

Abstract
The purpose of this study was to determine whether exogenous zinc prevents cardiac reperfusion injury by targeting the mitochondrial permeability transition pore (mPTP) via glycogen synthase kinase-3β (GSK-3β). The treatment of cardiac H9c2 cells with ZnCl2 (10 μM) in the presence of zinc ionophore pyrithione for 20 min significantly enhanced GSK-3β phosphorylation at Ser9, indicating that exogenous zinc can inactivate GSK-3β in H9c2 cells. The effect of zinc on GSK-3β activity was blocked by the phosphatidylinositol 3-kinase (PI3K) inhibitor LY-294002 but not by the mammalian target of rapamycin (mTOR) inhibitor rapamycin or the PKC inhibitor chelerythrine, implying that PI3K but not mTOR or PKC accounts for the action of zinc. In support of this interpretation, zinc induced a significant increase in Akt but not mTOR phosphorylation. Further experiments found that zinc also increased mitochondrial GSK-3β phosphorylation. This may indicate an involvement of the mitochondria in the action of zinc. The effect of zinc on mitochondrial GSK-3β phosphorylation was not altered by the mitochondrial ATP-sensitive K+ channel blocker 5-hydroxydecanoic acid. Zinc applied at reperfusion reduced cell death in cells subjected to simulated ischemia/reperfusion, indicating that zinc can prevent reperfusion injury. However, zinc was not able to exert protection in cells transfected with the constitutively active GSK-3β (GSK-3β-S9A-HA) mutant, suggesting that zinc prevents reperfusion injury by inactivating GSK-3β. Cells transfected with the catalytically inactive GSK-3β (GSK-3β-KM-HA) also revealed a significant decrease in cell death, strongly supporting the essential role of GSK-3β inactivation in cardioprotection. Moreover, zinc prevented oxidant-induced mPTP opening through the inhibition of GSK-3β. Taken together, these data suggest that zinc prevents reperfusion injury by modulating the mPTP opening through the inactivation of GSK-3β. The PI3K/Akt signaling pathway is responsible for the inactivation of GSK-3β by zinc.
Keywords: h9c2 cell, oxidant stress, simulated ischemia
as a transition metal found in most tissues, zinc is fundamental to the structure and function of many proteins, enzymes, and transcriptional factors (7). Zinc has also been demonstrated to play an important role in cellular signaling by modulating signal recognition, second messenger metabolism, protein kinase, and phosphatase activities (8, 27). Recent studies have shown that exogenous zinc can regulate activities of several important intracellular signaling elements such as phosphatidylinositol 3-kinase (PI3K) (5), Akt/PKB (2), p70S6 kinase (1, 25), mammalian target of rapamycin (mTOR) (29), extracellular signal-regulated kinase (ERK) (2), and glycogen synthase kinase-3β (GSK-3β) (19). Since these signaling kinases have been demonstrated to be involved in the mechanism underlying cardioprotection against reperfusion injury, the treatment of cardiac cells with zinc may lead to cardioprotection against reperfusion injury. An early study showed that zinc protects isolated rat hearts from ischemia/reperfusion injury by reducing oxidant stress (36). It has also been reported that exogenous zinc suppresses apoptosis in cardiac allografts in a dose-dependent manner (26). Recently, Karagulova et al. (23) documented that the treatment of isolated rat hearts at reperfusion with zinc ionophore pyrithione induces cardioprotection against reperfusion injury by preserving PKC isoforms. Moreover, we have recently demonstrated that zinc can prevent oxidant-induced mitochondrial damage in cardiomyocytes (20). Thus these studies strongly suggest that the addition of zinc might be a useful intervention to prevent ischemia/reperfusion injury. However, it should be noted that the above studies provide very limited information on the signaling mechanism underlying zinc-induced cardioprotection in the setting of ischemia/reperfusion.
A number of intracellular signaling elements have been proposed to be involved in cardioprotection against ischemia/reperfusion injury. Recent studies have revealed that GSK-3β plays a critical role in cardioprotection. The inactivation of GSK-3β is involved in the mechanism of ischemic preconditioning (40) and is also reported to be critical for opioid-induced cardioprotection against reperfusion injury (15). The inhibition of GSK-3β leads to prevention of mitochondrial permeability transition pore (mPTP) opening, which serves as an essential mechanism by which the kinase mediates the cardioprotective effects induced by pharmacological preconditioning (22), bradykinin (34), and N6-(3-iodobenzyl)-9-[5-(methylcarbamoyl)-β-d-ribofuranosyl]adenine (IB-MECA) (33). The mPTP opening plays an important role in myocardial ischemia/reperfusion injury, and the blockade of the pore opening leads to cardioprotection (38). A previous study (20) from our laboratory has shown that exogenous zinc could prevent oxidant-induced mPTP opening in rat cardiomyocytes. Furthermore, zinc has been reported to inhibit GSK-3β in Hela and SH-SY5Y cells (1, 5). Thus it is reasonable to hypothesize that zinc reduces cardiac cell death by targeting the mPTP through the inactivation of GSK-3β in the setting of ischemia/reperfusion.
In this study, we first examined whether exogenous zinc could inactivate GSK-3β through N-terminal phosphorylation of Ser9 in H9c2 cells. We then investigated the signaling mechanism by which zinc inactivates GSK-3β. Finally, we tested whether zinc can protect H9c2 cells from simulated ischemia/reperfusion injury via a GSK-3β-dependent mechanism.
MATERIALS AND METHODS
Cell culture.
The rat heart tissue-derived H9c2 cardiac myoblast cell line was purchased from American Type Culture Collection (Manassas, VA). Cells were cultured in Dulbecco's modified Eagle's medium, supplemented with 10% fetal bovine serum and 100 units penicillin-streptomycin at 37°C in a humidified 5% CO2-95% room air atmosphere.
GSK-3β plasmid DNA.
Two GSK-3β mutants, catalytically inactive GSK-3β (GSK-3β-KM-HA) and constitutively active GSK-3β (GSK-3β-S9A-HA) mutant plasmids containing HA tag, were kindly provided by Dr. Morris Birnbaun (University of Pennsylvania School of Medicine). After we purified the plasmid DNA using an Endofree Maxi kit (Qiagen), transient transfections were performed on 12-well plates using Fugene6 with 2 μg DNA according to the manufacturer's instructions (Roche). Briefly, H9c2 cells were seeded in a 12-well plate at a 50% confluency. Two hours after cell seeding, cells were transfected with β-galactosidase (pCDNA-His-LacZ) or the GSK-3β mutants using Fugene6 transfection reagents (DNA-to-reagent ratio = 1:3). Cells were replaced with fresh medium 24 h after the transfection. All experiments were done 48 h after transfection. The transfection efficiency test using β-galactosidase assay kit (Invitrogen) revealed that over 80% of cells expressed β-galactosidase (>80% transfection; data not shown). To test the protein expression levels of the mutant genes, the total GSK-3β protein levels were analyzed with Western blot analysis. Since both mutants include HA tags, the expressed constructs could be distinguished from the endogenous GSK-3β (31). Figure 1 shows that both the GSK-3β-KM-HA and GSK-3β-S9A-HA constructs were expressed in H9c2 cells.
Fig. 1.

Expression of endogenous (endo) and the mutant GSK-3β (GSK-3β-HA) in H9c2 cells. Cells were harvested 24 h after transfection.
Mitochondrial isolation.
Mitochondria and cytosolic fractions were isolated by differential centrifugation as previously described (3). Cells were homogenized in a buffer containing (in mM) 250 sucrose, 10 Tris·HCl (pH 7.4), 1 EDTA, 1 Na3VO4, and 1 NaF and protease inhibitor cocktail. The homogenate was centrifuged at 1,000 g for 10 min to remove nuclei and debris. The supernatant was centrifuged at 10,000 g for 30 min. The resultant supernatant was subsequently centrifuged at 10,000 g for 1 h to yield the cytosolic fraction. The 10,000-g pellet, corresponding to the mitochondrial fraction, was resuspended and centrifuged again at 10,000 g for 30 min. Mitochondria were then resuspended and homogenized.
Cell viability assay.
The cell viability was assessed by propidium iodide fluorometry using a fluorescence reader (SpectraMax, Molecular Devices, Sunnyvale, CA). Fluorescence intensity was measured at the excitation and emission wavelengths of 540 and 590 nm, respectively. Cells in 12-well plates coated with laminin were incubated in standard Tyrode solution containing (in mM) 140 NaCl, 6 KCl, 1 MgCl2, 1 CaCl2, 5 HEPES, and 5.8 glucose (pH 7.4) for 2 h before the experiments. The background fluorescence intensity (B) was measured 20 min after the addition of propidium iodide (30 μM). The cells were then subjected to 90 min of simulated ischemia followed by 30 min of reperfusion (see Experimental protocols). After 30 min of reperfusion, the fluorescence intensity (R) was measured again. Experiments were terminated by the addition of digitonin (300 μM). The final fluorescence intensity (F) was measured 20 min after the addition of digitonin. The cell viability was calculated by the following formula: 100(F − R)/(F − B) (in %).
Confocal imaging of mitochondrial membrane potential.
Mitochondrial membrane potential (ΔΨm) was measured using confocal microscopy as previously reported (20). Briefly, the cardiac cells cultured in a specific temperature-controlled culture dish were incubated with tetramethylrhodamine ethyl ester (TMRE, 100 nM) in standard Tyrode solution containing (in mM) 140 NaCl, 6 KCl, 1 MgCl2, 1 CaCl2, 5 HEPES, and 5.8 glucose (pH 7.4) for 10 min. The cells were then mounted on the stage of an Olympus FV 500 laser-scanning confocal microscope. The red fluorescence was excited with a 543-nm line of argon-krypton laser line and imaged through a 560-nm long-path filter. The temperature was maintained at 37°C with Delta T Open Dish Systems (Bioptechs, Butler, PA). The images recorded on a computer were quantified using ImageJ.
Western blot analysis.
An equal amount of protein lysates (whole cell or mitochondria or cytosol) was loaded and electrophoresed on SDS-polyacrylamide gel and transfected to a polyvinylidene difluoride membrane. The membranes were probed with primary antibodies that recognize the phosphorylation of GSK-3β, mTOR, and p70s6K. Each primary antibody binding was detected with a secondary antibody and visualized by the enhanced chemiluminescence method. An equal loading of samples was confirmed by reprobing the membranes with antibodies that recognize total proteins.
Experimental protocols.
Cultured cells were washed twice with PBS and then incubated in Tyrode solution for 2 h before the experiments. To examine the effect of extracellular zinc on GSK-3β (or mTOR and p70s6K) phosphorylation at Ser9, the cells were exposed to 10 μM ZnCl2 together with 4 μM zinc ionophore pyrithione for 20 min. The inhibitors were applied 20 min before the exposure to ZnCl2. To test the effect of exogenous zinc on cardiac ischemia/reperfusion injury, the cells were exposed to a simulated ischemia solution (glucose-free Tyrode solution containing 10 mM 2-deoxy-d-glucose and 10 mM sodium dithionite; see Ref. 18) for 90 min followed by 30 min of reperfusion with the normal Tyrode solution. ZnCl2 (10 μM) plus 4 μM pyrithione were applied at the onset of reperfusion for 30 min or during ischemia (90 min) only. In the study evaluating the effect of exogenous zinc on ΔΨm, the cells were exposed to 600 μM H2O2 for 20 min to cause mitochondrial oxidant damage. ZnCl2 (10 μM) plus 4 μM pyrithione were given 20 min before an exposure to H2O2.
Statistical analysis.
Data are expressed as means ± SE and obtained from at least six experiments. Statistical significance was determined using a one-way ANOVA followed by Tukey's test. A value of P < 0.05 was considered as statistically significant.
RESULTS
To test whether exogenous zinc can inactivate GSK-3β in H9c2 cells, we determined the effect of ZnCl2 on GSK-3β phosphorylation at Ser9 in total cell extracts. Preliminary studies showed that 10 μM of ZnCl2 was much more effective to phosphorylate GSK-3β than 1 μM ZnCl2 (349% vs. 165% of control). However, there was no further significant increase in GSK-3β phosphorylation by 100 μM (355% of control) ZnCl2. Therefore, we treated cells with 10 μM ZnCl2 in all experiments. As shown in Fig. 2, ZnCl2 (10 μM) dramatically enhanced GSK-3β phosphorylation (349 ± 55% of the control) in the presence of zinc ionophore pyrithione (4 μM), indicating that exogenous zinc can inactivate GSK-3β in H9c2 cells. The effect of zinc on GSK-3β phosphorylation was blocked by LY-294002, an inhibitor of PI3K, implying a role of the PI3K/Akt pathway in the action of zinc. The effect of zinc was not altered by either the mTOR inhibitor rapamycin or the PKC inhibitor chelerythrine, indicating mTOR and PKC may not be involved in the action of exogenous zinc on GSK-3β phosphorylation (Fig. 2). Figure 3 shows that zinc significantly enhanced the phosphorylation of Akt and p70s6K but not mTOR, confirming the above observation that the PI3K/Akt pathway but not mTOR is responsible for zinc-induced GSK-3β phosphorylation. In addition, zinc also increased p70s6K phosphorylation.
Fig. 2.

Western blot analysis of GSK-3β phosphorylation at Ser9 in cardiac H9c2 cells. H9c2 cells were treated with ZnCl2 (Zn2+, 10 μM) for 20 min. ZnCl2 (10 μM) significantly enhanced GSK-3β phosphorylation in H9c2 cells, an effect that was reversed by the phosphatidylinositol 3-kinase (PI3K) inhibitor LY-294002 (LY, 15 μM). The effect of zinc was not altered by either the mammalian target of rapamycin (mTOR) inhibitor rapamycin (Rapa, 5 nM) or the PKC inhibitor chelerythrine (Chel, 5 μM). Bars are means ± SE of at least 6 experimental observations each. *P < 0.05 vs. control; #P < 0.05 vs. Zn2+.
Fig. 3.

Western blot analysis of Akt (Ser473), mTOR (Ser2448), p70s6K (Thr389), and GSK-3β (Ser9) phosphorylation in cardiac H9c2 cells. H9c2 cells were exposed to ZnCl2 (Zn2+, 10 μM) for 20 min. Zinc significantly enhanced phosphorylation of Akt and p70s6K but not mTOR. Bars are means ± SE of at least 6 experimental observations each. *P < 0.05 vs. control.
Since zinc can prevent oxidant-induced mPTP opening (20) and an increase in GSK-3β phosphorylation within mitochondria was responsible for the suppression of the mPTP opening and cardioprotection afforded by ischemic precondition plus erythropoietin (32), we tested whether zinc can translocate GSK-3β to mitochondria and whether zinc can alter mitochondrial GSK-3β activity. As shown in Fig. 4, under physiological conditions (control), the amount of phosphorylated GSK-3β was extremely low compared with that of the total GSK-3β. When cells were treated with 10 μM ZnCl2, GSK-3β phosphorylation was markedly increased both in cytosol and in mitochondria (Fig. 4). Figure 4 also shows that zinc did not increase the mitochondrial GSK-3β amount compared with the control, implying that GSK-3β seems not to be translocated to mitochondria by zinc. To investigate the mechanism by which zinc phosphorylates mitochondrial GSK-3β, we tested whether the mitochondrial ATP-sensitive K+ (mitoKATP) channel blocker 5-hydroxydecanoic acid (5-HD) can prevent the action of zinc. As shown in Fig. 5, 5-HD (0.25 and 0.5 mM) did not alter the effect of zinc.
Fig. 4.

Western blot analysis of GSK-3β phosphorylation at Ser9 in cytosolic and mitochondrial fractions of H9c2 cells. H9c2 cells were exposed to Zn2+ (10 μM) for 20 min. Zinc markedly increased GSK-3β phosphorylation both in cytosol and in mitochondria. Bars are means ± SE of at least 6 experimental observations each. *P < 0.05 vs. control.
Fig. 5.

Western blot analysis of GSK-3β phosphorylation at Ser9 in the mitochondrial fraction of H9c2 cells. H9c2 cells were exposed to Zn2+ (10 μM) for 20 min. The mitochondrial ATP-sensitive K+ channel blocker 5-hydroxydecanoic acid (5-HD, 0.25 and 0.5 mM) did not alter the effect of zinc on mitochondrial GSK-3β phosphorylation. Bars are means ± SE of at least 6 experimental observations each. *P < 0.05 vs. control.
Given the important role of GSK-3β in cardioprotection, exogenous zinc may induce cardioprotection against ischemia/reperfusion injury via a GSK-3β-dependent mechanism. To test the effect of exogenous zinc on ischemia/reperfusion injury, H9c2 cells were subjected to 90 min of simulated ischemia followed by 30 min of reperfusion. Figure 6 shows that simulated ischemia/reperfusion significantly reduced cell viability to 31.3 ± 3.5%. ZnCl2 (10 μM) given during ischemia but not during reperfusion failed to improve cell viability (35.1 ± 4.9%). In contrast, ZnCl2 (10 μM) given only at the onset of reperfusion for 30 min dramatically increased cell viability to 69.1 ± 2.8%, indicating that zinc is cardioprotective during reperfusion rather than during ischemia and thus may have a potential to save myocardium from reperfusion injury. To examine whether the protective effect of zinc was mediated by the suppression of GSK-3β, the cells were transfected with the constitutively active GSK-3β (GSK-3β-S9A) mutant plasmid and then treated with ZnCl2 (10 μM) at reperfusion. As shown in Fig. 6, zinc was not able to improve cell viability (29.2 ± 2.9%), suggesting that exogenous zinc protects cardiac cells from (simulated ischemia) reperfusion injury by inactivating GSK-3β. Figure 6 also shows that cells transfected with the catalytically inactive GSK-3β (GSK-3β-KM) mutant plasmid were resistant to reperfusion injury (62.5 ± 3.7% in cell viability), an effect that was similar to that observed when naive cells were treated with zinc, further supporting a role of GSK-3β inactivation in the action of zinc. To examine whether exogenous zinc prevents reperfusion injury by targeting the mPTP opening, we tested the effect of zinc on the oxidant-induced loss of ΔΨm in H9c2 cells using confocal imaging technique. As shown in Fig. 7, 500 μM H2O2 caused a marked decrease in TMRE fluorescence (37.6 ± 2.2 of baseline), indicating a dissipation of ΔΨm. Since the loss of ΔΨm is an index of the mPTP opening, this result suggests that oxidant stress caused the mPTP opening. The treatment of cells with 10 μM ZnCl2 for 20 min in the presence of pyrithione prevented the loss of TMRE fluorescence (81.4 ± 2.3% of baseline), indicating that exogenous zinc can prevent the mPTP opening. In contrast, ZnCl2 was not able to preserve TMRE fluorescence (43.6 ± 3.1% of baseline) in cells transfected with the constitutively active GSK-3β (GSK-3β-S9A), strongly suggesting that zinc prevents the mPTP opening through inactivation of GSK-3β.
Fig. 6.

Cell viability assay in H9c2 cells subjected to 90 min simulated ischemia followed by 30 min of reperfusion. ZnCl2 (10 μM) applied during ischemia only did not improve cell viability [Zn2+(I)], compared with the ischemia/reperfusion control (I/R). Zinc given upon reperfusion for 30 min significantly increased cell viability, indicating that zinc can prevent reperfusion injury [Zn2+(R)]. Zinc given at reperfusion failed to increase cell viability in cells transfected with the constitutively active GSK-3β mutant (S9A), indicating that zinc prevents reperfusion injury through inactivation of GSK-3β. The transfection itself did not affect cell viability (S9A). Cell viability was improved without the treatment with zinc in cells transfected with the catalytically inactive GSK-3β mutant (KM). Bars are means ± SE of at least 6 experimental observations each. *P < 0.05 vs. control; #P < 0.05 vs. I/R.
Fig. 7.

Summarized data for tetramethylrhodamine ethyl ester (TMRE) fluorescence intensity measured with confocal microscopy 20 min after exposure to H2O2. Confocal fluorescence images of TMRE were collected at baseline and 20 min after exposure to H2O2 in H9c2 cells. When compared with control, ZnCl2 (10 μM) in the presence of pyrithione prevented TMRE fluorescence reduction, indicating that exogenous zinc can prevent the mitochondrial permeability transition pore (mPTP) opening. The preventive effect of ZnCl2 on the mPTP opening was lost in cells transfected with the constitutively active GSK-3β mutant (S9A), suggesting that zinc may prevent the mPTP opening by inactivating GSK-3β. The transfection itself (S9A control) did not alter TMRE fluorescence. Bars are means ± SE of at least 6 experimental observations each. *P < 0.05 vs. controls; #P < 0.05 vs. Zn2+.
DISCUSSION
In the present study, we have demonstrated for the first time that exogenous zinc prevents reperfusion (after simulated ischemia) injury by targeting the mPTP through inactivation of GSK-3β in cardiac cells. Zinc inactivates GSK-3β through the PI3K/Akt signaling pathway but not through mTOR or PKC.
Identified originally as a regulator of glycogen metabolism, GSK-3β is now a well-established cellular component contributing to gene expression, protein synthesis, cell proliferation, cell differentiation, and apoptosis (12, 21). The activity of GSK-3β is regulated by the phosphorylation of Ser9 and Tyr216 residues. Phosphorylation of Ser9 decreases but Tyr216 phosphorylation increases GSK-3β activity. Different from many other protein kinases, GSK-3β has a very high basal activity due to the phosphorylation of its Tyr216. Some intracellular signals such as PI3K/Akt, mTOR/p70s6K, PKC, and MAPKs inactivate GSK-3β through phosphorylation of Ser9 (11). ZnCl2 (15 μM) was shown to inhibit GSK-3β in vitro (19). It has also been reported that the treatment of SH-SY5Y cells with ZnSO4 (100 μM) for 4 h significantly enhanced GSK-3β phosphorylation at both Ser9 and Tyr216 sites (1). In this study, we found that a low dose of exogenous zinc rapidly (10 min) phosphorylates GSK-3β at Ser9 in cardiac H9c2 cells, suggesting that zinc can inactivate GSK-3β. Although we could not detect the changes in GSK-3β activity in this study, data from the experiment using the GSK-3β gene mutants (Fig. 6) strongly support that zinc acts through the inactivation of GSK-3β.
GSK-3β is inactivated by the PI3K/Akt pathway, PKC, PKA, mTOR/p70s6K, and MAPK (11, 21). Zinc can stimulate the PI3K/Akt signaling (5, 6, 25, 42) and has been reported to increase GSK-3β phosphorylation at Ser9 mainly through the PI3K pathway (1). In human lung epithelial cells, zinc prevents death receptor-mediated apoptotic cell death through the activation of the PI3K/Akt signaling pathway (4). In the present study, the PI3K inhibitor LY-294002 abolished the effect of zinc on GSK-3β phosphorylation at Ser9 and zinc dramatically increased Akt phosphorylation at Ser473, suggesting that zinc inactivates GSK-3β through the PI3K/Akt signaling pathway in cardiac H9c2 cells. In contrast to the inhibitory effect of LY-294002, either the mTOR inhibitor rapamycin or the PKC inhibitor chelerythrine failed to reverse the action of zinc on GSK-3β phosphorylation. In addition, we also found that zinc did not alter mTOR phosphorylation. These results indicate that both mTOR and PKC may not be involved in the event in which zinc inhibits GSK-3β. In this study, zinc increased p70s6K phosphorylation at Thr389, which supports previous reports that zinc phosphorylates p70s6K in Swiss 3T3 cells (24) and in SH-SY5Y cells (1). Although p70s6K is a well-known upstream regulator of GSK-3β activity, it remains unclear whether p70s6k activation plays a role in zinc-induced GSK-3β phosphorylation as an upstream signal.
In addition to regulation by phosphorylation, the action of GSK-3β is also regulated by its subcellular localization (21). Although GSK-3β is predominantly present in cytosol, it is also found in mitochondria and nuclei, where it is highly activated compared with cytosolic GSK-3β (9, 21). Mitochondria have been proposed as a critical regulator of cell death and life, since a rapid increase in permeability of the inner membrane due to the mPTP opening induces both apoptosis and necrosis (28). The mPTP opening plays a critical role in myocardial reperfusion injury, and the suppression of the pore opening leads to cardioprotection against reperfusion injury (16). A recent study has demonstrated that ischemic preconditioning plus erythropoietin infusion induces cardioprotection by inhibiting the mPTP opening through increased mitochondrial phospho-GSK-3β binding to adenine nucleotide translocase, a key component of the mPTP (32). Thus it is worthwhile to test whether zinc can also increase the level of phospho-GSK-3β in mitochondria. Our data showed that GSK-3β is mainly located in cytosol and that the phosphorylation levels of GSK-3β are very low in cytosol and mitochondria in untreated cells (Fig. 4). When treated with exogenous zinc, the GSK-3β phosphorylation was markedly increased not only in cytosol but also in mitochondria. Obviously, this will result in an inhibition of the mPTP opening, if mitochondrial phospho-GSK-3β can suppress the pore opening by interacting with mPTP components.
Zinc has been reported to activate KATP channels (37), and the opening of KATP channel with diazoxide could increase GSK-3β phosphorylation in cardiac mitochondria (22). Yet, it is well known that mitoKATP channel opening leads to cardioprotection against ischemia/reperfusion injury (10). Thus we examined whether zinc enhances mitochondrial GSK-3β phosphorylation by the opening of mitoKATP channels. Our data showed that the selective mitoKATP blocker 5-HD was not able to alter the effect of zinc on mitochondrial GSK-3β phosphorylation, indicating that zinc-induced mitochondrial GSK-3β phosphorylation is not mediated by the opening of mitoKATP. Further studies are required to understand the exact mechanism underlying zinc-induced mitochondrial GSK-3β phosphorylation.
The suppression of GSK-3β activity has been demonstrated to be an important mechanism for cardioprotection induced by ischemic preconditioning or by some cardioprotectants that were applied at reperfusion. The suppression of GSK-3β is proposed to protect myocardium by inhibiting the mPTP opening (22). Since the mPTP opens upon reperfusion but not during ischemia (14) and since many cardioprotective interventions applied at reperfusion can prevent the mPTP opening (17), the inactivation of GSK-3β during reperfusion is critical for cardioprotection against reperfusion injury. In the present study, we found that zinc was cardioprotective when applied upon reoxygenation but not during simulated ischemia, suggesting that exogenous zinc induces cardioprotection against reperfusion injury. This finding also suggests that zinc may have a great clinical potential since patients with acute myocardial infarction mainly present after the onset of coronary occlusion. Our finding also supports a recent report stating that zinc applied at reperfusion protects isolated rat hearts subjected to ischemia/reperfusion (23). Zinc has been demonstrated to prevent apoptosis in both in vitro and in vivo models (30, 39). Zinc can also suppress apoptosis in cardiac allografts in a dose-dependent manner (26). The antioxidant effect and a direct suppression of caspase-3 were proposed to contribute to the antiapoptotic effect of zinc (35, 41). However, a recent report documented that zinc can prevent apoptosis through the activation of ERK in SH-SY5Y neuroblastoma cells (2), implying that exogenous zinc may protect cells through intracellular signaling transduction. Recently, our group has demonstrated that exogenous zinc prevents oxidant-induced mPTP opening in cardiomyocytes, suggesting that zinc can protect the myocardium by targeting mitochondria (20). Our data show that zinc was not able to reduce reperfusion injury in cells transfected with the constitutively active GSK-3β (GSK-3β-S9A) mutant and that zinc can phosphorylate GSK-3β at Ser9 (inactivation), indicating that zinc protects cells from reperfusion injury by inactivating GSK-3β. We further found that cells transfected with the catalytically inactive GSK-3β (GSK-3β-KM) mutant in the absence of zinc revealed cardioprotection that was similar to that observed when cells were treated with zinc, further suggesting the importance of GSK-3β suppression in cardioprotection against ischemia/reperfusion injury.
Since the inactivation of GSK-3β is necessary for the cardioprotection of zinc in the present study, it is interesting to probe the cellular mechanism by which GSK-3β mediates the protective effect of zinc. Juhaszova et al. (22) have proposed that the convergence of various cardioprotective signaling pathways via inactivation of GSK-3β on mPTP is the general mechanism of cardiomyocyte protection. In addition, Gomez et al. (13) have recently reported that postconditioning failed to prevent the mPTP opening upon reperfusion in transgenic GSK-3β-S9A mice, suggesting that GSK-3β inactivation by postconditioning is required for the prevention of the mPTP opening during reperfusion. In previous studies from our laboratory (33, 34), bradykinin and IB-MECA prevented reperfusion injury by targeting mPTP through the inactivation of GSK-3β. The present study has shown that exogenous zinc can prevent oxidant-induced mPTP opening by inactivating GSK-3β, indicating an essential role of mPTP in zinc-induced cardioprotection. Therefore, we propose that zinc may prevent reperfusion injury by targeting the mPTP via the inactivation of GSK-3β. However, it should be mentioned that we determined the mPTP opening by monitoring changes in the mitochondrial membrane potential with TMRE and that a loss of the mitochondrial membrane potential may not always exclusively indicate the mPTP opening.
In summary (Fig. 8), exogenous zinc inactivates GSK-3β via the PI3K/Akt signaling pathway. The inactivation of GSK-3β leads to cardioprotection against reperfusion injury, which might be obtained through the inhibition of the mPTP opening. It is, therefore, reasonable to propose that the treatment of the heart with zinc might be of great benefit to patients with acute myocardial infarction. However, it should be mentioned that the cardioprotective effect of zinc were evaluated in cultured cardiac H9c2 cells subjected to simulated ischemia/reperfusion, rather than in isolated cardiomyocytes or perfused heart models. Obviously, further studies using perfused or open chest animal hearts are required to corroborate the current findings in the “real” setting of myocardial ischemia/reperfusion.
Fig. 8.
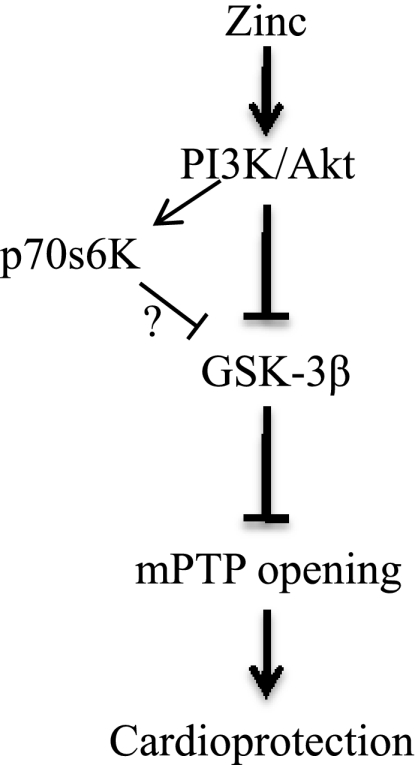
Signal transduction pathway leading to the cardioprotection of zinc at reperfusion.
GRANTS
This work was supported by National Heart, Lung, and Blood Institute Grant R01-HL-08336.
Acknowledgments
We are grateful to Dr. Morris Birnbaun (University of Pennsylvania School of Medicine) for providing the plasmids.
The costs of publication of this article were defrayed in part by the payment of page charges. The article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
REFERENCES
- 1.An WL, Bjorkdahl C, Liu R, Cowburn RF, Winblad B, Pei JJ. Mechanism of zinc-induced phosphorylation of p70 S6 kinase and glycogen synthase kinase 3β in SH-SY5Y neuroblastoma cells. J Neurochem 92: 1104–1115, 2005. [DOI] [PubMed] [Google Scholar]
- 2.An WL, Pei JJ, Nishimura T, Winblad B, Cowburn RF. Zinc-induced anti-apoptotic effects in SH-SY5Y neuroblastoma cells via the extracellular signal-regulated kinase 1/2. Brain Res Mol Brain Res 135: 40–47, 2005. [DOI] [PubMed] [Google Scholar]
- 3.Baines CP, Zhang J, Wang GW, Zheng YT, Xiu JX, Cardwell EM, Bolli R, Ping P. Mitochondrial PKCepsilon and MAPK form signaling modules in the murine heart: enhanced mitochondrial PKCepsilon-MAPK interactions and differential MAPK activation in PKCepsilon-induced cardioprotection. Circ Res 90: 390–397, 2002. [DOI] [PubMed] [Google Scholar]
- 4.Bao S, Knoell DL. Zinc modulates airway epithelium susceptibility to death receptor-mediated apoptosis. Am J Physiol Lung Cell Mol Physiol 290: L433–L441, 2006. [DOI] [PubMed] [Google Scholar]
- 5.Barthel A, Ostrakhovitch EA, Walter PL, Kampkötter A, Klotz LO. Stimulation of phosphoinositide 3-kinase/Akt signaling by copper and zinc ions: mechanisms and consequences. Arch Biochem Biophys 463: 175–182, 2007. [DOI] [PubMed] [Google Scholar]
- 6.Basuki W, Hiromura M, Sakurai H. Insulinomimetic Zn complex [Zn(opt)2] enhances insulin signaling pathway in 3T3-L1 adipocytes. J Inorg Biochem 101: 692–699, 2007. [DOI] [PubMed] [Google Scholar]
- 7.Berg JM, Shi Y. The galvanization of biology: a growing appreciation for the roles of zinc. Science 271: 1081–1085, 1996. [DOI] [PubMed] [Google Scholar]
- 8.Beyersmann D, Haase H. Functions of zinc in signaling, proliferation and differentiation of mammalian cells. Biometals 14: 331–341, 2001. [DOI] [PubMed] [Google Scholar]
- 9.Bijur GN, Jope RS. Glycogen synthase kinase-3β is highly activated in nuclei and mitochondria. Neuroreport 14: 2415–2419, 2003. [DOI] [PubMed] [Google Scholar]
- 10.Cohen MV, Baines CP, Downey JM. Ischemic preconditioning: from adenosine receptor to KATP channel. Annu Rev Physiol 62: 79–109, 2000. [DOI] [PubMed] [Google Scholar]
- 11.Cohen P, Frame S. The renaissance of GSK3. Nat Rev Mol Cell Biol 2: 769–776, 2001. [DOI] [PubMed] [Google Scholar]
- 12.Frame S, Cohen P. GSK3 takes centre stage more than 20 years after its discovery. Biochem J 359: 1–16, 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gomez L, Paillard M, Thibault H, Derumeaux G, Ovize M. Inhibition of GSK3β by postconditioning is required to prevent opening of the mitochondrial permeability transition pore during reperfusion. Circulation 117: 2761–2768, 2008. [DOI] [PubMed] [Google Scholar]
- 14.Griffiths EJ, Halestrap AP. Mitochondrial non-specific pores remain closed during cardiac ischaemia, but open upon reperfusion. Biochem J 307: 93–98, 1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gross ER, Hsu AK, Gross GJ. Opioid-induced cardioprotection occurs via glycogen synthase kinase β inhibition during reperfusion in intact rat hearts. Circ Res 94: 960–966, 2004. [DOI] [PubMed] [Google Scholar]
- 16.Halestrap AP, Clarke SJ, Javadov SA. Mitochondrial permeability transition pore opening during myocardial reperfusion—a target for cardioprotection. Cardiovasc Res 61: 372–385, 2004. [DOI] [PubMed] [Google Scholar]
- 17.Hausenloy DJ, Maddock HL, Baxter GF, Yellon DM. Inhibiting mitochondrial permeability transition pore opening: a new paradigm for myocardial preconditioning? Cardiovasc Res 55: 534–543, 2002. [DOI] [PubMed] [Google Scholar]
- 18.Ho JC, Wu S, Kam KW, Sham JS, Wong TM. Effects of pharmacological preconditioning with U50488H on calcium homeostasis in rat ventricular myocytes subjected to metabolic inhibition and anoxia. Br J Pharmacol 137: 739–748, 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ilouz R, Kaidanovich O, Gurwitz D, Eldar-Finkelman H. Inhibition of glycogen synthase kinase-3β by bivalent zinc ions: insight into the insulin-mimetic action of zinc. Biochem Biophys Res Commun 295: 102–106, 2002. [DOI] [PubMed] [Google Scholar]
- 20.Jang Y, Wang H, Xi J, Mueller RA, Norfleet EA, Xu Z. NO mobilizes intracellular Zn2+ via cGMP/PKG signaling pathway and prevents mitochondrial oxidant damage in cardiomyocytes. Cardiovasc Res 75: 426–433, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jope RS, Johnson GVW. The glamour and gloom of glycogen synthase kinase-3. Trends Biochem Sci 29: 95–102, 2004. [DOI] [PubMed] [Google Scholar]
- 22.Juhaszova M, Zorov DB, Kim SH, Pepe S, Fu Q, Fishbein KW, Ziman BD, Wang S, Ytrehus K, Antos CL, Olson EN, Sollott SJ. Glycogen synthase kinase-3β mediates convergence of protection signaling to inhibit the mitochondrial permeability transition pore. J Clin Invest 113: 1535–1549, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Karagulova G, Yue Y, Moreyra A, Boutjdir M, Korichneva I. Protective role of intracellular zinc in myocardial ischemia/reperfusion is associated with preservation of protein kinase C isoforms. J Pharmacol Exp Ther 321: 517–525, 2007. [DOI] [PubMed] [Google Scholar]
- 24.Kim S, Jung Y, Kim D, Koh H, Chung J. Extracellular zinc activates p70 S6 kinase through the phosphatidylinositol 3-kinase signaling pathway. J Biol Chem 275: 25979–25984, 2000. [DOI] [PubMed] [Google Scholar]
- 25.Kim S, Jung Y, Kim D, Koh H, Chung J. Extracellular zinc activates p70s6 kinase through the phosphatidylinositol 3-kinase signaling pathway. J Biol Chem 275: 25979–25984, 2000. [DOI] [PubMed] [Google Scholar]
- 26.Kown MH, Van der Steenhoven T, Blankenberg FG, Hoyt G, Berry GJ, Tait JF, Strauss HW, Robbins RC. Zinc-mediated reduction of apoptosis in cardiac allografts. Circulation 102: III228–III232, 2000. [DOI] [PubMed] [Google Scholar]
- 27.Krezel A, Hao Q, Maret W. The zinc/thiolate redox biochemistry of metallothionein and the control of zinc ion fluctuations in cell signaling. Arch Biochem Biophys 463: 188–200, 2007. [DOI] [PubMed] [Google Scholar]
- 28.Kroemer G, Dallaporta B, Resche-Rigon M. The mitochondrial death/life regulator in apoptosis and necrosis. Annu Rev Physiol 60: 619–642, 1998. [DOI] [PubMed] [Google Scholar]
- 29.Lynch CJ, Patson BJ, Goodman SA, Trapolsi D, Kimball SR. Zinc stimulates the activity of the insulin- and nutrient-regulated protein kinase mTOR. Am J Physiol Endocrinol Metab 281: E25–E34, 2001. [DOI] [PubMed] [Google Scholar]
- 30.Matsushita K, Kitagawa K, Matsuyama T, Ohtsuki T, Taguchi A, Mandai K, Mabuchi T, Yagita Y, Yanagihara T, Matsumoto M. Effect of systemic zinc administration on delayed neuronal death in the gerbil hippocampus. Brain Res 743: 362–365, 1996. [DOI] [PubMed] [Google Scholar]
- 31.Meares GP, Jope RS. Resolution of the nuclear localization mechanism of glycogen synthase kinase-3: functional effects in apoptosis. J Biol Chem 282: 16989–17001, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nishihara M, Miura T, Miki T, Tanno M, Yano T, Naitoh K, Ohori K, Hotta H, Terashima Y, Shimamoto K. Modulation of the mitochondrial permeability transition pore complex in GSK-3β-mediated myocardial protection. J Mol Cell Cardiol 43: 564–570, 2007. [DOI] [PubMed] [Google Scholar]
- 33.Park SS, Zhao H, Jang Y, Mueller RA, Xu Z. N6-(3-Iodobenzyl)-adenosine-5′-N-methylcarboxamide confers cardioprotection at reperfusion by inhibiting mitochondrial permeability transition pore opening via glycogen synthase kinase 3β. J Pharmacol Exp Ther 318: 124–131, 2006. [DOI] [PubMed] [Google Scholar]
- 34.Park SS, Zhao H, Mueller R, Xu Z. Bradykinin prevents reperfusion injury by targeting mitochondrial permeability transition pore through glycogen synthase kinase 3β. J Mol Cell Cardiol 40: 708–716, 2006. [DOI] [PubMed] [Google Scholar]
- 35.Perry DK, Smyth MJ, Stennicke HR, Salvesen GS, Duriez P, Poirier GG, Hannun YA. Zinc is a potent inhibitor of the apoptotic protease, caspase-3. A novel target for zinc in the inhibition of apoptosis. J Biol Chem 272: 18530–18533, 1997. [DOI] [PubMed] [Google Scholar]
- 36.Powell SR, Hall D, Aiuto L, Wapnir RA, Teichberg S, Tortolani AJ. Zinc improves postischemic recovery of isolated rat hearts through inhibition of oxidative stress. Am J Physiol Heart Circ Physiol 266: H2497–H2507, 1994. [DOI] [PubMed] [Google Scholar]
- 37.Prost AL, Bloc A, Hussy N, Derand R, Vivaudou M. Zinc is both an intracellular and extracellular regulator of KATP channel function. J Physiol 559: 157–167, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Suleiman MS, Halestrap AP, Griffiths EJ. Mitochondria: a target for myocardial protection. Pharmacol Ther 89: 29–46, 2001. [DOI] [PubMed] [Google Scholar]
- 39.Thomas DJ, Caffrey TC. Lipopolysaccharide induces double-stranded DNA fragmentation in mouse thymus: protective effect of zinc pretreatment. Toxicology 68: 327–337, 1991. [DOI] [PubMed] [Google Scholar]
- 40.Tong H, Imahashi K, Steenbergen C, Murphy E. Phosphorylation of glycogen synthase kinase-3β during preconditioning through a phosphatidylinositol-3-kinase-dependent pathway is cardioprotective. Circ Res 90: 377–379, 2002. [DOI] [PubMed] [Google Scholar]
- 41.Truong-Tran AQ, Carter J, Ruffin RE, Zalewski PD. The role of zinc in caspase activation and apoptotic cell death. Biometals 14: 315–330, 2001. [DOI] [PubMed] [Google Scholar]
- 42.Walter PL, Kampkötter A, Eckers A, Barthel A, Schmoll D, Sies H, Klotz LO. Modulation of FoxO signaling in human hepatoma cells by exposure to copper or zinc ions. Arch Biochem Biophys 454: 107–113, 2006. [DOI] [PubMed] [Google Scholar]