

Fluorescence labeling of biomacromolecules and cells has become one of the major tools to study structural organization and intra- and intermolecular processes within biological systems.1 Indeed, both highly fluorescent dyes and semiconductive quantum dots (QDs) are widely employed as fluorescent tags.2 Dual-color fluorescent tags, especially those that can be reversibly photoswitched, are particularly desirable because they can distinguish sites of interest from false positive signals generated by adventitious fluorescent biomolecules.3 Herein, we report a class of polymer nanoparticles containing both fluorescent dyes and photoswitchable chromophores within its hydrophobic core. Reversible photoisomerization of the second chromophore toggles it between forms that can or cannot quench the photoexcited fluorescent dye via energy transfer. The photoexcited acceptor is also emissive, so that the energy of the emitted photon, i.e., the identity of the fluorophore, is dependent upon the isomeric state of the second photoswitchable chromophore.
The specific synthetic approach uses an emulsion polymerization method to polymerize N-isopropylacrylamide (NIPAM) in the presence of other polymerizable monomers, including styrene (St), divinylbenzene (DVB), and the acrylatelinked dyes perylene diimide (PDI) and spiropyran (SP) (Scheme 1). When using a water-soluble initiator, NIPAM initially polymerizes because the other monomeric units are not appreciably water-soluble. Poly(N-isopropylacrylate) (PNIPAM), however, undergoes a hydrophilic to hydrophobic transition above its low critical transition temperature (LCST), Tt = 31 °C.4 As a result, PNIPAM collapses into hydrophobic spheres at elevated temperatures with the assistance of the surfactant Tween 20. This transition then allows the incorporation of the solubilized hydrophobic monomers into the growing polymer nanospheres.
Scheme 1.
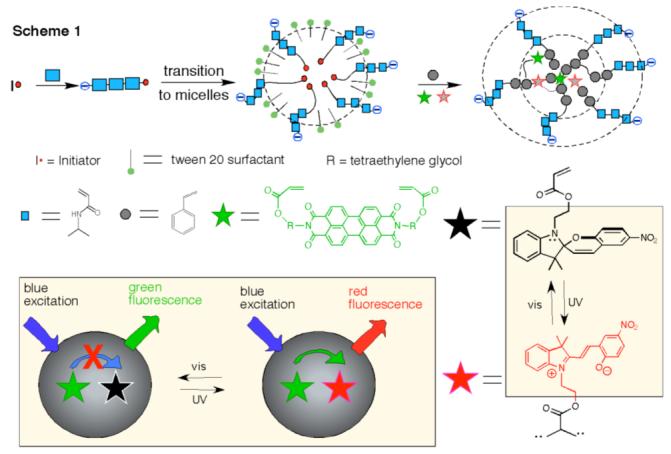
The as-prepared nanoparticles are spherical with mean diameters varying from 50 nm to 110 nm depending on the feed ratio of monomers. The particles shown in Figure 1A are nearly monodisperse and have an average size of ∼56 nm, as characterized by transmission electron microscopy (TEM). They also have negatively charged carboxyl functional groups on their surfaces; hence, they can be easily dispersed in water and are stable in many biological buffers and extracellular fluids both above and below their LCST. When the photoswitchable dye is in its ring-closed (spiro) form, the perylene chromophore located in the hydrophobic core strongly emits a distinctive green fluorescence (λmax ∼535 nm, with a strong overtone at 575 nm; quantum yield (φ≈ 0.9)).5 However, UV-induced ring opening converts the spiro-compound to its open-ring form (merocyanine)6 whose visible absorption band (λmax ∼588 nm) nearly coincides with the perylene emission bands. Consequently, the perylene emission is strongly quenched by fluorescence resonance energy transfer (FRET).7 Because the spiro form has no visible absorption with energy lower than the π→π* transition of perylene dye, it cannot quench the perylene fluorescence by this mechanism. Slow thermal ring closing of the mero form regenerates the spiro form in a reaction that is accelerated by visible light; in either case, this conversion is accompanied by the appearance of the intrinsic green fluorescence of the perylene chromophore.
Figure 1.
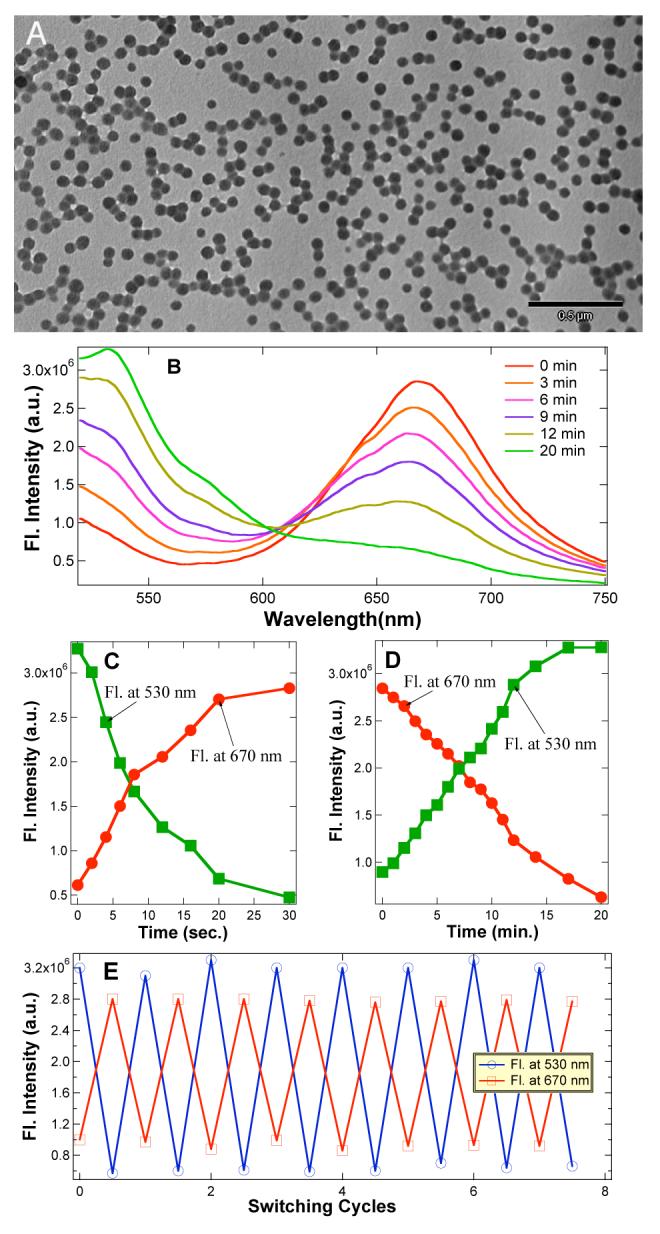
(A) TEM images of polymer nanoparticles (PNPs) (∼56 nm) containing active perylene diimide dye and spiropyran dye. (B) 50-nm polymer nanoparticles (7.0 nM) fluoresce intense red while exciting at 420 nm; red-fluorescence gradually diminishes with concomitant increase in green fluorescence upon exposure to visible light (λ > 515 nm) at 8-10 × 10-9 Einstein. (C) Plot of a single cycle, in which the particle fluorescence switches from green to red in ∼30 sec under UV light 2.8 × 10-9 Einstein. (D) Plot of a single reverse cycle, in which the particle fluorescence switches from red to green. Particle size and concentration is same as (B). (E) Fluorescence switching between red color and green color was achieved while alternating UV and visible light illumination followed by fluorescence measurements at 420 nm, respectively. The UV light illumination period is 2 min and visible light illumination period is 20 min while all other parameters are same as B, C, and D.
Neither spiro nor mero forms of the dye fluoresce appreciably in water, although the mero form possesses weak fluorescence in polar organic solvents and within self-assembled films.8 However, in the hydrophobic cavities of water-soluble nanoparticles, the mero-form acquires strong red fluorescence (600 to 750 nm, with φ = 0.18 estimated by comparison to a rhodamine B standard).9 Consequently, FRET converts the green emission from perylene to red emission from merocyanine when the spiro-containing nanoparticles are photochemically switched to mero-containing nanoparticles. This effect is illustrated in Figure 1B, where exposure to UV-radiation at 365 nm switches green-fluorescence off and turns on strong red fluorescence. Conversely, excitation with visible light at 488 nm switches the fluorescence from red to green.
Because UV light induces the spiro → mero transition and visible light reverses the process, there must be a critical wavelength where net switching is zero, i.e., where the relative mero- and spiro-forms of the dye remain constant. This critical wavelength is ∼ 420 nm, at which spiro-nanoparticles and mero-nanoparticles are found to emit steady green and red fluorescence, respectively. This wavelength can be used to interrogate the particles while they undergoing photoinitiated switching by exposure to light at higher or lower energies (Figures 1C,D).
The photoluminescence switching speed depends on a number of parameters such as photon wavelength, radiation intensity, sample concentration, and penetration depth. For 7.0 nM spironanoparticles and a photon flux of 2.8 × 10-9 Einstein (260 nm > λ > 390 nm), it takes ∼30 sec to switch a 1-cm cuvette from green fluorescence to red fluorescence (Figure 1C); the reverse process under photon flux of 8-10 × 10-9 Einstein (λ > 515 nm) requires ∼20 min. Photoisomerization quantum yields are generally ≤ 0.6;10 therefore photoswitching can be fast under the optimum conditions. Indeed, optically switching of single spiro- or mero-nanoparticles is very fast and only requires low photon intensity (vide infra). The wavelength at which the particle fluorescence is rapidly switched from green to red is 350 ± 10 nm, corresponding to the low-energy absorption maximum of the spiropyran. Conversely, excitation into the absorption band of merocyanine at 588 nm causes effectively switching from red to green fluorescence.
In addition to enhancing merocyanine fluorescence, incorporating the spiropyran dye into the hydrophobic core of the nanoparticle substantially enhances its photochemical stability. As is evident from the data in Figure 1E, optical switching of fluorescence can be repeated numerous times without any apparent “fatigue” effects, or photobleaching, that are thought to be caused primarily by bimolecular degradative reactions, involving the triplet-excited spiropyran.11 Immobilization of the dye within the protective hydrophobic nanoenvironment significantly impedes these bimolecular reactions.
A key question is what is the fundamental switching unit, i.e., does the macroscopic ensemble-averaged fluorescence accurately reflect the emissive properties of the individual nanoparticles? As illustrated in Figure 2, spectra obtained from single particles demonstrate that the fundamental switching units do indeed reside within individual particles. For these measurements, samples were pre-illuminated at 365 nm with a mercury lamp and were subsequently excited using a 488 nm Ar ion laser. Pre-illumination at 365 nm converted the spiropyran dye to its mero-form and therefore the initial particle fluorescence was red. Since 488-nm light induces mero → spiro photoisomerization, the fluorescence color subsequently switched from red to green under continuous illumination from the laser beam.
Figure 2.
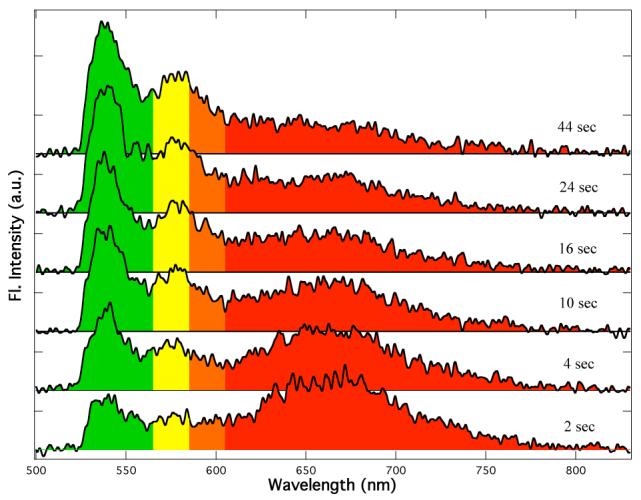
Fluorescence spectra obtained upon 488 nm excitation of a single polymer nanoparticle after pre-illumination of the sample with a UV lamp for 2 min. As time elapses, the red fluorescence converts to green fluorescence.
Fluorescence color switching within the particles was almost instantaneous under laser powers normally used for single particle imaging. To obtain the spectra shown in Figure 2, it was necessary to reduce the laser power from normal 0.8 kW/cm2 to 0.4 kW/cm2 in order to slow down the photoisomerization to a rate at which the process could be monitored. Even at this low power, switching was already evident in the 2s acquisition time required to obtain the first spectral trace (Figure 2). Similar to ensemble measurements, fluorescence color switching from green to red was >10 times faster than from red to green and its rate could not be monitored with the available equipment.
At the dye loading levels used in the 50 nm particles, the average separation distance between dyes is ∼ 6 nm, assuming that they are uniformly dispersed within the hydrophobic core of the nanoparticles; this distance falls within the usual range of Förster distances (Ro), which are typically 2-6 nm.12 This close proximity of donor and acceptor molecules, coupled with the high degree of spectral overlap between the perylene emission and merocyanine absorption bands probably causes FRET to be highly efficient. This efficiency can be modulated by altering the number density of spiropyran dye molecules, i.e., the distance between energy donor and acceptor, within the nanoparticles; thus, for example, in cases where spectral overlaps are not optimal, partial compensation can be achieved by increasing the relative amount of acceptor. For example, at SP/PDI feed ratio of 44, particle fluorescence cannot be switched completely to red because of unquenched green fluorescence. The critical SP/PDI ratio, which ensure complete switching from green fluorescence to red fluorescence and vice versus, is estimated to be 100-200. Thus, adjusting the dye ratio during the polymerization is central to achieving nanoparticles with good color switching properties.
One paramount issue is to achieve green <=> red fluorescence switching in the cytosolic media. Because the fluorophores are protected by the polymer shells, fluorescent nanospheres should be much better protected than single fluorophores from collisional quenching or cell milieu-induced nonradiative decay. At culture temperature 37 °C, greater than LCST, colloidal polymer particles are stable in the extracellular fluid and no aggregation was observed.
Dual-color polymer nanoparticles were delivered into human embryonic kidney (HEK-293) cells by gently mixing dilute suspensions of nanoparticles with LipofectamineTM 2000 (Invitrogen), followed by incubation with the cells (Figure 3). Fluorescence images were acquired using a 488-nm laser and a liquid-nitrogen cooled CCD detector. The nanoparticles delivered into the cells contained the ring-closed spiropyran and therefore gave strong green fluorescence (Figure 3B). Brief exposure to UV illumination with a hand held lamp converted the spiropyran to its mero-form, resulting in red fluorescence emission (Figure 3C); this fluorescence color switching was reversible in the cytoplasmic cell compartment (Figures 3D,E). These results confirm the potential usefulness of dual color nanoparticles as fluorescent probes in biological systems exhibiting high background autofluorescence or possessing fluorophores that interfere in one wavelength region. Thus, the dual-color nanoparticles can be selectively highlighted with either green or red fluorescence.
Figure 3.
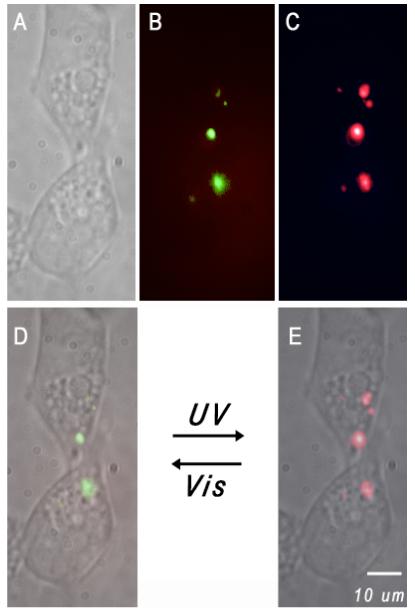
Dual color fluorescent nanoparticles were transported into HEK-293 cells using liposomes as delivery vehicles; these internalized polymer nanoparticles can be selectively highlighted with either green or red fluorescence. (A) White light image of the cells under studies. Fluorescence imaging of the same cells is shown when the nanoparticles emit green (B) or red color (C), respectively. Images (D) and (E) are overlays of (A) and (B) or (A) and (C), respectively. A short UV pulse switches the highlighted green spots (D) to vivid red fluorescence (E) while visible light reverses the process.
In conclusion, photoswitchable spiropyran dyes can be integrated with fluorescent perylene diimide into the hydrophobic core of hydrophilic polymer nanoparticles to generate optically addressable two-color fluorescent systems that exhibit high photoluminescence and superior resistance to photobleaching. Relative intensities of the red and green fluorescent components can be controlled by the feed ration used during formation by emulsion polymerization. These high-contrast, highly intense dual-color photoluminescent nanoparticles appear particularly suited to applications in tracking and labeling components of complex biological systems.
Supplementary Material
Acknowledgment
We acknowledge the support of National Institute of General Medical Sciences (Grant GM065306 to ADQL) and the U.S. Department of Energy, Office of Basic Energy Sciences, Divisions of Chemical Sciences (Grant DEFG03-99ER14943 to JKH) and Materials Science and Engineering (to ADQL). ADQL is a Beckman Young Investigator (BYI).
Footnotes
Supporting Information Available: Experimental methods.
References
- (1) (a).Michalet X, Pinaud FF, Bentolila LA, Tsay JM, Doose S, Li JJ, Sundaresan G, Wu AM, Gambhir SS, Weiss S. Science. 2005;307:538–544. doi: 10.1126/science.1104274. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Weissleder R. Ntziachristos. V. Nat. Med. 2003;9:123–128. doi: 10.1038/nm0103-123. [DOI] [PubMed] [Google Scholar]
- (2) (a).Zimmer M. Chem. Rev. 2002;3:759–781. doi: 10.1021/cr010142r. [DOI] [PubMed] [Google Scholar]; (b) Medintz IL, Uyeda HT, Goldman ER, Mattoussi H. Nat. Mater. 2005;4:435–446. doi: 10.1038/nmat1390. [DOI] [PubMed] [Google Scholar]
- (3) (a).Chudakov DM, Verkhusha VV, Staroverov DB, Souslova EA, Lukyanov S, Lukyanov KA. Nat. Biotechnol. 2004;11:1435–1439. doi: 10.1038/nbt1025. [DOI] [PubMed] [Google Scholar]; (b) Wiedenmann J, Ivanchenko S, Oswald F, Schmitt F, Rocker C, Salih A, Spindler KD, Nienhaus GU. Proc. Natl. Acad. Sci. USA. 2004;101:15905–15910. doi: 10.1073/pnas.0403668101. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Ando R, Mizuno H, Miyawaki A. Science. 2004;306:1370–1373. doi: 10.1126/science.1102506. [DOI] [PubMed] [Google Scholar]
- (4).Schild HG. Prog. Polym. Sci. 1992;17:163–249. [Google Scholar]
- (5).Sadrai M, Hadel L, Sauers RR, Husain S, Krogh-Jespersen K, Westbrook JD, Bird GR. J. Phys. Chem. 1992;96:7988–7996. [Google Scholar]
- (6) (a).Crano JC, Guglielmetti RJ, editors. Organic Photochromic and Thermochromic Compounds. Plenum Press; New York: 1999. [Google Scholar]; (b) Berkovic G, Krongauz V, Weiss V. Chem. Rev. 2000;100:1741–1754. doi: 10.1021/cr9800715. [DOI] [PubMed] [Google Scholar]; (d) Minkin VI. Chem Rev. 2004;104:2751–2776. doi: 10.1021/cr020088u. [DOI] [PubMed] [Google Scholar]
- (7) (a).Giordano L, Jovin TM, Irie M, Jares-Erijman EA. J. Am. Chem. Soc. 2002;124:7481–7489. doi: 10.1021/ja016969k. [DOI] [PubMed] [Google Scholar]; (b) Norsten TB, Branda NR. J. Am. Chem. Soc. 2001;123:1784–1785. doi: 10.1021/ja005639h. [DOI] [PubMed] [Google Scholar]; (c) Zhu L, Zhu M-Q, Hurst JK, Li AD. Q. J. Am. Chem. Soc. 2005;127:8968–8970. doi: 10.1021/ja0423421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8) (a).Bahr JL, Kodis G, de la Garza L, Lin S, Moore AL, Moore TA, Gust D. J. Am. Chem. Soc. 2001;123:7124–7133. doi: 10.1021/ja010058t. [DOI] [PubMed] [Google Scholar]; (b) Minami T, Tamai N, Yamazaki T, Yamazaki I. J. Phys. Chem. 1991;95:3988–3993. [Google Scholar]
- (9).Zhu M-Q, Zhu L, Han JJ, Wu W, Hurst JK, Li AD. Q. J. Am. Chem. Soc. 2006;128:4303–4309. doi: 10.1021/ja0567642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Tamai N, Matsuhara H. Chem. Phys. Lett. 1992;191:189–194. [Google Scholar]
- (11) (a).Matsushima R, Nishiyama M, Doi M. J. Photochem. Photobiol. A: Chem. 2001;139:63–69. [Google Scholar]; (b) Sakuragi M, Aoki K, Tamaki T, Ichimura K. Bull. Chem. Soc. Jpn. 1990;63:74–79. [Google Scholar]; (c) Baillet G, Giusti G, Guglielmetti R. J. Photochem. Photobiol. A. 1993;70:157–161. [Google Scholar]; (d) Baillet G, Campredon M, Guglielmetti R, Giusti G, Aubert C. J. Photochem. Photobiol. A. 1994;83:147–151. [Google Scholar]
- (12).Miyawaki A, Tsien RY. Methods Enzymol. 2000;327:472–500. doi: 10.1016/s0076-6879(00)27297-2. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.