System (Syngene, Frederick, MD) using a UV-light transilluminator.
Northern analyses and reverse-transcriptase polymerase chain reactions
Total RNA was isolated from cell lines with TRIzol reagent (Life Technologies, Rockville, MD) according to the manufacturer’s specifications. For northern assays, RNA was electrophoresed, blotted and probed as described previously [21]. For RT-PCR, cDNA from each genotype was synthesized using total RNA and SuperScript II (Invitrogen) according to the manufacturer’s instructions. Primers for gp91phox and β-actin were previously described [21]. PCR reactions were performed in parallel for each genotype using standard conditions for 21, 24 and 27 cycles (gp91phox), or for 18, 21 and 27 cycles (β-actin). Reaction products were electrophoresed in a 0.8% agarose gel that contained EtBr and then digitally photographed using the Genius BioImaging System.
Electron micrographs
Cells were washed in PBS containing Complete (Roche) and PMSF (Sigma-Aldrich) protease inhibitors. Pellets of cells were fixed in freshly prepared 2.0% glutaraldehyde plus 2.0% formaldehyde in 0.1 M sodium cacodylate buffer (pH 7.2) for 1 hour at room temperature, washed 3 times in cacodylate buffer, fixed in 1% OsO4 for 1 hour at room temperature and washed 3 times in cacodylate buffer. The pellets were embedded in 2% low melting agarose, allowed to solidify, cut into ~0.5mm cubes, washed in cacodylate buffer, dehydrated in an ethanol series, in propylene oxide, and then in propylene oxide/epon mixtures (vol/vol) as follows: 20/80, 15 minutes; 50/50, 1 hour and 80/20 on a rotator overnight. Propylene oxide was allowed to evaporate and the cell cubes were transferred to 100% epon. The cubes were placed in capsules filled with epon and baked at 60°C for 48 hours. Polymerized blocks were thin-sectioned, stained with uranyl acetate and lead citrate, and visualized in a Zeiss 10CA electron microscope equipped with an AMT CCD camera.
Growth assays
Proliferation assays were performed by diluting cells to 5 × 105 cells in 5 mL of medium, and then quantifying viable cell numbers using trypan blue exclusion and visual inspection with a hemacytometer. For each data point, four fields were counted with a minimum of two separate cell counts, and cells were analyzed in triplicate wells. Cytokine concentration-dependent proliferative responses were assessed using the CellTiter 96 Aqueous Non-Radioactive Cell Proliferation Assay (Promega, Madison, WI). Cells (50µL at 3 × 105 cells/mL) were dispensed into wells of a 96-well plate that contained 50 µL of medium with decreasing concentrations of the appropriate cytokine (starting concentrations of 200 ng/mL and 22 ng/mL of SCF and GM-CSF, respectively). After 2 days of growth, reagents were added according to the manufacturer’s specifications, and each absorbance was read at 490 nm using a MRX Microplate Reader (Dynatech Laboratories, Chantilly, VA).
Function assays of differentiated cell lines
Prior to all functional assays, each EPRO cell line was induced with ATRA for 5 days and morphologic maturation was confirmed by Wright-Giemsa staining of cytospin smears. Assays for the respiratory burst, chemotaxis and phagocytosis were performed essentially as previously described.[21] Chemotaxis assays were performed using Corning Transwell 24 well plates with 3.0 µm pore polycarbonate membranes (Corning Life Sciences, Acton, MA), using medium alone or 0.3 µg/mL of either mouse chemoattractant, keratinocyte chemokine (KC) or macrophage-inflammatory protein-2 (MIP-2, R & D Systems, Minneapolis, MN). The respiratory burst response was measured using luminol-enhanced chemiluminescence provided by Diogenes reagent (National Diagnostics, Atlanta, GA), and cells were activated with either phorbal myristate acetate (PMA, 3.2 µM, Sigma) or serum-opsonized Zymosan A (Sigma, 4 µg/mL). Phagocytosis was performed using cells (1 × 106) incubated with 107 serum-opsonized fluoroscein-conjugated zymosan particles (Molecular Probes, Eugene, OR) for 30 min at 37°C. The number of cells with internalized particles was counted by visual inspection using a Zeiss Axiovert 200M microscope at 20X power and fluorescent light.
Western analyses of NADPH oxidase proteins
For whole cell lysates, cells (1 × 107) were washed in PBS supplemented with Complete Proteinase Inhibitor (Roche Applied Science, Indianapolis, IN), phenylmethanesulfonyl fluoride (1 mM, Sigma) and diisopropyl fluorophosphate (1 µM, Sigma) and then lysed in 10% glycerol, 3% sodium dodecyl sulfate, 62.5 mM Tris (pH6.8), and 50 mM DTT. Equal volumes (20 µL) were then electrophoresed using 4–12% Bis-Tris precast gels (Invitrogen) and probed as previously described [25]. Antibodies for gp91phox, p47phox and p67phox were all from BD Transduction Laboratories (San Diego, CA). Antibodies against actin were from Santa Cruz Biotechnologies (Santa Cruz, CA).
Statistical analysis
Statistical data was generated using Excel (Microsoft, Redmond, WA) software with the Student’s two sample t-test.
Results
Abnormal development of neutrophils lacking LBR expression
The original wild-type EML cells (EML-+/+) described by Tsai et al were generated from the bone marrow of BDF1 male mice by transducing the harvested cells with a dominant negative RARα [23]. Cells cultured with SCF, IL-3 and 10µM ATRA for three days were then shown to form myeloid progenitors (CFU-GMs), many of which were developmentally arrested promyelocytes that proliferated continuously in GM-CSF. Additional studies demonstrated that ATRA-induction of these promyelocytes (EPRO-+/+ cells) caused terminal differentiation into mature neutrophils that express multiple neutrophil-specific genes and exhibit normal functional responses [21,26]. Here we generated EML-like cells using the bone marrow of an ichthyosis (ic/ic) mouse and a phenotypically normal littermate, and transducing both types of cells with the same dominant negative RARα. The genotype of each derived cell line was confirmed by PCR amplification of the Lbr gene locus using primers that flank the ichthyosis mutation site [20]. As expected, genomic DNA isolated from the original EML-+/+ cells generated a single band corresponding to the normal Lbr gene (Fig. 1A). In contrast, products generated from EML-ic/ic cells yielded only 121 bp fragments, indicative of the CC insertion in C57BL/6J-Lbric-J/Lbric-J mice. DNA from the littermate control yielded two fragments, indicating that these cells are heterozygous for the ic mutation, hereafter termed EML-+/ic cells.
Figure 1. Mutation of the LBR locus in EML/EPRO-ic/ic cells inhibits LBR transcriptional activation during neutrophil differentiation.
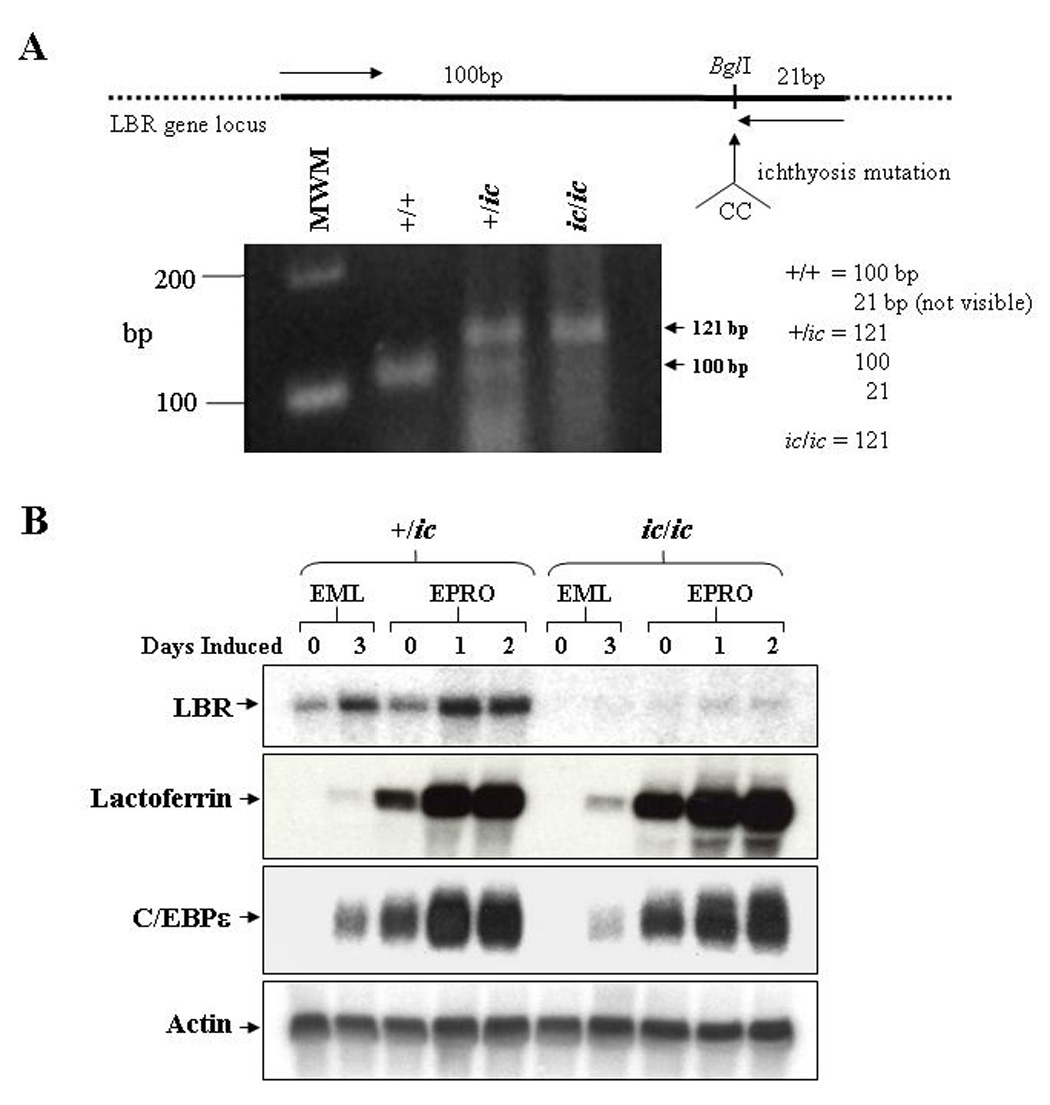
(A) Genomic analysis of the derived EML cell lines identifies the ichthyosis mutation. Genomic DNA from each genotype was isolated and subjected to PCR amplification using primers (indicated by arrows in the upper diagram) specific to the LBR gene but that create a BglI site at the 3’ end of the fragments. This site is disrupted by the ichthyosis mutation (CC insertion). Digestion of the amplified 121bp fragments from wild-type cells with BglI yield 100 and 21bp fragments, cells with the ichthyosis mutation yield only 121bp fragments, and cells heterozygous for ic yield all three fragments. Shown below the diagram are the amplified products from each genotype that were electrophoresed and stained with ethidium bromide. (B) Expression of LBR and neutrophil-specific genes in differentiated ic/ic cells versus +/ic cells were analyzed by Northern blot hybridizations. Total RNA (10 µg) from undifferentiated vs. ATRA-induced EML/EPRO cells of each genotype was electrophoresed, blotted and sequentially probed with labeled cDNA for lactoferrin, C/EBPε and actin as a loading control. The changes in LBR expression shown in each genotype are representative of at least three separate inductions and northern analyses.
Changes in Lbr gene expression were then compared between EML-+/ic versus -ic/ic cells upon induction of differentiation. LBR transcription in EML-+/ic cells was found to increase during two stages of induction, first as EML cells differentiated into EPRO cells, and second as EPRO cells differentiated into mature neutrophils (Fig. 1B, upper panel). Similar results were observed in EML-+/+ and EPRO-+/+ cells (data not shown). These results are consistent with previous studies of human LBR transcription in ATRA-induced HL-60 cells, and confirm that Lbr mRNA expression increases during murine neutrophil maturation [18,19]. In contrast to +/ic cells, Lbr transcription was undetectable in uninduced EML-ic/ic cells and barely detectable in either ATRA/IL-3-induced EML-ic/ic cells or uninduced EPRO-ic/ic cells. Lbr mRNAs were then detected in ATRA-induced EPRO-ic/ic cells, but levels were significantly less than those observed in differentiated +/ic cells. These results support the conclusion that the ic/ic cells are homozygous for a hypomorphic allele that yields very low Lbr transcript expression. Furthermore, absent protein expression in these cells is reported in the subsequent studies by Zwerger et al [22]. Interestingly, the expression of genes that encode either the secondary granule protein lactoferrin or the neutrophil-specific transcriptional regulator C/EBPε was not affected by the ichthyosis mutation in ATRA-induced EPRO-ic/ic cells (Fig. 1B, lower panels); similar data was found for neutrophil gelatinase (data not shown).
We next examined the nuclear morphology of differentiated EPRO-+/+, -+/ic and -ic/ic cells (Fig. 2A). Differentiated EPRO-+/ic cells contained lobulated or ring-shaped nuclei that were indistinguishable from EPRO-+/+ cells. In contrast, most differentiated EPRO-ic/ic cells contained a single condensed nucleus that was either round or kidney-shaped: only a small percentage of cells were bilobed or had a ring appearance. Less than 3% of cells had lobulated nuclei. These morphologic anomalies are essentially identical to those seen in neutrophils from ic/ic mice, and confirm that the EPRO-ic/ic cells recapitulate the ichthyosis neutrophil phenotype. The ultrastructural features of the nuclei from heterozygous and homozygous ic EPRO cells were examined by electron microscopy (Fig. 2B). Consistent with the stained cytospin smears, ATRA-induced EPRO-+/+ and -+/ic cells displayed ring-shaped, lobulated nuclei with multiple regions of condensed chromatin localized to the nuclear envelope, while induced EPRO-ic/ic cells contained single ovoid or indented nuclei, many with large regions of condensed chromatin displaced from the nuclear envelope. To quantify the extent of lobulation in differentiated cells of each genotype, the numbers of cells with multilobed, bilobed or kidney/round shaped nuclei were determined (Fig. 2C). Consistent with previous studies of heterozygous +/ic mice, we did identify a few hypolobulated +/ic neutrophils but the numbers were not significantly different from those found in +/+ neutrophils ([17] and LD Shultz, unpublished observations). By contrast, there was a dramatic difference in the number of kidney/round shaped nuclei in differentiated ic/ic cells.
Figure 2. Severe hypolobulation of nuclei in differentiated EPRO-ic/ic cells.
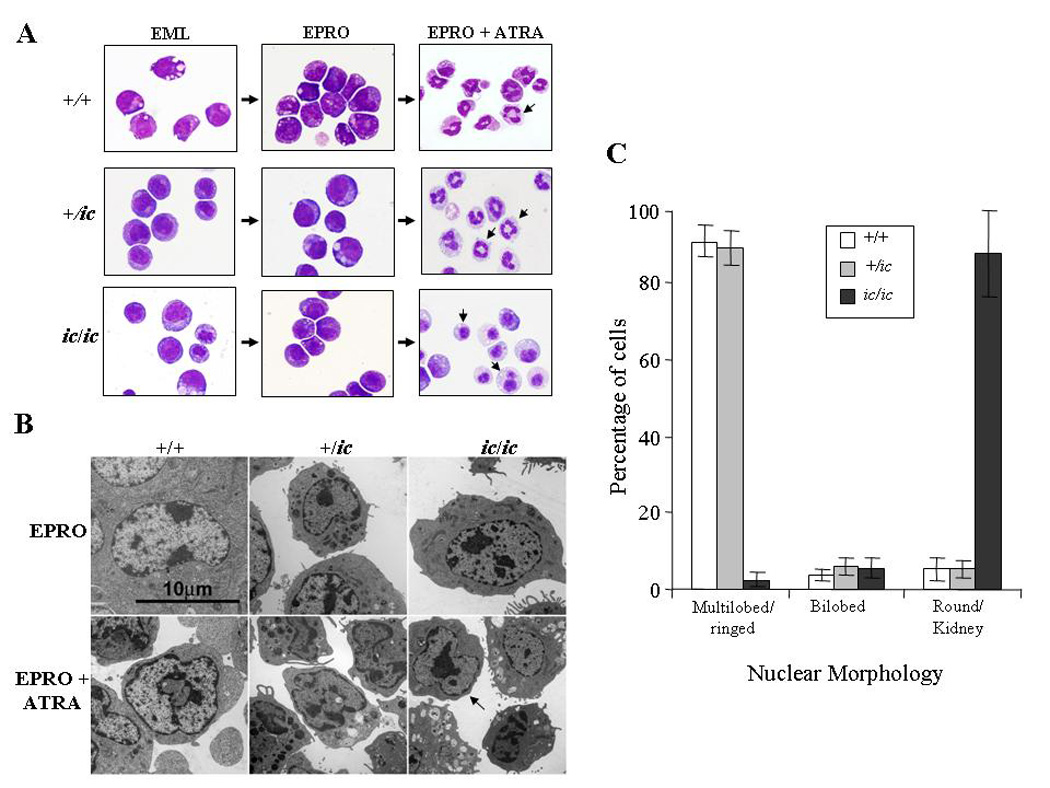
EML-+/+, -+/ic and −ic/ic cells were induced with ATRA (10 µM) plus IL-3 to differentiate into promyelocytic EPRO cells and then further differentiated into mature neutrophils with a second dose of ATRA. (A) Cytospin smears of control +/+ and +/ic vs. ic/ic cells were stained with Wright-Giemsa and then photographed at X 60 magnification using an Olympus BX41 microscope and a DP71 digital camera with accompanying software. Shown are random representations of cells from each stage of development. Arrows in pictures of ATRA-induced EPRO-+/+, -+/ic cells and −ic/ic cells indicate differences in lobulation between each genotype. (B) Electron micrographs of EPRO and ATRA-induced cells from +/+ vs. +/ic and ic/ic cells. (C) Percentages of ATRA-induced EPRO cells with different nuclear morphologies. At least 5 separate inductions of each genotype were performed, and the number of cells exhibiting each type of nuclear morphology was counted at X 40 magnification using an Olympus BX41 microscope. Shown are the average counts of approximately 100 analyzed cells from each induction.
Loss of LBR expression disrupts GM-CSF-induced promyelocytic growth
We next characterized the growth characteristics of EML/EPRO-ic/ic and -+/ic cells. Both cell lines demonstrated factor dependence, with most cells of either genotype dying within 24 hours of cytokine deprivation (data not shown). Both genotypes as EML cells also proliferated well in SCF and in routine passage appeared to be equivalent to +/+ cells, although EML-ic/ic cells did exhibit a small loss of growth response to SCF as compared to EML-+/ic cells (Fig. 3A, left panel). By comparison, EPRO-ic/ic cells maintained in GM-CSF demonstrated a pronounced growth defect compared to EPRO-+/ic cells (Fig. 3A, right panel). A nonradioactive proliferation assay was also used to examine responses of each cell line to increasing concentrations of the appropriate cytokine. Similar to the growth profiles, EML-ic/ic cells showed little difference in response to SCF at all concentrations (Fig. 3B, left panel), but exhibited less response to low concentrations (< 1 ng/mL) of GM-CSF as compared to EPRO-+/ic cells (Fig. 3B, right panel).
Figure 3. Proliferation of EPRO-ic/ic cells is inhibited by the loss of LBR expression.
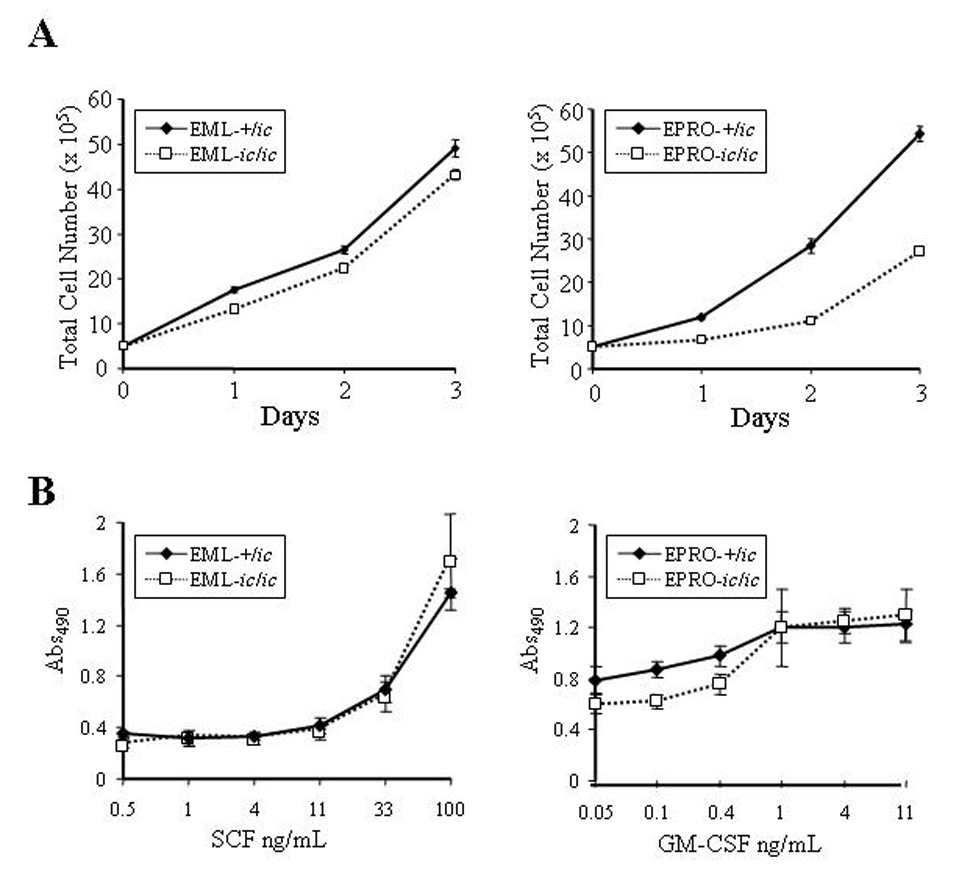
(A) Expansion of viable cells was assessed by visual inspection using trypan blue exclusion. Cells of each genotype were initiated at 1 × 105 cells/mL in EML (left panel) or EPRO (right panel) growth medium, for a total of 5 × 105 cells at day 0. Increases in absolute cell numbers were then determined at 24 hour intervals. Data shown are from 3 independent assays ± SD. P values for growth of EML-+/ic vs. −ic/ic cells are: Day 1, p = 0.0003; Day 2, p = 0.006; Day 3, p = 0.012. P values for EPRO+/ic vs. −ic/ic cells are: Day 1, p = 0.001; Day 2, p < 0.0001; Day 3, p < 0.0001. (B) Proliferation responses to increasing concentrations of cytokines were examined using MTS reduction reagent and absorbances were measured. Each cell type was diluted to equivalent starting concentrations (1 × 105 cells/mL) and increasing concentrations of rmSCF (left panel) or rmGM-CSF (right panel) were used to stimulate growth of EML and EPRO cells, respectively. Shown are data from 3 independent assays ± SD. Differences in responses of EPRO-+/ic vs. −ic/ic to concentrations of GM-CSF below 1 ng/mL were statistically significant (p < 0.0005).
Neutrophils lacking LBR expression exhibit deficient functional responses
The functional responses of ATRA-induced EPRO-ic/ic cells were next examined using well-established assays that test for chemotaxis, the respiratory burst and phagocytosis [21]. Chemotaxis exhibited by ATRA-induced cells was measured using transwell plates with 3.0 µm membranes that separated the cells from medium containing either KC or MIP-2, both potent murine chemoattractants. As shown in Figure 4A, EPRO-+/ic cells demonstrated significant migration of cells in response to either KC (33.8 ± 7.5 × 104 cells) or MIP-2 (47.7 ± 6.5 × 104 cells). In contrast, responses of EPRO-ic/ic to either chemokine were dramatically reduced (KC: 2.5 ± 2 × 104; MIP-2: 18.2 ± 1 × 104 cells). EPRO-ic/ic cells in medium alone also showed less random migration into the bottom chamber. However, some hypolobulated cells migrated into the bottom chamber under all conditions (Fig. 4B), suggesting that aberrant nuclear lobulation in these neutrophils impeded their progress but did not completely abrogate their capacity to migrate through the 3.0 µm membrane.
Figure 4. Neutrophil chemotaxis is severely inhibited by the loss of LBR expression.
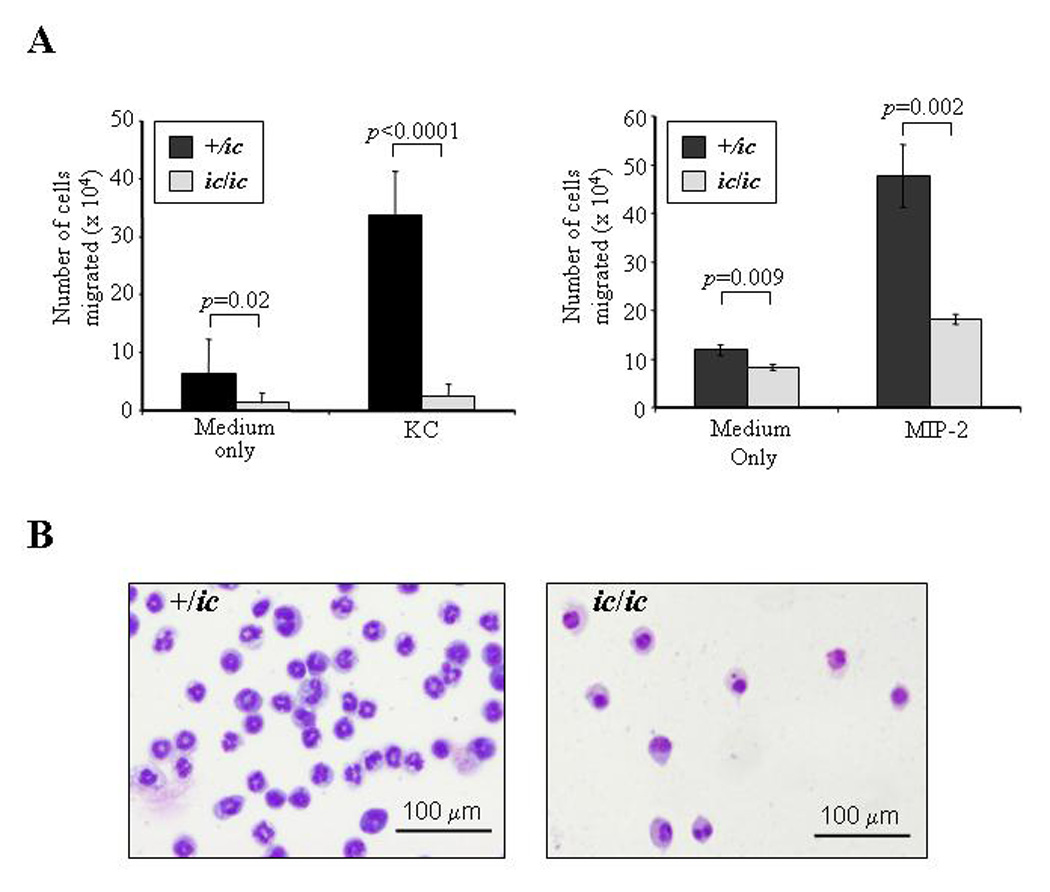
(A) Chemotaxis in response to KC and MIP-2 is reduced in differentiated EPRO-ic/ic vs. -+/ic cells. ATRA-induced cells were incubated in transwell plates in which each chemoattractant was placed in the lower chamber and cells (1 ×106) were placed in the upper chamber, each separated by a 3.0 µm polycarbonate membrane. The chambers were incubated for 2 hours and the number of cells that migrated into the bottom chamber was counted by trypan blue exclusion. P values for comparisons between genotypes are shown above each bar set. (B) Cells isolated from the bottom chambers after KC-induced chemotaxis were Wright-Giemsa-stained and photographed as described in Figure 2. Shown is a representation of the differentiated cells that migrated after 2 hours of incubation.
Activation of the respiratory burst was examined using a luminol-enhanced chemiluminescence assay and two different stimuli. EPRO-+/ic cells produced a strong respiratory burst in response to either PMA or opsonized zymosan (OZ) to levels previously observed by wild-type EPRO cells (Fig. 5A) [21]. In contrast, EPRO-ic/ic cells produced a dramatically reduced respiratory burst in response to either stimulus. Phagocytosis of OZ particles by either genotype was also examined, but the percentage of positive cells and numbers of internalized particles per cell were similar (Fig. 5B). Thus, although loss of LBR expression dramatically disrupted chemotaxis and the respiratory burst, the mutation has little effect on phagocytosis.
Figure 5. Loss of LBR expression severely disrupts the respiratory burst but not phagocytosis.
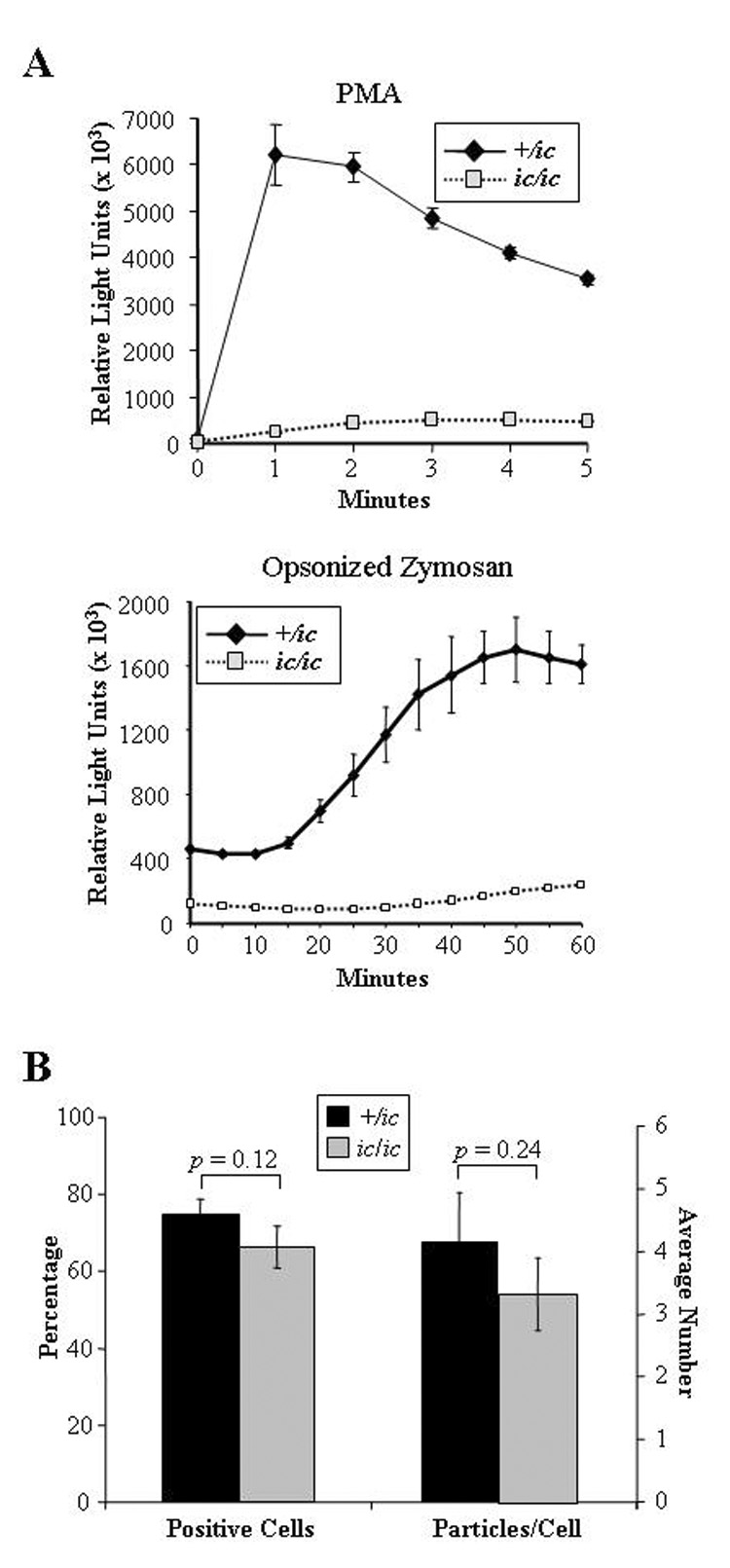
(A) Respiratory burst responses of EPRO-ic/ic cells were reduced compared to +/ic cells. The respiratory burst was activated by the addition of PMA (3.2 µM, upper panel) or opsonized zymosan (4 µg, lower panel) and respiratory burst was measured using luminol-enhanced chemiluminescence. For each assay, triplicate samples were assayed in parallel with errors as SD, and the shown responses are representative of at least three independent experiments. (B) EPRO-ic/ic cells produce significant levels of phagocytosis. Shown are the percentages of EPRO-+/ic cells vs. −ic/ic cells that internalized OZ particles (left bars) and the average number of particles internalized per cell (right bars). Shown above each bar set are the p values that indicate the differences are below the threshold of significance (p > 0.05).
Loss of LBR expression disrupts gp91phox expression
The NADPH oxidase complex is comprised of both membrane-bound and cytoplasmic proteins that are rapidly assembled upon neutrophil activation. Genetic mutations that affect the expression of any one of these components impair function of the oxidase complex, leading to chronic granulomatous disease (recently reviewed in [27,28]). We therefore assessed whether the loss of respiratory burst in EPRO-ic/ic cells was due to abnormal expression of an NADPH oxidase component. Protein expression of three oxidase components was examined: the membrane spanning portion of cytochrome b558, gp91phox, and two cytoplasmic proteins, p47phox and p67phox. We found that expression of p47phox and p67phox were upregulated by ATRA induction in both EPRO-+/ic and −ic/ic cells (Figs. 5A and Fig. 5B). Expression of gp91phox was also increased, but the levels in EPRO-ic/ic cells were significantly less than those of EPRO-+/ic cells. To determine if EPRO-ic/ic cells also displayed deficient transcription of the gene encoding gp91phox, semi-quantitative RT-PCR was performed on cDNA generated from ATRA-induced EPRO-+/+, -+/ic, and −ic/ic cells (Fig. 5C). Although gp91phox transcripts were detectable in ATRA-induced EPRO-ic/ic cells, levels were significantly less than those detected in differentiated +/+ and +/ic cells. Together these results indicate that the loss of oxidase activity in EPRO-ic/ic is due to insufficient gp91phox expression, and demonstrate that deficient LBR expression affects both neutrophil nuclear morphology and the activation of at least one gene essential to neutrophil functional responses.
Discussion
The segmented nucleus of the neutrophil has been postulated to provide increased fluidity that augments its capacity to navigate past endothelial cells and within interstitial spaces. Previous studies of neutrophils with the Pelger-Huët anomaly (caused by heterozygous LBR mutations) have tested this hypothesis, but these studies yielded conflicting results [6–9]. Here we have characterized a novel cell line, EML/EPRO-ic/ic, which lacks LBR protein expression due to homozygous hypomorphic Lbr alleles. Upon ATRA induction, EPRO-ic/ic cells differentiated into mature neutrophils that expressed secondary granule protein genes and C/EBPE, but lacked nuclear lobulation (Fig. 1 and Fig. 2). In fact, most of the differentiated EPRO-ic/ic cells displayed a single nuclear lobe as compared to kidney or bilobed nuclei. When tested for chemotaxis, significantly fewer differentiated EPRO-ic/ic cells as compared to control cells migrated through the 3.0 µm pore-size membrane in response to the CXC chemokines KC or MIP-2 (Fig. 4A). In addition, fewer EPRO-ic/ic cells in medium alone migrated through the membrane. The overall lack of EPRO-ic/ic cell migration could be caused by several mechanisms, the simplest being that deficient nuclear lobulation reduced cell fluidity and impeded the progress of the mutant neutrophils through the membrane. Alternatively, the membrane may have excluded most hypolobulated neutrophils, such that only those with some nuclear fluidity, i.e. those with a bilobed morphology, were capable of passing through the membrane. Since we did observe some EPRO-ic/ic cells that successfully migrated, most of which displayed a single large nuclear lobe (see Fig. 4B), this suggests that migration itself is intact, but that the rate of migration was simply impeded.
Neutrophil chemotaxis is primarily mediated by the activation of G-protein-coupled receptors. Upon binding the chemokine, the activated receptor releases the Gβγ heterodimer from associated trimeric G-proteins, which can then promote the activation of phosphatidylinositol 3-kinase-γ (PI3Kγ), the production of phosphatidylinositol 3,4,5 trisphosphate and the activation of protein kinase B (akt/PKB). Activation of this signaling cascade is thought to play a pivotal role during both human and mouse neutrophil migration by stimulating F-actin polymerization [29–32]. However, the lack of PI3Kγ in knockout mice does not appear to affect random neutrophil migration, even in the presence of a chemoattractant (e.g. chemokinesis) [33]. In differentiated EPRO-ic/ic cells, both chemokine-stimulated chemotaxis and random migration in medium alone were reduced. Furthermore, phagocytosis is also dependent on F-actin formation, but this response was not significantly deficient in ic/ic neutrophils. Thus, while it is formally possible that loss of LBR expression disrupts G-protein-regulated signaling pathways, it is unlikely to completely explain the severely deficient chemotaxis exhibited by ic/ic neutrophils. Future analyses of these and other pathways that may also regulate neutrophil chemotaxis, such as GTPases and calcium-dependent protein kinases [34,35], in the ic/ic neutrophils may help determine whether the defective chemotaxis is due strictly to nuclear structural defects. Since the concentrations used in the chemotaxis assays were most likely maximal doses, an examination of dose responses, migration rates and degrees of shape changes will also help to further characterize this important functional defect in the ic/ic neutrophils.
In addition to the observed effects on chemotaxis, loss of LBR expression also inhibited GM-CSF-dependent growth of EPRO cells while only marginally affecting SCF-stimulated growth of EML cells. However, ATRA-induced maturation of EML cells and the expression of secondary granule protein genes in EPRO cells were intact. These observations suggest that the loss of LBR expression affects a signaling pathway specific to GM-CSF-induced proliferation. GM-CSF stimulates proliferative responses primarily by activating the JAK2/STAT5 signaling pathway, although PI3-kinase and Ras/Raf/MAP kinase pathways have also been implicated in myeloid cell growth (reviewed in [36]). We have performed a preliminary analysis that indicates that the level of STAT5 phoshorylation is normal in EPRO-ic/ic cells when stimulated with GM-CSF (unpublished observations). It is therefore unclear whether the observed defects in promyelocyte growth are due to abnormal JAK/STAT signaling or due to defects in an alternative pathway downstream of GM-CSF. Interestingly, recent studies have shown that inhibition of PI3-kinase disrupts the generation of myeloid progenitors but enhances the hematopoietic potential of embryonic stem cells [37]. Thus the lack of LBR expression may be disrupting one or more pathways that converge on the PI3-kinase signaling pathway, which in turn causes a disruption to both promyelocytic cell growth and chemotaxis but not to SCF-stimulated responses. Further studies of the signaling pathways that are active in EPRO-ic/ic cells during growth, differentiation and chemotaxis are required to determine whether the phenotypes are related.
One of the most striking and unexpected abnormalities found in ATRA-induced EPRO-ic/ic cells was the severe loss of respiratory burst generated by either PMA or opsonized zymosan (see Figure 5). Much of this loss is likely due to the deficient expression of gp91phox, a critical component of the NADPH oxidase complex (Fig. 6). How might loss of LBR affect the expression of gp91phox but not other oxidase components? One possibility is that loss of LBR expression directly affects the activities of a transcription factor that regulates gp91phox gene expression. Several recent studies of other inner nuclear envelope proteins lend support to this notion; the Xenopus homolog of mammalian MAN1, XMAN1, was shown to block transcriptional regulation by bone morphogenetic protein, and LAP2β repressed the transcriptional activity of several transcription factors, including E2F family members p53 and NF-kB [38–40]. Multiple transcriptional regulators have been identified to positively regulate gp91phox expression, including NF-kB, PU.1, C/EBPε and HoxA9 [41–44]. It is therefore conceivable that the loss of LBR expression specifically affects the ability of one or more of these factors to properly initiate gp91phox gene activation. A caveat to this notion is that several of these transcriptional regulators also affect the expression of at least one other NADPH oxidase component that was found to be normally expressed in EPRO-ic/ic cells.
Figure 6. Loss of LBR expression inhibits gp91phox expression in differentiated EPRO-ic/ic cells.
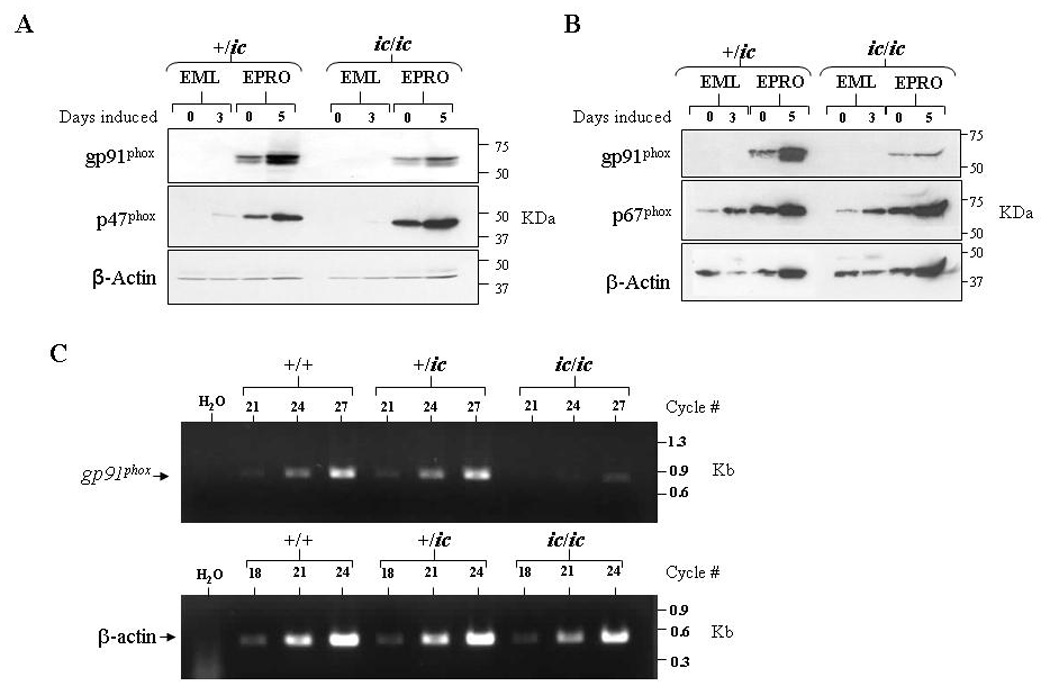
Western blots were generated from uninduced and induced +/ic and ic/ic cells, and then sequentially probed with antibodies for (A) gp91phox and p47phox or (B) gp91phox and p67phox. Both blots were also probed for β-actin expression, and equivalent, total numbers of cells were used for each lysate. (C) Semi-quantitative RT-PCR analysis reveals reduced gp91phox transcript expression in ATRA-induced EPRO-ic/ic as compared to +/+ and +/ic cells. Shown are ethidium bromide-stained gels containing RT-PCR amplified products from each genotype using increasing numbers of cycles, using primers specific to mouse gp91phox (top) and mouse β-actin (bottom) as a loading control.
Deficient gp91phox expression may diminish the respiratory burst, but this cannot solely explain the dramatic phenotype since EPRO-ic/ic cells still express significant amounts of gp91phox protein (Fig. 6). It is therefore possible that loss of LBR expression may cause other abnormalities that affect the function of NADPH oxidases. The Lbr gene encodes two distinct domains, a nucleoplasmic domain at the N-terminus that functions in binding lamin B and chromatin proteins, and a membrane-spanning, C-terminal domain that is highly homologous to sterol reductases and can function in cholesterol biosynthesis [45–47]. Studies of a human with homozygous LBR mutations suggested that the cause of Greenberg/HEM dysplasia was due to disrupted sterol Δ14 reductase activity [16]. Interestingly, differentiated HL-60 cells depleted in cholesterol have a decreased respiratory burst response to both PMA and OZ, and show defective cell shape changes in response to a chemotactic peptide [48,49]. Thus a loss of sterol Δ14 reductase activity caused by the ic mutation may contribute to the deficient respiratory burst and chemotaxis in ATRA-induced EPRO-ic/ic cells. It should be noted, however, that a second gene encodes sterol reductase activity in humans and mice (DHCR14), and a very recent study has indicated that Greenberg/HEM dysplasia may be due to laminopathies caused by deficient LBR expression rather than a loss of sterol reductase activity [50].
The specificity of our observed phenotypes may seem surprising given the essential roles that nuclear envelope proteins and associated lamins are thought to play in regulating nuclear structural integrity throughout the cell cycle, but many mutations that disrupt nuclear envelope or lamin protein expression also cause rather specific phenotypes in humans and mice (recently reviewed in [51]). Mutations in LBR cause abnormalities specific to skeletal and skin development and nuclear abnormalities in specific hematopoietic lineages [16,20]. Loss of emerin expression results in X-linked Emery-Dreifuss muscular dystrophy, a specific disorder of muscle development [52,53]. Mutation of the lamin B1 gene, Lmb1, in mice disrupts both skeletal and lung development, leads to nuclear abnormalities, and disrupts the proliferative capacity of primary embryonic fibroblasts [54]. Mutations in the human LMNA gene cause laminopathies that affect at least five distinct developmental pathways, and cells overexpressing a mutant lamin A protein display abnormal exit from the cell cycle [55,56]. In all cases, the mutations fail to lead to widespread cell death. Thus our findings that the lack of LBR expression in EPRO-ic/ic cells inhibits distinct neutrophil functions and the expression of a single component of the NADPH oxidase are not unusual and may implicate the function of nuclear membrane components in transcriptional regulation. Interestingly, our subsequent study presents evidence that the lack of LBR expression in EPRO-ic/ic cells correlates with increased expression of lamin A/C [22].
Our studies of the EML/EPRO-ic/ic cells have revealed several new functions for LBR and/or nuclear segmentation in mediating neutrophil functional responses and perhaps gene expression, but these studies may have wider implications for LBR in hematopoietic diseases. Diseases that disrupt the myeloid differentiation program, such as myelodysplastic syndrome (MDS) and acute myelogenous leukemia (AML), profoundly affect the most distal events in neutrophil maturation and often cause the appearance of circulating neutrophils with hyposegmented nuclei [57,58]. This abnormality has also been identified in transplant recipients receiving immunosuppressive drugs [59,60]. The abnormality is termed pseudo or acquired PHA, based on its similarity to familial PHA. While the morphologic features of these anomalies have been well characterized, our knowledge of the factors or molecular lesions that cause the anomalies is very limited. Future studies of the EML/EPRO-ic/ic cells may broaden our understanding of the molecular mechanisms that control nuclear segmentation, and may reveal how LBR acts through lamin B and chromatin to regulate both this complex process and the reorganization of the nucleus that must occur during every cell cycle.
Acknowledgments
This work was supported by grants from the National Institutes of Health to P.G. (KO1-DK60565 and R15-HL089933) and to D.E.O. (R15-HL075809).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Zhelev DV, Alteraifi A. Signaling in the motility responses of the human neutrophil. Ann Biomed Eng. 2002;30:356–370. doi: 10.1114/1.1477446. [DOI] [PubMed] [Google Scholar]
- 2.Niggli V. Signaling to migration in neutrophils: importance of localized pathways. Int J Biochem Cell Biol. 2003;35:1619–1638. doi: 10.1016/s1357-2725(03)00144-4. [DOI] [PubMed] [Google Scholar]
- 3.Dao C, Metcalf D, Zittoun R, Bilski-Pasquier G. Normal human bone marrow cultures in vitro: cellular composition and maturation of the granulocytic colonies. Br J Haematol. 1977;37:127–136. [PubMed] [Google Scholar]
- 4.Sanchez JA, Wangh LJ. New insights into the mechanisms of nuclear segmentation in human neutrophils. J Cell Biochem. 1999;73:1–10. doi: 10.1002/(sici)1097-4644(19990401)73:1<1::aid-jcb1>3.0.co;2-s. [DOI] [PubMed] [Google Scholar]
- 5.Jandl JH. Blood. A Textbook of Hematology. 2nd ed. New York, NY: Little, Brown and Co.; 1996. Leukocyte anomalies; pp. 788–789. [Google Scholar]
- 6.Park BH, Dolen J, Snyder B. Defective chemotactic migration of polymorphonuclear leukocytes in Pelger-Huët anomaly. Proc Soc Exp Biol Med. 1977;155:51–54. doi: 10.3181/00379727-155-39743. [DOI] [PubMed] [Google Scholar]
- 7.Repo H, Vuopio P, Leirisalo M, Jansson SE, Kosunen TU. Impaired neutrophil chemotaxis in Pelger-Huët anomaly. Clin Exp Immunol. 1979;36:326–333. [PMC free article] [PubMed] [Google Scholar]
- 8.Matsumoto T, Harada Y, Yamaguchi K, Matsuzaki H, Sanada I, Yoshimura T, et al. Cytogenetic and functional studies of leukocytes with Pelger-Huët anomaly. Acta Haematol. 1984;72:264–273. doi: 10.1159/000206399. [DOI] [PubMed] [Google Scholar]
- 9.Johnson CA, Bass DA, Trillo AA, Snyder MS, DeChatelet LR. Functional and metabolic studies of polymorphonuclear leukocytes in the congenital Pelger-Huët anomaly. Blood. 1980;55:466–469. [PubMed] [Google Scholar]
- 10.Oosterwijk JC, Mansour S, van Noort G, Waterham HR, Hall CM, Hennekam RC. Congenital abnormalities reported in Pelger-Huët homozygosity as compared to Greenberg/HEM dysplasia: highly variable expression of allelic phenotypes. J Med Genet. 2003;40:937–941. doi: 10.1136/jmg.40.12.937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Stobbe H, Jorke D. Findings in homozygous carriers of Pelger's anomaly. Schweiz Med Wochenschr. 1965;95:1524–1529. [PubMed] [Google Scholar]
- 12.Aznar J, Vaya A. Homozygous form of the Pelger-Huët leukocyte anomaly in man. Acta Haematol. 1981;66:59–62. doi: 10.1159/000207095. [DOI] [PubMed] [Google Scholar]
- 13.Siegert E, Beier L, Grabner H. Homozygotic Pelger-Huët anomaly. Kinderarztl Prax. 1983;51:164–169. [PubMed] [Google Scholar]
- 14.Erice JG, Perez JM, Pericas FS. Homozygous form of the Pelger-Huët anomaly. Haematologica. 1999;84:748. [PubMed] [Google Scholar]
- 15.Hoffmann K, Dreger CK, Olins AL, Olins DE, Shultz LD, Lucke B, et al. Mutations in the gene encoding the lamin B receptor produce an altered nuclear morphology in granulocytes (Pelger-Huët anomaly) Nat Genet. 2002;31:410–414. doi: 10.1038/ng925. [DOI] [PubMed] [Google Scholar]
- 16.Waterham HR, Koster J, Mooyer P, Noort Gv G, Kelley RI, Wilcox WR, et al. Autosomal recessive HEM/Greenberg skeletal dysplasia is caused by 3 beta-hydroxysterol delta 14-reductase deficiency due to mutations in the lamin B receptor gene. Am J Hum Genet. 2003;72:1013–1017. doi: 10.1086/373938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Buendia B, Courvalin JC, Collas P. Dynamics of the nuclear envelope at mitosis and during apoptosis. Cell Mol Life Sci. 2001;58:1781–1789. doi: 10.1007/PL00000818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Olins AL, Herrmann H, Lichter P, Olins DE. Retinoic acid differentiation of HL-60 cells promotes cytoskeletal polarization. Exp Cell Res. 2000;254:130–142. doi: 10.1006/excr.1999.4727. [DOI] [PubMed] [Google Scholar]
- 19.Olins AL, Olins DE. Cytoskeletal influences on nuclear shape in granulocytic HL-60 cells. BMC Cell Biol. 2004;5:30. doi: 10.1186/1471-2121-5-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shultz LD, Lyons BL, Burzenski LM, Gott B, Samuels R, Schweitzer PA, et al. Mutations at the mouse ichthyosis locus are within the lamin B receptor gene: a single gene model for human Pelger-Huët anomaly. Hum Mol Genet. 2003;12:61–69. doi: 10.1093/hmg/ddg003. [DOI] [PubMed] [Google Scholar]
- 21.Gaines P, Chi J, Berliner N. Heterogeneity of functional responses in differentiated myeloid cell lines reveals EPRO cells as a valid model of murine neutrophil functional activation. J Leukoc Biol. 2005;77:669–679. doi: 10.1189/jlb.1004567. [DOI] [PubMed] [Google Scholar]
- 22.Zwerger M, Herrmann H, Gaines P, Olins AL, Olins DE. Granulocyte nuclear differentiation of Lamin B Receptor deficient mouse EPRO cells. Experimental Hematology. 2008 doi: 10.1016/j.exphem.2008.03.003. (this issue) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tsai S, Bartelmez S, Sitnicka E, Collins S. Lymphohematopoietic progenitors immortalized by a retroviral vector harboring a dominant-negative retinoic acid receptor can recapitulate lymphoid, myeloid, and erythroid development. Genes Dev. 1994;8:2831–2841. doi: 10.1101/gad.8.23.2831. [DOI] [PubMed] [Google Scholar]
- 24.Tsai S, Bartelmez S, Heyman R, Damm K, Evans R, Collins SJ. A mutated retinoic acid receptor-alpha exhibiting dominant-negative activity alters the lineage development of a multipotent hematopoietic cell line. Genes Dev. 1992;6:2258–2269. doi: 10.1101/gad.6.12a.2258. [DOI] [PubMed] [Google Scholar]
- 25.Maun NA, Gaines P, Khanna-Gupta A, Zibello T, Enriquez L, Goldberg L, Berliner N. GCSF signaling can differentiate promyelocytes expressing a defective retinoic acid receptor: evidence for divergent pathways regulating neutrophil differentiation. Blood. 2004;103:1693–1701. doi: 10.1182/blood-2002-10-3247. [DOI] [PubMed] [Google Scholar]
- 26.Lawson ND, Krause DS, Berliner N. Normal neutrophil differentiation and secondary granule gene expression in the EML and MPRO cell lines. Exp.Hematol. 1998;26:1178–1185. [PubMed] [Google Scholar]
- 27.Roos D, van Bruggen R, Meischl C. Oxidative killing of microbes by neutrophils. Microbes Infect. 2003;5:1307–1315. doi: 10.1016/j.micinf.2003.09.009. [DOI] [PubMed] [Google Scholar]
- 28.Goebel WS, Dinauer MC. Gene therapy for chronic granulomatous disease. Acta Haematol. 2003;110:86–92. doi: 10.1159/000072457. [DOI] [PubMed] [Google Scholar]
- 29.Li Z, Jiang H, Xie W, Zhang Z, Smrcka AV, Wu D. Roles of PLC-beta2 and -beta3 and PI3Kgamma in chemoattractant-mediated signal transduction. Science. 2000;287:1046–1049. doi: 10.1126/science.287.5455.1046. [DOI] [PubMed] [Google Scholar]
- 30.Naccache PH, Levasseur S, Lachance G, Chakravarti S, Bourgoin SG, McColl SR. Stimulation of human neutrophils by chemotactic factors is associated with the activation of phosphatidylinositol 3-kinase gamma. J Biol Chem. 2000;275:23636–23641. doi: 10.1074/jbc.M001780200. [DOI] [PubMed] [Google Scholar]
- 31.Hirsch E, Katanaev VL, Garlanda C, Azzolino O, Pirola L, Silengo L, et al. Central role for G protein-coupled phosphoinositide 3-kinase gamma in inflammation. Science. 2000;287:1049–1053. doi: 10.1126/science.287.5455.1049. [DOI] [PubMed] [Google Scholar]
- 32.Sasaki T, Irie-Sasaki J, Jones RG, Oliveira-dos-Santos AJ, Stanford WL, Bolon B, et al. Function of PI3Kgamma in thymocyte development, T cell activation, and neutrophil migration. Science. 2000;287:1040–1046. doi: 10.1126/science.287.5455.1040. [DOI] [PubMed] [Google Scholar]
- 33.Hannigan M, Zhan L, Li Z, Ai Y, Wu D, Huang CK. Neutrophils lacking phosphoinositide 3-kinase gamma show loss of directionality during N-formyl-Met-Leu-Phe-induced chemotaxis. Proc Natl Acad Sci U S A. 2002;99:3603–3608. doi: 10.1073/pnas.052010699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Verploegen S, van Leeuwen CM, van Deutekom HW, Lammers JW, Koenderman L, Coffer PJ. Role of Ca2+/calmodulin regulated signaling pathways in chemoattractant induced neutrophil effector functions. Comparison with the role of phosphotidylinositol-3 kinase. Eur J Biochem. 2002;269:4625–4234. doi: 10.1046/j.1432-1033.2002.03162.x. [DOI] [PubMed] [Google Scholar]
- 35.Chodniewicz D, Zhelev DV. Chemoattractant receptor-stimulated F-actin polymerization in the human neutrophil is signaled by 2 distinct pathways. Blood. 2003;101:1181–1184. doi: 10.1182/blood-2002-05-1435. [DOI] [PubMed] [Google Scholar]
- 36.Barreda DR, Hanington PC, Belosevic M. Regulation of myeloid development and function by colony stimulating factors. Dev.Comp.Immunol. 2004;28:509–554. doi: 10.1016/j.dci.2003.09.010. [DOI] [PubMed] [Google Scholar]
- 37.Bone HK, Welham MJ. Phosphoinositide 3-kinase signalling regulates early development and developmental haemopoiesis. J.Cell.Sci. 2007;120:1752–1762. doi: 10.1242/jcs.003772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Osada S, Ohmori SY, Taira M. XMAN1, an inner nuclear membrane protein, antagonizes BMP signaling by interacting with Smad1 in Xenopus embryos. Development. 2003;130:1783–1794. doi: 10.1242/dev.00401. [DOI] [PubMed] [Google Scholar]
- 39.Nili E, Cojocaru GS, Kalma Y, Ginsberg D, Copeland NG, Gilbert DJ, et al. Nuclear membrane protein LAP2beta mediates transcriptional repression alone and together with its binding partner GCL (germ-cell-less) J Cell Sci. 2001;114:3297–3307. doi: 10.1242/jcs.114.18.3297. [DOI] [PubMed] [Google Scholar]
- 40.Somech R, Shaklai S, Geller O, Amariglio N, Simon AJ, Rechavi G, et al. The nuclear-envelope protein and transcriptional repressor LAP2beta interacts with HDAC3 at the nuclear periphery, and induces histone H4 deacetylation. J Cell Sci. 2005;118:4017–4025. doi: 10.1242/jcs.02521. [DOI] [PubMed] [Google Scholar]
- 41.Anrather J, Racchumi G, Iadecola C. NF-kappaB regulates phagocytic NADPH oxidase by inducing the expression of gp91phox. J Biol Chem. 2006;281:5657–5667. doi: 10.1074/jbc.M506172200. [DOI] [PubMed] [Google Scholar]
- 42.Mazzi P, Donini M, Margotto D, Wientjes F, Dusi S. IFN-gamma induces gp91phox expression in human monocytes via protein kinase C-dependent phosphorylation of PU.1. J Immunol. 2004;172:4941–4947. doi: 10.4049/jimmunol.172.8.4941. [DOI] [PubMed] [Google Scholar]
- 43.Lekstrom-Himes JA, Dorman SE, Kopar P, Holland SM, Gallin JI. Neutrophil-specific granule deficiency results from a novel mutation with loss of function of the transcription factor CCAAT/enhancer binding protein epsilon. J Exp Med. 1999;189:1847–1852. doi: 10.1084/jem.189.11.1847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bei L, Lu Y, Eklund EA. HOXA9 activates transcription of the gene encoding gp91Phox during myeloid differentiation. J Biol Chem. 2005;280:12359–12370. doi: 10.1074/jbc.M408138200. [DOI] [PubMed] [Google Scholar]
- 45.Holmer L, Pezhman A, Worman HJ. The human lamin B receptor/sterol reductase multigene family. Genomics. 1998;54:469–476. doi: 10.1006/geno.1998.5615. [DOI] [PubMed] [Google Scholar]
- 46.Silve S, Dupuy PH, Ferrara P, Loison G. Human lamin B receptor exhibits sterol C14-reductase activity in Saccharomyces cerevisiae. Biochim Biophys Acta. 1998;1392:233–244. doi: 10.1016/s0005-2760(98)00041-1. [DOI] [PubMed] [Google Scholar]
- 47.Holmer L, Worman HJ. Inner nuclear membrane proteins: functions and targeting. Cell Mol Life Sci. 2001;58:1741–1747. doi: 10.1007/PL00000813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Vilhardt F, van Deurs B. The phagocyte NADPH oxidase depends on cholesterol-enriched membrane microdomains for assembly. EMBO J. 2004;23:739–748. doi: 10.1038/sj.emboj.7600066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Niggli V, Meszaros AV, Oppliger C, Tornay S. Impact of cholesterol depletion on shape changes, actin reorganization, and signal transduction in neutrophil-like HL-60 cells. Exp Cell Res. 2004;296:358–368. doi: 10.1016/j.yexcr.2004.02.015. [DOI] [PubMed] [Google Scholar]
- 50.Wassif CA, Brownson KE, Sterner AL, Forlino A, Zerfas PM, Wilson WK, et al. HEM dysplasia and ichthyosis are likely laminopathies and not due to 3beta-hydroxysterol Delta14-reductase deficiency. Hum Mol Genet. 2007;16:1176–1187. doi: 10.1093/hmg/ddm065. [DOI] [PubMed] [Google Scholar]
- 51.Worman HJ, Bonne G. "Laminopathies": a wide spectrum of human diseases. Exp Cell Res. 2007;313:2121–2133. doi: 10.1016/j.yexcr.2007.03.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bione S, Maestrini E, Rivella S, Mancini M, Regis S, Romeo G, et al. Identification of a novel X-linked gene responsible for Emery-Dreifuss muscular dystrophy. Nat Genet. 1994;8:323–327. doi: 10.1038/ng1294-323. [DOI] [PubMed] [Google Scholar]
- 53.Nagano A, Koga R, Ogawa M, Kurano Y, Kawada J, Okada R, et al. Emerin deficiency at the nuclear membrane in patients with Emery-Dreifuss muscular dystrophy. Nat Genet. 1996;12:254–259. doi: 10.1038/ng0396-254. [DOI] [PubMed] [Google Scholar]
- 54.Vergnes L, Peterfy M, Bergo MO, Young SG, Reue K. Lamin B1 is required for mouse development and nuclear integrity. Proc Natl Acad Sci U S A. 2007;101:10428–10433. doi: 10.1073/pnas.0401424101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Capell BC, Collins FS. Human laminopathies: nuclei gone genetically awry. Nat Rev Genet. 2006;7:940–952. doi: 10.1038/nrg1906. [DOI] [PubMed] [Google Scholar]
- 56.Favreau C, Higuet D, Courvalin JC, Buendia B. Expression of a mutant lamin A that causes Emery-Dreifuss muscular dystrophy inhibits in vitro differentiation of C2C12 myoblasts. Mol Cell Biol. 2004;24:1481–1492. doi: 10.1128/MCB.24.4.1481-1492.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kuriyama K, Tomonaga M, Matsuo T, Ginnai I, Ichimaru M. Diagnostic significance of detecting pseudo-Pelger-Huët anomalies and micro-megakaryocytes in myelodysplastic syndrome. Br J Haematol. 1986;63:665–669. doi: 10.1111/j.1365-2141.1986.tb07550.x. [DOI] [PubMed] [Google Scholar]
- 58.Young NS, Sloand EM, Barrett J. Myelodysplastic Syndrome. In: Young NS, Gerson SL, High KA, editors. Clinical Hematology. Vol. 2006. Pennsylvania, PA: Mosby Elsevier; 2006. pp. 203–217. [Google Scholar]
- 59.Asmis LM, Hadaya K, Majno P, Toso C, Triponez F, Starobinski M. Acquired and reversible Pelger-Huet anomaly of polymorphonuclear neutrophils in three transplant patients receiving mycophenolate mofetil therapy. Am J Hematol. 2003;73:244–248. doi: 10.1002/ajh.10359. [DOI] [PubMed] [Google Scholar]
- 60.Etzell JE, Wang E. Acquired Pelger-Huet anomaly in association with concomitant tacrolimus and mycophenolate mofetil in a liver transplant patient: a case report and review of the literature. Arch Pathol Lab Med. 2006;130:93–96. doi: 10.5858/2006-130-93-APAIAW. [DOI] [PubMed] [Google Scholar]