

Abstract
We report here the design and synthesis of a series of π-conjugated fluorescent dyes with D-A-D (D: donor; A: Acceptor), D-π-D, A-π-A, and D-π-A for applications as the signaling motif in biological-synthetic hybrid foldamers for DNA detection. Horner-Wadsworth-Emmons (HWE) reaction and Knoevenagel condensation were demonstrated as the optimum ways for construction of long π-conjugated systems. Such rod-like chromophores have distinct advantages, as their fluorescence properties are not quenched by the presence of DNA. To be incorporated into the backbone of DNA, the chromophores need to be reasonably soluble in organic solvent for solid-phase synthesis, and therefore a strategy of using flexible tetra(ethylene glycol) (TEG) linkers at either end of these rod-like dyes were developed. The presence of TEG facilitates the protection of the chain-growing hydroxyl group with DMTrCl (dimethoxy trityl chloride) as well as the activation of the coupling step with phosphoramidite chemistry on an automated DNA synthesizer. To form fluorescence resonance energy transfer (FRET) pairs, six synthetic chromophores with blue to red fluorescence have been developed and those with orthogonal fluorescent emission were chosen for incorporation into DNA-chromophore hybrid foldamers.
Introduction
Syntheses of deoxyribonucleic acid (DNA) and dye conjugates (1–3) have enabled many optical probes; these conjugate probes provide a means to study biomolecules within living cells in details via fluorescence imaging (4,5) and single molecules fluorescence spectroscopy (6–8). However, introducing multiple fluorescent dyes (N > 2) into the backbone of a single DNA chain remains largely an unsolved challenge for organic chemistry. To study complex biochemical assemblies and reactions, it is necessary in some systems to have multiple fluorescence probes on a single DNA chain. One such interesting architectural motif in supramolecular chemistry is to use the self-assembling amphiphilic properties of the dyes to induce folded nanostructures with alternating single strand DNA looping around the folded cores (9). Using this principle, hybrid biological and synthetic foldamers have been readily developed for DNA detection (10). The strategy is that organic fluorescence chromophores function as an optical reporter while the biological domain, in this case, a specific DNA sequence, functions as a high fidelity molecular recognition element. The rationale here is that when the built-in DNA sequences in the hybrid foldamer complement with the target DNA in completely or partially matched situation, the folded nanostructure of the foldamer will be converted to an extended one, a process in which fluorescence color of the foldamer will change dramatically because of FRET between two adjacent chromophores (11). Different mismatched DNA sequences lead to various degrees of emission color changes due to different levels of FRET between the organic chromophores at a given time. From the structural skeleton point of view, molecular beacons usually have organic dyes at both ends of a single DNA sequence and therefore their termini are dead end for incorporation into long DNA sequences (12–16), the hybrid foldamers have the organic dyes inserted between multiple DNA sequences, which allow further biological manipulation such that the DNA ends can be cut by endonucleases and incorporated into genes with a DNA polymerase or ligase. These special structural features make the hybrid foldamers unique and suitable candidates for studying biological mechanisms.
Since the foldamer probe with alternating hydrophobic and hydrophilic structures are constructed on solid-phase synthesis using an automated DNA synthesizer (Applied Biosystem 8909) (9), it is imperative to develop fluorescent building blocks with an activated linker which can be attached to the growing end of the DNA chain and a temporary blocking group to prevent non-desired coupling reactions and permit further chain extension after deprotection. The resulting hybrid foldamers have multiple fluorescent chromophores and its emission color is controlled by the distance between the chromophores separated by the DNA sequences (Figure 1). For completely mismatched sequences, no hybridization occurs between the foldamer and the mismatched target; therefore the flexible single stranded DNA chains allow individual chromophores to come to close proximity due to the hydrophobic and π-stacking effect and the net result is that fluorescence emission is in the long wavelength region because of the high degree of FRET between individual organic dyes. For perfect matched sequences, however, hybridization turns flexible single stranded DNA into rigid double helixes, which in turn separates chromophores, and reduces the level of FRET between the organic dyes. In this case, the emission color from the foldamer will depend on the short wavelength chromophore. Therefore, we have designed and synthesized a series of fluorescent chromophores with emission wavelength ranging from blue to red; all fluorescent compounds bear tetraethylene glycol side chains with –OH termini, which are amenable to DMTr protection and phosphoramidite chemistry.
Figure 1.
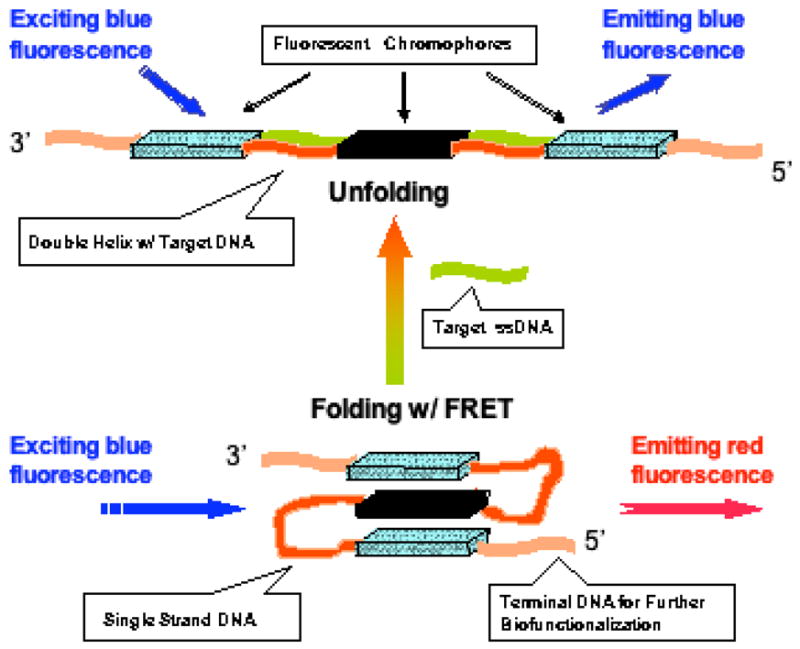
Incorporation of highly fluorescent chromophores into DNA main chains enables many biotechnologies, including DNA monitoring and detection.
As a class of bis(styryl)benzene derivatives, the donor-π-donor (D-π-D), acceptor-π-acceptor (A-π-A), donor-acceptor-donor (D-A-D), and acceptor-donor-acceptor (A-D-A) structural motifs have received a great deal of attention because they exhibit exceptional large two-photon absorption cross sections, up to ~400 times that of trans-stilbene (17). Compared to our prior perylene tetracarboxylic diimide unit, these new chromophores have desired features such as higher fluorescence quantum yield when incorporated into DNA, and adjustable excitation and emission wavelength via structural variations on similar molecular architectures. Meanwhile, a red fluorescence dye based on the symmetric structure of (2,6-dimethyl-4H-pyran-4-ylidene)malononitrile (DCM laser dye) has been developed according to a recent report (18), the large dipole moment and high decomposition temperature of this Y-shaped structure make it an attractive candidate for red fluorescence signaling reporter. Consequently, we have developed a series of organic chromophores with different fluorescent colors as molecular building blocks (Figure 2), which are suitable for construction of new hybrid foldamers as molecular probes (10).
Figure 2.

Chemical structures of highly fluorescent dye molecules that are amenable to the general strategy developed here: one hydroxyl group is protected with a dimethoxy trityl (DMTr) group and the other hydroxy group is activated with phosphoramidite.
Experimental Section
General methods: Solvents and reagents were purified where necessary using literature methods (31). In particular, N,N-dimethylformamide (DMF) was distilled from 4Å MS (Molecular Sieve) under reduced pressure and stored under argon. Acetonitrile (MeCN) was heated under reflux over calcium hydride and distilled under argon. MALDI Mass Spectra was obtained with an ABVS-2025 spectrometer. 1H NMR spectra were recorded with a Mercury 300 (300 MHz) spectrometer for solutions in CDCl3 (CD3OD) at ambient temperature. 13C NMR spectra were recorded at 75.48 MHz with a Mercury 300 spectrometer for solutions in CDCl3 (CD3OD) adopting 77.23 ppm for the central line of CDCl3. 31P-NMR chemical shifts were reported in ppm using 85% H3PO4 as an external reference. Reactions were monitored by thin-layer chromatography (TLC) on a precoated plate of silica gel 60 F254 (EM Science). Column chromatography was performed on silica gel 60 (230–400 mesh, EM Science).
Compounds in tetraethylene chain optimization and modification: 4-(2-(2-(2-Hydroxy-ethoxy)-ethoxy)-ethoxy)-benzaldehyde (7)
A mixture of 2-(2-(2-Chloroethoxy)ethoxy)ethanol (commercial) (1.0 g, 5.9 mmol), 4-hydroxybenzaldehyde (0.8 g, 6.5 mmol, 1.1 equiv), and K2CO3 (1.8 g, 2.2 equiv) in dry N,N-dimethylformamide (DMF) (10 mL) was heated to 120 °C (oil bath 110 °C) under argon. After the stirring was continued at this temperature for 16 h the reaction mixture was cooled to room temperature. The solvent (DMF) was evaporated in vacuum, and the residue was diluted with CHCl3. The obtained suspension was suction-filtered and the filtrate was collected. After dried over Na2SO4, the organic phase was concentrated, and the residue was purified by chromatography on a silica gel column eluted with EtOAc/MeOH (20:1) to give the title product (950 mg, yield, 63%) as a yellow oil: Rf 0.33 (EtOAc/MeOH 20/1). ‘H NMR (CDCl3) δ (ppm): 9.89 (s, 1H, -CHO), 7.82 (dt, 2H, J1 = 9.0Hz, J2 = 2.7Hz, benzaldehyde), 7.02 (dt, 2H, J1 = 9.0Hz, J2 = 2.7Hz, benzaldehyde), 4.21 (t, 2H, J = 4.8Hz, triethylene glycol chain), 3.89 (t, 2H, J = 4.8Hz, triethylene glycol chain), 3.75–3.67 (m, 6H, triethylene glycol chain), 3.61 (bt, 2H, J = 4.8Hz, triethylene glycol chain), 2.40 (bt, 1H, J = 6.0Hz, -OH).
4-(2-(2-(2-Trityloxy-ethoxy)-ethoxy)-ethoxy)-benzaldehyde (8)
A mixture of 7 (0.5 g, 2.0 mmol), Trityl chloride (1.2 g, 4.3 mmol, 2.2 equiv), and a catalytic amount of 4-(Dimethylamino)pyridine (DMAP) in dry pyridine (5 mL) was heated to 40–50 °C under argon. After the stirring was continued at this temperature for 24 h the reaction mixture was cooled to room temperature. The solvent was removed in vacuum, and the residue was diluted with EtOAc. The obtained suspension was suction-filtered and the filtrate was collected. After dried over Na2SO4, the organic phase was concentrated, and the residue was purified by chromatography on a silica gel column eluted with Cyclohexane/EtOAc/Pyridine (300/100/4, v/v) to give the title product (770 mg, yield 99%): Rf 0.31 (300/100/4, v/v). ‘H NMR (CDCl3) δ (ppm): 9.87 (s, 1H, -CHO), 7.80 (dt, 2H, J1 = 8.7Hz, J2 = 2.1Hz, benzaldehyde), 7.48-7.43 (m, 6H, benzene ring of trityl group), 7.46-7.18 (m, 9H, benzene ring of trityl group), 6.98 (dt, 2H, J1 = 8.7Hz, J2 = 2.1Hz, benzaldehyde), 4.20 (bt, 2H, J = 4.8Hz, triethylene glycol chain), 3.92 (bt, 2H, J = 4.8Hz, triethylene glycol chain), 3.78-3.67 (m, 6H, triethylene glycol chain), 3.24 (t, 2H, J = 4.8Hz, triethylene glycol chain).
4-[2-[2-[2-(2-hydroxyethoxy)ethoxy]ethoxy]ethoxy]-benzaldehyde (10)
A mixture of 2-(2-(2-(2-Chloroethoxy)ethoxy)ethoxy)ethanol (9) (19) (2.0 g, 9.4 mmol), 4-Hydroxybenzaldehyde (1.4 g, 1.2 equal), and K2CO3 (2.5 g, 2 equal) in dry N,N-dimethylformamide (DMF) (10 mL) was heated to 100 °C (oil bath 110 °C) under argon. The stirring was continued at this temperature for 72 h. After the reaction mixture was cooled to room temperature, the solvent (DMF) was evaporated in vacuum and the residue was diluted with CHCl3. The obtained suspension was suction-filtered and the filtrate was collected. After dried over Na2SO4, the organic phase was concentrated and the residue was purified by chromatography on a silica gel column eluted with EtOAc/MeOH (20:1) to give the title product (2.25 g, yield 80%) as a yellow oil: Rf 0.3 (EtOAc/MeOH 20/1). ‘H NMR (CDCl3) δ (ppm): 9.87 (s, 1H, -CHO), 7.82 (dt, 2H, J1 = 9.0Hz, J2 = 2.7Hz, benzaldehyde), 7.01 (dt, 2H, J1 = 9.0Hz, J2 = 2.7Hz, benzaldehyde), 4.22 (bt, 2H, J = 4.8Hz, tetraethylene glycol chain), 3.89 (bt, 2H, J = 4.8Hz, tetraethylene glycol chain), 3.77-3.59 (m, 12H, tetraethylene glycol chain), 2.53 (t, 1H, J = 6.0Hz, -OH); 13C NMR (CDC13) δ (ppm): 190.9, 163.9, 132.1, 130.1, 115.0, 72.65, 71.07, 70.86, 70.78, 70.53, 69.68, 67.92, 61.95. MS (MALDI-TOF): 299.01 [M+H]+, 320.99 [M+Na]+.
4-(2-(2-(2-(2-(Bis-(4-methoxy-phenyl)-phenyl-methoxy)-ethoxy)-ethoxy)-ethoxy)-ethoxy)-benzaldehyde (11)
A mixture of 10 (0.5 g, 1.7 mmol), DMTrCl (1.2 g, 3.5 mmol, 2 equiv), and a catalytic amount of 4-(Dimethylamino)pyridine (DMAP) in dry Pyridine (10 mL) was stirred at room temperature under argon. After 6 h, the reaction solvent was removed in vacuum and the residue was directly subject to a silica gel column eluted with Cyclohexane/EtOAc (gradient elution with 1% Pyridine: 500/150 → 300/150 → 150/150, v/v) to give the title product (1.056 g, yield 96%): Rf 0.42 (Cyclohexane/EtOAc 1/1, v/v). ‘H NMR (CDCl3) δ (ppm): 9.87 (s, 1H, -CHO), 7.79 (dt, 2H, J1 = 8.7Hz, J2 = 2.1Hz, benzaldehyde), 7.45 (m, 2H, benzene ring of DMTr group), 7.36-7.15 (m, 3H, benzene ring of DMTr group), 7.34 (dt, 4H, J1 = 9.0Hz, J2 = 2.7Hz, methoxybenzene ring of DMTr group), 6.97 (dt, 2H, J1 = 8.7Hz, J2 = 2.1Hz, benzaldehyde), 6.81 (dt, 4H, J1 = 9.0Hz, J2 = 2.7Hz, methoxybenzene ring of DMTr group) 4.15 (bt, 2H, J = 4.8Hz, tetraethylene glycol chain), 3.87 (bt, 2H, J = 4.8Hz, tetraethylene glycol chain), 3.77 (s, 6H, -OCH3), 3.77-3.64 (m, 10H, tetraethylene glycol chain), 3.22 (t, 2H, J = 4.8Hz, tetraethylene glycol chain); 13C NMR (CDC13) δ (ppm): 191.0, 164.0, 158.5, 145.3, 136.5, 132.1, 130.3, 130.2, 128.4, 127.9, 126.8, 123.9, 115.1, 113.2, 86.2, 71.2, 71.1, 71.0, 69.7, 68.0, 63.4, 55.5, 27.3.
2-(2-(2-(2-N-n-Butylaminoethoxy)ethoxy)ethoxy)ethanol (12)
To a solution of 2-(2-(2-(2-Chloroethoxy)ethoxy)ethoxy)ethanol (2 g, 9.4 mmol) in n-Butylamine 10 mL at room temperature was added K2CO3 (1 g, 0.77 equal). The reaction mixture was heated to reflux for 24 h. Then another 10 mL of n-Butylamine was added into the mixture and the reaction was continued to reflux another 12 h. The reaction mixture was cooled to room temperature and the n-butylamine was removed in vacuum. The residue was diluted with CHCl3 and the suspension was suction-filtered. The filtrate was collected and concentrated to give the title product as yellow syrup in quantitative yield, which could be used directly to the next step without further purification. ‘H NMR (CDCl3) δ (ppm):3.55-3.51 (m, 14H, tetraethylene glycol chain), 2.66 (bt, 2H, J = 5.1Hz, tetraethylene glycol chain), 2.47 (t, 2H, J = 7.2Hz, n-butyl chain), 1.43-1.31 (m, 2H, n-butyl chain), 1.30-1.16 (m, 2H, n-butyl chain), 0.80 (t, 3H, J = 7.2Hz, n-butyl chain); 13C NMR (CDC13) δ (ppm): 72.9, 70.6, 70.4, 70.3, 70.2, 61.3, 49.5, 49.2, 32.1, 20.6, 14.1. MS (MALDI-TOF): 249.92 [M+H]+, 271.92 [M+Na]+, 287.86 [M+K]+.
4-N-n-Butyl-N-[2-[2-[2-(2-hydroxyethoxy)ethoxy]ethoxy]ethyl]amino benzaldehyde (13)
A mixture of 2-[2-[2-(2-N-n-Butylaminoethoxy)ethoxy]ethoxy]ethanol (12) (2.3 g, 9.4 mmol), 4-Fluorobenzaldehyde (1.0 mL, 9.4 mmol), and K2CO3 (1.5 g, 1.1 equal) in dry N,N-dimethylformamide (DMF) (10 mL) was heated with stirring to 120 °C (oil bath 128 °C) under argon. The stirring was continued at this temperature for 72 h, and the reaction mixture was cooled to room temperature. After the solvent (DMF) was evaporated in vacuum, the residue was diluted with CHCl3. The obtained suspension was washed with brine (2× 200 mL) and dried over Na2SO4. The organic layer was concentrated and the residue was purified by chromatography on a silica gel column eluted with EtOAc/MeOH (20:1) to give the title product (1.2 g, 36% in two steps based on 9) as a yellow oil: Rf 0.4 (EtOAc/MeOH 20/1). ‘H NMR (CDCl3) δ (ppm): 9.56 (s, 1H, -CHO), 7.57 (bd, 2H, J = 9.3Hz, aromatic ring), 6.58 (bd, 2H, J = 8.7Hz, aromatic ring), 3.65-3.40 (m, 16H, tetraethylene glycol chain), 3.29 (t, 2H, J = 7.8Hz, n-butyl chain), 1.59-1.40 (m, 2H, n-butyl chain), 1.30-1.18 (m, 2H, n-butyl chain), 0.85 (t, 3H, J = 7.5Hz, n-butyl chain); 13C NMR (CDC13) δ (ppm): 189.7, 152.4, 131.9, 124.5, 110.6, 72.4, 70.5, 70.4, 70.3, 70.1, 68.1, 61.4, 51.1, 50.3, 28.9, 20.0, 13.9. MS (MALDI-TOF): 353.23 [M]+.
4-((2-(2-(2-(2-(Bis-(4-methoxy-phenyl)-phenyl-methoxy)-ethoxy)-ethoxy)-ethoxy)-ethyl)-butyl-amino)-benzaldehyde (14)
A mixture of 13 (475 mg, 1.35 mmol), DMTrCl (912 mg, 2.7 mmol, 2 equiv), and a catalytic amount of 4-(Dimethylamino)pyridine (DMAP) in dry Pyridine (10 mL) was stirred at room temperature under argon. After the stirring was continued overnight, the reaction solvent was removed in vacuum and the residue was directly subject to a silica gel column eluted with Cyclohexane/EtOAc (gradient elution with 1% Pyridine: 500/150 → 300/150 → 150/150, v/v) to give the title product (800 mg, yield 90%): Rf 0.19 (Cyclohexane/EtOAc 1/1, v/v). ‘H NMR (CDCl3) δ (ppm): 9.69 (s, 1H, -CHO), 7.68 (bd, 2H, J = 9.0Hz, benzaldehyde), 7.46 (m, 2H, benzene ring of DMTr group), 7.32-7.15 (m, 3H, benzene ring of DMTr group), 7.34 (dt, 4H, J1 = 9.0Hz, J2 = 3.0Hz, methoxybenzene ring of DMTr group), 6.81 (dt, 4H, J1 = 9.0Hz, J2 = 3.0Hz, methoxybenzene ring of DMTr group), 6.66 (bd, 2H, J = 9.0Hz, benzaldehyde), 3.77 (s, 6H, -OCH3), 3.68-3.53 (m, 14H, tetraethylene glycol chain), 3.37 (bt, 2H, J = 7.9Hz, -NCH2CH2CH2CH3), 3.22 (t, 2H, J = 5.2Hz, tetraethylene glycol chain), 1.62-1.52 (m, 2H, -NCH2CH2CH2CH3), 1.40-1.28 (m, 2H, -NCH2CH2CH2CH3), 0.95 (t, 3H, J = 7.2Hz, -NCH2CH2CH2CH3).
Compounds in preparation of asymmetric pyridinium bistyrylbenzene: 4-(2-(2-(2-(2-(tert-Butyl-dimethyl-silanyloxy)-ethoxy)-ethoxy)-ethoxy)-ethoxy)-benzaldehyde (20)
A mixture of 10 (1.4 g, 4.7 mmol), tert-Butyldimethylsilyl chloride (TBDMSCl) (0.85 g, 5.6 mmol, 1.2 equiv), and N,N-Diisopropylethylamine (DIPEA) (1.2 mL, 1.5 equiv) in dry CH2Cl2 (15 mL) was stirred at room temperature under argon. After 5 h, half of the starting material had gone. Another batch of DIPEA and TBDMSCl as well as a catalytic amount of DMAP was added. The reaction mixture was stirred overnight. The solvent (CH2Cl2) was evaporated in vacuum, and the residue was purified by chromatography on a silica gel column eluted with Cyclohexane/EtOAc (1/1) to give the title product (1.5 g, yield 77%): Rf 0.44 (Cyclohexane/EtOAc, 1/1). ‘H NMR (CDCl3) δ (ppm): 9.88 (s, 1H, -CHO), 7.83 (dt, 2H, J1 = 9.0Hz, J2 = 2.1Hz, benzaldehyde), 7.02 (dt, 2H, J1 = 9.0Hz, J2 = 2.1Hz, benzaldehyde), 4.22 (t, 2H, J = 5.1Hz, tetraethylene glycol chain), 3.89 (t, 2H, J = 5.1Hz, tetraethylene glycol chain), 3.75 (bt, 2H, J = 5.1Hz, tetraethylene glycol chain), 3.71-3.59 (m, 8H, tetraethylene glycol chain), 3.49 (bt, 2H, J = 5.1Hz, tetraethylene glycol chain), 0.83 (s, 9H, -Si(CH3)2C(CH3)3), 0.06 (s, 6H, -Si(CH3)2C(CH3)3); 13C NMR (CDC13) δ (ppm): 190.9, 164.0, 132.1, 130.2, 115.1, 72.9, 71.2, 71.0, 70.99, 70.94, 69.7, 68.0, 63.0, 26.3, 18.7, −4.9.
4-(2-(4-(2-(2-(2-(2-(tert-Butyl-dimethyl-silanyloxy)-ethoxy)-ethoxy)-ethoxy)-ethoxy)-phenyl)-vinyl)-benzonitrile (22)
A solution of Diisopropylamine (3.2 mL, 22.8 mmol, 4.3 equiv) in THF (40 mL) at room temperature under argon was cooled to −78 °C, and n-BuLi (12.5 mL, 1.6 M in Hexanes, 3.8 equiv) was added via syringe. The mixture was warmed to 0 °C for 30 min, and re-cooled to −78 °C. A solution of 4-Cyanobenzylphosphonic acid diethyl ester 21 (2.3 g, 9 mmol) in THF (15 mL) was pre-cooled, and transferred via cannula into the LDA solution at −78 °C, then the reaction mixture was warmed up to 0 °C. Another solution of 20 (2.2 g, 5.3 mmol) in THF (15 mL) was added into the reaction via cannula at 0 °C, and the reaction was warmed up to room temperature. After 24 h of stirring at room temperature, the reaction mixture was diluted with CH2Cl2 and the organic phase was washed with aqueous NaHCO3 twice, then brine. The solvent (CH2Cl2) was evaporated in vacuum, and the residue was purified by chromatography on a silica gel column eluted with CH2Cl2/EtOAc (5/1, v/v) to give the title product (2.1 g, yield 84%): Rf 0.66 (DCM/EtOAc, 5/1). ‘H NMR (CDCl3) δ (ppm): 7.61 (dt, 2H, J1 = 8.7Hz, J2 = 1.8Hz, styryl benzene), 7.55 (dt, 2H, J1 = 8.7Hz, J2 = 1.8Hz, styryl benzene), 7.45 (dt, 2H, J1 = 8.7Hz, J2 = 2.1Hz, ethoxybenzene), 7.14 (d, 2H, J = 16.2Hz, double bond trans linkage), 6.94 (d, 2H, J = 16.2Hz, double bond trans linkage), 6.93 (dt, 2H, J1 = 8.7Hz, J2 = 2.1Hz, ethoxybenzene), 4.16 (t, 2H, J = 4.8Hz, tetraethylene glycol chain), 3.88 (t, 2H, J = 4.8Hz, tetraethylene glycol chain), 3.76 (bt, 2H, J = 4.8Hz, tetraethylene glycol chain), 3.76-3.64 (m, 8H, tetraethylene glycol chain), 3.56 (bt, 2H, J = 4.8Hz, tetraethylene glycol chain), 0.89 (s, 9H, -Si(CH3)2C(CH3)3), 0.06 (s, 6H, -Si(CH3)2C(CH3)3); 13C NMR (CDC13) δ (ppm): 159.5, 142.4, 132.7, 132.1, 129.4, 128.4, 126.8, 124.8, 119.4, 115.2, 110.3, 72.9, 71.2, 71.0, 70.99, 70.95, 70.0, 67.8, 63.0, 26.3, 18.7, −4.7.
4-(2-(4-(2-(2-(2-(2-(tert-Butyl-dimethyl-silanyloxy)-ethoxy)-ethoxy)-ethoxy)-ethoxy)-phenyl)-vinyl)-benzaldehyde (23)
A solution of 22 (710 mg, 1.47 mmol) in Et2O (40 mL) was cooled to −78 °C. DIBALH (2.9 mL, in Hexane 1 M, 2 equiv) was added to the reaction solution dropwise via syringe in two equal portions. The reaction was stirred for 7 h at −78 °C, then for 30 min at 0 °C. The reaction mixture was transferred via cannula into 10% AcOH aqueous solution (150 mL) with stirring. The obtained solution was stirred vigorously till organic layer was separated from the aqueous layer. The organic layer was washed twice with aqueous NaHCO3 (sat). Then the solvents were evaporated in vacuum, and the residue was purified by chromatography on a silica gel column eluted with CH2Cl2/EtOAc (5/1, v/v) to give the title product (428 mg, yield 57%): Rf 0.50 (CH2Cl2/EtOAc, 5/1). ‘H NMR (CDCl3) δ (ppm): 9.98 (s, 1H, -CHO), 7.85 (bd, 2H, J = 8.4Hz, benzaldehyde), 7.62 (bd, 2H, J = 8.4Hz, benzaldehyde), 7.47 (dt, 2H, J1 = 8.7Hz, J2 = 1.8Hz, ethoxybenzene), 7.19 (d, 2H, J = 16.2Hz, double bond trans linkage), 7.02 (d, 2H, J = 16.2Hz, double bond trans linkage), 6.93 (dt, 2H, J1 = 8.7Hz, J2 = 1.8Hz, ethoxybenzene), 4.16 (t, 2H, J = 4.8Hz, tetraethylene glycol chain), 3.88 (t, 2H, J = 4.8Hz, tetraethylene glycol chain), 3.76 (bt, 2H, J = 5.1Hz, tetraethylene glycol chain), 3.76-3.66 (m, 8H, tetraethylene glycol chain), 3.56 (bt, 2H, J = 5.1Hz, tetraethylene glycol chain), 0.89 (s, 9H, -Si(CH3)2C(CH3)3), 0.06 (s, 6H, -Si(CH3)2C(CH3)3); 13C NMR (CDC13) δ (ppm): 191.7, 159.3, 144.0, 135.1, 131.9, 130.4, 129.6, 128.3, 126.7, 125.3, 115.1, 72.9, 71.1, 71.0, 70.93, 70.88, 69.9, 67.7, 62.9, 26.2, 18.7, −4.9.
4-(2-(4-(2-(2-(2-(2-Hydroxy-ethoxy)-ethoxy)-ethoxy)-ethoxy)-phenyl)-vinyl)-benzaldehyde (24)
To a solution of 23 (100 mg, 0.19 mmol) in THF (10 mL) was added Tetrabutylammonium fluoride (TBAF) (0.2 mL, 1.1 equiv, 1.0 M in THF) slowly at room temperature. After 5 min, another 0.1 mL TBAF was added. Then several drops of 10% AcOH/THF was added to quench the reaction. The solvent was evaporated in vacuum, and the crude product was purified by flash chromatography on a silica gel column eluted with CH2Cl2/MeOH (20/1) to give the title product (78 mg, yield 100%). From 1H NMR the isomers (trans/cis double bond linkages) were observed. The isomerization was conducted: the isomers were dissolved in DMF (12 mL), and a catalytic amount of I2 was added. The system was heated to 140 °C for 36 h to give the trans product (ee ≥ 95%). Rf 0.26 (CH2Cl2/MeOH, 20/1). ‘H NMR (CDCl3) δ (ppm): 9.93 (s, 1H, -CHO), 7.80 (bd, 2H, J = 8.4Hz, benzaldehyde), 7.57 (bd, 2H, J = 8.4Hz, benzaldehyde), 7.43 (bd, 2H, J = 9.0Hz, ethoxybenzene), 7.15 (d, 2H, J = 16.5Hz, double bond trans linkage), 6.96 (d, 2H, J = 16.5Hz, double bond trans linkage), 6.90 (bd, 2H, J = 9.0Hz, ethoxybenzene), 4.12 (t, 2H, J = 4.8Hz, tetraethylene glycol chain), 3.84 (t, 2H, J = 4.8Hz, tetraethylene glycol chain), 3.74-3.62 (m, 10H, tetraethylene glycol chain), 3.60-3.55 (m, 2H, tetraethylene glycol chain).
4-(2-(4-(2-(2-(2-(2-(Bis-(4-methoxy-phenyl)-phenyl-methoxy)-ethoxy)-ethoxy)-ethoxy)-ethoxy)-phenyl)-vinyl)-benzaldehyde (25)
A mixture of 24 (180 mg, 0.45 mmol), DMTrCl (0.5 g, 1.5 mmol, 3 equiv), and a catalytic amount of 4-(Dimethylamino)pyridine (DMAP) in dry pyridine (5 mL) was stirred at room temperature under argon. After the stirring was continued overnight, the reaction solvent was removed in vacuum and the residue was directly subject to a silica gel column eluted with CH2Cl2/EtOAc/pyridine (100/10/1, v/v) to give the title product (210 mg, yield 66%): Rf 0.36 (CH2Cl2/EtOAc 10/1, v/v). ‘H NMR (CDCl3) δ (ppm): 9.98 (s, 1H, -CHO), 7.85 (bd, 2H, J = 8.4Hz, benzaldehyde), 7.62 (bd, 2H, J = 8.4Hz, benzaldehyde), 7.46 (m, 2H, benzene ring of DMTr group), 7.45 (bd, 2H, J = 8.7Hz, ethoxybenzene), 7.34 (dt, 4H, J1 = 9.0Hz, J2 = 2.4Hz, methoxybenzene ring of DMTr group), 7.32-7.15 (m, 4H, benzene ring of DMTr group and double bond trans linkage), 7.00 (d, 1H, J = 16.2Hz, double bond trans linkage), 6.91 (dt, 2H, J1 = 8.7Hz, J2 = 2.1Hz, ethoxybenzene), 6.81 (dt, 4H, J1 = 9.0Hz, J2 = 2.4Hz, methoxybenzene ring of DMTr group), 4.12 (bt, 2H, J = 4.8Hz, tetraethylene glycol chain), 3.86 (bt, 2H, J = 4.8Hz, tetraethylene glycol chain), 3.77 (s, 6H, -OCH3), 3.74-3.65 (m, 10H, tetraethylene glycol chain), 3.22 (t, 2H, J = 5.1Hz, tetraethylene glycol chain).
4-(2-(4-(2-(2-(2-(2-(Bis-(4-methoxy-phenyl)-phenyl-methoxy)-ethoxy)-ethoxy)-ethoxy)-ethoxy)-phenyl)-vinyl)-phenyl)-vinyl)-1-(2-(2-(2-(2-hydroxy-ethoxy)-ethoxy)-ethoxy)-ethyl)-pyridinium chloride (26)
A mixture of 25 (200 mg, 0.28 mmol), 1-(2-(2-(2-(2-Hydroxy-ethoxy)-ethoxy)-ethoxy)-ethyl)-4-methyl-pyridinium chloride (90 mg, 0.29 mmol, 1 equiv), and a catalytic amount of Piperidine in absolute EtOH (30 mL) was heated to 80 °C for 12 h. After the system was cooled to room temperature, the reaction solvent was removed in vacuum and the residue was directly subject to a silica gel column eluted with CH2Cl2/MeOH/Pyridine/H2O (400/60/5/5, v/v) to give the title product (85 mg, yield 30%): Rf 0.32 (CH2Cl2/MeOH/Pyridine/H2O, 400/60/5/5, v/v). ‘H NMR (CDCl3) δ (ppm): 9.27 (d, 2H, J = 6.6Hz, pyridinium), 7.91 (d, 2H, J = 6.6Hz, pyridinium), 7.64 (d, 1H, J = 16.8Hz, double bond trans linkage), 7.60 (d, 2H, J = 8.4Hz, styryl benzene), 7.53 (d, 2H, J = 8.4Hz, styryl benzene), 7.47-7.42 (m, 2H, benzene ring of DMTr group), 7.45 (bd, 2H, J = 8.7Hz, ethoxybenzene), 7.34 (dt, 4H, J1 = 9.0Hz, J2 = 2.1Hz, methoxybenzene ring of DMTr group), 7.28-7.15 (m, 3H, benzene ring of DMTr group), 7.16 (d, 1H, J = 16.5Hz, double bond trans linkage), 7.12 (d, 1H, J = 16.8Hz, double bond trans linkage), 6.97 (d, 1H, J = 16.5Hz, double bond trans linkage), 6.89 (bd, 2H, J = 8.7Hz, ethoxybenzene), 6.81 (dt, 4H, J1 = 9.0Hz, J2 = 2.1Hz, methoxybenzene ring of DMTr group), 5.07 (bt, 2H, tetraethylene glycol chain), 4.12 (bt, 4H, J = 4.8Hz, tetraethylene glycol chain), 3.85 (bt, 2H, J = 4.8Hz, tetraethylene glycol chain), 3.77 (s, 6H, -OCH3), 3.74-3.55 (m, 22H, tetraethylene glycol chain), 3.22 (t, 2H, J = 5.1Hz, tetraethylene glycol chain).
Detritylation of 26 (34)
To a solution of 26 (20 mg, 0.02 mmol) in CHCL3 (5 mL) was added Cl2HCOOH (1 drops with a 9′′-Pasteur pipet). After the completion of the reaction, the solvent was removed in vacuum and the residue was directly subject to a silica gel column eluted with CH2Cl2/MeOH/Pyridine/H2O (400/60/5/5, v/v) to give the detritylated product 34 in quantitative yield: Rf 0.23 (CH2Cl2/MeOH/Pyridine/H2O, 400/60/5/5, v/v). ‘H NMR (CDCl3) δ (ppm): 9.11 (bs, 2H, pyridinium), 7.92 (bs, 2H, pyridinium), 7.60 (d, 1H, J = 16.5Hz, double bond trans linkage), 7.55 (d, 2H, J = 8.4Hz, styryl benzene), 7.47 (d, 2H, J = 8.4Hz, styryl benzene), 7.42 (d, 2H, J = 8.7Hz, ethoxybenzene), 7.10 (d, 1H, J = 16.2Hz, double bond trans linkage), 7.06 (d, 1H, J = 16.5Hz, double bond trans linkage), 6.91 (d, 1H, J = 16.2Hz, double bond trans linkage), 6.89 (d, 2H, J = 8.7Hz, ethoxybenzene), 4.91 (bs, 2H, tetraethylene glycol chain), 4.15 (bt, 2H, J = 4.8Hz, tetraethylene glycol chain), 4.02 (bs, 2H, tetraethylene glycol chain), 3.86 (bt, 2H, J = 4.8Hz, tetraethylene glycol chain), 3.78-3.55 (m, 24H, tetraethylene glycol chain).
1-(2-(2-(2-(2-Hydroxy-ethoxy)-ethoxy)-ethoxy)-ethyl)-4-methyl-pyridinium tetraphenylborate (27)
A mixture of Picoline (1 g, 11 mmol) and 2-(2-(2-(2-Chloro-ethoxy)-ethoxy)-ethoxy)-ethanol (1.5 g, 7.1 mmol) was heated to 140 °C for 4–6 h under Argon. After the system was cooled to room temperature, the excess picoline was removed in vacuum. The obtained brown oil was dissolved in Acetonitrile and Tetraphenylboron sodium (2.5 g, 7.3 mmol) was added into the solution. The mixture was refluxed for 20 min and concentrated to a residue which was directly subject to a silica gel column eluted with CH2Cl2/MeOH (10/1, v/v) to give the title product (4.2 g) in a quantitative yield. Rf 0.42 (CH2Cl2/MeOH, 10/1, v/v). ‘H NMR (CDCl3) δ (ppm): 7.52-7.44 (m, 8H, benzene ring of BPh4), 6.96 (t, 8H, J = 7.2Hz, benzene ring of BPh4), 6.80 (bt, 4H, J = 7.2Hz, benzene ring of BPh4), 6.63 (d, 2H, J = 7.5Hz, pyridinium), 6.60 (d, 2H, J = 7.5Hz, pyridinium), 3.67-3.34 (m, 12H, tetraethylene glycol chain), 3.22-3.16 (m, 4H, tetraethylene glycol chain), 2.49 (t, 1H, J = 6.0Hz, -OH), 2.21 (s, 3H, -CH3); 13C NMR (CDC13) δ (ppm): 165.2, 164.5, 163.9, 163.2, 158.2, 143.2, 136.2, 128.0, 126.2, 126.1, 122.3, 72.6, 70.7, 70.5, 70.3, 70.27, 68.8, 61.8, 59.9, 22.2.
1-(2-(2-(2-(2-(Bis-(4-methoxy-phenyl)-phenyl-methoxy)-ethoxy)-ethoxy)-ethoxy)-ethyl)-4-methyl-pyridinium tetraphenylborate (28)
A mixture of 27 (510 mg, 0.87 mmol), DMTrCl (600 mg, 1.77 mmol, 2 equiv), and a catalytic amount of 4-(Dimethylamino)pyridine (DMAP) in dry Pyridine (10 mL) was stirred at room temperature under argon. After 12 h, the reaction solvent was removed in vacuum and the residue was directly subject to a silica gel column eluted with CH2Cl2/MeOH/Pyridine (750/25/7, v/v) to give the title product (450 mg, yield 58%): Rf 0.33 (CH2Cl2/MeOH/Pyridine 750/25/7, v/v). ‘H NMR (CDCl3) δ (ppm): 7.52-7.44 (m, 10H, benzene ring of BPh4 and DMTr groups), 7.32 (bd, 4H, J = 9Hz, methoxybenzene ring), 7.34-7.15 (m, 3H, benzene ring of DMTr group) 6.96 (t, 8H, J = 7.2Hz, benzene ring of BPh4), 6.80 (bt, 4H, J = 7.2Hz, benzene ring of BPh4), 6.79 (bd, 4H, J = 9Hz, methoxybenzene ring), 6.67 (bd, 2H, J = 6.9Hz, pyridinium), 6.63 (bd, 2H, J = 6.9Hz, pyridinium), 3.75 (s, 3H, -OCH3), 3.67-3.60 (m, 4H, tetraethylene glycol chain), 3.58-3.52 (m, 2H, tetraethylene glycol chain), 3.52-3.44 (m, 2H, tetraethylene glycol chain), 3.44-3.36 (m, 2H, tetraethylene glycol chain), 3.24-3.14 (m, 6H, tetraethylene glycol chain), 2.17 (s, 3H, -CH3); 13C NMR (CDC13) δ (ppm): 165.2, 164.6, 163.9, 163.3, 158.6, 158.1, 145.2, 143.2, 136.3, 136.2, 130.3, 128.4, 128.00, 127.97, 127.3, 127.0, 126.2, 126.1, 122.3, 113.3, 86.2, 71.0, 70.9, 70.88, 70.7, 70.5, 68.8, 63.4, 59.9, 55.6, 22.1.
1-(2-(2-(2-(2-(Bis-(4-methoxy-phenyl)-phenyl-methoxy)-ethoxy)-ethoxy)-ethoxy)-ethyl)-4-(2-(4-(2-4-(2-(2-(2-(2-hydroxy-ethoxy)-ethoxy)-ethoxy)-ethoxy)-phenyl)-vinyl)-phenyl)-vinyl)-pyridinium tetraphenylborate (29)
A mixture of 28 (230 mg, 0.26 mmol), 24 (105 mg, 0.26 mmol, 1 equiv), and a catalytic amount (6 drops, 9′′-Pasteur pipet) of Piperidine in absolute EtOH (30 mL) was heated to 100 °C for 12 h. After the reaction was cooled to room temperature, the reaction solvent was removed in vacuum and the residue was directly subject to a silica gel column eluted with CH2Cl2/MeOH/Pyridine/H2O (800/60/4/3, v/v) to give the title product (73 mg, yield 22%): Rf 0.37 (CH2Cl2/MeOH/Pyridine/H2O, 400/60/5/5, v/v). ‘H NMR (CDCl3) δ (ppm): 7.56-7.43 (m, 16H, benzene ring of BPh4 (8H), benzene ring of DMTr group (2H), styryl benzene (6H)), 7.36-7.18 (m, 4H, benzene ring of DMTr group (3H) and double bond trans linkage (1H)), 7.32 (bd, 4H, J = 9Hz, methoxybenzene ring), 7.17 (d, 1H, J = 15.9Hz, double bond trans linkage), 7.06 (d, 2H, J = 8.7Hz, pyridinium), 7.02 (t, 8H, J = 7.5Hz, benzene ring of BPh4), 6.98 (d, 1H, J = 15.9Hz, double bond trans linkage), 6.94 (d, 2H, J = 8.7Hz, pyridinium), 6.85 (bt, 4H, J = 7.5Hz, benzene ring of BPh4), 6.78 (bd, 4H, J = 9Hz, methoxybenzene ring), 6.74 (d, 1H, J = 16.2Hz, double bond trans linkage), 4.18 (bt, 2H, J = 4.8Hz, tetraethylene glycol chain), 3.88 (t, 2H, J = 4.8Hz, tetraethylene glycol chain), 3.73 (s, 6H, -OCH3), 3.78-3.58 (m, 18H, tetraethylene glycol chain), 3.58-3.52 (m, 2H, tetraethylene glycol chain), 3.52-3.47 (m, 2H, tetraethylene glycol chain), 3.46-3.36 (m, 4H, tetraethylene glycol chain), 3.23 (bt, 2H, J = 4.8Hz, tetraethylene glycol chain); 13C NMR (CDC13) δ (ppm): 158.2, 152.5, 149.5, 144.8, 143.7, 141.1, 135.9, 134.5, 132.8, 130.3, 129.9, 129.5, 128.6, 128.0, 127.9, 127.6, 126.9, 126.7, 125.8, 125.7, 125.2, 123.6, 122.8, 121.9, 114.8, 112.9, 85.8, 72.4, 70.7, 70.6, 70.5, 70.3, 70.2, 69.6, 68.8, 67.4, 63.0, 61.7, 55.1.
2-(2-(2-(2-(4-(E-2-(4-(E-2-Pyridin-4-yl-vinyl)-phenyl)vinyl)-phenoxy)-ethoxy)-ethoxy)-ethoxy)-ethanol (30)
A mixture of 24 (180 mg, 0.45 mmol), 4-Picoline (1 mL, 10.3 mmol, 23 equiv), Acetic anhydride (1.3 mL), and Acetic acid (0.6 mL) was heated to 100 °C for 12 h. A complex was then obtained after chromatography. The improved procedure is as follows: a mixture of 24 (60 mg, 0.15 mmol), 4-Picoline (1.8 mL, 18 mmol), and 37% HCl (0.72 mL, 8.8 mmol) gave a yellow solution. The reaction mixture was heated to 100 °C for 48 h and cooled to room temperature. The reaction was diluted with CHCl3, and washed with aqueous NaHCO3, extracted with CHCl3. The organic layer was collected, dried over Na2SO4, concentrated, and the residue was subject to a silica gel column eluted with CH2Cl2/MeOH (20/1, v/v) to give the title product (18 mg, yield 25%): Rf 0.28 (CH2Cl2/MeOH, 20/1, v/v). ‘H NMR (CDCl3) δ (ppm): 8.57 (d, 2H, J = 6.0Hz, pyridinium), 7.51 (bs, 4H, styryl benzene), 7.44 (dt, 2H, J1 = 8.7Hz, J2 = 1.8Hz, ethoxybenzene), 7.37 (bd, 2H, J = 6.0Hz, pyridinium), 7.28 (d, 1H, J = 16.2Hz, double bond trans linkage), 7.10 (d, 1H, J = 16.5Hz, double bond trans linkage), 7.02 (d, 1H, J = 16.5Hz, double bond trans linkage), 6.98 (d, 1H, J = 16.2Hz, double bond trans linkage), 6.92 (dt, 2H, J1 = 8.7Hz, J2 = 1.8Hz, ethoxybenzene), 4.16 (bt, 2H, J = 4.8Hz, tetraethylene glycol chain), 3.87 (t, 2H, J = 4.8Hz, tetraethylene glycol chain), 3.77-3.64 (m, 10H, tetraethylene glycol chain), 3.61 (bt, 2H, J = 5.1Hz, tetraethylene glycol chain); 13C NMR (CDC13) δ (ppm): 158.8, 150.2, 145.0, 138.4, 135.1, 133.1, 130.3, 129.1, 128.0, 127.6, 126.9, 126.1, 125.6, 121.0, 115.1, 72.8, 71.1, 71.0, 70.9, 70.6, 70.0, 67.7, 62.1.
2-(2-(2-(2-(Bis-(4-methoxy-phenyl)-phenyl-methoxy)-ethoxy)-ethoxy)-ethoxy)-ethylene chloride (32)
A mixture of 2-(2-(2-(2-Chloro-ethoxy)-ethoxy)-ethoxy)-ethanol (1.5 g, 4.7mmol), DMTrCl (3.18 g, 9.4 mmol, 2 equiv), and a catalytic amount of 4-(Dimethylamino)pyridine (DMAP) in dry Pyridine (40 mL) was stirred at room temperature under argon. After 12 h, the reaction solvent was removed in vacuum. The reaction mixture was diluted with CH2Cl2 and washed with aqueous NaHCO3 and brine. The organic layer was collected, dried over Na2SO4, concentrated, and the residue was subject to a silica gel column eluted with Cyclohexane/EtOAC (3/1 w/1% Pyridine, v/v) to give the title product in a quantitative yield. Rf 0.35 (Cyclohexane/EtOAc 3/1, v/v). ‘H NMR (CDCl3) δ (ppm): 7.44 (bd, 2H, J = 7.2Hz, benzene ring of DMTr group), 7.36 (dt, 4H, J1 = 9.0Hz, J2 = 2.1Hz, methoxybenzene ring of DMTr group), 7.28 (bt, 2H, J = 7.2Hz, benzene ring of DMTr group), 7.20 (tt, 1H, J1 = 7.2Hz, J2 = 2.1Hz, benzene ring of DMTr group), 6.83 (dt, 4H, J1 = 9.0Hz, J2 = 2.4Hz, methoxybenzene ring of DMTr group), 3.78 (s, 6H, -OCH3), 3.74 (bt, 2H, J = 5.1Hz, tetraethylene glycol chain), 3.71-3.66 (m, 10H, tetraethylene glycol chain), 3.60 (bt, 2H, J = 5.7Hz, tetraethylene glycol chain), 3.24 (t, 2H, J = 5.1Hz, tetraethylene glycol chain); 13C NMR (CDC13) δ (ppm): 158.5, 145.3, 136.5, 130.3, 128.4, 128.0, 126.9, 113.3, 86.2, 71.6, 71.1, 71.0, 70.99, 63.4, 55.5, 43.1, 27.3.
2-(2-(2-(2-(Bis-(4-methoxy-phenyl)-phenyl-methoxy)-ethoxy)-ethoxy)-ethoxy)-ethylene iodide (33)
A mixture of 32 (506 mg, 0.98 mmol), KI (1.4 g, 8.4 mmol, 8.6 equiv), and NaI (1.2 g, 8 mmol, 8 equiv) in dry Acetone (25 mL) was stirred at room temperature. During the reaction, another two batches of NaI (3 g) were added. After 72 h, the reaction solvent was removed in vacuum. The reaction mixture was diluted with CH2Cl2 and washed with aqueous NaHCO3 and brine. The organic layer was collected, dried over Na2SO4, concentrated, and the residue was subject to a silica gel column eluted with Cyclohexane/EtOAc (3/1 w/1% Pyridine, v/v) to give the title product in a 90% yield. Rf 0.38 (Cyclohexane/EtOAc, 3/1, v/v). ‘H NMR (CDCl3) δ (ppm): 7.46 (bd, 2H, J = 7.2Hz, benzene ring of DMTr group), 7.35 (dt, 4H, J1 = 9.0Hz, J2 = 2.4Hz, methoxybenzene ring of DMTr group), 7.28 (bt, 2H, J = 7.2Hz, benzene ring of DMTr group), 7.20 (tt, 1H, J1 = 7.2Hz, J2 = 2.4Hz, benzene ring of DMTr group), 6.82 (dt, 4H, J1 = 9.0Hz, J2 = 2.4Hz, methoxybenzene ring of DMTr group), 3.78 (s, 6H, -OCH3), 3.74 (bt, 2H, J = 6.9Hz, tetraethylene glycol chain), 3.71-3.66 (m, 10H, tetraethylene glycol chain), 3.23 (bt, 2H, J = 6.9Hz, tetraethylene glycol chain); 13C NMR (CDC13) δ (ppm): 158.5, 145.3, 136.5, 130.3, 128.4, 128.0, 126.9, 113.3, 86.2, 71.6, 71.1, 71.0, 70.99, 63.4, 55.5, 43.1, 27.3.
1-(2-(2-(2-(2-(Bis-(4-methoxy-phenyl)-phenyl-methoxy)-ethoxy)-ethoxy)-ethoxy)-ethyl)-4-(2-(4-(2-(4-(2-(2-(2-(2-hydroxy-ethoxy)-ethoxy)-ethoxy)-ethoxy)-phenyl)-vinyl)-phenyl)-vinyl)-pyridinium iodide (31)
A mixture of 30 (18 mg, 0.038 mmol) and 33 (60 mg, 0.1 mmol, 2.6 equiv) in anhydrous CH2Cl2 (10 mL) was heated to 60 °C for 24 h. After the system was cooled to room temperature, the reaction solvent was removed in vacuum and the residue was directly subject to a silica gel column eluted with CH2Cl2/MeOH/Pyridine/H2O (400/60/5/5, v/v) to give the title product (20 mg, yield 49%): Rf 0.5 (CH2Cl2/MeOH/Pyridine/H2O, 400/60/5/5, v/v). ‘H NMR (CDCl3) δ (ppm): 9.07 (bs, 2H, pyridinium), 7.89 (bs, 2H, pyridinium), 7.52 (s, 4H, styryl benzene), 7.51 (d, 1H, J = 16.2Hz, double bond trans linkage), 7.46 (d, 2H, J = 9.0Hz, ethoxybenzene), 7.48-7.42 (m, 2H, benzene ring of DMTr group), 7.33 (dt, 4H, J1 = 9.0Hz, J2 = 2.1Hz, methoxybenzene ring of DMTr group), 7.28-7.15 (m, 3H, benzene ring of DMTr group), 7.16 (d, 1H, J = 15.3Hz, double bond trans linkage), 6.98 (d, 1H, J = 16.2Hz, double bond trans linkage), 6.97 (d, 1H, J = 15.3Hz, double bond trans linkage), 6.93 (bd, 2H, J = 9.0Hz, ethoxybenzene), 6.78 (dt, 4H, J1 = 9.0Hz, J2 = 2.1Hz, methoxybenzene ring of DMTr group), 4.17 (bt, 2H, J = 4.8H, tetraethylene glycol chain), 4.05 (bs, 2H, tetraethylene glycol chain), 3.88 (bt, 4H, J = 4.8Hz, tetraethylene glycol chain), 3.75 (s, 6H, -OCH3), 3.76-3.55 (m, 22H, tetraethylene glycol chain), 3.24 (bt, 2H, J = 4.8Hz, tetraethylene glycol chain).
Compounds in preparation of E,E-1,4-Bis-4,4′-(2-(2-(2-(2-hydroxyethoxy)ethoxy)ethoxy)ethoxy)styryl benzene Derivatives: E,E-1,4-Bis-4,4′-(2-(2-(2-tritylethoxy)ethoxy)ethoxy)styryl benzene (15 trans)
To a solution of 1,4-Phenylenebis(methylene)bis(chlorotriphenyl) phosphorane (500 mg, 0.715 mmol) in THF (5 mL) was added n-BuLi (0.89 mL, 1.4 mmol, 1.6 M in Hexanes) at 0 °C under argon. The mixture was stirred at room temperature for 0.5 h and cooled to −78 °C. A solution of 8 (770 mg, 1.55 mmol) in THF (4 mL) was pro-cooled and added dropwise to the cooled mixture via syringe. After 0.5 h the reaction mixture was warmed up to room temperature and stirred for another 1 h. The reaction solvents were removed in vacuum and the reaction mixture was diluted with CHCl3. The organic phase was washed with brine, dried over Na2SO4, concentrated. Then the title product was precipitated with ethanol or methanol as a yellow solid. Isomerization: the solid was dissolved in DMF with a catalytic amount of I2 and the mixture was refluxed overnight. After evaporation of the solvent, the residue was purified by chromatography on a silica gel column eluted with Cyclohexane/EtOAc (2/1, v/v) to give the double bond trans linked product in a yield of 17%. Rf 0.38 (Cyclohexane/EtOAc, 2/1). ‘H NMR (CDCl3) δ (ppm): 7.51-7.46 (m, 16H, benzene ring of styryl benzene and trityl group), 7.44 (d, 4H, J = 8.7Hz, ethoxybenzene), 7.30 (bt, 12H, J = 6.9Hz, benzene ring of trityl group), 7.23 (bt, 6H, J = 7.2Hz, benzene ring of trityl group), 7.07 (d, 2H, J = 16.2Hz, double bond trans linkage), 6.97 (d, 2H, J = 16.2Hz, double bond trans linkage), 6.91 (d, 4H, J = 8.7Hz, ethoxybenzene), 4.16 (bt, 4H, J = 4.8Hz, tetraethylene glycol chain), 3.91 (t, 4H, J = 4.8Hz, tetraethylene glycol chain), 3.80-3.68 (m, 20H, tetraethylene glycol chain), 3.26 (bt, 2H, J = 5.1Hz, tetraethylene glycol chain). For the double bond cis linked product: Z, Z-1,4-Bis-4,4′-(2-(2-(2-tritylethoxy)ethoxy)ethoxy)styryl benzene (15 cis) ‘H NMR (CDCl3) δ (ppm): 7.48-7.44 (m, 12H, benzene ring of trityl group), 7.26 (bt, 12H, J = 6.9Hz, benzene ring of trityl group), 7.19 (bt, 3H, J = 6.9Hz, benzene ring of trityl group), 7.15 (bd, 4H, J = 9.0Hz, ethoxybenzene), 7.12 (bs, 4H, styryl benzene), 6.74 (bd, 4H, J = 9.0Hz, ethoxybenzene), 6.48 (d, 2H, J = 12.3Hz, double bond cis linkage), 6.42 (d, 2H, J = 12.3Hz, double bond cis linkage), 4.07 (bt, 4H, J = 4.8Hz, tetraethylene glycol chain), 3.85 (bt, 4H, J = 4.8Hz, tetraethylene glycol chain), 3.76-3.64 (m, 20H, tetraethylene glycol chain), 3.23 (bt, 2H, J = 5.1Hz, tetraethylene glycol chain).
E,E-1,4-Bis-4,4′-(2-(2-(2-(2-(Bis-(4-methoxy-phenyl)-phenyl-ethoxy)ethoxy)ethoxy) ethoxy)ethoxy)styryl benzene (17)
To a solution of bis-ylide precursor, 1,4-Phenylenebis(methylene)bis(chlorotriphenyl) phosphorane (550 mg, 0.79 mmol), in THF (5 mL) was added n-BuLi (1 mL, 1.6 mmol, 1.6 M in Hexanes) dropwise at 0 °C under argon. The mixture was stirred at room temperature for 0.5 h and cooled to −78 °C. A solution of 11 (1 g, 1.6 mmol) in THF (5 mL) was pre-cooled and added dropwise to the cooled mixture via syringe. After 0.5 h, the reaction mixture was warmed up to room temperature and stirred for another 4 h. The reaction solvents were removed in vacuum and the reaction mixture was diluted with CHCl3. The organic phase was washed with brine, dried over Na2SO4, concentrated. Then the residue was dissolved with CHCl3 and precipitated with ethanol. A yellow solid was collected as the title product with pure double bond trans linkages (150 mg, yield 15%). Rf 0.5 (Cyclohexane/EtOAc 1/2). Isomerization: the impure part (trans-cis double bond linkages) was deprotected (DMTr-group off) via Cl2HCOOH in CH2Cl2. The obtained solid was dissolved in DMF with a catalytic amount of I2 and the mixture was refluxed for 4 days. After evaporation of the solvent, the residue was purified by chromatography on a silica gel column eluted with CH2Cl2/MeOH (400/30, v/v) to give the double bond trans linked product (190 mg) in a yield of 35%. Rf 0.31 (CH2Cl2/MeOH, 20/1.5). For 17: ‘H NMR (CDCl3) δ (ppm): 7.49-7.43 (m, 8H, benzene ring of DMTr group and styryl benzene), 7.42 (d, 4H, J = 8.7Hz, ethoxybenzene), 7.34 (bd, 8H, J = 9.0Hz, methoxybenzene ring of DMTr group), 7.31-7.23 (m, 4H, benzene ring of DMTr group), 7.19 (bt, 2H, J = 7.2Hz, benzene ring of DMTr group), 7.05 (d, 2H, J = 16.2Hz, double bond trans linkage), 6.96 (d, 2H, J = 16.2Hz, double bond trans linkage), 6.89 (d, 4H, J = 8.7Hz, ethoxybenzene), 6.81 (bd, 4H, J = 9.0Hz, methoxybenzene ring of DMTr group), 4.12 (bt, 4H, J = 4.8Hz, tetraethylene glycol chain), 3.85 (bt, 4H, J = 4.8Hz, tetraethylene glycol chain), 3.77 (s, 12H, -OCH3), 3.75-3.64 (m, 20H, tetraethylene glycol chain), 3.22 (t, 4H, J = 5.1Hz, tetraethylene glycol chain); 13C NMR (CDC13) δ (ppm): 158.7, 158.6, 145.3, 136.8, 136.5, 130.5, 130.3, 128.4, 128.1, 128.0, 127.9, 126.9, 126.8, 126.5, 115.1, 113.3, 86.2, 71.2, 71.1, 71.06, 70.0, 67.7, 63.5, 55.5.
E,E-1,4-Bis-4,4′-((2-(2-(2-(2-(Bis-(4-methoxy-phenyl)-phenyl-methoxy)-ethoxy)-ethoxy)-ethoxy)-ethyl)-butyl-amino)-styrylbenzene (18)
To a solution of bis-ylide precursor, 1,4-Phenylenebis(methylene)bis(chlorotriphenyl) phosphorane (430 mg, 0.62 mmol), in THF (5 mL) was added n-BuLi (0.76 mL, 1.2 mmol, 1.6 M in Hexanes) dropwise at 0 °C under argon. The mixture was stirred at room temperature for 0.5 h and cooled to −78 °C. A solution of 14 (800 mg, 1.2 mmol) in THF (7 mL) was pre-cooled and added dropwise to the cooled mixture via syringe. After 0.5 h, the reaction mixture was warmed up to room temperature and stirred for another 12 h. The reaction solvents were removed in vacuum and the reaction mixture was diluted with CHCl3. The organic phase was washed with brine, dried over Na2SO4, concentrated, and the residue was purified by chromatography on a silica gel column eluted with Cyclohexane/EtOAc/Pyridine (300/200/5, v/v) to give the double bond trans linked product (220 mg, yield 26%). Rf 0.32 (Cyclohexane/EtOAc, 3/2). Isomerization: the impure part (trans-cis double bond linkages) was detritylated (DMTr-group off) via Cl2HCOOH in CH2Cl2 within 3 h. The de-protection reaction then was diluted with CHCl3, washed with NaHCO3 aqueous solution/brine, and concentrated. The obtained solid was dissolved in DMF with a catalytic amount of I2 and the mixture was refluxed for 12 h. After evaporation of the solvent, the residue was purified by chromatography on a silica gel column eluted with CH2Cl2/MeOH (400/20, v/v) to give the double bond trans linked product 2 (27 mg) in a yield of 5.6%. Rf 0.31 (CH2Cl2/MeOH, 20/1). For 18: ‘H NMR (CDCl3) δ (ppm): 7.56-7.50 (m, 4H, benzene ring of DMTr group), 7.48-7.38 (m, 8H, styryl benzene and ethoxybenzene, 7.42 (bd, 8H, J = 9.0Hz, methoxybenzene ring of DMTr group), 7.36-7.20 (m, 6H, benzene ring of DMTr group), 7.06 (d, 2H, J = 16.2Hz, double bond trans linkage), 6.92 (d, 2H, J = 16.2Hz, double bond trans linkage), 6.86 (bd, 4H, J = 9.0Hz, methoxybenzene ring of DMTr group), 6.70 (d, 4H, J = 8.7Hz, methoxybenzene), 3.78 (s, 12H, -OCH3), 3.76-3.64 (m, 24H, tetraethylene glycol chain), 3.61-3.54 (bt, 4H, J = 5.4Hz, tetraethylene glycol chain), 3.40-3.31 (m, 4H, -NCH2CH2CH2CH3), 3.29 (bt, 4H, J = 5.1Hz, tetraethylene glycol chain), 1.68-1.56 (m, 4H, -NCH2CH2CH2CH3), 1.46-1.31 (m, 4H, -NCH2CH2CH2CH3), 0.997 (t, 6H, J = 7.2Hz, -NCH2CH2CH2CH3); 13C NMR (CDC13) δ (ppm): 158.6, 147.7, 145.4, 136.9, 136.6, 130.3, 128.5, 128.4, 128.0, 126.9, 126.5, 125.3, 124.0, 113.3, 112.0, 86.2, 71.1, 71.09, 68.9, 63.5, 55.5, 51.6, 50.9, 29.7, 21.0, 14.5.
HWE reagent tetraethyl 1,4-xylylenediphosphonate was obtained according to literatures (32,33).
E,E-1,4-Bis-4,4′-(2-(2-(2-(2-hydroxyethoxy)ethoxy)ethoxy)ethoxy)styryl benzene (5)
A mixture of tetraethyl 1,4-xylylenediphosphonate (1.05 g, 2.78 mmol) and 3 equivalent of 4-[2-[2-[2-(2-hydroxyethoxy)ethoxy]ethoxy]ethoxy]-benzaldehyde (10) (2.45g, 8.22 mmol) was dried in vacuum overnight and dissolved in anhydrous DMF (25 mL) under argon. To the stirred solution of bisphosphonate and aldehyde at 0 °C was added dropwise a solution of potassium tert-butoxide (KOtBu) (922 mg, 3 equal) in anhydrous DMF (15 mL) over 15 min by syringe. After stirring for 0.5 h at 0 °C, the reaction mixture was slowly warmed up to room temperature and stirred for another12 h, the reaction was quenched with water. The crude product was collected by suction filtration and the filtrate was evaporated to dryness. The residue was resolved with CHCl3 and washed with water (2 × 100 mL). The organic layer was dried over Na2SO4 and concentrated to dryness. The crude product was then purified by being washed with tetrahydrofuran (THF) twice to give 1.69 g (91%) of the title compound (5). ‘H NMR (CDCl3) δ (ppm): 7.47 (s, 4H, aromatic ring), 7.45 (d, 4H, J = 8.7Hz, aromatic ring), 7.06 (d, 2H, J = 16.2Hz, double bond trans-linkage), 6.97 (d, 2H, J = 16.2Hz, double bond trans-linkage), 6.92 (d, 4H, J = 8.7Hz, aromatic ring), 4.16 (bt, 4H, J = 4.8Hz, tetraethylene glycol chain), 3.87 (bt, 4H, J = 4.8Hz, tetraethylene glycol chain), 3.78-3.58 (m, 24H, tetraethylene glycol chain); 13C NMR (CDC13/CD3OD, 0.3mL/0.2mL) δ 15(ppm):8.2, 136.5, 130.3, 127.6, 127.5, 126.4, 126.2, 114.6, 72.5, 70.55, 70.50, 70.38, 70.02, 69.63, 67.3, 61.2. MS (MALDI-TOF): m/z 666.8 [M]+, 667.8 [M+1]+, 689.7 [M+Na]+.
Monotritylation of E,E-1,4-Bis-4,4′-(2-(2-(2-(2-hydroxyethoxy)ethoxy)ethoxy)ethoxy)styryl benzene (40)
E,E-1,4-Bis-4,4′-(2-(2-(2-(2-hydroxyethoxy)ethoxy)ethoxy)ethoxy)styryl benzene (5) (375 mg, 0.56 mmol) was dissolved in dry Pyridine (50 mL), followed by addition of DMTrCl (415 mg, 1.23 mmol) and DMAP (~10 mg). The mixture was stirred at room temperature under argon overnight, and TLC monitoring (10/0.75, CH2Cl2/MeOH) showed the formation of monosubstituted (Rf 0.32) and disubstituted (Rf 0.74) products as well as the starting material (Rf 0.14). The reaction mixture was concentrated in vacuum and the residue was subject to a silica gel column with CH2Cl2/MeOH/Pyridine (100/5/0.5) as eluents affording the title product (160 mg, yield 30%) as a yellow powder. Meanwhile, disubstituted fraction was also collected and further monodetritylation was carried out as the following: A detritylation solution was made by mixing ZnCl2 (1.5 g) into 110 mL of CH2Cl2/MeOH (10/1, v/v). Then the disubstituted compound (150 mg, 0.12 mmol) was dissolved by 50 mL of the detritylation solution and the reaction mixture was monitored by TLC (20/1, CH2Cl2/MeOH). Once the appearance of the spot corresponding to starting material (E,E-1,4-Bis-4,4′-(2-(2-(2-(2-hydroxyethoxy)ethoxy)ethoxy)ethoxy)styryl benzene) on the TLC plate, the reaction was quenched by pouring into a saturated aq. NaHCO3 solution. The mixture was extracted by CHCl3, and the organic layer was washed with brine, collected, dried over Na2SO4, concentrated, and subject to a silica gel column (100/5/0.5, CH2Cl2/MeOH/Pyridine) to give the title product 40 (48 mg, yield 42 %). Based on the starting material, the total yield of the monosubstituted product is 38 %. The product is stored with a stabilizer (Diisopropylethylamine) under argon at −80 °C. ‘H NMR (CDCl3) δ (ppm): 7.46 (s, 4H, aromatic ring of styryl benzene), 7.49-7.38 (m, 6H, aromatic rings of styryl benzene and DMTr), 7.34 (dt, 4H, J1 = 8.7Hz, J2 = 2.3Hz, methoxylbenzene ring), 7.31-7.15 (m, 3H, benzene ring of DMTr), 7.06 (d, 1H, J = 16.2Hz, double bond trans-linkage), 7.05 (d, 1H, J = 16.2Hz, double bond trans-linkage), 6.96 (d, 1H, J = 16.2Hz, double bond trans-linkage), 6.95 (d, 1H, J = 16.2Hz, double bond trans-linkage), 6.94-6.84 (m, 4 H, aromatic ring of styryl benzene), 6.81 (dt, 4H, J1 = 8.7Hz, J2 = 2.3Hz, methoxylbenzene ring), 4.19-4.08 (m, 4H, tetraethylene glycol chain), 3.89-3.82 (m, 4H, tetraethylene glycol chain), 3.80-3.58 (m, 28H, CH3O and tetraethylene glycol chain), 3.22 (t, 2H, J = 5.1Hz, DMTrOCH2); 13C NMR (CDC13) δ (ppm): 158.57, 158.55, 158.43, 145.2, 136.74, 136.70, 136.4, 130.47, 130.40, 130.2, 128.3, 128.00, 127.96, 127.87, 127.79, 126.77, 126.67, 126.44, 126.38, 115.0, 113.2, 86.1, 72.7, 71.09, 71.04, 70.98, 70.95, 70.88, 70.81, 70.56, 69.95, 67.64, 63.4, 62.0, 55.4.
The Phosphoramidite of Monotritylated E,E-1,4-Bis-4,4′-(2-(2-(2-(2-hydroxyethoxy)ethoxy)ethoxy)ethoxy)styryl benzene (43)
To a solution of monotritylated E,E-1,4-Bis-4,4′-(2-(2-(2-(2-hydroxyethoxy)ethoxy)ethoxy)ethoxy)styryl benzene (40) (86 mg, 0.089 mmol) in 15 mL CH2Cl2 (dry) was added 5 equivalent of diisopropylethylamine (0.08 mL). Then Chloro-N,N-diisopropylaminocyanoethoxyphosphane (0.04 mL, 0.18 mmol, ~2 equal) was added dropwise at room temperature under argon. After 20 min of stirring under argon at room temperature, the reaction mixture was diluted with 100 mL of CH2Cl2/Et3N (300/15, v/v), and the organic phase was washed with a saturated aq. NaHCO3 solution and brine. The organic layer was dried over Na2SO4, filtered, and evaporated to dryness. The residue was subject to a silica gel column (CH2Cl2/EtOAc/Et3N, 3/6/1) to give the title product (88 mg, yield 85%) as a yellow powder, which should be used freshly for the next phosphotriester step in order to achieve a higher coupling yield.
E,E-1,4-Bis-4,4′-((2-(2-(2-(2-hydroxyethoxy)-ethoxy)-ethoxy)-ethyl)-butyl-amino)-styrylbenzene (2)
To a solution of tetraethyl 1,4-xylylenediphosphonate (1.5 g, 3.9 mmol) and 4-N-n-Butyl-N-[2-[2-[2-(2-hydroxyethoxy)ethoxy]ethoxy]ethyl]amino benzaldehyde (13) (4 g, 11 mmol, 3 equiv) in anhydrous DMF (20 mL) was added KOBut (1.2 g, 11 mmol, 3 equiv) pre-dissolved in DMF (10 mL) dropwise via syringe at 0 °C under argon. After stirring for 0.5 h at 0 °C, the reaction mixture was slowly warmed up to room temperature and stirred for another12 h, then the reaction was quenched with water. The crude product was collected by suction filtration and the filtrate was evaporated to dryness. The residue was re-dissolved with CHCl3 and washed with brine (2 × 100 mL). The organic layer was dried over Na2SO4 and concentrated to dryness. The crude product was then purified by chromatography on a silica gel column eluted with EtOAc/MeOH (20/1, v/v) to give the double bond trans linked product (1.54 g, yield 51%). Rf 0.33 (EtOAc/MeOH, 20/1). ‘H NMR (CDCl3) δ (ppm): 7.42 (s, 4H, styryl benzene), 7.36 (bd, 4H, J = 8.7Hz, ethoxybenzene), 7.02 (d, 2H, J = 16.2Hz, double bond trans linkage), 6.88 (d, 2H, J = 16.2Hz, double bond trans linkage), 6.66 (d, 4H, J = 8.7Hz, ethoxybenzene), 3.76-3.51 (m, 28H, tetraethylene glycol chain), 3.34 (bt, 4H, J = 7.5Hz, -NCH2CH2CH2CH3), 1.65-1.52 (m, 4H, -NCH2CH2CH2CH3), 1.44-1.28 (m, 4H, -NCH2CH2CH2CH3), 0.96 (t, 6H, J = 7.5Hz, -NCH2CH2CH2CH3); 13C NMR (CDC13) δ (ppm): 147.7, 136.8, 128.3, 127.9, 126.4, 125.3, 124.0, 111.9, 72.8, 71.0, 70.9, 70.88, 70.6, 68.8, 62.1, 51.5, 50.7, 29.6, 20.6, 14.4. MS (MALDI-TOF): 776.6 [M]+.
E, 1-(4-((2-(2-(2-(2-(Bis-(4-methoxy-phenyl)-phenyl-methoxy)-ethoxy)-ethoxy)-ethoxy)-ethyl)-butyl-amino)-styryl)-E, 4-(4-((2-(2-(2-(2-(Bis-(4-methoxy-phenyl)-phenyl-methoxy)-ethoxy)-ethoxy)-ethoxy)-ethyl)-butyl-amino)-styryl) benzene (39)
E,E-1,4-Bis-4,4′-((2-(2-(2-(2-hydroxyethoxy)-ethoxy)-ethoxy)-ethyl)-butyl-amino)-styrylbenzene (2) (1.116 g, 1.44 mmol) was dissolved in dry Pyridine (50 mL), followed by addition of DMTrCl (1.55 g, 4.3 mmol, 3 equiv) and DMAP (~10 mg). The mixture was stirred at room temperature under argon overnight, and TLC monitoring (CH2Cl2/MeOH, 400/15, v/v) showed the formation of monosubstituted (Rf 0.64) and disubstituted (Rf 0.88) products as well as the starting material (Rf 0.31). The reaction mixture was concentrated in vacuum and the residue was subject to a silica gel column with CH2Cl2/MeOH/pyridine (400/15/2) as eluents to give the title product (390 mg, yield 25%) as a yellow powder. Meanwhile, disubstituted fraction was also collected and further monodetritylation was carried out as the following: a detritylation solution was made by mixing ZnCl2 (1.5 g) with 110 mL of CH2Cl2/MeOH (10/1, v/v). Then the disubstituted compound (1150 mg, 0.83 mmol) was dissolved by 50 mL of the detritylation solution and the reaction mixture was monitored by TLC (CH2Cl2/MeOH, 400/15). Once the appearance of the spot corresponding to starting matrerial 2 on the TLC plate, the reaction was quenched by pouring into a saturated aq. NaHCO3 solution. The mixture was extracted by chloroform, and the organic layer was washed with brine, collected, dried over Na2SO4, concentrated, and subject to a silica gel column (CH2Cl2/MeOH/Pyridine, 400/15/2) to give the title product (470 mg, yield 53 %). Based on the starting material, the total yield of the monosubstituted product is 55 %. The product is stored with a stabilizer (Diisopropylethylamine) under argon at −80 °C. ‘H NMR (CDCl3) δ (ppm): 7.48-7.44 (m, 2H, benzene ring of DMTr group), 7.42 (bs, 4H, styryl benzene), 7.40-7.32 (m, 4H, butylaminobenzene), 7.34 (bd, 4H, J = 9.0Hz, methoxybenzene ring of DMTr group), 7.31-7.24 (m, 2H, benzene ring of DMTr group), 7.19 (bt, 1H, J = 7.2Hz, benzene ring of DMTr group), 7.02 (d, 1H, J = 16.2Hz, double bond trans linkage), 7.01 (d, 1H, J = 15.9Hz, double bond trans linkage), 6.88 (d, 2H, J = 16.2Hz, double bond trans linkage), 6.87 (d, 1H, J = 15.9Hz, double bond trans linkage), 6.81 (bd, 4H, J = 9.0Hz, methoxybenzene ring of DMTr group), 6.67 (d, 2H, J = 8.7Hz, butylaminobenzene), 6.64 (d, 2H, J = 8.7Hz, butylaminobenzene), 3.77 (s, 6H, -OCH3), 3.76-3.50 (m, 30H, tetraethylene glycol chain), 3.38-3.26 (m, 4H, -NCH2CH2CH2CH3), 3.22 (bt, 2H, J = 5.1Hz, tetraethylene glycol chain), 1.64-1.52 (m, 4H, -NCH2CH2CH2CH3), 1.42-1.28 (m, 4H, -NCH2CH2CH2CH3), 0.96 (t, 3H, J = 7.2Hz, -NCH2CH2CH2CH3), 0.94 (t, 3H, J = 7.2Hz, -NCH2CH2CH2CH3); 13C NMR (CDC13) δ (ppm): 158.5, 147.7, 145.3, 136.9, 136.9, 136.5, 130.3, 128.4, 128.2, 128.2, 127.94, 127.91, 126.9, 126.4, 125.3, 125.3, 123.9, 123.9, 113.2, 111.92, 111.89, 86.2, 72.8, 71.1, 71.0, 70.98, 70.95, 70.88, 70.6, 68.8, 63.4, 62.1, 55.5, 51.5, 50.8, 29.6, 20.6, 14.4.
The Phosphoramidite of E, 1-(4-((2-(2-(2-(2-(Bis-(4-methoxy-phenyl)-phenyl-methoxy)-ethoxy)-ethoxy)-ethoxy)-ethyl)-butyl-amino)-styryl)-E, 4-(4-((2-(2-(2-(2-(Bis-(4-methoxy-phenyl)-phenyl-methoxy)-ethoxy)-ethoxy)-ethoxy)-ethyl)-butyl-amino)-styryl) benzene (41)
To a solution of E, 1-(4-((2-(2-(2-(2-(Bis-(4-methoxy-phenyl)-phenyl-methoxy)-ethoxy)-ethoxy)-ethoxy)-ethyl)-butyl-amino)-styryl)-E, 4-(4-((2-(2-(2-(2-(Bis-(4-methoxy-phenyl)-phenyl-methoxy)-ethoxy)-ethoxy)-ethoxy)-ethyl)-butyl-amino)-styryl) benzene (39) (770 mg, 0.71 mmol) in 80 mL CH2Cl2 (dry) was added 7 equivalent of diisopropylethylamine (0.8 mL). Then Chloro-N,N-diisopropylaminocyanoethoxyphosphane (0.47 mL, 2.1 mmol, ~3 equal) was added dropwise at room temperature under argon. After 20 min of stirring under argon at room temperature, the reaction mixture was diluted with 100 mL of CH2Cl2/Et3N (300/15, v/v), and the organic phase was washed with a saturated aq. NaHCO3 solution and brine. The organic layer was dried over Na2SO4, filtered, and evaporated to dryness. The residue was subject to a silica gel column (CH2Cl2/EtOAc/Et3N, 3/6/1) to give the title product (870 mg, yield 96%) as a yellow powder, which should be used freshly for the next phosphotriester step in order to achieve a higher coupling yield. ‘H NMR (CDCl3) δ (ppm): 7.48-7.44 (m, 2H, benzene ring of DMTr group), 7.42 (bs, 4H, styryl benzene), 7.40-7.32 (m, 4H, butylaminobenzene), 7.34 (bd, 4H, J = 9.0Hz, methoxybenzene ring of DMTr group), 7.31-7.15 (m, 3H, benzene ring of DMTr group), 7.01 (d, 1H, J = 16.2Hz, double bond trans linkage), 7.00 (d, 1H, J = 16.2Hz, double bond trans linkage), 6.88 (d, 2H, J = 16.2Hz, double bond trans linkage), 6.87 (d, 1H, J = 16.2Hz, double bond trans linkage), 6.81 (bd, 4H, J = 9.0Hz, methoxybenzene ring of DMTr group), 6.67 (d, 2H, J = 8.7Hz, butylaminobenzene), 6.64 (d, 2H, J = 8.7Hz, butylaminobenzene), 3.77 (s, 6H, -OCH3), 3.90-3.75 (m, 2H, -OCH2CH2CN), 3.70-3.48 (m, 32H, -N(CH(CH3)2)2 and tetraethylene glycol chain), 3.38-3.26 (m, 4H, -NCH2CH2CH2CH3), 3.22 (bt, 2H, J = 5.1Hz, tetraethylene glycol chain), 2.63 (bt, 2H, J = 6.5Hz, -OCH2CH2CN), 1.64-1.52 (m, 4H, -NCH2CH2CH2CH3), 1.42-1.28 (m, 4H, -NCH2CH2CH2CH3), 1.19 (d, 6H, J = 6.8Hz, -CH(CH3)2), 1.17 (d, 2H, J = 6.8Hz, -CH(CH3)2), 0.96 (t, 3H, J = 7.2Hz, -NCH2CH2CH2CH3), 0.94 (t, 3H, J = 7.2Hz, -NCH2CH2CH2CH3); 13C NMR (CDC13) δ (ppm): 158.6, 147.7, 145.3, 136.9, 136.6, 130.3, 128.4, 128.3, 128.3, 127.98, 127.95, 126.9, 126.4, 125.37, 125.32, 124.0, 123.95, 113.3, 111.9, 86.1, 71.5, 71.4, 71.0, 70.98, 70.94, 70.90, 70.8, 68.8, 63.4, 62.9, 62.7, 58.9, 58.6, 55.4, 51.4, 50.7, 43.4, 43.2, 29.6, 24.93, 24.86, 24.83, 24.77, 20.6, 20.5, 14.4, 14.3; 31P NMR (CDC13) δ (ppm): 149.6.
(4-(2-(4-(Butyl-(2-(2-(2-(2-hydroxy-ethoxy)-ethoxy)-ethoxy)-ethyl)-amino)-phenyl)-vinyl)-benzyl)-phosphonic acid diethyl ester (19)
To a solution of Tetraethyl 1,4-xylylenediphosphonate (466 mg, 1.2 mmol, 1.2 equiv) and 4-N-n-Butyl-N-[2-[2-[2-(2-hydroxyethoxy)ethoxy]ethoxy]ethyl]amino benzaldehyde (13) (360 mg, 1.1 mmol) in anhydrous DMF (15 mL) was added KOBut (123 mg, 1.1 mmol, 1 equiv) pre-dissolved in DMF (4 mL) dropwise via syringe at 0 °C under argon. After stirring for 0.5 h at 0 °C, the reaction mixture was slowly warmed up to room temperature and stirred for another12 h. The crude product was then purified by chromatography on a silica gel column eluted with EtOAc/MeOH (20/1, v/v) to give the double bond trans linked product 2 (130 mg, yield 31%) and the title product (trans/cis 3/1) in a yield of 45% (281 mg). Rf 0.25 (EtOAc/MeOH, 20/1). ‘H NMR (CDCl3) δ (ppm): double bond trans/cis linkage (3/1): 7.40 (d, 2H (3/4), J = 8.1Hz, aromatic ring), 7.35 (d, 2H (3/4), J = 8.7Hz, aromatic ring), 7.28 (d, 2H (1/4), J = 8.4Hz, aromatic ring), 7.24 (dd, 2H (3/4), J1 = 8.1Hz, J2 = 2.4Hz, aromatic ring), 7.16 (dd, 2H (1/4), J = 8.4Hz, aromatic ring), 7.10 (d, 2H (1/4), J = 8.7Hz, aromatic ring), 7.00 (d, 1H (3/4), J = 16.5Hz, double bond trans linkage), 6.84 (d, 1H (3/4), J = 16.5Hz, double bond trans linkage), 6.65 (d, 2H (3/4), J = 8.7Hz, aromatic ring), 6.48 (d, 2H (1/4), J = 8.7Hz, aromatic ring), 6.42 (d, 1H (1/4), J = 12.3Hz, double bond cis linkage), 6.33 (bd, 1H (1/4), J = 12.3Hz, double bond cis linkage), 4.06-3.95 (m, 4H, -OCH2CH3), 3.74-3.46 (m, 16H, tetraethylene glycol chain), 3.35 (bt, 2H (3/4), J = 7.5Hz, -NCH2CH2CH2CH3), 3.27 (bt, 2H (1/4), J = 7.5Hz, -NCH2CH2CH2CH3), 3.17 (bs, 1H (3/4), benzyl), 3.16 (bs, 1H (1/4), benzyl), 3.10 (bs, 1H (3/4), benzyl), 3.08 (bs, 1H (1/4), benzyl), 1.63-1.50 (m, 2H, -NCH2CH2CH2CH3), 1.40-1.28 (m, 2H, -NCH2CH2CH2CH3), 1.24 (t, 6H, J = 7.1Hz, -OCH2CH3), 0.95 (t, 3H (3/4), J = 7.2Hz, -NCH2CH2CH2CH3), 0.92 (t, 3H (1/4), J = 7.2Hz, -NCH2CH2CH2CH3). MS (MALDI-TOF): 576.6 [M]+.
E, 1-(4-((2-(2-(2-(2-hydroxyethoxy)-ethoxy)-ethoxy)-ethyl)-butyl-amino)-styryl)-E, 4-(4-(2-(2-(2-(2-hydroxyethoxy)-ethoxy)-ethoxy)-ethoxy)styryl) benzene (3)
To a solution of 19 (87 mg, 0.15 mmol) and 10 (90 mg, 0.3 mmol, 2 equiv) in anhydrous DMF (8 mL) was added KOBut (35 mg, 0.31 mmol, 2 equiv) pre-dissolved in DMF (4 mL) dropwise via syringe at 0 °C under argon. After stirring for 0.5 h at 0 °C, the reaction mixture was slowly warmed up to room temperature and stirred for another 12 h. Water was added to quench the reaction. After evaporation of the solvent, the crude product was purified by chromatography on a silica gel column eluted with EtOAc/MeOH (10/1, v/v) to give the title product (60 mg) in a yield of 55%. Rf 0.39 (EtOAc/MeOH, 10/1). ‘H NMR (CDCl3) δ (ppm): 7.45 (bs, 4H, styryl benzene), 7.44 (bd, 2H, J = 9.0Hz, ethoxybenzene), 7.38 (d, 2H, J = 8.7Hz, butylaminobenzene), 7.04 (d, 1H, J = 16.2Hz, double bond trans linkage), 7.03 (d, 1H, J = 16.2Hz, double bond trans linkage), 6.97 (d, 1H, J = 16.2Hz, double bond trans linkage), 6.92 (bd, 2H, J = 9.0Hz, ethoxybenzene), 6.88 (d, 1H, J = 16.2Hz, double bond trans linkage), 6.66 (d, 2H, J = 8.7Hz, butylaminobenzene), 4.16 (bt, 2H, J = 4.8Hz, tetraethylene glycol chain), 3.87 (bt, 2H, J = 4.8Hz, tetraethylene glycol chain), 3.76-3.51 (m, 28H, tetraethylene glycol chain), 3.34 (bt, 2H, J = 7.5Hz, -NCH2CH2CH2CH3), 2.53 (bt, 1H, J = 6.9Hz, -OH), 2.47 (bt, 1H, J = 6.9Hz, -OH), 1.65-1.52 (m, 2H, -NCH2CH2CH2CH3), 1.44-1.28 (m, 2H, -NCH2CH2CH2CH3), 0.96 (t, 3H, J = 7.5Hz, -NCH2CH2CH2CH3); 13C NMR (CDC13) δ (ppm): 158.5, 147.7, 137.5, 136.1, 130.7, 128.7, 127.97, 127.8, 127.6, 126.7, 126.4, 125.2, 123.8, 115.0, 111.9, 72.4, 72.4, 70.7, 70.6, 70.56, 70.50, 70.2, 69.6, 68.4, 67.3, 61.7, 51.1, 50.3, 29.2, 20.2, 14.0. MS (MALDI-TOF): 721.3 [M]+. HWE reagent Tetraethyl-2,5-dicyano-1,4-xylylenediphosphonate was obtained according to literatures (27, 28).
2,5-Dicyano-E,E-1,4-Bis-4,4′-((2-(2-(2-(2-hydroxy-ethoxy)-ethoxy)-ethoxy)-ethyl)-butyl-amino)-styrylbenzene (4)
To a solution of Tetraethyl-2,5-dicyano-1,4-xylylenediphosphonate (186 mg, 0.43 mmol) and 4-N-n-Butyl-N-[2-[2-[2-(2-hydroxyethoxy)ethoxy]ethoxy]ethyl]amino benzaldehyde (13) (460 mg, 1.3 mmol, 3 equiv) in anhydrous DMF (10 mL) was added KOBut (155 mg, 1.3 mmol, 3 equiv) pre-dissolved in DMF (10 mL) dropwise via syringe at 0 °C under argon. After stirring for 0.5 h at 0 °C, the reaction mixture was slowly warmed up to room temperature and stirred for another12 h, then the reaction was quenched with water. The crude product was collected by suction filtration and the filtrate was evaporated to dryness. The residue was re-dissolved with CHCl3 and washed with brine (2 × 100 mL). The organic layer was dried over Na2SO4 and concentrated to dryness. The crude product was then purified by chromatography on a silica gel column eluted with EtOAc/MeOH (20/1, v/v) to give the double bond trans linked product (233 mg, yield 65%). Rf 0.23 (EtOAc/MeOH, 20/1). ‘H NMR (CDCl3) δ (ppm): 7.94 (bs, 2H, styryl benzene), 7.44 (d, 4H, J = 8.7Hz, butylaminobenzene), 7.19 (d, 2H, J = 16.2Hz, double bond trans linkage), 7.10 (d, 2H, J = 16.2Hz, double bond trans linkage), 6.68 (d, 4H, J = 8.7Hz, butylaminobenzene), 3.74 (bt, 4H, J = 4.5Hz, tetraethylene glycol chain), 3.71-3.55 (m, 28H, tetraethylene glycol chain), 3.37 (bt, 2H, J = 7.5Hz, -NCH2CH2CH2CH3), 2.62 (bs, 2H, -OH), 1.65-1.52 (m, 2H, -NCH2CH2CH2CH3), 1.37 (sext, 2H, J = 7.5Hz, -NCH2CH2CH2CH3), 0.97 (t, 3H, J = 7.5Hz, -NCH2CH2CH2CH3); 13C NMR (CDC13) δ (ppm): 149.1, 138.8, 134.6, 129.2, 129.1, 123.4, 117.4, 116.9, 114.2, 111.9, 72.7, 71.0, 70.89, 70.81, 70.6, 68.7, 62.0, 51.4, 50.6, 29.4, 20.5, 14.2. MS (MALDI-TOF): 827.0 [M]+.
2,5-Dicyano-E,E-1,4-bis-4,4′-(2-(2-(2-(2-hydroxy-ethoxy)ethoxy)ethoxy)ethoxy) styryl benzene (1)
To a solution of Tetraethyl-2,5-dicyano-1,4-xylylenediphosphonate (200 mg, 0.47 mmol) and 10 (418 mg, 1.4 mmol, 3 equiv) in anhydrous DMF (15 mL) was added KOBut (160 mg, 1.4 mmol, 3 equiv) pre-dissolved in DMF (10 mL) dropwise via syringe at 0 °C under argon. After stirring for 0.5 h at 0 °C, the reaction mixture was slowly warmed up to room temperature and stirred for another 12 h, then the reaction was quenched with water. The crude product was collected by suction filtration and the filtrate was evaporated to dryness. The residue was re-dissolved with CHCl3 and washed with brine (2 × 100 mL). The organic layer was dried over Na2SO4 and concentrated to dryness. The crude product was then purified by chromatography on a silica gel column eluted with EtOAc/MeOH (10/1, v/v) to give the double bond trans linked product (201 mg, yield 60%). Rf 0.20 (EtOAc/MeOH, 20/1). ‘H NMR (CDCl3) δ (ppm): 7.97 (s, 2H, styryl benzene), 7.50 (bd, 4H, J = 8.7Hz, ethoxybenzene), 7.23 (d, 2H, J = 16.2Hz, double bond trans linkage), 7.18 (d, 2H, J = 16.2Hz, double bond trans linkage), 6.95 (d, 4H, J = 8.7Hz, ethoxybenzene), 4.16 (bt, 4H, J = 4.8Hz, tetraethylene glycol chain), 3.87 (bt, 4H, J = 4.8Hz, tetraethylene glycol chain), 3.78-3.64 (m, 20H, tetraethylene glycol chain), 3.60 (bt, 4H, J = 4.8Hz, tetraethylene glycol chain), 2.95 (bs, 2H, -OH); 13C NMR (CDC13) δ (ppm): 160.1, 138.9, 134.6, 129.6, 129.0, 128.7, 119.7, 117.0, 115.3, 114.8, 72.8, 71.0, 70.85, 70.77, 70.5, 69.8, 67.7, 61.9. MS (MALDI-TOF): 739.9 [M+Na]+.
Compounds in preparation of E,E-4-Dicyanomethylene-2,6-bis-p-N,N′-n-butyl-N, N′-(2-(2-(2-(2-hydroxyethoxy)ethoxy)ethoxy)ethyl)-aminostyryl pyran Derivatives: E,E-4-Dicyanomethylene-2,6-bis-p-N,N′-n-butyl-N,N′-(2-(2-(2-(2-hydroxyethoxy)ethoxy)ethoxy)ethyl)-aminostyryl pyran (6)
A mixture of 2,6-Dimethyl-4-(dicyanomethylene) pyran (220 mg, 1.28 mmol), 4-N-n-Butyl-N-[2-[2-[2-(2-hydroxyethoxy)ethoxy]ethoxy]ethyl]aminobenzaldehyde (1.0 g, 2.8 mmol), and Piperidine (1 mL, 10 mmol) in toluene (25 mL) was heated to reflux with a Dean-Stark trap under argon overnight. Then the reaction was cooled and the solvent was evaporated to dryness in vacuum. CHCl3 was added to dilute the residue, and the organic layer was washed with water (100 mL) and brine (100 mL). After dried over Na2SO4, the organic phase was concentrated and the residue was purified by chromatography on a silica gel column eluted with EtOAc/MeOH (20/1) to give the title product (538 mg, yield 50%) as a red powder: Rf 0.14 (EtOAc/MeOH 20/1). ‘H NMR (CDCl3) δ (ppm): 7.42 (d, 4H, J = 9.0Hz, aromatic ring), 7.41 (d, 2H, J = 16.2Hz, double bond trans-linkage), 6.69 (d, 4H, J = 9.0Hz, aromatic ring), 6.51 (s, 2H, pyran ring), 6.47 (d, 2H, J = 16.2Hz, double bond trans-linkage), 3.55-3.51 (m, 32H, tetraethylene glycol chain), 3.39 (t, 4H, J = 7.5Hz, n-butyl chain), 1.70-1.52 (m, 4H, n-butyl chain), 1.46-1.30 (m, 4H, n-butyl chain), 0.98 (t, 6H, J = 7.5Hz, n-butyl chain); 13C NMR (CDC13) δ (ppm): 159.4, 156.2, 149.8, 138.2, 129.9, 122.1, 116.5, 112.8, 111.7, 105.3, 72.6, 70.9, 70.8, 70.7, 70.4, 68.5, 61.8, 51.4, 50.5, 29.4, 20.4, 14.2. MS (MALDI-TOF): m/z 844.2 [M+1]+.
E,E-4-Dicyanomethylene-2,6-bis-4.4′-(2-(2-(2-(2-hydroxy-ethoxy)ethoxy)ethoxy) ethyl)styryl pyran (37)
A mixture of 2,6-Dimethyl-4-(dicyanomethylene) pyran (206 mg, 1.2 mmol), 10 (800 mg, 2.68 mmol), Acetic acid (0.5 mL, 8.5 mmol), and Piperidine (1 mL, 10 mmol) in Toluene (25 mL) was heated to reflux with a Dean-Stark trap under argon for 36 h. Then the reaction was cooled and the solvent was evaporated to dryness in vacuum. CHCl3 was added to dilute the residue, and the organic layer was washed with brine (2×100 mL). After dried over Na2SO4, the organic phase was concentrated and the residue was purified by chromatography on a silica gel column eluted with EtOAc/MeOH (10:1) to give the title product (130 mg, yield 15%): Rf 0.23 (EtOAc/MeOH 20/1). ‘H NMR (CDCl3) δ (ppm): 7.52 (bd, 4H, J = 9.0Hz, ethoxybenzene), 7.46 (d, 2H, J = 16.2Hz, double bond trans linkage), 6.98 (d, 4H, J = 9.0Hz, ethoxybenzene), 6.64 (s, 2H, pyran), 6.63 (d, 2H, J = 16.2Hz, double bond trans linkage), 4.20 (bt, 4H, J = 4.8Hz, tetraethylene glycol chain), 3.89 (bt, 4H, J = 4.8Hz, tetraethylene glycol chain), 3.78-3.64 (m, 20H, tetraethylene glycol chain), 3.62 (bt, 4H, J = 4.8Hz, tetraethylene glycol chain); 13C NMR (CDC13) δ (ppm): 161.0, 158.8, 137.8, 129.7, 127.7, 116.5, 115.7, 115.5, 106.7, 72.7, 71.1, 70.88, 70.81, 70.5, 69.8, 67.8, 62.0. MS (MALDI-TOF): 731.7 [M]+, 787.8 [M+K+H2O]+.
Monotritylation of E,E-4-Dicyanomethylene-2,6-bis-p-N,N′-n-butyl-N,N′-(2-(2-(2-(2-hydroxyethoxy)ethoxy)ethoxy)ethyl)-aminostyryl pyran (38)
E,E-4-Dicyanomethylene-2,6-bis-p-N,N′-n-butyl-N,N′-(2-(2-(2-(2-hydroxyethoxy)ethoxy)ethoxy)ethyl)-aminostyryl pyran (6) (538 mg, 0.64 mmol) was dissolved in dry Pyridine (50 mL), followed by addition of 3 equivalence of DMTrCl (684 mg) and DMAP (~10 mg). The mixture was stirred at room temperature under argon for 5 days, and TLC monitoring (20/1, CH2Cl2/MeOH) showed the formation of monosubstituted (Rf 0.25) and disubstituted (Rf 0.98) products as well as the starting material (Rf 0.09). The reaction mixture was concentrated in vacuum and the residue was directly subject to a silica gel column using the gradient elution technique with CH2Cl2/MeOH/Pyridine (500/10/3 to 500/20/3) as eluents affording the monosubstituted product (250 mg, yield 35%); disubstituted product, and recovered starting material (123 mg). Meanwhile, disubstituted fraction was collected and further monodetritylation was carried out according to the following procedure: A detritylation solution was made by mixing ZnCl2 (1.5 g) with 110 mL of CH2Cl2/MeOH (10/1, v/v). Then the crude disubstituted compound (~900 mg) was dissolved by 50 mL of the detritylation solution and the reaction mixture was monitored by TLC (20/1, CH2Cl2/MeOH). Once the appearance of the spot corresponding to the starting material (E,E-4-Dicyanomethylene-2,6-bis-p-N,N′-n-butyl-N,N′-(2-(2-(2-(2-hydroxyethoxy)ethoxy)ethoxy)ethyl)-aminostyryl pyran) on the TLC plate, the reaction was quenched by addition of a saturated aq. NaHCO3 solution (100 mL). The mixture was extracted by chloroform, and the organic layer was washed with brine, collected, dried over Na2SO4, concentrated, and subject to a silica gel column (500/10/3, CH2Cl2/MeOH/Pyridine) to give the title product 38 (128 mg, yield 17.5 %). If the recovered starting material was counted, the total yield of the monotritylation would be 67% based on the consumed starting material. ‘H NMR (CDCl3) δ (ppm): 7.49-7.38 (m, 8H, aromatic rings and double bond trans-linkage), 7.34 (dt, 4H, J1 = 2.7Hz, J2 = 9Hz, methoxylbenzene ring), 7.31-7.15 (m, 3H, benzene ring), 6.81 (dt, 4H, J1 = 2.7Hz, J2 = 9Hz, methoxylbenzene ring), 6.69 (d, 2H, J = 8.7Hz, benzene ring), 6.66 (d, 2H, J = 9.0Hz, benzene ring), 6.54 (d, 1H, J = 2.1Hz, pyran ring), 6.52 (d, 1H, J = 2.1Hz, pyran ring), 6.48 (d, 1H, J = 15.9Hz, double bond trans-linkage), 6.46 (d, 1H, J = 15.9Hz, double bond trans-linkage), 3.77 (s, 6H, CH3O), 3.56-3.50 (m, 30H, tetraethylene glycol chain), 3.44-3.31 (m, 4H, n-butyl chain), 3.22 (t, 2H, J = 5.1Hz, tetraethylene glycol chain), 1.69-1.49 (m, 4H, n-butyl chain), 1.48-1.42 (m, 4H, n-butyl chain), 0.97 (t, 3H, J = 7.5Hz, n-butyl chain), 0.96 (t, 3H, J = 7.5Hz, n-butyl chain). 13C NMR (CDC13) δ (ppm): 159.5, 158.4, 156.2, 149.8, 145.1, 138.2, 136.4, 130.2, 129.9, 128.3, 127.8, 126.7, 122.2, 122.1, 116.5, 113.1, 112.94, 112.88, 111.8, 111.7, 105.4, 86.1, 72.7, 71.0, 70.92, 70.84, 70.76, 70.5, 68.6, 63.3, 61.9, 56.1, 55.4, 51.4, 50.6, 29.4, 20.5, 14.3. MS (ESI): m/z 1145.6 [M]+.
The Phosphoramidite of Monotritylated E,E-4-Dicyanomethylene-2,6-bis-p-N,N′-n-butyl-N,N′-(2-(2-(2-(2-hydroxyethoxy)ethoxy)ethoxy)ethyl)-aminostyryl pyran (42)
To a solution of monotritylated E,E-4-Dicyanomethylene-2,6-bis-p-N,N′-n-butyl-N,N′-(2-(2-(2-(2-hydroxyethoxy)ethoxy)ethoxy)ethyl)-aminostyryl pyran (38) (200 mg, 0.17 mmol) in dry CH2Cl2 (20 mL) was added 2.7 equivalent of diisopropylethylamine (0.08 mL). Then chloro-N,N-diisopropylaminocyanoethoxyphosphane (0.05 mL, ~1.2 equal) was added dropwise at room temperature under argon. After 20 min of stirring under argon at room temperature, the reaction mixture was diluted with CH2Cl2/Et3N (300/15, v/v) 100 mL and the organic phase was washed with a saturated aq. NaHCO3 solution and brine. The organic layer was dried over Na2SO4, filtered, and evaporated to dryness. The residue was immediately subject to a silica gel column (CH2Cl2/EtOAc/Et3N, 3/6/1) to give the title product (200 mg, yield 88%) as a red powder, which should be used freshly for the next phosphotriester step in order to achieve a higher coupling yield. ‘H NMR (CDCl3) δ (ppm): 7.48-7.38 (m, 8H, aromatic rings and double bond trans-linkage), 7.34 (dt, 4H, J1 = 2.4Hz, J2 = 9Hz, methoxylbenzene ring), 7.31-7.15 (m, 3H, benzene ring of DMTr group), 6.81 (dt, 4H, J1 = 2.4Hz, J2 = 9Hz, methoxylbenzene ring), 6.69 (d, 2H, J = 8.7Hz, benzene ring), 6.67 (d, 2H, J = 9.0Hz, benzene ring), 6.56 (d, 1H, J = 2.1Hz, pyran ring), 6.54 (d, 1H, J = 2.1Hz, pyran ring), 6.50 (d, 1H, J = 15.9Hz, double bond trans-linkage), 6.48 (d, 1H, J = 15.6Hz, double bond trans-linkage), 3.91-3.70 (m, 4H, -CH2CH2CN, -CH(CH3)2), 3.77 (s, 6H, CH3O), 3.70-3.50 (m, 30H, tetraethylene glycol chain), 3.44-3.31 (m, 4H, -NCH2CH2CH2CH3), 3.22 (t, 2H, J = 5.4Hz, tetraethylene glycol chain), 2.64 (t, 2H, J = 6.5Hz, -CH2CH2CN), 1.70-1.52 (m, 4H, -NCH2CH2CH2CH3), 1.48-1.30 (m, 4H, -NCH2CH2CH2CH3), 1.18 (d, 6H, J = 6.9Hz, -CH(CH3)2), 1.17 (d, 6H, J = 6.6Hz, -CH(CH3)2), 0.97 (t, 3H, J = 7.5Hz, -NCH2CH2CH2CH3) 0.96 (t, 3H, J = 7.5Hz, -NCH2CH2CH2CH3). 13C NMR, (CDC13) δ (ppm): 159.2, 158.1, 155.9, 149.5, 144.9, 137.9, 136.1, 129.9, 129.6, 128.0, 127.5, 126.5, 121.9, 121.8, 117.6, 116.2, 112.8, 112.7, 111.5, 105.1, 85.8, 71.2, 71.1, 70.7, 70.6, 70.53, 70.49, 68.3, 63.0, 62.6, 62.4, 58.5, 58.3, 55.9, 55.1, 51.1, 50.3, 46.1, 43.1, 42.9, 29.1, 24.65, 24.58, 24.55, 24.49, 20.3, 20.24, 20.18, 14.0.; 31P NMR (CDC13) δ (ppm): 149.5.
Solid phase synthesis of DNA-chromophore conjugates
The solid phase syntheses were carried out on an Expedite Nucleic Acid Syntheses System. The DNA bases were synthesized on either glass beads or polystyrene spheres using software protocol supplied by PerSeptive Biosystems (Framingham, MA) and the phosphoramidite reagents of DNA bases were obtained from Applied Biosystems. The phosphoramidite reagents of organic chromophores were synthesized according to procedures described in this report and their solid phase syntheses were carried out using custom protocol in order to improve the reaction yields. Typically, the phosphoramidite concentrations of the organic dyes used for the solid phase syntheses were greater than 40 mg/ml. Standard DNA base coupling delivers the phosphoramidite reagent with activator to the solid phase column immediately and allows for 47 sec for coupling reaction. This reaction time is not adequate for coupling chromophoric phosphoramidite to DNA bases and hence chromophoric coupling protocol was modified by delivering the phosphoramidite reagents to the column over 4 sec period (instead of immediately) and the reaction time was extended to 140 sec (instead of 47 sec). For high yield of coupling, phosphoramidite reagents must be prepared and purified with silica gel columns immediately before carrying out the solid phase syntheses. Using the modified protocol described here, the yield of the coupling reaction is typically greater than 80% as indicated by the trityl data.
Results and Discussion
The strategy for incorporating rigid dyes, which typically have poor solubility, into biomolecules is to functionalize them with very soluble ethylene glycol chains. In Scheme 1, all precursor compounds with flexible ethylene glycol chains were synthesized (19, 20) as the appropriate benzaldehyde 7 – 14, which can be further condensed with ylids to make large π-conjugated systems. The experimental procedures for the synthesis are described in the experimental section.
Scheme 1.

Our initial attempt was to use commercially available monochlorotriethylene glycol as the precursor for introducing soluble groups. Accordingly, in Scheme 2, compound 15 was synthesized using conventional Wittig reaction with poor yield and its detritylated derivative 16 is insoluble in organic solvents. The poor solubility of the final product 16 hinders further chemical manipulation and promotes us to make longer tetraethylene glycol chain for better soluble amphiphilic chromophores. Again, Wittig reaction was used in the initial coupling reaction between the desired aldehyde 11 and the bis-ylid precursor. The reaction yielded only 15% of all-trans 17, mainly due to the formation of cis-trans isomers of 17 at low temperature (−78 °C). Detritylation of 17 yielded the final product 5, which could be dissolved in chloroform or dichloromethane. Following the above procedure, soluble compound 2 was also synthesized. Comparing the solubility difference between 16 and 2 or 5, we have concluded that tetraethylene glycol was an excellent flexible spacer for enhancing solubility of rigid chromophores. Furthermore, tetraethylene glycol proved to be excellent hinges when inserted into the DNA sequence and the influence from the spacers to the folding or unfolding behaviors of resulting hybrid foldamers was minimum.
Scheme 2.

However, the above method for synthesizing all trans double bond π-conjugates still needs improvement. Thus, Horner-Wadsworth-Emmons (HWE) reaction was used instead of conventional Wittig reaction and all three bis(styryl)benzene derivatives were synthesized again by condensing phosphonate ylid with corresponding benzaldehyde derivatives as shown in Scheme 3. The HWE reagent (phosphonate ylid) was readily prepared from α,α′-dibromo-p-xylene according to the reported procedure (21). In dried DMF, 3 equiv of desired aldehyde 10 or 13 was reacted with phosphonate ylid using KOBut as the promoter at room temperature to give the final products 2 or 5 in a high yield. Even with the bulky tetraethylene glycol chain, the HWE condition still favored the formation of all trans double bond configuration. To prepare polar chromophore 3, intermediate 19 was prepared following the same procedure for 2 and 5 except that 1 equiv of benzaldehyde 10 was used. Then final step-wise coupling reaction of 19 and 13 afforded the asymmetric π-conjugate 3.
Scheme 3.

It was reported (22) that different donor or acceptor substituents on the ends of bis(styryl)benezene π-conjugate could significantly change their emission colors because of the electron donating or withdrawing effects. The oxygen substitute on both ends of the π-conjugate in 5 can be regarded as weak electron donors, involving in the symmetric electron transfer from both ends to the middle of the conjugated system; compound 5 has bright blue fluorescence. In 2, n-butyl amino groups were used to end the π-conjugate instead of an oxygen atom as the electron donor to make a stronger donor-π-donor system, which gives green fluorescence. The trend appears that stronger electron donating groups will red-shift fluorescence emission. Following this trend, the featured asymmetric compound 3 has a fluorescence emission wavelength between 2 and 5. The fact that asymmetric compound 3 does not have a fluorescence emission longer than the symmetric compound 2 may suggest that an acceptor is required in the asymmetric compound in order to have yellow to red fluorescence. With efficient blue fluorophores in hand, our efforts have shifted to prepare similar bis(styryl)benzene structures with red fluorescent emission; the strategy is to vary the structure of middle π-conjugated bridge and tune the electronic properties of the end substituents. In Scheme 4, three asymmetric bis(styryl)benzene structures were synthesized with the pyridinium ion as an extremely strong electron acceptor in the π-conjugated structure (20–33); the resulting D-π-A structures have yellow-orange emissions (23–26). However, the phosphoramidite products of all three precursors, 26, 29, 31, were not isolated because of low mobility on silica column despite the fact that three different counter ions with various polarity and DMTr protection were attempted (experimental section).
Scheme 4.

Scheme 5 shows the syntheses of the chromophore blocks with red-orange fluorescence based on a donor-acceptor-donor (D-A-D) structure with electron rich-poor-rich distribution. Phosphonate ylid 36 was synthesized as reported previously (27,28). The same HWE procedures as described for preparation of 2 and 5 were carried out except that tetraethyl 1,4-xylylenediphosphonate was replaced by 36, consequently 1 and 4 were synthesized in good yields. Because of the electron withdrawing properties of the di-cyano groups, the hydrogen atoms on the methyl group of (2,6-dimethyl-4H-pyran-4-ylidene)malononitrile are acidic and in the presence of base the methyl groups can directly react with desired aldehyde in a Knoevenagel condensation. The simplicity of Knoevenagel condition leads to compounds 6 and 37 in one step (18). While compound 37 has low optical density and weak fluorescence properties, compound 6 emits impressive red fluorescence with peak maximum at 612 nm in organic solvents.
Scheme 5.

One important requirement for these fluorescent chromophores is to have asymmetric substitutions at either end of the rod-like or Y-shaped structures. This way, they can be protected with DMTr group on one end and activated with phosphoramidite group on the other. Such DMTr protected and phosphoramidite activated chromophores are suitable for incorporation into automated DNA synthesis.
Therefore one of the key steps is the phosphoramidite activation and its efficiency will depend on the yield and purity of its precursor—monotritylated derivative of the chromophoric building blocks. To improve the yield of monotritylated product, we have developed an efficient route as shown in Scheme 6. In a typical monotritylation process, two products were formed: monotritylated product and bistritylated product. The monotritylated product was purified as the desired product while the bistritylated product can be further converted to the monotritylated product using a weak Lewis acid zinc chloride as the detritylation reagent (29).
Scheme 6.

A typical detritylation solution consists of ZnCl2 (1.5 g) with 110 mL of CH2Cl2/MeOH (10/1, v/v). The detritylation procedure involves treating the bistritylated product in the detritylation solution. The reaction can be monitored with TLC and typically detritylation finishes within an hour. Once the spot corresponding to the original, non-substituted chromophore appeared on the TLC plate, the reaction was quenched by pouring its content into an aqueous solution saturated with NaHCO3. After chromatography, the yield of the monotritylated product exceeded 40% based on the bistritylated compound. While combining with the direct monotritylation product, the total yield of the mono-tritylated product had been significantly improved (from ~30% to 56–77%). This method can be applied to large-scale preparation of monotritylated derivatives of chromophoric building blocks and the resulting mono-substituted products can be kept in −80 °C freezer under basic atmosphere for several months without noticeable decomposition.
Finally, a few fluorescent chromophores, which have orthogonal fluorescence emission, were chosen for phosphoramidite activation. They are compound 38, the precursor 6 of which emits red fluorescence with an intensity maximum at 612 nm, compound 40, the precursor 5 of which emits blue fluorescence with an emission maximum at 430 nm, and compound 39, the precursor 2 of which emits (at 473 nm) in blue-green region (Table 1). The fluorescence spectra of the red-blue or red-blue/green FRET pairs have little overlap, hence orthogonal in fluorescent emissions. For efficient FRET pairs, the absorption maximum of the red-fluorescence chromophore (488 nm) should overlap perfectly with the fluorescent emission of the blue-fluorescence donor.
Table 1.
UV and fluorescence data. The solvent used is chloroform; the concentrations of various chromophores are ~10 μM
| Chromophores | Absorbance (λmax) Wavelength (nm) | Excitation Wavelength (nm) | Emission (λmaxEM) Wavelength (nm) |
|---|---|---|---|
| 1 | 379 | 379 | 469 |
| 2 | 398 | 399 | 473 |
| 3 | 369 | 384 | 454 |
| 4 | 477 | 477 | 559 |
| 5 | 359 | 360 | 409, 430 |
| 6 | 488 | 490 | 612 |
| 29 | 433 | 418 | 581 |
The phosphoramidite derivatives were made according to our prior report (30). It is worth mentioning that methylene chloride or methylene chloride/acetonitrile (1/1) was employed as reaction solvents to ensure the maximum solubility of the chromophore precursor, as is the key point to reach the highest yield of the phosphoramidite derivatives. Also 5–10% triethylamine in the system was necessary to stabilize the phosphoramidite compounds. The procedure describing the synthesis of phosphoramidites is presented in the experimental section. The yields of crude phosphoramidites from the monotritylated derivatives usually exceeded 90%, suitable for solid-state synthesis of phosphoramidite chemistry. The stability of these phosphoramidite chromophores was not optimum in solutions such that the phosphoramidite reagents must be prepared immediately before solid-phase synthesis. Because of the presence of tetra ethylene glycol units, the phosphoramidite reagents usually cannot be purified with crystallization, but flash chromatography was effectively for their final purification before usage. Additionally, the dumb-bell shaped spots on TLC plates, which correspond to the two steric isomers due to the chiral phosphorus atom in the phosphoramidite of normal DNA bases, were hardly observed in these phosphoramidite chromophores.
Solid phase synthesis was employed to incorporate the final chromophoric candidates into single strand DNA backbones as shown in Scheme 7. Our ability to synthesize asymmetrically substituted chromophoric building blocks has established a firm foundation for preparation of foldamers with a controlled molecular orientation, well-defined biological and synthetic sequences. All fluorescent chromophores have been functionalized with hydroxyl groups at their termini. Furthermore, one hydroxyl group will be protected with DMTr and the other will be activated with the β-cyanoethyl phosphoramidite group.
Scheme 7.

Foldamer synthesis employs the same strategy of DNA synthesis, i.e., one end of the chromophore is activated with phosphoramidite group for attacking the hydroxyl group at the growing chain terminal and the other end can be regenerated for chain growth by removing the DMTr protected hydroxyl group. The essential steps are coupling between a chain terminal hydroxyl group and a phosphoramidite group followed by detritylation to regenerate the chain terminal hydroxyl group for chain growth.
Unlike DNA synthesis that uses phosphoramidite on the secondary hydroxyl group of deoxyribose, the chromophoric blocks use phosphoramidite groups on primary hydroxyl groups, which have completely different inherent activities. Therefore, their stability and activity towards hydroxyl group at chain termini are critical to the success of solid phase synthesis. Nonetheless, we have achieved coupling yields as high as 95% by increasing the phosphoramidite concentration, reaction time, and by repeating the coupling step. The solid-phase synthesis of foldamer is compatible with DNA synthesis; fluorescent chromophores can be incorporated anywhere in the sequence, for example a juxtaposition of blue and red chromophores (41-42-41) embedded in the DNA main chains as shown in Figure 3 (51-mer). Using the trityl monitor reading of the five bases before the three chromophore sequence and five bases after the chromophore sequence, we obtained a total yield for coupling three chromophores consecutively is 92%, which corresponds to 97% yield per chromophore coupling. When the chromophore is coupled to a DNA base, followed by another DNA base (57-mer), the yield is also very high. For example, at the 48th position, the chromophore 42 is introduced and the yield of 95% is obtained using the three bases immediately before and after this position. Although trityl absorption does not provide extremely accurate yields for coupling reactions, but it should be a good indicator to gauge these high yield coupling reactions.
Figure 3.
A, T, C, and G stands for single DNA bases linked by phosphotriester and number “5” or “6” stands for synthetic chromophores. Specifically, number “6” here is the red emitting chromophore with TEG chains linked by phosphotriester (compound 42) with either DNA bases or chromophores, and number “5” represents the blue emitting chromophore with TEG chains linked by phosphotriester (compound 41) with other DNA bases or chromophores. Note the coupling yields for the two chromophores to DNA bases are almost as high as those of the coupling efficiency of DNA bases. Conversely, coupling of DNA bases to the chromophores are equally high.
51-mer: 5′-GGC CAA CTC CAT CAC TAG GGG TTC CT-41-42-41-GGA GGG GTG GAG TCG TGA CGT G-3′
57-mer: 5′-TTT GAT GCT-42-AGT CCA TTT GTC AGT GCT-41-ACC AGA GAT GAG TTG ATG-42-TAC TGC CGA-3′
39-mer: 5′-AA-42-AA-42-AA-42-AA-42-AA-42-AA-42-AA-42-AA-42-AA-42-AA-42-AA-42-AA-42-AAG-3′
Another example of our state-of-the-art coupling syntheses is shown in Figure 3 with alternating sequences of 42 and double A bases (39-mer); again the yield of each individual steps are high. For example, using the ratio of the two bases before and after position 13 (42), we obtained a yield of 92%. Because of such high coupling efficiency, the overall yield is at 40% at 39mer. In the finished sequence, all together 12 fluorescent dyes are introduced into the DNA backbone; this demonstrates the power of synthetic organic chemistry in biological applications.
As long as the dye number density in the DNA sequence is not too high, the resulting hybrid sequence behaves frequently like oligo-DNA. For example, the dye-DNA conjugates can be cleaved from the solid support using concentrated ammonium hydroxide and the crude product can be further purified with HPLC as shown in Figure 4. Monitoring at the absorption (λmax ~485 nm) of the red-fluorescence dye (42), the major isolated product is, typically, the desired full-length dye-DNA conjugates. The HPLC-purified dye-DNA conjugates have good purity and are suitable for most biological assays.
Figure 4.

HPLC purification of a DNA 40 mer containing one orange-fluorescence dye (compound 42) and two blue-fluorescence dyes (compound 43) in the sequence. Full length DNA sequence containing three organic dyes has an elution time of approximate 28 min while using a linear gradient of H2O and acetonitrile containing 0.1 M TEAA (triethylamine acetate).
Conclusion
In conclusion, we have developed a new method for converting normal organic fluorescent dyes into asymmetrically functionalized phosphoramidite reagents, which has enabled bio-conjugation of many fluorescent chromophores at multiple desired positions of DNA backbones. Using Horner-Wadsworth-Emmons (HWE) reaction and Knoevenagel condensation, a series of highly fluorescent amphiphilic compounds was constructed; these compounds are suitable for labeling DNA since they can be conveniently protected with DMTr and activated with phosphoramidite and their fluorescence is not quenched by the presence of DNA bases.
Acknowledgments
The authors acknowledge the support of National Institute of General Medical Sciences (Grant GM065306) and the Arnold and Mabel Beckman Foundation. ADQL is a Beckman Young Investigator (BYI). We thank LBB2 for the Mass measurement and Maggie Tam for the valuable suggestion.
References
- 1.Letsinger RL, Wu TF. Use of a stilbenedicarboxamide bridge in stabilizing, monitoring, and photochemically altering folded conformations of oligonucleotides. J Am Chem Soc. 1995;117:7323–7328. [Google Scholar]
- 2.Lewis FD, Wu TF, Burch EL, Bassani DM, Yang JS, Schneider S, Jager W, Letsinger RL. Hybrid oligonucleotides containing stilbene units - excimer fluorescence and photodimerization. J Am Chem Soc. 1995;117:8785–8792. [Google Scholar]
- 3.Bevers S, O’Dea TP, McLaughlin LW. Perylene- and naphthalene-based linkers for duplex and triplex stabilization. J Am Chem Soc. 1998;120:11004–11005. [Google Scholar]
- 4.Xu XHN, Brownlow W, Huang S, Chen J. Single-molecule detection of efflux pump machinery in Pseudomonas aeruginosa. Biochem Biophys Res Commun. 2003;305:79–86. doi: 10.1016/s0006-291x(03)00692-2. [DOI] [PubMed] [Google Scholar]
- 5.Martin RM, Leonhardt H, Cardoso MC. DNA labeling in living cells. Cytometry Part A. 2005;67A:45–52. doi: 10.1002/cyto.a.20172. [DOI] [PubMed] [Google Scholar]
- 6.Hards A, Zhou CQ, Seitz M, Brauchle C, Zumbusch A. Simultaneous AFM manipulation and fluorescence imaging of single DNA strands. ChemPhysChem. 2005;6:534–540. doi: 10.1002/cphc.200400515. [DOI] [PubMed] [Google Scholar]
- 7.Ishikawa M, Maruyama Y, Ye JY, Futamata M. Single-molecule imaging and spectroscopy using fluorescence and surface-enhanced Raman scattering. J Biol Phys. 2002;28:573–585. doi: 10.1023/A:1021222318740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Garcia-Parajo MF, Veerman JA, van Noort SJT, de Grooth BG, Greve J, van Hulst NF. Near-field optical microscopy for DNA studies at the single molecular level. Bioimaging. 1998;6:43–53. [Google Scholar]
- 9.Wang W, Wan W, Zhou HH, Niu S, Li ADQ. Alternating DNA and π-conjugated sequences. Thermophilic foldable polymers. J Am Chem Soc. 2003;125:5248–5249. doi: 10.1021/ja0341900. [DOI] [PubMed] [Google Scholar]
- 10.Wang W, Wan W, Stachiw A, Li ADQ. Foldamers with hybrid biological and synthetic sequences as selective DNA fluorescent probes. Biochemistry. 2005;44:10751–10756. doi: 10.1021/bi050945z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schleifenbaum A, Stier G, Gasch A, Sattler M, Schultz C. Genetically encoded FRET probe for PKC activity based on pleckstrin. J Am Chem Soc. 2004;126:11786–11787. doi: 10.1021/ja0460155. [DOI] [PubMed] [Google Scholar]
- 12.Fujimoto K, Shimizu H, Inouye M. Unambiguous detection of target DNAs by excimer-monomer switching molecular beacons. J Org Chem. 2004;69:3271–3275. doi: 10.1021/jo049824f. [DOI] [PubMed] [Google Scholar]
- 13.Du H, Disney MD, Miller BL, Krauss TD. Hybridization-based unquenching of DNA hairpins on Au surfaces: Prototypical “molecular beacon” biosensors. J Am Chem Soc. 2003;125:4012–4013. doi: 10.1021/ja0290781. [DOI] [PubMed] [Google Scholar]
- 14.Fang XH, Li JJ, Tan WH. Using molecular beacons to probe molecular interactions between lactate dehydrogenase and single-stranded DNA. Anal Chem. 2000;72:3280–3285. doi: 10.1021/ac991434j. [DOI] [PubMed] [Google Scholar]
- 15.Tsourkas A, Behlke MA, Xu Y, Bao G. Spectroscopic features of dual fluorescence/luminescence resonance energy-transfer molecular beacons. Anal Chem. 2003;75:3697–3703. doi: 10.1021/ac034295l. [DOI] [PubMed] [Google Scholar]
- 16.Antony T, Thomas T, Sigal LH, Shirahata A, Thomas TJ. A molecular beacon strategy for the thermodynamic characterization of triplex DNA: Triplex formation at the promoter region of cyclin D1. Biochemistry. 2001;40:9387–9395. doi: 10.1021/bi010397z. [DOI] [PubMed] [Google Scholar]
- 17.Albota M, Beljonne D, Brédas JL, Ehrlich JE, Fu JY, Heikal AA, Hess SE, Kogej T, Levin MD, Marder SR, McCord-Maughon D, Perry JW, Röckel H, Rumi M, Subramaniam G, Webb WW, Wu X, Xu C. Design of organic molecules with large two-photon absorption cross sections. Science. 1998;281:1653–1656. doi: 10.1126/science.281.5383.1653. [DOI] [PubMed] [Google Scholar]
- 18.Moylan CR, Ermer S, Lovejoy SM, McComb IH, Leung DS, Wortmann R, Krdmer P, Twieg RJ. (Dicyanomethylene)pyran derivatives with C-2v symmetry: An unusual class of nonlinear optical chromophores. J Am Chem Soc. 1996;118:12950–12955. [Google Scholar]
- 19.Amabilino DB, Ashton PR, Brown CL, Cordova E, Godinez LA, Goodnow TT, Kaifer AE, Newton SP, Pietraszkiewicz M, Philp D, Raymo FM, Reder AS, Rutland MT, Slawin AMZ, Spencer N, Stoddart JF, Williams DJ. Molecular meccano. 2 Self-assembly of [eta]catenanes. J Am Chem Soc. 1995;117:1271–1293. [Google Scholar]
- 20.Maeda H, Furuyoshi S, Nakatsuji Y, Okahara M. Intramolecular cyclization of N, N-bis(oligooxyethylene)amines: a new synthesis of monoaza crown ethers. Tetrahedron. 1982;38:3359–3362. [Google Scholar]
- 21.Pond SJK, Tsutsumi O, Rumi M, Kwon O, Zojer E, Brédas JL, Marder SR, Perry JW. Metal-ion sensing fluorophores with large two-photon absorption cross sections: Aza-crown ether substituted donor-acceptor-donor distyryl benzenes. J Am Chem Soc. 2004;126:9291–9306. doi: 10.1021/ja049013t. [DOI] [PubMed] [Google Scholar]
- 22.Bartholomew GP, Bazan GC. Synthesis, characterization, and spectroscopy of 4,7,12,15-[2.2]paracyclophane containing donor and acceptor groups: Impact of substitution patterns on through-space charge transfer. J Am Chem Soc. 2002;124:5183–5196. doi: 10.1021/ja0121383. [DOI] [PubMed] [Google Scholar]
- 23.For TBDMS manipulation: Lombardo L. Diisopropylethylamine: an effective catalyst for the introduction of the tert-butyldimethylsilyl group. Tetrahedron Lett. 1984;25:227–228.
- 24.Corey EJ, Venkateswarlu A. Protection of hydroxyl groups as tert-butyldimethylsilyl derivatives. J Am Chem Soc. 1972;94:6190–6191. [Google Scholar]
- 25.For ion replacement: Brimacombe JS, Kabir AKMS. Higher-carbon sugars. Part 10 The synthesis and osmylation of 7,8-dideoxy-1,2:3,4-di-O-isopropylidene-b-L-glycero-D-galacto-oct-7-enopyranose and related studies. Car Res. 1988;174:37–45.
- 26.For condensation: Maekawa Y, Kato S, Saigo K, Hasegawa M, Ohashi Y. Crystallographic interpretation of the topochemical behavior of alkyl alpha-cyano-4-[2-(4-pyridyl)ethenyl]cinnamates in the crystalline state - enhancement of photopolymerizability by complex-formation. Macromolecules. 1991;24:2314–2322.
- 27.Wenseleers W, Stellacci F, Meyer-Friedrichsen T, Mangel T, Bauer CA, Pond SJK, Marder SR, Perry JW. Five orders-of-magnitude enhancement of two-photon absorption for dyes on silver nanoparticle fractal clusters. J Phys Chem B. 2002;106:6853–6863. [Google Scholar]
- 28.Ngola SM, Kearney PC, Mecozzi S, Russell K, Dougherty DA. A selective receptor for arginine derivatives in aqueous media. Energetic consequences of salt bridges that are highly exposed to water. J Am Chem Soc. 1999;121:1192–1201. [Google Scholar]
- 29.Ding XL, Wang W, Kong FZ. Detritylation of mono- and di-saccharide derivatives using ferric chloride hydrate. Carbohydr Res. 1998;303:445–448. [Google Scholar]
- 30.Wang W, Li LS, Helms G, Zhou HH, Li ADQ. To fold or to assemble? J Am Chem Soc. 2003;125:1120–1121. doi: 10.1021/ja027186h. [DOI] [PubMed] [Google Scholar]
- 31.Perrin DD, Armarego WLF, editors. Purification of Laboratory Chemicals. 3. Pergamon Press; [Google Scholar]
- 32.Stuhr-Hansen N, Christensen JB, Harrit N, Bjrnholm T. Novel synthesis of protected thiol end-capped stilbenes and oligo(phenylenevinylene)s (OPVs) J Org Chem. 2003;68:1275–1282. doi: 10.1021/jo0263770. [DOI] [PubMed] [Google Scholar]
- 33.Schwöppe D, Meier H. Crown ether substituted 1,4-distyrylbenzene and their complexation with alkali picrates. J Prakt Chem. 2000;342 (5):459–464. [Google Scholar]