

Abstract
Emulsion polymerization yields 40–400 nm diameter polymer nanoparticles with spiropyran-merocyanine dyes incorporated into their hydrophobic cavities; in contrast to their virtually nonfluorescent character in most environments, the merocyanine forms of the encapsulated dyes are highly fluorescent. Spiro-mero photoisomerization is reversible, allowing the fluorescence to be switched “on” and “off” by alternating UV and visible light. Immobilizing the dye inside hydrophobic pockets of nanoparticles also improves its photostability, rendering it more resistant than the same dyes in solution to fatigue effects arising from photochemical switching. The photophysical characteristics of the encapsulated fluorophores differ dramatically from those of the same species in solution, making nanoparticle-protected hydrophobic fluorophores attractive materials for potential applications such as optical data storage and switching and biological fluorescent labeling. To evaluate the potential for biological tagging, these optically addressable nanoparticles have been delivered into living cells and imaged with a liquid nitrogen cooled CCD.
Introduction
Photochromic materials have been the focus of intensive investigations for several decades1–3 because they have high potential for applications to optically rewritable data storage,4 optical switching,5,6 and chemical sensing.7 Like other stimuli-responsive materials such as heat and pH-sensitive materials,8,9 photochromic devices require that either the nanostructure or the molecular structure be responsive to an external stimulus —in this case, light. Consequently, organic functional groups with stimuli-responsive photochromic units have been widely applied to the construction of photoswitchable materials such as polymer thin films and organic-inorganic hybrids.10,11 Herein, we report that polymer nanoparticles formed by emulsion polymerization have photochromic effects; in particular, the fluorescence of these photochromic nanoparticles can be optically switched “on” and “off” with specific wavelengths of light.
Optical switching of nanoparticle photoluminescence is important because it opens new possibilities for developing individually addressable nanoscale devices with light. For biological applications, fluorescence switching should be reversible and high contrast so that it can be used to selectively highlight cells, organelles, or proteins. In this context, dual color fluorescent marker proteins such as cyan fluorescent protein (PS-CFP) and EosPF have recently attracted attention as biochemical markers. These proteins can be photoconverted from cyan to green (PS-CFP)12 or from 516 nm to 581 nm (EosPF)13 after exposure to near-UV irradiation at 405 or 390 nm, respectively, confirming their presence at labeled sites. These processes are not reversible, however, which limits their applicability;12,13 in fact, only several examples of nanoparticles that undergo reversible fluorescence switching have been reported,14,15 and rarely photoswitches fluoresce by itself.
Many photoresponsive solid films have been developed; for example, surfactants containing acetylene moieties that are polymerized into thin films simultaneously acquire irreversible photoluminescence.16 However, these films usually lack reversibility, as well as fast response, upon application of pulses of external stimuli.17 Consequently, to date, there have been few studies on practical applications of nanoparticles capable of luminescence switching. Ideally, it would also seem preferable to develop devices that could function in environmentally benign aqueous systems.18,19
Experimental Section
Sample preparation
Reagents
All reagents and solvents were purchased from Aldrich Chemical Co. and used as received except as stated otherwise. 2,2′-N-isopropylacrylamide (NIPAM) and 4,4′-azobis(4-cyano-valeric acid) (ABVA) were recrystallized from n-hexane and methanol, respectively. Styrene (St, 99%), butyl acrylate and methylmethacrylate (MMA) were distilled from CaH2 under reduced pressure.
Characterization
1H NMR spectra were recorded in CDCl3 using a JEOL JNM-AL300 instrument. Dynamic light scattering (DLS) measurements were carried out on a Beckman-Coulter N4 instrument at fixed scattering angles of 62.6 and 90° with the 632.8 line of a He-Ne laser as excitation source; standard polystyrene microspheres were used for instrument calibration. The average particle sizes and size distributions were obtained from the autocorrelation decay functions by CONTIN analysis using standard software package supplied by Beckman-Coulter. A JEOL 1010 transmission electron microscope (TEM) operated at 100 kV was employed to obtain TEM images. The microscope sample was prepared by placing a drop of the polymer dispersion on a carbon-coated Cu grid, followed by solvent evaporation at room temperature.
Synthesis of 5-(1,3-dihydro-3,3-dimethyl-6-nitrospiro[2H-1-benzopyran-2,2′-(2H)-indole])ethyl acrylate (SP)
5-(1,3-dihydro-3,3-dimethyl-6-nitrospiro[2H-1-benzopyran-2,2′-(2H)-indole]) ethanol (SP-ethanol) was synthesized according to procedures described in the literature.20 Yield: 31%. 1H NMR (300 MHz, CDCl3) δ 1.14 and 1.35 (3H each, s, C(CH3)2), 3.53 (2H, m, COOCH2), 4.36(2H, m, NCH2), 5.87 and 6.14 (2H and 1H, m, CH2=CH), 6.36 (1H, d, CCH=CHPh), 6.78 (1H, d, 7′-H of indoline), 6.87 (1H, d, CCH=CHPh), 6.95 (2H, m,, 4′ and 5′-H of indoline), 7.17 (1H, d, 6′-H of indoline), 7.20 (1H, m, 4-H of benzopyran), 8.0 (2H, m, 5 and 7-H of benzopyran).
Emulsion polymerization
Emulsion polymerization was carried out according to the literature procedure with minor modifications as reported elsewhere.21 The percentage of spiropyran comonomer incorporated into the nanoparticles is > 96% based on UV-analysis of the organic phase (CHCl3) extracted from the emulsion mixture. The TEM characterization and dynamic light scattering of the as-prepared polymer nanoparticles are presented in the results and discussion section.
Cell growth and nanoparticle surface modification
Transformed human embryonic kidney (HEK 293) cells were grown in 24-well culture plates until cell populations reached ~105 cells per well. A 1 pmol nanoparticle suspension was prepared in 50 μL DMEM (Dulbecco’s Modified Eagle Medium). In a separate vial, 0.5 μL Lipofectamine™ 2000 reagent was mixed with 50 μL DMEM and incubated for 5 minutes at RT, after which the dilute nanoparticle suspension was added followed by an additional 20 minutes incubation at RT.
Delivery of nanoparticles into living cells
Nanoparticles were delivered into cells by adding 100 μL of the nanoparticle-Lipofectamine™ 2000 suspension to each cell-containing well in the culture plate. After incubating for 5 hours at 37°C in a CO2 incubator, the cells were washed with 500 μL DMEM and resuspended in 500 μL DMEM containing 10% FBS (Fetal Bovine Serum).
Sample preparation for microscopy and spectroscopy
Cover glasses (Gold Seal No. 1, Fisher) were cleaned by a protocol comprising sonication in 2% Micro 90™ for 30 minutes, rinsing thoroughly with 18 MΩ water, drying in an oven, soaking in Chromerge™ for one hour, rinsing thoroughly with 18 MΩ water, and finally heating gently in a methane flame to dryness. After cooling slowly, the cover glasses were stored in a dust-free container. Nanoparticle samples were prepared by spin-coating (4000 rpm) ~30 μL of a dilute suspension of nanoparticles in spectroscopic grade ethanol onto a cover glass. Samples were diluted sequentially until the density of nanoparticles on the cover glass was suitable for single particle measurements. For cell samples, cover glasses were modified by gluing o-rings onto one side and filling the resulting wells with a DMEM solution. Washed cells were transferred into the well and allowed to settle before inspection.
Microscopy and spectroscopy
Microscope design
Samples were placed on a custom-built stage of an inverted microscope (Zeiss Axiovert 200) equipped with a high numerical aperture oil immersion objective (Zeiss, 100X, 1.3 NA) and an X-Y nanopositioner stage (Mad City Labs). Connected to the side port of the microscope was a spectrometer (Acton Research Corp.) coupled to a liquid nitrogen cooled CCD detector (Princeton Instruments, Roper Scientific). The spectrometer was equipped with both a mirror for imaging and a grating for spectroscopy. An avalanche photo diode (APD) was used to collect photons from the bottom microscope port. Excitation entering the back port of the microscope was redirected by an appropriate filter set into the back aperture of the objective. Emission light was collected through the same objective and directed to the side or bottom port of the microscope.
Wide field nanoparticle imaging
For wide field imaging of nanoparticles a mercury vapor short arc lamp illuminator (Zeiss HBO 100) was mounted to the back illumination port of the microscope. Two filter sets, each consisting of an excitation filter (ex), dichroic beamsplitter (bs), and emission filter (em), were used to isolate either the 365 nm (Zeiss, filter set 2, ex365; bs295; em420) or 488 nm (Zeiss, filter set 16, ex485/20; bs510; em515) excitation lines from the mercury lamp spectrum by changing positions on the filter carousel. Excitation light was not attenuated.
Samples were first illuminated with 365 nm light to convert the photoswitches to their strongly emitting mero forms, thereby facilitating focusing of the nanoparticles via CCD imaging. After achieving focus, the 365 nm light was blocked and a new area on the cover glass was moved into the illumination area via the nanopositioner. The new area was illuminated with 365 nm light for 3–4 seconds. After this period, illumination at 488 nm and image acquisition were simultaneously started. Fluorescence emission was guided through a red long pass filter (620 nm) before being directed onto the spectrometer mirror, which reflected the image onto the CCD chip. Wide field images were recorded until fluorescence from the individual nanoparticles dropped below background levels of the CCD (typically 4–5 minutes), indicating that the majority of photoswitches had reverted to the spiro form. All images were collected using a three second integration time in WinSpec/32 software. Two-dimensional black and white CCD images were converted into 3-D color surface plots in Igor Pro software (Wavemetrics).
Cell imaging
To minimize heating effects, an argon ion laser (488 nm) and hand-held UV lamp were used instead of the mercury lamp for these studies. In a typical cell imaging procedure, cells were first focused and positioned with an overhead incandescent light, then exposed to UV light (365 nm) for a few seconds; intracellular nanoparticles were then focused using the 488 nm laser line and exposed to UV light for 10 seconds, after which fluorescence switching was monitored under 488 nm laser excitation. The last two steps were repeated as required for data collection. Laser power (15 mW) was attenuated to 0.9 mW prior to entering microscope optics. All emission was directed through a red long pass filter as described above.
Single nanoparticle spectroscopy
Samples were first focused as described for wide field nanoparticle imaging. After a new area on the cover glass was moved into place and illuminated with 365 nm light for 3–4 seconds, the mercury lamp was unmounted and the 488 nm line from an argon ion laser was directed into the back port of the microscope. Prior to entering the microscope, the laser beam was collimated and expanded to slightly overfill the back aperture of the objective with parallel rays in order to achieve a diffraction limited spot. Filter set 16 (see above) was used for all single particle measurements.
To collect single particle spectra, an image of the nanoparticles was first obtained by scanning a small area (typically 10×10 μm) of the sample over the diffraction limited laser spot using the nanopositioner. Emission from this type of scan was directed to the bottom microscope port and onto the APD. The resulting image showing individual particles (bright spots) was used to park the laser on a specific nanoparticle for spectral acquisition. The entire scanning and image generation routine was achieved using custom LabVIEW software. The typical step size and dwell (integration) times for each pixel was 200 nm and 100 ms, respectively. Once a specific nanoparticle was chosen, the light (emission) train was switched to the spectrometer/CCD, this time using the spectrometer’s grating instead of the mirror. Spectra were acquired through WinSpec/32 software using a typical dwell time of 1–2 seconds. Acquisition continued until the fluorescence emission was no longer discernable from the baseline. Typical laser powers used for single particle spectral acquisition were ~800 W/cm2 at the cover glass surface.
Results and Discussion
As photochromic materials, colorless spiropyrans undergo photoinduced ring opening reactions under ultraviolet irradiation to yield their isomeric merocyanine forms, which have strong solvatochromic visible absorption bands in the range of 500–600 nm.22 This band arises from the extended π-conjugation that develops between the indolene and the pyran rings following rupture of the C (spiro)-O bond. Back-conversion from the mero forms of the dyes to the spiro forms occurs thermally at relatively slow rates (typically, t1/2 ~ 103 s), but is accelerated by excitation into the merocyanine visible band.23
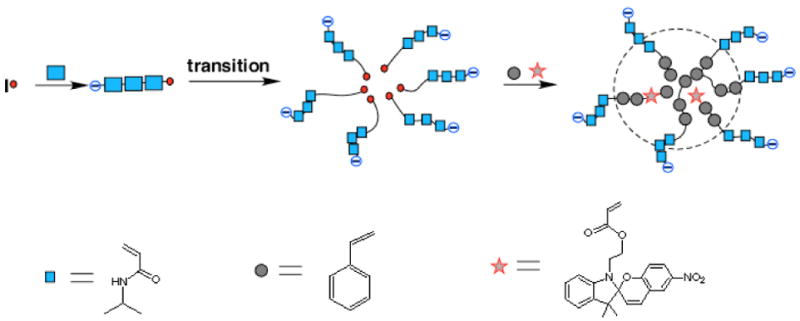
Our approach to constructing a reversibly switchable particulate system was to embed spiropyrans within the hydrophobic cavities of polymeric nanoparticles (Scheme 1). These were prepared by using N-isopropylacrylamide (NIPAM), styrene (St) as the primary monomeric units in combination with minor amounts of divinyl benzene (DVB) as the cross-linker and 5-(l,3-dihydro-3,3-dimethyl-6-nitrospiro[2H-1-benzopyran-2,2′-(2H)-indole])ethyl acrylate (SP) as the optically active unit; occasionally small amounts of butylacrylate (BA) were also added to lower the nanoparticle glass transition temperature (Tg). The specific technique employed here is based on a modified emulsion polymerization method.21 In a typical experiment, the water-soluble compound, 4,4′-azobis(4-cyano-valeric acid), is used to initiate polymerization and therefore the monomer that polymerizes first is water-soluble NIPAM. Since poly(N-isopropylacrylamide), PNIPAM, is thermosensitive and exhibits hydrophobic properties at temperatures above its low critical transition temperature (Tt = 31 °C),24 the NIPAM chains become increasingly hydrophobic as polymerization progresses at 90 °C and self-organize into micelles with the assistance of the surfactent Tween 20. After this transition from hydrophilic chains to hydrophobic polymerizing chains and formation of micelle structures, hydrophobic monomers (St, SP, and DVB) begin to participate in the polymerization and are incorporated into the growing chains. The net result is formation of polymer nanoparticles with hydrophilic NIPAM shells and hydrophobic styrene and spiropyran cores that are cross-linked with divinylbenzene. Because the shells are hydrophilic, the resulting nanoparticles are easily suspended in water, but because the polymer chains are cross-linked, individual particles remain insoluble, i.e., retain their identities.
Scheme 1.
The as-prepared nanoparticles were characterized by transmission electron microscopy (TEM) and dynamic light scattering (DLS). Plots of DLS measurements carried out at scattering angles of 62.6 and 90 degrees showed that the polymer nanoparticles are spherical with average diameters of 68 nm and a relatively narrow polydispersity of 0.098 (Figure 1A). The same polymer nanoparticles measured with TEM (Figure 1B) gave a somewhat smaller mean size diameter of 60 nm, although the size distribution was again quite uniform. Since the DLS measurements were carried out in solution and the TEM measurements were made on dry solid under high vacuum, this difference is probably attributable to solvent-induced swelling of the soft polymer nanoparticles. Polymer nanoparticles with average diameters ranging from 40 nm to 400 nm were readily prepared by varying the feed ratios of monomers and/or surfactants. Specifically, at a ~2:1 molar ratio of hydrophobic and hydrophilic monomers, smaller polymer nanoparticles (~40 nm) generally formed, whereas larger nanoparticles were usually obtained when the ratio of hydrophobic monomers (e.g., St) to the hydrophilic monomer (NIPAM) was increased.
Figure 1.
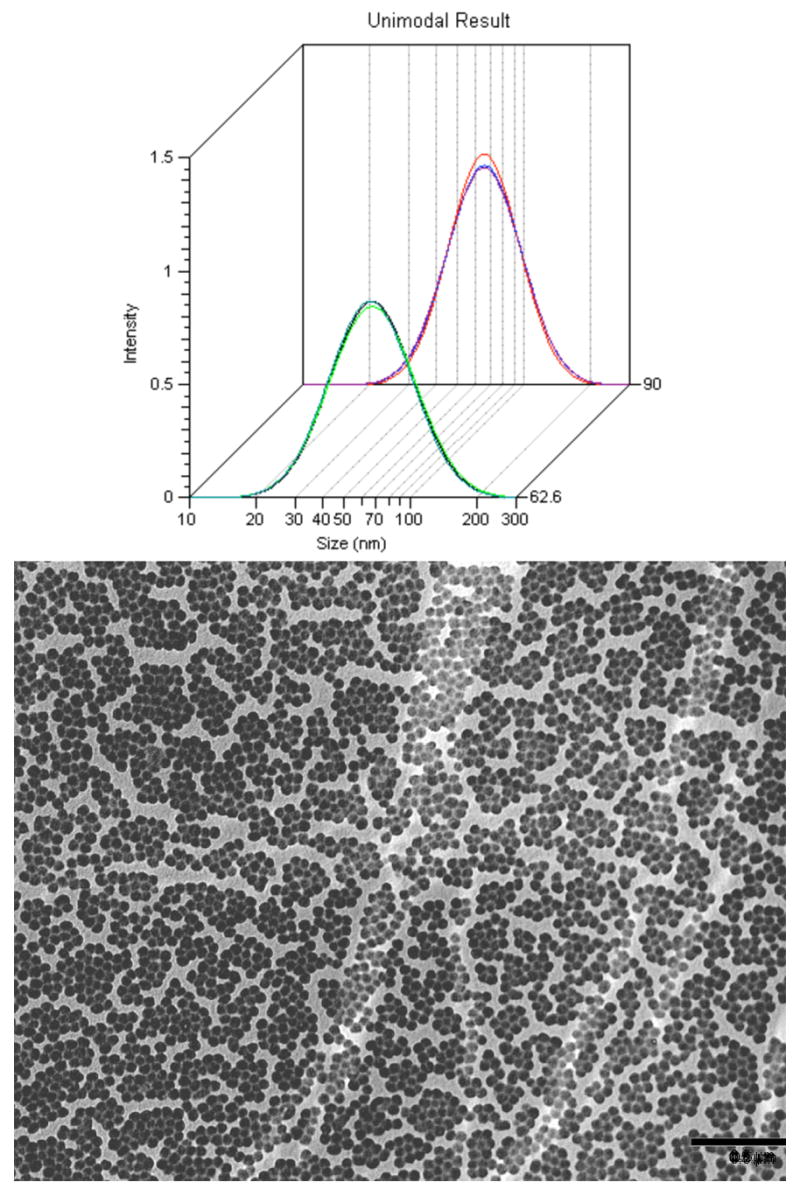
Light scattering and TEM. (A) Dynamic light scattering carried out 62.6° and 90° angles on 1.4 nM photochromic polymer nanoparticle solutions. (B) Micrograph from electron transmission microscope. Scale bar is 500 nm.
Neither spiro nor mero forms of the dye fluoresce appreciably in water, although the mero form possesses detectable fluorescence in polar organic solvents25 and within self-assembling films.26 The emission spectra and dynamics are complicated by the existence of several geometrical isomers of the merocyanine, which have different fluorescence quantum yields and lifetimes and apparently interconvert on the same time scale as fluorescence decay,25 and, in highly doped films, by energy transfer among monomeric and various aggregated species.26 In our studies, the mero form of the dye incorporated into the hydrophobic core of polymer nanoparticles was highly luminescent (Scheme 2). The photoisomerization quantum yield from spiro- to mero-form is 28% in the hydrophobic nanoparticles, whereas the photoisomerization quantum yield of spiropyran in methanol is 42%. As shown in Figure 2A, the mero-nanoparticles fluoresce strongly in the red region of 640 to 780 nm in aqueous solutions. The same water-soluble merocyanine dye dissolved in aqueous solution gave no observable fluorescence under comparable conditions. Based upon measured intensities, a merocyanine molecule residing in a hydrophobic nanoparticle was at least 200 times more strongly fluorescent than in aqueous solutions. Likewise, only weak fluorescence was observed when the same amount of merocyanine dye molecules were incorporated into polymer chains which do not form micellar structures (Figure 2A). This spiro-containing polymer was prepared by AIBN-initiated radical polymerization of styrene and the spiro-monomer at 90°C for 8 hours. The mero-nanoparticles also fluoresced strongly in organic solvents and when incorporated into solid thin films, whereas the fluorescence intensity of the free merocyanine dye and the merocyanine-containing polymer remained relatively nonfluorescent in these environments. These results underscore the importance of the nanoparticle microphase environment to gaining fluorescence intensity.
Scheme 2.
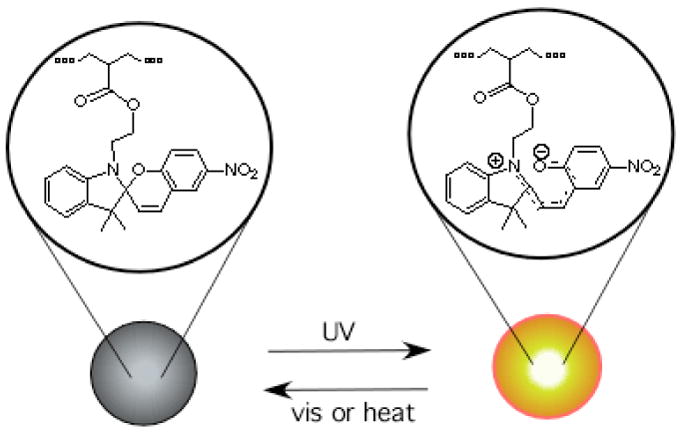
Figure 2.
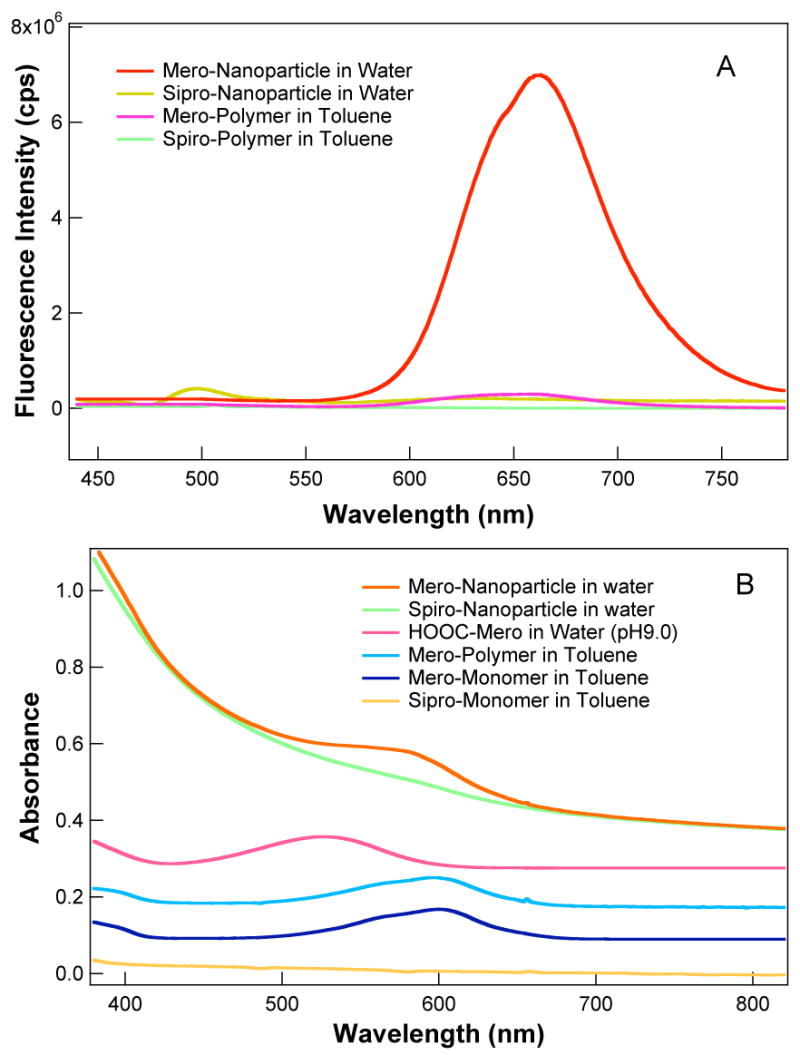
Fluorescence (A). Merocyanine has no fluorescence in the visible region. When merocyanine is incorporated into non-micelle-forming polymers, a weak fluorescence is observed (pink curve). However, a strong red fluorescence (λmax ~660 nm) is observed (red curve) when merocyanine is incorporated into hydrophobic cores of the polymer nanoparticles. The concentration of the nanoparticles is 7.0 nM and all solutions contain 0.3 mM photoswitchable dyes. Absorption (B). Optical absorption spectra of nanoparticles, polymers, and monomers, which are offset for display purposes. The spiropyran concentrations are the same as in panel A.
The fluorescence quantum yield for the merocyanine-containing particle was determined to be ϕ = 0.24 by comparing its fluorescence intensity to that of a rhodamine B standard (ϕ = 0.48).27 These observations support the general rule-of-thumb that fluorophores tend to emit strongly when situated in hydrophobic pockets. In the present case, several factors may contribute to this phenomenon. The fluorophores confined within the hydrophobic cavities with protective shells are isolated from nonradiative decay pathways or electron transfer pathways generated by collisions with solution components. Additionally, the high quantum yield could be due to the fact that molecules trapped in the polymer environment have restricted conformational flexibility, which may minimize nonradiative relaxation through internal motions of the excited molecules.
Optical absorption data are also presented in Figure 2B. The merocyanine visible band is solvatochromic, absorbing maximally at 525 nm in water and at 600 nm in non-polar organic solvents such as toluene. The corresponding band maximum for the mero-particles in water appears at 588 nm, indicating that the merocyanine is situated in a hydrophobic environment within the nanoparticle core.
In these particles, the light energies required for fluorescence excitation and photoisomerization of the dye are spectrally well resolved. For both spiro- and mero-nanoparticles, fluorescence excitation was carried out at 420 nm, a wavelength where neither spiro- nor mero-form has appreciable absorption. Consequently, interference arising from photoinduced interconversion of mero-and spiro-forms is negligible. In contrast, excitation of the spiro-nanoparticles below 400 nm converted them to mero-nanoparticles and excitation of mero-nanoparticles with visible light or wavelength above 450 nm converted them to spiro-nanoparticles.
In fact, photo-excited state can lead to either fluorescence or photochemical conversion. The photochemical quantum yield and fluorescence quantum yield together governs which pathway is favored. Our experiments indicate that photochemical quantum yield at 420 nm excitation must be considerably lower than that of fluorescence quantum yield, thus this wavelength is an ideal to elicit fluorescence with minimum effects on photo-switching.
For spiropyran dyes, “fatigue” effects caused by photodegradation of the dyes under UV irradiation are commonly observed. Both O2-dependent and O2-independent mechanisms have been identified, as discussed extensively elsewhere.28–31 Cyclic photoswitching of the polymer nanoparticle, however, exhibits better reversibility when compared to other environments, including when spiropyran molecules are covalently attached to nanoparticle surfaces (Figure 3).31 When spiropyran are attached to nanoparticle surfaces, the reversibility, as gauged by optical absorption at 580 nm, was 83% at 2000 s and 50% at 4000 s, whereas the polymer nanoparticles with spiropyran inside are 95% and 75% reversible at 2000 s and 4000 s, respectively. One factor contributing to this stability may be that incorporation minimizes bimolecular degradative reactions that involve the triplet-excited dye. Specifically, based on the monomer feed ratio, each 68 nm particle contains on average ~6400 spiropyran molecules. This calculation assumes all styrene and spiropyran monomers are polymerized and the density of polystyrene is 1.05 g/cm3. The corresponding volume occupied by each spiropyran molecules is ~25 nm3 or, on average, the spiropyrans are separated by ~2.9 nm, effectively blocking bimolecular interactions.
Figure 3.
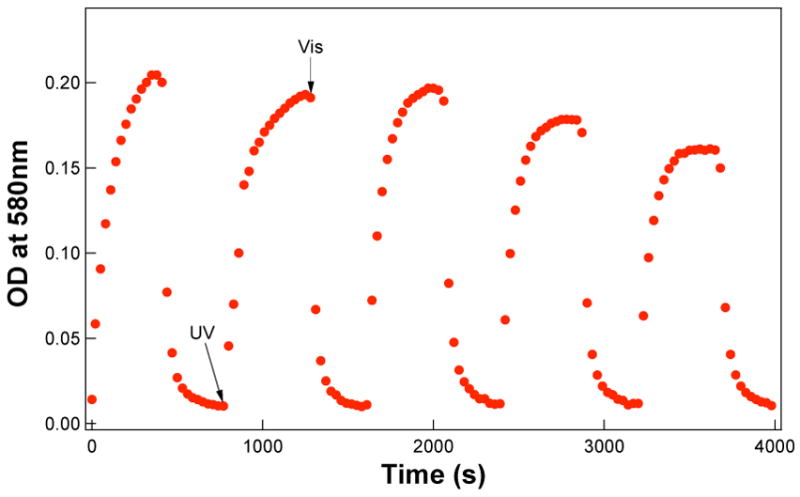
Reversible nanoparticle photochromism. 68 nm-nanoparticles (7.0 nM) are blue after UV irradiation and become colorless after exposure to visible light.
Under the prevailing conditions, 4–6 min UV irradiation at 0.97 mW (photon flux: 2.8 × 10−9 einstein) is required to complete spiro-to-mero switching in the nanoparticle core; this rate is considerably slower than that of switching of spiropyran molecules on the surface of nanocrystals under the same photon flux, which requires only ~5 sec.31 Typically, at approximately 3.5 min, spiro-nanoparticles are switched to ~90% of their equilibrium value. The slower switching speed apparently reflects the solid-state environment of the nanoparticles, where configurational switching must overcome energetic barriers imposed by the closely packed organic molecules. In solution, however, the barriers posed by fluidic solvent molecules may be much less than that of solid phase core of the nanoparticles. Although the switching speed slows somewhat in polymer nanoparticles, the reversibility of photoswitching improves as side reactions are suppressed. Consequently, the optical density at 588 nm, which is a quantitative measure of the mero-form, can be almost completely recovered to the original level in repetitive cycles (Figure 3).
Mero-spiro back-conversion requires ~ 6 min under 16–20 mW illumination (8–10 × 10−9 einstein) with visible light. However, most of mero-nanoparticles (~90%) are isomerized to spiro-nanoparticles within ~2 min; this rate is again somewhat slower than mero-to-spiro conversion of photoswitching of merocyanine molecules bound to the surface of nanocrystals (~90 sec) under the same light illumination intensity.31 What is remarkable is that forward and backward fluorescence switching of the photochromic nanoparticles have essentially the same rates. Unlike photoswitching of dye molecules in solution or on the surface of nanocrystals, fluorescence switching of the photochromic nanoparticles seems to be rate-limited by polymer dynamics in a manner that minimizes the influence of the photodynamic process itself. Molecules trapped in nanoparticles have restricted conformational flexibility, thus causing rate differences between forward and backward photoisomerization to effectively disappear.
Under simultaneous excitation of the particles at 420 nm and optical switching with alternating UV (λ < 400 nm) and visible (λ > 450 nm) light, fluorescence switching is observed. As shown in Figure 4, fluorescence switching with UV and visible light is reversible, at least for several cycles. The wavelength at which fluorescence is most rapidly switched on is 350 nm, the absorption maximum of the spiropyran, although nanoparticle fluorescence can be induced at other wavelengths throughout the UV region. Similarly, excitation into the absorption band of the merocyanine at 588 nm causes most rapid quenching of fluorescence. These excitation energies are well resolved—therefore clean separation between photoinduced nanoparticle switching and fluorescence excitation can be achieved.
Figure 4.
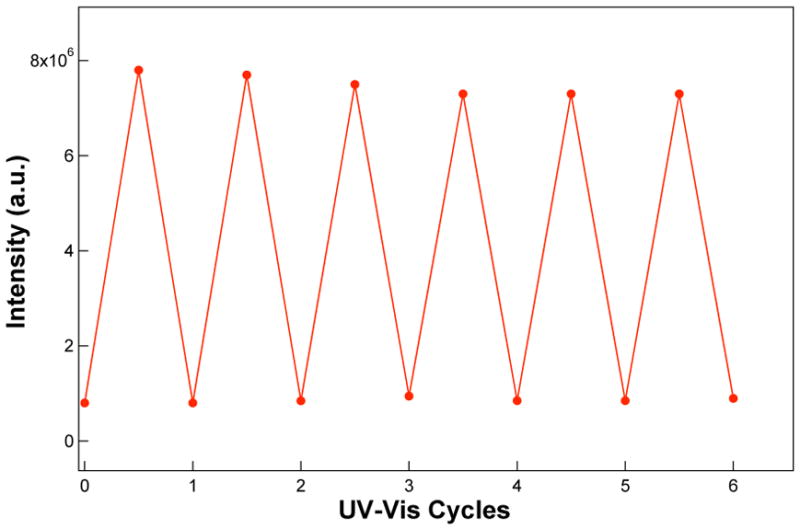
Fluorescence switching. Fluorescence of 7.0 nM 68 nm photochromic nanoparticles under 420 nm excitation is switched on and off with alternating UV and visible irradiation, as described in the text. The fluorescence intensity reaches maximal intensity in 20 s under 2.8 × 10−9 einstein excitation, whereas it takes 20 min to switch off fluorescence at ambient conditions and this can be accelerated with stronger visible light illumination (see text).
To complement the solution measurements of fluorescence switching in ensembles of polymer nanoparticles, fluorescence switching in single nanoparticles was carried out (Figure 5). In the single particle fluorescence measurements, samples that had been preilluminated at 365 nm were excited using the 488 nm, from either an argon laser or a mercury lamp; this is not an optimal wavelength for fluorescence, but was dictated by equipment limitations. As shown in Figure 5, the mero-nanoparticles were fluorescent under laser excitation at 488 nm, whereas the spiro-nanoparticles remained dark. It took ~2 min to completely switch from bright mero-nanoparticles to dark spiro-nanoparticles. Since each nanoparticle contains many spiropyran dye molecules, the fluorescence switching is not a sharp on-and-off process; rather it is gradual, even for single particles. The importance of single nanoparticles fluorescence measurements is that it identifies the smallest fluorescence-switching unit, which resides within each individual nanoparticles, rather than a solution mixture.
Figure 5.
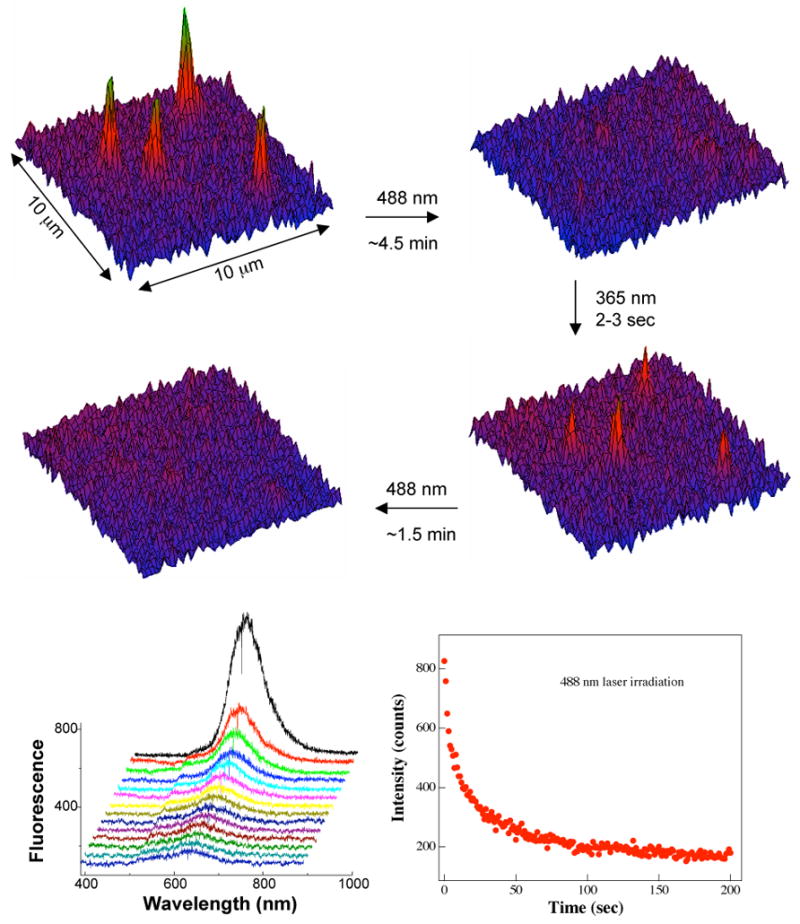
Single particle fluorescence switching. Fluorescence is switched on (top left) after 2–3 sec UV irradiation and off (top right) with visible 488 nm light illumination while viewing a ~10 × 10 μm2 area; these on-and-off processes are reversible (middle). A single 68-nm particle containing 6400 photoswitching molecules has a single fluorescence maximum (bottom left), which is slightly blue-shifted from that of ensemble spectra. The kinetics of fluorescence decay (bottom right) indicates a 2-min mero-to-spiro switching time on the nanoparticles under the prevailing conditions.
Polymer nanoparticles containing spiropyran-merocyanine photoswitches employ the whole particle as a switching unit. The photochromism of the resulting polymer nanoparticles is obvious to the naked eye when the samples are irradiated with a hand-held UV-light source. The nanoparticle is colorless in the spiropyran form and turns to dark blue within several seconds under 365 nm UV irradiation. The dark blue mero-nanoparticles also emit a strong red fluorescence. Upon standing in ambient light, the fluorescence of the polymer nanoparticles became visibly dimmer and finally disappeared after about 30 min. This process was quite reversible and could be repeated many times. These results showed that through UV (< 400 nm) and visible radiation (> 450 nm) both photochromic and on-and-off fluorescence properties can be controlled with specific frequencies of light, suggesting potential applications as optical digital data storage units based on either absorption or fluorescence read-out.
Photoswitchable nanoparticles could find additional applications. For example, one might use these nanoparticles to label and highlight specific sites of interest in biological systems. The fluorescence switching capabilities would provide confirmation that binding of the labeled nanoparticles has occurred, rather than a false positive signal of an interfering fluorophore. Alternatively, the capabilities for reversible display of red fluorescence could be exploited to develop “smart” inks from the water-soluble nanoparticles, e.g., photoswitchable inks to protect materials against counterfeit, where matters are deemed important. To illustrate this possibility, a spotted pattern was made by placing nanoparticles within selected wells of a 96-well microplate (Figure 6). Under UV-irradiation, the red luminescent photopattern is vividly displayed. Upon standing for 30 min under visible room light, the pattern completely disappeared. The photoerased pattern returned when the object was exposed to UV irradiation. This fluorescence is readily detected visually after only a few seconds of UV illumination (Figures 5 and 6), although optical absorption studies indicate that it takes 2–3 min for the majority of spiro-nanoparticles to convert to mero-nanoparticles (Figure 3). Thus, in projected applications, complete conversion of isomeric forms is not essential to achieving functional systems and constraints associated with switching times are relaxed.
Figure 6.

Optical read-and-erase. Patterning with photochromic nanoparticles contained within selected microplate wells (aqueous solutions) under UV and visible light illumination. Red fluorescence is observed after UV irradiation and is erased with visible light irradiation. This on-and-off cycle can be repeated many times and dried films display similar fluorescence on-off phenomena.
To demonstrate the feasibility of using these nanoparticles in biological imaging, we have investigated the possibilities of delivering them into living cells and effecting in vivo optical switching. Because the nanoparticles have hydrophilic shells consisting of NIPAM moieties and the water-soluble carboxylic groups from the initiator at the polymer chain termini, they are suitable for incorporation into the polar cavities of liposomes. Using liposomes as a delivery vehicle, we have successfully introduced optically switchable nanoparticles into HEK-293 cells, as shown by the images in Figure 7. Paramount issues in these studies were the extent to which the cytosolic media would quench fluorescence and whether reversibly spiro-mero photoisomerization could be achieved at reasonable switching speeds.
Figure 7.

Fluorescence imaging of optically switchable nanoparticles in a single HEK-293 cell. Red fluorescence (bright spots) of mero-particles is elicited with a 10 s UV light pulse, followed by fluorescence imaging using 488 nm excitation and a liquid N2-cooled CCD detector. The fluorescence is slowly switched off by visible 488 nm light and the bright spots fade with increasing exposure time (top panel with time labels). The kinetic profiles for fluorescence fading are given in the lower left panel. Application of a second 10 s UV pulse regenerates the red fluorescence (middle panel with time labels), which again fades under 488 nm illumination with identical kinetics (bottom right curve). White light imaging of the same cell under studies (a) shows more details of the cellular structures, along with another HEK cell (b) having its nucleus identified at the upper left corner.
As shown in Figure 7, a short UV (365 nm) pulse switched on polymer nanoparticle fluorescence inside living HEK-293 cells when excited with a 488 Ar laser. Thus, as in aqueous solutions, the fluorophores are protected in the hydrophobic core of the particle and their fluorescence is unlikely to be quenched by components of the biological milieu. The mechanism of internalization of the liposome-encapsulated nanoparticles is presently unknown, but we clearly observed many weak emitters in addition to bright spots within the cell, indicating that, following internalization, the nanoparticles have become widely distributed throughout the cell.
Because the 488-nm excitation line from the laser source converts the merocyanine to non-fluorescent spiropyran, the red fluorescence from the particles becomes weaker with increasing exposure time so that by 5 s the intensity is severely attenuated (Figure 7). It is therefore evident that within the cell fast fluorescence-off switching can be achieved. The fluorescence-on process is also very fast; red fluorescence can be strongly elicited within only 10 s illumination of a hand-held UV lamp (Figure 7). Within such a short UV pulse, no apparent damage occurs to the cell. The fluorescence-on process can be accelerated if stronger light source such as a laser is used. These experiments also demonstrate that intracellular fluorescence switching is reversible. As shown in Figure 7, application of the second UV pulse regenerates a nearly identical fluorescence pattern within the cell with nearly the same integrated fluorescence intensity as originally measured. As before, the intensity fades under continuous 488 nm illumination with the same kinetic decay profile. Overall, these data demonstrate that fluorescence switching is reversible and quantitatively repeatable.
Conclusion
We have synthesized polymer nanoparticles capable of undergoing reversible fluorescence photoswitching and demonstrated that on-and-off switching approximating digital response can be elicited with alternating UV (< 400 nm) and visible (> 450 nm) light. Using liposomes, these photoswitchable nanoparticles can be delivered into living cells and their presence confirmed by fluorescence imaging. Alternating illumination with UV-and-visible light switches the fluorescence of the nanoparticles on and off, confirming that such nanoparticles could be developed into optically addressable or verifiable fluorophores. These high-contrast, photoswitchable nanoparticles have potential utility as smart inks, time sensitive displays, and biological markers.
Acknowledgments
We acknowledge the support of National Institute of General Medical Sciences (Grant GM065306 to ADQL)) and the Arnold and Mabel Beckman Foundation, and the U.S. Department of Energy (DOE), Office of Basic Energy Sciences, Division of Chemical Science (Grant DE-FG03-99ER14943 to JKH) and Division of Materials Science (ADQL with PNNL). ADQL is a Beckman Young Investigator (BYI).
References
- 1.Crano JC, Guglielmetti RJ. Organic Photochromic and Thermochromic Compounds. Plenum Press; New York: 1999. [Google Scholar]
- 2.Berkovic O, Krongauz V, Weiss V. Chem Rev. 2000;100:1741–1754. doi: 10.1021/cr9800715. [DOI] [PubMed] [Google Scholar]
- 3.Guglielmetti R. In: Photochromism: (Revised Ed) Dürr H, Bouas-Laurent TH, editors. Elsevier; Amsterdam: 2003. pp. 855–878. [Google Scholar]
- 4.Irie M. Chem Rev. 2000;100:1685–1716. doi: 10.1021/cr980069d. [DOI] [PubMed] [Google Scholar]
- 5.Irie M, Fukaminato T, Sasaki T, Tamai N, Kawai T. Nature. 2002;420:759–760. doi: 10.1038/420759a. [DOI] [PubMed] [Google Scholar]
- 6.Feringa BL, editor. Molecular Switches. Wiley-VCH; Weinheim: 2001. [Google Scholar]
- 7.de Silva AP, Gunaratne HQN, Gunnlaugsson T, Huxley AJM, McCoy CP, Rademacher JT, Rice TE. Chem Rev. 1997;97:1515–1566. doi: 10.1021/cr960386p. [DOI] [PubMed] [Google Scholar]
- 8.Zhu MQ, Wang LQ, Exarhos GJ, Li ADQ. J Am Chem Soc. 2004;126:2656–2657. doi: 10.1021/ja038544z. [DOI] [PubMed] [Google Scholar]
- 9.Kim JH, Lee TR. Chem Mater. 2004;16:3647–3651. [Google Scholar]
- 10.Evans RA, Hanley TL, Skidmore MA, Davis TP, Such GK, Yee LH, Ball GE, Lewis DA. Nature Mater. 2005;4:249–253. doi: 10.1038/nmat1326. [DOI] [PubMed] [Google Scholar]
- 11.Liu NG, Chen Z, Dunphy DR, Jiang YB, Assink RA, Brinker CJ. Angew Chem Int Ed. 2003;42:1731–1734. doi: 10.1002/anie.200250189. [DOI] [PubMed] [Google Scholar]
- 12.Chudakov DM, Verkhusha VV, Staroverov DB, Souslova EA, Lukyanov S, Lukyanov KA. Nature Biotech. 2004;22:1435–1439. doi: 10.1038/nbt1025. [DOI] [PubMed] [Google Scholar]
- 13.Wiedenmann J, Ivanchenko S, Oswald F, Schmitt F, Rocker C, Salih A, Spindler KD, Nienhaus GU. Proc Natl Acad Sci USA. 2004;101:15905–15910. doi: 10.1073/pnas.0403668101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.(a) Medintz IL, Trammell SA, Mattoussi H, Mauro JM. J Am Chem Soc. 2004;126:30–31. doi: 10.1021/ja037970h. [DOI] [PubMed] [Google Scholar]; (b) Lim SJ, An BK, Jung SD, Chung MA, Park SY. Angew Chem Int Ed. 2004;43:6346–6350. doi: 10.1002/anie.200461172. [DOI] [PubMed] [Google Scholar]
- 15.(a) Ando R, Mizuno H, Miyawaki A. Science. 2004;306:1370–1373. doi: 10.1126/science.1102506. [DOI] [PubMed] [Google Scholar]; (b) Jares-Erijman E, Giordano L, Spagnuolo C, Lidke K, Jovin TM. Mol Cryst Liq Cryst. 2005;430:257–265. [Google Scholar]
- 16.Yang Y, Lu Y, Lu M, Huang J, Haddad R, Xomeritakis G, Liu N, Malanoski AP, Sturmayr D, Fan H, Sasaki DY, Assink RA, Shelnutt JA, van Swol R, Lopez GP, Burns AR, Drinker CJ. J Am Chem Soc. 2003;125:1269–1277. doi: 10.1021/ja027332j. [DOI] [PubMed] [Google Scholar]
- 17.Such G, Evans RA, Yee LH, Davis TP. J Macromol Sci-Polym Rev. 2003;C43:547–579. [Google Scholar]
- 18.Gaynor S, Qiu M, Matyjaszewski K. Advancing Sustainability Through Green Chemistry And Engineering. 2002;823:113–126. ACS Symposium Series, 2002. [Google Scholar]
- 19.Hiller J, Mendelsohn JD, Rubner MF. Nature Mater. 2002;1:59–63. doi: 10.1038/nmat719. [DOI] [PubMed] [Google Scholar]
- 20.Raymo FM, Giordani S. J Am Chem Soc. 2001;123:4651–4652. doi: 10.1021/ja005699n. [DOI] [PubMed] [Google Scholar]
- 21.(a) Chen MQ, Kishida A, Akashi M. J Polym Sci Part A: Polym Chem. 2000;34:2213–2220. [Google Scholar]; (b) Li ADQ, Wang W, Wang LQ. Chem-Eur J. 2003;9:4594. [Google Scholar]; (c) Wang W, Wan W, Zhou HH, Niu SQ, Li ADQ. J Am Chem Soc. 2003;125:5248. doi: 10.1021/ja0341900. [DOI] [PubMed] [Google Scholar]; (d) Wang W, Han JJ, Wang LQ, Li LS, Shaw WJ, Li ADQ. Nano Lett. 2003;3:455. [Google Scholar]; (e) Wang W, Li LS, Helms G, Zhou HH, Li ADQ. J Am Chem Soc. 2003;125:1120. doi: 10.1021/ja027186h. [DOI] [PubMed] [Google Scholar]; (f) Ishizu K, Yamashita M, Ichimura A. Polymer. 1997;38:5471–5474. [Google Scholar]
- 22.Menju A, Hayashi K, Irie M. Macromolecules. 1981;14:755–758. [Google Scholar]
- 23.Minkin VI. Chem Rev. 2004;104:2751–2776. doi: 10.1021/cr020088u. [DOI] [PubMed] [Google Scholar]
- 24.Schild HG. Prog Polym Sci. 1992;17:163–249. [Google Scholar]
- 25.Bahr JL, Kodis G, de la Garza L, Lin S, Moore AL, Moore TA, Gust D. J Am Chem Soc. 2001;123:7124–7133. doi: 10.1021/ja010058t. [DOI] [PubMed] [Google Scholar]
- 26.Minami T, Tamai N, Yamazaki T, Yamazaki I. J Phys Chem. 1991;95:3988–3993. [Google Scholar]
- 27.Karstens T, Kobs K. J Phys Chem. 1980;84:1871–1872. [Google Scholar]
- 28.Li XL, Li JL, Wang YM, Matsuura T, Meng JB. J Photochem Photobiol A. 2004;161:201–213. [Google Scholar]
- 29.Baillet G, Giusti G, Guglielmetti R. J Photochem Photobiol A. 1993;70:157–161. [Google Scholar]
- 30.Baillet G, Campredon M, Guglielmetti R, Giusti G, Aubert C. J Photochem Photobiol A. 1994;83:147–151. [Google Scholar]
- 31.Zhu L, Zhu MQ, Hurst JK, Li ADQ. J Am Chem Soc. 2005;127:8968–8970. doi: 10.1021/ja0423421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Albert B, Johnson A, Lewis J, Raff M, Roberts K, Walter P, editors. Molecular Biology of the Cell. 4. Garland Science Group, Taylor & Francis; New York: 2002. [Google Scholar]