

Abstract
Schistosoma mansoni NAD(P)+ catabolizing enzyme (SmNACE) is a new member of the ADP-ribosyl cyclase family. In contrast to all the other enzymes which are involved in the production of metabolites that elicit Ca2+ mobilization, SmNACE is virtually unable to transform NAD+ into the second messenger cyclic ADP-ribose (cADPR). Sequence alignments revealed that one of four conserved residues within the active site of these enzymes was replaced in SmNACE by a histidine (His103) instead of the highly conserved tryptophan. To find out whether the inability of SmNACE to catalyze the canonical ADP-ribosyl cyclase reaction is linked to this change we have replaced His103 with a tryptophan. The H103W mutation in SmNACE was indeed found to restore ADP-ribosyl cyclase activity as cADPR amounts for 7% of the reaction products, i.e., a value larger than observed for other members of this family such as CD38. Introduction of a Trp103 residue provides some of the binding characteristics of mammalian ADP-ribosyl cyclases such as increased affinity for Cibacron blue and slow-binding inhibition by araF-NAD+. Homology modeling of wild-type and H103W mutant three-dimensional structures, and docking of substrates within the active sites, provide new insight into the catalytic mechanism of SmNACE. Both residue side chains share similar roles in the nicotinamide-ribose bond cleavage step leading to an E.ADP-ribosyl reaction intermediate. They diverge however in the evolution of this intermediate; His103 provides a more polar environment favoring the accessibility to water and hydrolysis leading to ADP-ribose at the expense of the intramolecular cyclization pathway resulting in cADPR.
CD38, which is expressed by cells isolated from many mammalian species, is a ‘multifunctional’ bitopic ecto-enzyme that catalyzes the transformation of NAD+ into ADPR1 (NAD+glycohydrolase activity) and cADPR (ADP-ribosyl cyclase activity) (1, 2). In addition, CD38 also hydrolyzes cADPR into ADPR (cADPR hydrolase activity) and produces pyridinium analogues of NAD(P)+, such as NAADP+, by a transglycosidation reaction (reviewed in 2, 3). CD38 is remarkable in that it transforms NAD(P)+ into one or more calcium-mobilizing metabolites (4). Like CD38, the GPI-anchored ecto-enzyme CD157 can catalyze similar metabolic reactions (5). However, neither CD38 nor CD157 are particularly efficient cyclases as they produce only low levels of cADPR (about 1-3% of the reaction products). In contrast to these membrane-bound mammalian enzymes, the hydrosoluble ADP-ribosyl cyclase isolated from Aplysia californica, produces almost entirely cADPR from NAD+ (6) and makes, under standard conditions, very little ADPR (7). Despite the apparent differences in the catalytic properties of CD38, CD157 and the Aplysia cyclase, an unifying partitioning reaction mechanism has been suggested for this family of enzymes (Figure 1). This mechanism proposes that the cleavage of the nicotinamide-ribose bond of NAD+ leads to the formation of a common E·ADP-ribosyl intermediate. This intermediate then evolves to the reaction products through competing pathways: i) a macroscopically irreversible intermolecular reaction with water yielding ADPR, and ii) a reversible intramolecular reaction that gives cADPR (8, 9). In the presence of an excess of alternative acceptors such as pyridines or methanol, transglycosidation and methanolysis can also be observed (3).
FIGURE 1.

(A) Minimal kinetic mechanism for reactions catalyzed by ADP-ribosyl cyclase family members. (B) Molecular mechanism of the reaction catalyzed by SmNACE. This hypothetical mechanism was adapted from studies on mammalian CD38. The reaction involves the cleavage of the nicotinamide-ribosyl bond via a late transition state leading to the formation of either a covalent acylal ADP-ribosyl intermediate or a carboxylate-stabilized oxocarbenium ion intermediate. In either case the intermediate is then transformed via competing pathways : i) reaction with water yielding ADP-ribose, or ii) intramolecular cyclization to form cyclic ADP-ribose by reaction of N1 of the adenine ring with the C1’’ of the intermediate. The catalytic carboxylate corresponds to Glu202 in SmNACE. At a molecular/structural level, the precise mechanism by which the ‘signature’ Glu124, the second active site carboxylic group, controls the cyclization to hydrolysis ratio remains poorly understood. For a full discussion of the molecular mechanism of these enzymes, see (3).
We have previously proposed that the preference of the E·ADP-ribosyl intermediate to undergo hydrolysis/solvolysis or cyclization depends on the structure of the particular enzyme within the ADP-ribosyl cyclase family (3). Therefore, to better understand the mode of action of this family of enzymes at a molecular level it is of great interest to identify the active site residues that direct the transformation of NAD+ towards hydrolysis versus cyclization. The crystallographic structures of A. californica ADP-ribosyl cyclase (10), human CD157 (11) and human CD38 (12) are now known and, in combination with site-directed mutagenesis studies, they have yielded important clues regarding the role for particular residues within the active site. Overall, these enzymes have very similar three dimensional structures, including the architecture their active sites. Three key residues in the active site of human CD38 are involved in either catalysis (Glu226) (9) and/or in the binding/positioning of the substrate within the active site (Trp125 and Trp189) (13). These residues are conserved in all the different Aplysia ADP-ribosyl cyclases, CD38 and CD157 family members. A fourth residue, Glu146, was shown to influence the balance between cyclization and hydrolysis in human (14) and mouse CD38 (15). This residue is conserved in all CD38 and Aplysia cyclase family members, however it is replaced e.g. by a Ser in the human CD157 (Figure 2). The molecular mechanism of the reactions catalyzed by the ADP-ribosyl cyclases (Figure 1B), i.e. the chemical nature of the transition state of the scissile bond cleavage in NAD+ and of the ADP-ribosyl intermediate, and the role of the two Glu residues present in the active site of CD38 and Aplysia cyclases, has been discussed recently (3).
FIGURE 2.

Sequence alignment between S. mansoni NACE and representative members of the ADP-ribosyl cyclase family. The amino acid sequence alignment (Clustal W 1.82) of the active site domains reveals the conservation of the invariant catalytic Glu (Glu202 ; bold) and other important residues of the active site such as Glu124 (bold) and Trp165 (shaded). The second invariant Trp residue present in the active site of all other members of the cyclase family (grey shaded) is replaced by His103 in SmNACE. The ‘signature region’ TLEDTLLGY (shaded) is present in all known CD38 orthologs.
Recently we cloned the newest member of the ADP-ribosyl cyclase family from Schistosoma mansoni, that we named NACE (i.e., NAD(P)+ Catabolizing Enzyme). SmNACE showed significant structural and functional analogy to the other members of this enzyme family (16). However, in stark contrast to all these enzymes, SmNACE is almost exclusively a NAD+glycohydrolase and is essentially unable to catalyze the formation of cADPR (< 0.02 % of reaction products) from NAD+. This is despite the fact that SmNACE is able to convert the surrogate substrate NGD+ (17) into cGDPR in high yield (16). Analysis of the putative active site of this enzyme, obtained by homology modeling, revealed that three of the four key residues described above for CD38 and ADP-ribosyl cyclase were strictly conserved: Glu124, contained within the ‘signature region’, Trp165 and the catalytic Glu202 (Figure 2). However the second tryptophan (Trp125 in human CD38), which is highly conserved throughout this family of enzymes, is replaced by His103 in SmNACE. Furthermore, in a putative Schistosoma japonicum NACE (Accession No AY222890) (18), this highly conserved Trp residue is also replaced by a histidine (His96; Figure 2), suggesting that this exchange of residues might have a functional role in the schistosome enzymes. In the present work we produced a H103W mutant for S. mansoni NACE and studied the catalytic properties of this mutant to determine whether changing this residue would restore ADP-ribosyl cyclase activity in SmNACE.
EXPERIMENTAL PROCEDURES
Site-directed Mutagenesis of SmNACE
The H103W mutant was constructed by PCR (PfuTurbo polymerase; Stratagene) using the pPICZαA vector (Invitrogen) containing solNACE cDNA as template (16). SolNACE cDNA encodes the hydrosoluble ecto-domain of SmNACE (residues 20-275) which lacks the 5′ leader and 3′ GPI-anchor sequences of the original full length SmNACE (16). A myc-epitope fused to a His6-tag was introduced at the C-terminus of this construct to facilitate purification of the recombinant protein. The H in position 103 (CAC) was changed into a W (TGG) using the following oligonucleotide primers (mutated bases in bold): forward, 5′-AGCAGTACTTCTGGAGCCAGGTGATG-3′ and reverse 5′-CATCACCTGGCTCCAGAAGTACTGCT-3′. The technique used was adapted from the QuickChange multi-site directed mutagenesis kit (Stratagene). The mutation was verified by DNA sequencing of the clones using the 5′ and 3′ AOX primers (Invitrogen). The selected plasmid was then used to transform Pichia pastoris GS115 (his4) or SMD1163 (his4, pep4, prb1) strains (Invitrogen) according to the manufacturer’s instructions.
Expression and Purification of the Recombinant Proteins
Clones of yeast transformed with the expression plasmids (pPICZαA/SmNACE and those carrying the H103W mutation in the SmNACE sequence) were grown overnight at 30°C in 10 mL of BMGY (Invitrogen). The cells were then harvested and resuspended in 5 mL BMMY/casamino acids (BMGY containing 0.5% v/v methanol instead of glycerol and 1% w/v casamino acids) (19). For the mutant, this induction medium was supplemented with 1.0 mM EDTA and 0.2 mM PMSF. After 24 h, 0.5% methanol was again added and the cultures were stopped after 48 h. The cells were then harvested and the enzymatic activities were tested in the supernatants. The clone that gave the highest yields in recombinant enzyme was selected for a scale-up. To that end, a 5 mL overnight starter culture was inoculated to flasks containing 100 mL BMGY and grown until reaching an OD600 = 25. The cells were then harvested and resuspended in 50 mL BMMY/1% w/v casamino acids supplemented or not (see above) with 1.0 mM EDTA and 0.2 mM PMSF. After 24 h 0.5% v/v methanol was again added and induction was stopped after 48 h.
After centrifugation the supernatant was dialyzed against 10 mM potassium phosphate buffer, pH 7.4 (Buffer A) and the proteins were loaded on a 5 mL Blue Sepharose 6 Fast Flow CL-6B (Amersham Biosciences) column. The recombinant SmNACE was eluted with a 0–1.5 M NaCl linear gradient in Buffer A. This step yielded a wild-type enzyme of high purity (> 95%). The purification of the mutant, which was expressed at lower yield, necessitated an additional chromatography step on a Ni2+-affinity column. The enzyme, previously dialyzed against a 100 mM potassium phosphate buffer, pH 8.0, containing 300 mM NaCl (Buffer B), was loaded on a Hi-Trap™ Chelating HP (Amersham Biosciences) column (1 mL), pre-equilibrated with a 50 mM solution of NiSO4 in water followed by Buffer B. The column then was washed with Buffer B containing 10 mM imidazole and the H103W mutant of recombinant SmNACE containing the His6-sequence was eluted with a linear gradient of 10-500 mM imidazole in Buffer B. The collected fractions containing the enzyme were pooled and dialyzed against Buffer A. To avoid cross-contamination, separate affinity columns were used for WT and mutant SmNACE. The protein concentration was determined by the protein BCA assay (Pierce ) using BSA as standard.
SDS-PAGE Analysis and Immunoblotting
The purity of the protein preparations was estimated by SDS-PAGE using a Mini-Protean II electrophoresis cell (Bio-Rad) and silver (20) or Coomassie blue staining. The proteins separated by SDS-PAGE were electrophoretically transferred to a Immobilon-P membrane (Millipore). After blocking, the membrane was probed for 2 h at room temperature with the primary antibody anti-NACE, obtained by immunization (16), diluted to 1:500. After washing, the immunoblot was incubated with the HRP-anti-mouse IgG (1:10,000) (Jackson ImmunoResearch) for 1h at room temperature. The blot was developed with a chemiluminescence kit (Amersham Biosciences).
Enzymatic Assays
SmNACE catalytic activity was measured under saturating (400 μM) or limiting (10 μM) amounts of NAD+ in the presence of 2.5×105 dpm [adenosine-U-14C]NAD+ (Amersham Biosciences) as described before (16). Briefly, the enzyme was suspended in Buffer A and incubated at 37°C with substrate (200 μL final volume). At selected times, aliquots (50 μL) were removed and enzyme activity was stopped by adding ice-cold perchloric acid (2% v/v final concentration). After neutralization with 3.5 M K2CO3, the precipitated proteins were removed by centrifugation. Product formation was monitored by HPLC on 300 x 3.9 mm μBondapack C18 (Waters) or 250 x 4.6 mm Acclaim C18 5μm (Dionex) columns. The reaction products were eluted isocratically at a flow rate of 1 mL/min with a 10 mM ammonium phosphate buffer (pH 5.5) containing 0.8–1.2% (v/v) acetonitrile and detected by radiodetection (Flo-one, Packard Radiometric Instruments) when using [14C]NAD+ or by absorbance recording at 260nm. Kinetic parameters, Km and Vmax, were determined from the plots of the initial rates of product(s) (ADPR + cADPR) formation as a function of substrate concentration (5–400 μM NAD+, 8 data points) according to Michaelis-Menten kinetics, using a nonlinear regression program (GraphPad, Prism).
The pH-dependent activities of WT SmNACE and H103W mutant were determined using 1,N6-etheno-NAD+ (Sigma) as substrate. This continuous assay consists in measuring an increase (about 12-fold) of fluorescence (λem = 410 nm; λexc = 310 nm) resulting from the hydrolytic conversion of the dinucleotide to 1,N6-etheno-ADPR (21). Reactions were performed at 37°C in 10 mM sodium citrate or potassium phosphate buffers at pH 4.0–8.0 (1 mL final volume) in a thermostatically controlled fluorimeter cuvette (Shimadzu, RF-5301). The kinetic parameters were calculated as above from the initial rates. The specificity constants Vmax/Km were determined alternatively from progress curves by using an initial 1,N6-etheno-NAD+ concentration of 2.0 μM. Under these conditions where [S]0 ≪ Km, the values of Vmax/Km were directly obtained by fitting the curves to the equation: [S] = [S]0exp(-Kmt/Vmax) (22, 23). The apparent pKa values were calculated from plots of Vmax and Vmax/Km as a function of pH (24) with a nonlinear curve-fitting program.
Methanolysis reactions catalyzed by SmNACE were performed at 37°C in Buffer A in the presence of 50 μM NAD+, [adenosine-U-14C]NAD+ (2.5 x105 dpm ) and 0–3 M methanol (200 μl final volume). Aliquots, taken at different time points, were treated as described above and the reaction products were analyzed by HPLC (25). The transformation of NGD+ was followed fluorometrically (17). The assays were conducted, at 37°C, in Buffer A in the presence of 10–300 μM NGD+ and enzyme in a 2 mL final volume. The fluorescence changes were monitored at λem = 410 nm (λexc = 310nm) corresponding to the appearance of cGDPR. The effect of 0–3 M methanol on cyclization rates of 100 μM NGD+ was determined similarly and the relative proportions of the reaction products GDPR, cGDPR and β-methyl GDPR were determined by HPLC as described above; see also (7).
Inhibition Studies
Inhibition of the H103W mutant of SmNACE by araF-NAD+ was studied fluorometrically using 1,N6-etheno-NAD+ as substrate. Assays were performed in Buffer A in the presence of 60 μM 1,N6-etheno-NAD+ and 0–10 μM araF-NAD+ (26) (final volume 1 mL). The reaction was started by addition of the enzyme and followed over time for 20 min. The progress curves were analyzed as described previously (25, 26) by fitting the slow-binding inhibition to the equation :
Use of a nonlinear regression program yielded the different parameters, i.e. vo, initial rate; vs, steady-state rate; k, apparent first-order constant for reaching the steady-state enzyme-inhibitor complex; and F0, the initial fluorescence. The kinetic parameters koff, kon and Ki (= koff/kon ) were calculated from the plot of k against inhibitor concentration (6 data points) according to the equation:
A Km = 23.8 μM for 1,N6-etheno-NAD+ (H103W mutant) was used for these calculations.
The same protocol was used to study the inhibition of the enzyme by Cibacron blue F3GA. In this case classical inhibition was observed and the IC50 value, i.e. the concentration of inhibitor that reduces the activity of the enzyme by 50%, was determined by nonlinear regression curve fitting.
Molecular Modeling of WT and H103W Mutant SmNACE
Five homology models of SmNACE were generated using the Modeller 8.1 automodel protocol (27) with the crystallographic coordinates of human CD38 (1yh3), human CD157 (1isf) and Aplysia cyclase (1lbe) as templates. The N-terminal and C-terminal regions (1–11 and 285–303) were omitted in the model because of the lack of homologous template structures. The quality of the models was evaluated using Procheck and WHATIF (http://biotech.ebi.ac.uk). The best representative structure was optimized by energy minimization in implicit water using AMBER 8.0 (28) with the Parm99 parameter set and the Generalized-Born approach. The three-dimensional structure of the H103W mutant was obtained by the manual edition of WT SmNACE in SYBYL 7.1. The rotameric state of the Trp103 residue was chosen based on the conformations of Trp101, Trp109 and Trp125 observed in the crystal structures of Aplysia cyclase, human CD157/BST-1 and CD38, respectively. The region surrounding the mutation was relaxed by 1000 steps of Powell energy minimization in SYBYL 7.1 using Amber99 force field.
pKa Prediction of the Active Site Titratable Residues
Structures were prepared using PDB2PQR web server (29) and pKa calculations were performed by both PROPKA (http://propka.chem.uiowa.edu/) (30) and the Protein pKa Estimator (version 0.9, OpenEye Scientific Software, Inc.). The dielectric constant values used Poisson-Boltzmann equation (31) were set depending on the residue accessibility.
Conformational Analysis of cADPR and cGDPR
The crystal structure of free cADPR is published (32, 33). cGDPR structure was obtained by manual edition of cADPR followed by a rapid energy minimization (default parameters) in SYBYL 7.1. Atom and bond types were set according to the known tautomeric states. The coordinates of both molecules were stored in MOL2 files. Conformational analysis were set up in Maestro ver. 7 (Schroedinger Inc.). For each compound, a 500-step Monte Carlo conformational search was performed in MacroModel ver. 8 (Schroedinger Inc.). An energy minimization in a simulated water environment was then performed on the compounds in their ionized state using the truncated-Newton conjugate-gradient method (TNCG) and AMBER* force field. Conformers were selected using a 3 kcal/mol energy cutoff and clustered based on RMS difference between corresponding torsion angles in pairs of structures using Xcluster (Schroedinger Inc.). Representative structures were analyzed using SYBYL 7.1.
Flexible Docking of NAD+, NGD+, cADPR and cGDPR into WT and Mutant SmNACE active sites
Protein and ligand input files were prepared using SYBYL 7.1. Atom typing, stereochemistry and local geometry were checked and all hydrogen atoms were added and coordinates were saved in MOL2 format. The docking simulations were carried out using GOLD ver. 3.0. (34). Binding site was defined using a sphere of 12 Å centered on the centroïd of the atom subset composed of His(Trp)103 and Trp165 side chains. The default parameters set was chosen to run the genetic algorithm. The Goldscore scoring function was used to evaluate poses. The automatic placement of the ligand was biaised by one to three intermolecular distance restraints (see Supporting Information). Docked poses were assessed using Silver 2.3.4.
RESULTS
Expression of the H103 Mutant of SmNACE
SmNACE was subjected to site-directed mutagenesis to assess the functional significance of His103. In contrast to the wild-type recombinant enzyme (WT SmNACE), which was produced in fairly good yield (16 mg/L) by the methylotrophic yeast P. pastoris (16), mutation of the single active site His103 to Trp (H103W SmNACE) resulted in a poorly expressed protein (~ 1 mg/L). Nevertheless, the expressed H103W mutant was secreted into the medium by Pichia and bound efficiently to a Cibacron blue affinity gel indicating a native-like conformation for its NAD+ binding site (see below). After eluting the mutant H103W SmNACE from the column of immobilized Cibacron blue, a second step purification on a Ni2+-affinity gel was used to further purify the His-tagged protein. As found previously for the WT enzyme (16), the H103W SmNACE was expressed by Pichia as two broad protein bands of approximately 41 and 43 kDa (Figure 3A). The molecular weight of the soluble recombinant protein is larger than the expected molecular mass (32 kDa) because NACE is heavily glycosylated (16). Identical to what we previously observed with WT SmNACE (16), both protein bands from the yeast expressing H103W SmNACE were recognized by polyclonal antibodies raised in response to immunization with the WT SmNACE cDNA (Figure 3A).
FIGURE 3.

Recombinant soluble H103W SmNACE catalyzed transformation of NAD+. (A) Analysis by SDS-PAGE and Western blotting of purified recombinant H103W SmNACE mutant. Left lane: silver-stained SDS-PAGE gel (12%) – Right lane: immunoblot using anti-NACE antibody (16). The molecular weights (kDa) of the purified mutant are indicated. (B) Kinetics of NAD+ transformation catalyzed by H103W SmNACE mutant. Assays were carried out at 37°C in 10 mM potassium phosphate buffer, pH 7.4. Initial rates were determined at the given substrate concentrations and the data were fitted to the Michaelis-Menten equation. (C) Representative HPLC radiochromatograms of the reaction products observed after transformation of 14C-labeled NAD+ by WT (upper panel) and H103W mutant SmNACE (lower panel). The assays were performed at 37°C in 10 mM potassium phosphate buffer, pH 7.4, in the presence of [14C]NAD+ and the reaction products were monitored by radiodetection. (D) Representative HPLC elution profile of the products obtained by solvolysis of NAD+ catalyzed by the H103W SmNACE mutant in the presence of 3.0 M methanol. The methanolysis peak was identified by co-elution with an authentic sample of β-methyl ADPR (57).
The H103W Mutant of SmNACE has a Readily Measurable ADP-ribosyl Cyclase Activity
To determine the consequence of mutating the His103 active site residue, the catalytic properties of the WT and H103W SmNACE were compared. Replacing His103 by a Trp residue had little impact on the specific activity of SmNACE as the kinetic parameters (Km and Vmax) of the H103W mutant enzyme were very similar to that of the WT enzyme when NAD+ was used as the substrate (Figure 3B and Table 1). As we previously reported (16, 35), when WT SmNACE was incubated in the presence of [14C]NAD+, the formation of cADPR could not be detected. In sharp contrast, the mutant H103W SmNACE had clearly measurable ADP-ribosyl cyclase activity. As shown in Figure 3C, the cADPR peak was easily detected on HPLC profiles and it accounted for 6.8 ± 1.6 % (n =15) of the reaction products (Table 2). In fact, compared to the activity of the WT SmNACE enzyme, the replacement of His103 with Trp103 increased the ADP-ribosyl cyclase activity of SmNACE by 2 to 3 orders of magnitude. This remarkable result points to the competence of the active site residue-103 in the partitioning of the E·ADP-ribosyl intermediate between cyclization and hydrolysis. The data further indicates that reintroduction of a Trp residue that is conserved in all other members of cyclase enzyme family to SmNACE restores the ability of SmNACE to catalyze the hallmark reaction of this class of enzymes, namely the ability to transform NAD+ into cADPR. Comparatively, the H103W mutant of SmNACE is even a better cyclase than CD38 and CD157.
Table 1.
Kinetic parameters for WT and H103W SmNACE
| Substratea | SmNACE variant | Km (μM ) |
Vmax (μmol/min/mg) |
cyclizationb (%) |
|---|---|---|---|---|
| NAD+ | WTc | 38.5 ± 6.8 | 13.2 ± 2.6 | ~ 0.02 |
| H103W | 32.4 ± 5.4 | 13.3 ± 0.9 | 6.8 | |
| NGD+ | WTc | 23.4 ± 1.1 | 4.8 ± 0.1 | 85.0 |
| H103W | 47.7 ± 4.7 | 11.5 ± 0.4 | 90.0 |
All reactions were performed at 37 °C in 10 mM potassium phosphate buffer (pH 7.4) in the presence of purified recombinant WT and H103W SmNACE. The kinetic parameter values were calculated from a nonlinear regression fit to the Michaelis-Menten equation.
Formation of cADPR or cGDPR given as % of total reaction products
Data taken from (16).
Table 2.
Predicted pKa values of key residues in the active site of wild type and H103W mutant SmNACE and human CD38
| Residue | SmNACE | Human CD38 | ||||
|---|---|---|---|---|---|---|
| pKa (WT ) | pKa (H103W) | pKa | ||||
| Glu (signature)d | 0.96a | 0.00b | 5.22a | 1.45b | 5.78a | 1.55b |
| Glu (catalytic)e | 6.83a | 4.25b | 7.18a | 6.25b | 7.16a | 4.75b |
| His103 | 7.16a | 6.35b | n.a.c | n.a.c | n.a.c | n.a.c |
Empirical calculation using PROPKA.
Calculation based on electrostatic continuum model using dielectric constant values ranging from 2 for the catalytic Glu to 6 for His103 and the ‘signature’ Glu.
Not applicable.
Glu124 and Glu146 in SmNACE and human CD38, respectively, are part of the ‘signature region’ of the ADP-ribosyl cyclases.
Glu202 and Glu226 in SmNACE and human CD38, respectively.
Although WT SmNACE is an extremely poor ADP-ribosyl cyclase, we previously showed that it is an excellent GDP-ribosyl cyclase and efficiently transformed the surrogate substrate NGD+ into GDPR and cGDPR (16), with the cyclic compound accounting for approximately 85% of the reaction products (35). To determine whether the H103W mutant of SmNACE was a more efficient GDP-ribosyl cyclase than the WT SmNACE, we measured the kinetic parameters and the cyclization efficiency using using NGD+ as a substrate. Similar to our results with NAD+, the kinetic parameters for NGD+ were directly comparable with the H103W mutant and WT SmNACE (Table 1). Interestingly, the mutant was also a slightly better cyclase of NGD+ as cGDPR accounted for 90.0 ± 1.3 % (n = 5) of the reaction products when H103W SmNACE was incubated with NGD+ (Table 1).
Methanolysis of NAD+ and NGD+ Catalyzed by the H103W SmNACE Mutant
We wanted to verify whether the mutation of SmNACE altered the kinetic mechanism of the enzyme. First we confirmed that similar to WT SmNACE (16) and other ADP-ribosyl cyclases such as CD38 (25, 36, 37), methanol acted as an alternate acceptor in the transformation of NAD+which competed with water to form, with complete retention of configuration, β-methyl ADPR at the expense of ADPR (Figure 1). Moreover, for a cyclase such as the H103W SmNACE mutant, we predicted that the formation of cADPR as reaction product would also concomitantly decrease in the presence of methanol (8). This was borne out by experiment as the transformation of [14C]NAD+ in the presence of 3 M methanol yielded ADPR and β-methyl ADPR (2/3 and 1/3 of the reaction products, respectively) while cADPR was barely measurable (< 1% of reaction products)(Figure 3D). The partition ratio K (= [β-methyl ADPR]/[ADPR] x [H2O]/[CH3OH]), which indicates the relative reactivity of the E.ADP-ribosyl intermediate with water and methanol, was calculated to be 9.5 ± 1.5 (n = 12) which is somewhat higher than 5.0, the value found for WT SmNACE (16). Next, we checked whether methanol (0–3 M) had any effect on the turnover rate of the catalyzed reaction. In the presence of 3 M methanol, i.e. the highest concentration used which results in the formation of the methanolysis product in a 34 % proportion of reaction products, we could not measure an increased transformation rate of NAD+. A similar absence of acceleration was previously observed with the WT enzyme (16), indicating that the formation of the reaction intermediate is the rate limiting step in both WT and H103W SmNACE. Altogether these results indicate that H103W mutation does not drastically affect the selectivity of the reaction intermediate vs. acceptors in the solvolytic step and nor does it affect the rate limiting step of the reaction catalyzed by SmNACE.
We also studied the effect of methanol on the transformation of NGD+. As described above, this dinucleotide is overwhelmingly converted into cGDPR by WT SmNACE and the hydrolytic pathway yielding GDPR is not favored. However, as assessed by the fluorometric monitoring of the transformation of NGD+ into cGDPR catalyzed by the WT SmNACE, addition of methanol resulted in a marked inhibition of the cyclization rate (up to 23% in the presence of 3 M methanol) (Figure 4A). HPLC analysis of the reaction products revealed that this inhibition was accompanied, as expected, by a concomitant decreased formation of cGDPR and by the appearance of the methanolysis product β-methyl GDPR (not shown). This result is consistent with the occurrence of a methanolysis reaction at the expense of the cyclization reaction and with a preferential reactivity of the E.GDP-ribosyl intermediate with methanol compared to water. For example at 3 M methanol, the production of cGDPR by WT SmNACE was reduced to 60% of reaction products, whereas β-methyl GDPR accounted for 23%. The solvolysis/cyclization ratio (i.e. [GDPR+β-methyl GDPR]/[cGDPR]) was linearly correlated with the concentration of methanol (Figure 4B) – excluding a preferential binding of this solvent to the active site within this concentration range – and the partition ratio K was about 30 ± 4 (n = 6). Interestingly, methanol had a much lesser effect on the reaction catalyzed by the H103W SmNACE mutant (Figure 4A); this was borne out by HLPC analysis which showed that even at 3 M methanol, β-methyl GDPR accounted only for about 3 % of the reaction products. These results indicate that H103W mutation which results in an increased NGD+ cyclization efficiency, concomitantly also renders the solvolytic alternate pathway less competitive. The occurrence of such an inverse correlation is highly reminiscent of the results we have obtained previously with A. californica ADP-ribosyl cyclase. During the catalytic transformation of NGD+, this enzyme kinetically favors the cyclization of the GDP-ribosyl intermediate to such an extent that intermolecular reactions with water or methanol cannot compete with the intramolecular pathway (7).
FIGURE 4.

Effect of methanol, Cibacron blue F3GA and pH on reactions catalyzed by SmNACE. (A) Effect of increasing concentrations of methanol on the rate of transformation of 100 μM NGD+ into cGDPR catalyzed by WT (squares) and H103W mutant (triangles) of SmNACE. The assays were carried out at 37°C in 10 mM potassium phosphate buffer, pH 7.4, and the cyclization reaction progress monitored fluorometrically at λem = 410 nm (λexc = 310nm). Residual activities (means ± SD, n=4) are percent of the activity measured in the absence of solvent. (B) Effect of methanol on the solvolysis/cyclization ratio ( = [GDPR + β-methyl GDPR]/[cGDPR]) observed on transformation of 100 μM NGD+ catalyzed by WT SmNACE (means ± SD, n=3). The reaction products were analyzed by HPLC and their relative proportions were quantified as described previously (8). (C) Inhibition of WT and H103W SmNACE by Cibacron blue F3GA. The effect of the Cibacron blue on the initial rates of the transformation of 20 μM 1,N6-etheno-NAD+ catalyzed by SmNACE in 10 mM potassium phosphate buffer, pH 7.4, was followed fluorometrically (λem = 410 nm; λexc = 310 nm) at 37°C. The IC50 values were calculated from the plot of residual activity against log of dye concentration. WT (triangles) and H103W SmNACE mutant (squares). (D) pH dependence of WT SmNACE. The pH profile of Vmax, measured at 37°C, using 1,N6-etheno-NAD+ as substrate displayed a critical ionization with a pKa of 5.82 ± 0.07 which must be unprotonated for catalytic activity. (E) pH profiles of Vmax/Km values determined at 37°C using 1,N6-etheno-NAD+ as substrate (n = 3). WT (triangles) and H103W SmNACE mutant (squares). In (D) and (E) the solid lines represent the fit of the data (n = 3) to a single pKa model.
Comparison of the Inhibition of WT and H103W Mutant of SmNACE by AraF-NAD+ and Cibacron Blue F3GA
A very interesting feature of SmNACE is that this enzyme is much less sensitive to inhibition by molecules such as araF-NAD+ and Cibacron blue F3GA. This observation suggests that the differences within the active site of SmNACE might be exploited to design inhibitors that are specific for the schistosome enzymes. AraF-NAD+ is one of the most potent inhibitors (nM range) known for CD38 and it belongs to the class of slow-binding inhibitors (25, 26). In contrast, this molecule was found to be only a very modest inhibitor (IC50 ≫ 10 μM) of SmNACE (16). To test whether the residue at position 103 might play a crucial role in the mode of interaction of inhibitors to the active site of SmNACE, we studied the inhibition of the H103W mutant by these two molecules.
In the presence of araF-NAD+ progress curves typical of a slow-binding inhibition could be observed (not shown). Under our experimental conditions the initial rates v0 (see Experimental Procedures) were not affected by this NAD+ analogue, which is indicative of a slow interconversion between E and E.I, the enzyme.inhibitor complex (38). The following constants were calculated for the formation of E.I: kon = 1.88 x103 M−1s−1, koff = 1.5 x 10−3 s−1 (t1/2 = 7.7 min) and Ki = 0.80 μM. Thus, the presence of Trp103 seems to be instrumental in the binding affinity of araF-NAD+ which is at least 2 orders of magnitude higher than for the WT NACE active site.
Cibacron blue F3GA is a water soluble triazine dye which has been used as inhibitor of enzymes binding mononucleotides such as ATP (39) or dinucleotides such as NAD(P)+. The high affinity of Cibacron blue for the active site of this large variety of enzymes results from a complex combination of hydrophobic, electrostatic and hydrogen bonding interactions. Some structural studies have however revealed that the dye is primarily a mimic for the ADP moiety of the mono(di)nucleotides (40, 41). The immobilized form of the blue dye on solid supports has been widely exploited for protein purification by pseudo-affinity chromatography (42, 43), and has been used to purify cyclase family members, including CD38 (44) and SmNACE(16). In addition, Cibracon blue F3GA is a potent inhibitor of CD38 enzyme activity with an IC50 = 17.8 ± 4.4 nM and 0.190 ± 0.027 μM for recombinant bovine or human CD38, respectively (H. Muller-Steffner, unpublished). However, this dye is a less potent inhibitor of WT SmNACE for which a IC50 = 1.15 ± 0.14 μM was measured (Figure 4C). Interestingly, the H103W mutation increases the inhibitory power of Cibacron blue about 5-fold with a IC50 = 0.23 ± 0.05 μM (Figure 4C). This effect – although modest – suggests that residue-103 within the active site of SmNACE could be part of the Cibacron blue binding pocket and that the bulkier hydrophobic side chain of the Trp103 residue of the mutant is somewhat better able to provide nonpolar interactions with the aromatic rings of the dye than the WT His103 residue.
Modeling of the WT SmNACE and H103W Mutant Active Sites
To determine the impact on catalysis of the interactions between His103 and other critical residues, we modeled the active sites of WT and H103W mutant of SmNACE. The side chains of Glu124 and Glu202, which are two active site residues crucial for catalytic activity of SmNACE, are in close vicinity to the imidazole ring of His103 in the WT enzyme (Figure 5A). In the three-dimensional model, the distances measured between the carboxylate and imidazole groups are about 4.0 and 6.7 Å for Glu124 and Glu202, respectively. The replacement of the titratable His103 by a non-ionizable tryptophan results in significant modifications of the electrostatic environment of the two Glu (Figure 5B). Accordingly, we have predicted the pKa values of the Glu124 and Glu202 carboxylic groups using an empirical method and a method based on an electrostatic continuum models that numerically solves the linearized Poisson-Boltzmann equation. Although each approach yields somewhat different absolute values (Table 2), they both support the propensity of the two Glu residues to become less acidic upon replacement of His103 with a tryptophan. The most dramatic pKa shift is observed in the mutant for Glu124 (ΔpKa = + 1.45 to + 4.25). For comparison, pKa values observed in the H103W mutant are quite similar to those predicted for the equivalent residues in human CD38. Importantly, it should be stressed that Glu124 and Glu202 are the only titratable residues within the active site whose pKa’s can be affected by the H103W mutation.
FIGURE 5.
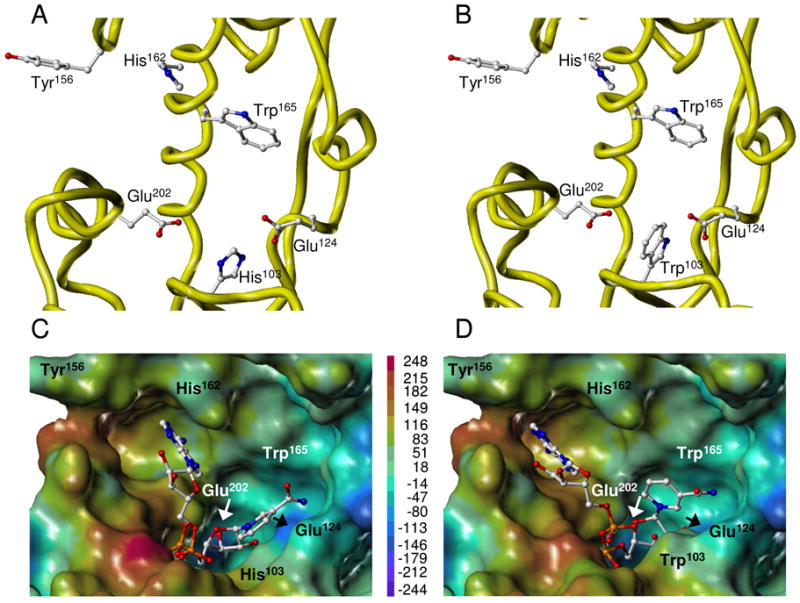
Three-dimensional structure of WT and H103W SmNACE active sites. Free WT (A) and mutant (B) enzymes. Yellow ribbons illustrate the secondary structure elements that form the catalytic cleft. Side chains of the key residues are depicted using ball-and-stick representation and colored by atom type. NAD+ docked into the active site of WT (C) and mutant (D) enzymes. The best GOLD poses (fitness values of 64 and 82, respectively) of NAD+ are shown in ball-and-stick and colored by atom type. The active site is represented as Connolly surface and colored according to the electrostatic potential computed using AMBER7 FF99 charges. The color ramp of electrostatic potential (in kcal/mol) ranges from red (most positive) to purple (most negative). The protein is always displayed in the same orientation. The rendering was performed using SYBYL ver. 7.1. Color code : white = C, red = O, dark blue = N, cyan = H and orange = P.
The values of these ionizable groups can also be approached experimentally by studying the dependence of the kinetic parameters of WT and H103W SmNACE on pH. The pH-activity profiles were determined using the substrate 1,N6-etheno-NAD+ and, in the range between 5.0 to 8.0, the plot of the values of Vmax versus pH for the wild-type enzyme reveals a single critical ionizable residue with a pKa value of 5.82 ± 0.07 (Figure 4D). In this pH range the Km values remained essentially constant and the pH profile of Vmax/Km vs. pH yielded a similar pKa of 6.05 ± 0.10 (Figure 4E). This residue, which must be unprotonated for activity, has an ionization constant consistent with the pKa expected for a histidine side-chain in a relatively non-polar environment. Interestingly, the experimental values obtained here are close to the pKa predicted for His103 using the electrostatic continuum model (Table 2). In the absence of His103, a more acidic group with a pKa value of 5.4 ± 0.1 was similarly found in the H103W mutant by studying the effect of the 4.0–7.5 pH range on the values of Vmax/Km (Figure 4E). The identity of this residue which is critical for the activity of the mutant is unknown; however catalytic Glu202 which is less acidic than Glu124 and whose pKa is somewhat shifted upwards in the H103W mutant (Table 2) could be an excellent candidate.
To assess the substrate binding mode, NAD+ was successfully docked into the active site of SmNACE using the program GOLD 3.0. The best poses obtained for WT and H103W mutant SmNACE are very similar (Figure 5C,D). The nicotinamide ring is buried within the active site pocket in an essentially apolar environment, making it likely to engage in a strong π-stacking interaction with Trp165 and to establish an edge-to-face aromatic interaction with His/Trp103. Interestingly, according to this model a neutral form of the imidazole ring of His103, as well as the 5-membered pyrrole ring part of Trp103, are also in favorable position for carbohydrate-aromatic interactions (45) involving the cluster of the C-H bonds on the β-face of the ribose linked to nicotinamide. A similar ribose-aromatic interaction has been observed recently in a crystal structure of A. californica ADP-ribosyl cyclase with a covalently bound ribose-5′-phosphate (46). The adenine moiety of NAD+ is positioned outside this cleft in an anti conformation with the base away from the ribose ring. In this conformation the distance between adenine N1 and nicotinamide-attached ribose C1″ is approximately 10 Å. Consequently, the formation of cADPR implies a rotation from this anti to a syn conformation of the adenine with respect to the N9-ribose bond.
Next, to provide a better understanding of the structural determinants which govern the cyclase vs. hydrolase reaction selectivity observed for NAD+ and NGD+, we first compared the three dimensional structures of cADPR and cGDPR. Conformational space explored by cADPR and cGDPR was sampled using the MacroModel program (Figure S1 in Supporting Information). The cADPR conformational search reproduced the available crystal structure of free cADPR (32, 33), thereby validating the efficiency of the method to predict reliable three-dimensional structures. Indeed, the adenine moiety of cADPR was found in syn conformation about the N9-C1′ ribose bond and in anti conformation about the N1-C1″ ribose bond. The exploration of cGDPR structure indicated that the guanine base is in anti conformation about the N9-C1′ ribose bond. Both syn and anti orientations were observed with respect to the N7-C1″-ribose of cGDPR. The superimposition of the ribose and pyrophosphate heavy atoms of cADPR and cGDPR (Figure 6A) results in a minimal base overlap and quite distinct distributions of donor and acceptor H-bonds. Moreover, the guanine ring contains more polar residues than adenine and is less aromatic. Together, all these features suggest different binding modes for adenine and guanine moieties during the cyclization reaction.
FIGURE 6.
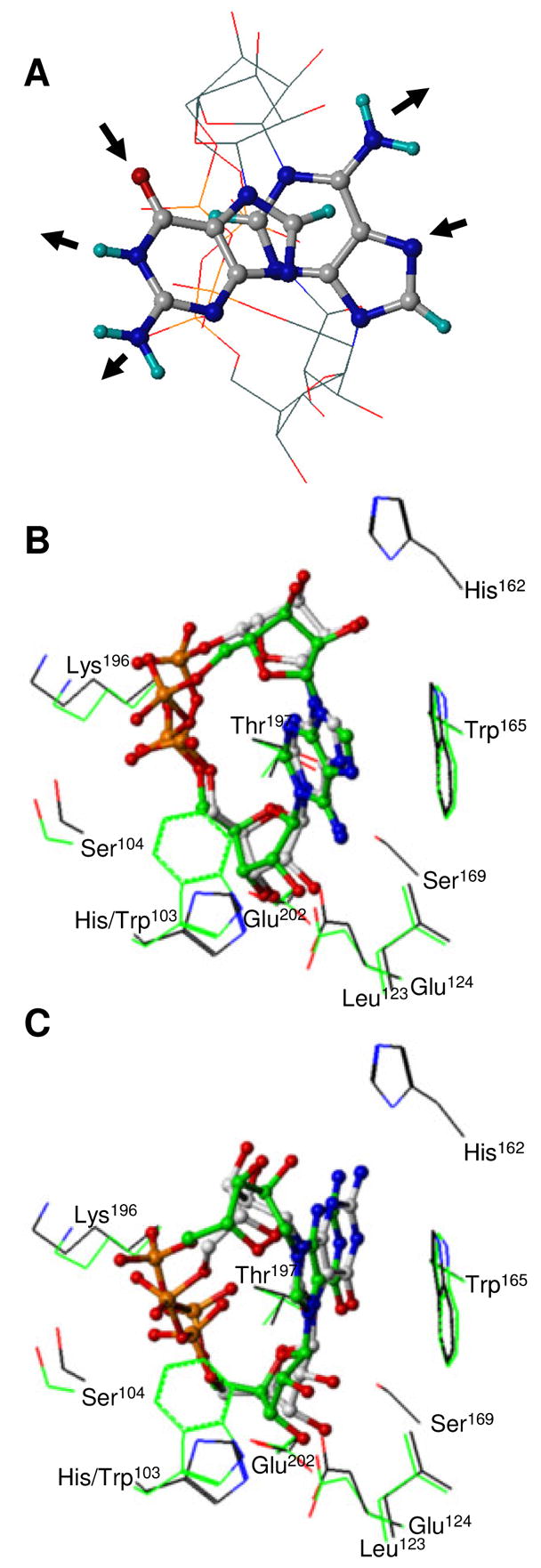
Views of cADPR and cGDPR three-dimensional structure. (A) Free cADPR and cGDPR. Best-fit superposition of the pyrophosphate and ribose heavy atoms of the MacroModel lowest energy conformers. For sake of clarity, ribose and pyrophosphate groups are depicted without hydrogen atoms using line representation whereas all atoms of adenine and guanine bases are shown using ball-and-stick representation. Molecules are colored by atom type as defined in Figure 5. H-bond donors and acceptors in the Watson Crick edge of the bases are indicated using arrows. cADPR (B) and cGDPR (C) docked into the WT and H103W SmNACE active site. Carbon atoms of the WT and mutant complexes are shown in grey and green respectively (the other atoms are colored as defined in Figure 4). All ligand heavy atoms are shown using ball-and-stick. Side chains of the key residues involved in ligand binding are depicted using a line representation and illustrate the surrounding enzyme structure. The rendering was performed using SYBYL 7.1.
If we make the reasonable assumption that reactions catalyzed by SmNACE occur according to molecular mechanisms similar to those advocated for the other ADP-ribosyl cyclases (3), the ribosyl moiety of the A(G)DP-ribosyl intermediate generated by the nicotinamide-ribose bond cleavage would be expected to be close to the carboxylate of Glu202. Based on this premise, we generated theoretical three-dimensional models of complexes between cADPR or cGDPR and WT or mutant SmNACE. As expected, different base positioning were observed for cADPR and cGDPR (Figure 6B,C). The binding of adenine into the SmNACE active site cleft may be principally mediated by π-stacking and Van der Waals interactions with His/Trp103 and Trp165 residues. These interactions are significantly favored in the H103W mutant. By contrast, guanine may preferentially interact with the active site using H-bonds and consequently, the H103W mutation is not predicted to have a dramatic effect on guanine binding.
DISCUSSION
In this study we demonstrated that replacement of His103 in the active site of S. mansoni NACE by a tryptophan – a residue conserved in all the other members of the ADP-ribosyl cyclase family – results in a dramatic increase (2 to 3 orders of magnitude) in the ADP-ribosyl cyclase activity of this enzyme. Thus, in contrast to the WT SmNACE, the transformation of NAD+ catalyzed by the H103W mutant yields a readily detectable proportion of cADPR which amounts to about 7% of the reaction products. Importantly, replacement of His103 with a tryptophan has no consequence on the kinetic parameters of SmNACE (Table 1) indicating that the increased efficiency of the cyclization step observed in the H103W mutant does not originate from changes in the ground-state binding of the substrate or from an overall change of the transition state energy of the catalyzed reaction. For H103W SmNACE, these data are consistent with a partitioning kinetic mechanism (Figure 1A) in which : i) the formation of the E·ADP-ribosyl intermediate is a rate-limiting step, as found for the WT SmNACE (16) and mammalian CD38 (3), and ii) the increased rate of the intramolecular reaction of the ADP-ribosyl intermediate, leading to cADPR, occurs at the expense of its reaction with water. In further support of this mechanism, trapping experiments with methanol which acts as a powerful nucleophilic acceptor in competition with water and the intramolecular cyclization process in the transformation of the common E·ADP-ribosyl intermediate, show that the catalyzed reaction turnover in not accelerated. Altogether, the key observation made in this study is that the introduction of tryptophan residue at position 103 of the active site unmasks an alternate pathway which diverts the ADP-ribosyl intermediate towards the intramolecular cyclization process.
Surprisingly, despite the importance of Trp103 in the SmNACE mutant, no role for the equivalent active site residue Trp125 in controlling the ADP-ribosyl cyclase activity was described in human CD38 (13). However, previous studies from our laboratory (15) and from the Lee laboratory (14) showed that the Glu residue in the ‘signature region’ (Glu150 and Glu146 in respectively mouse and human CD38) dictated whether cyclization or hydrolysis of NAD+ was the favored reaction pathway. Thus mutation of this active site residue resulted in a significant increase in the transformation of NAD+ into cADPR by murine CD38 which accounted for up to 25% of the reaction products (15). Likewise for the E146A mutant of human CD38, cADPR amounted to about 75 % of the total reaction products (14). However, unlike what we observed with the H103W mutant of SmNACE, mutation of the ‘signature’ Glu residue increased the cyclization at the expense of the reaction turnover which was reduced by more than 95% (15, 47). Interestingly, replacement of Glu146 in human CD38 by residues with small and neutral side chains favored the cyclization process, which has been suggested to be correlated with a reduced access of water molecules to the enzyme active site (14). Therefore, it appears that mutation of the active site ‘signature’ Glu residue, which seemingly controls the partitioning of the reaction intermediate in CD38, also affects the overall efficacy of its catalytic process. If the ‘signature’ Glu is responsible for controlling the partitioning of the reaction intermediate in CD38, why would mutation of His103 to a Trp alter the cyclization efficiency of SmNACE ? As we showed in Table 2, the ‘signature’ Glu124 in H103W SmNACE has an estimated pKa close to that of Glu146, its equivalent residue in human CD38, and both values are much higher than the calculated pKa of Glu124 in WT SmNACE. We believe that the increased polarity of the highly acidic carboxylic group of Glu124 in the WT SmNACE may explain, at least in part, the reaction specificity of SmNACE which, by increasing the solvation of the active site, might provide a more efficient activation of the water molecule involved in the nucleophilic attack of the reaction intermediate thus favoring the hydrolysis pathway. In support of this conclusion, our modeling studies indicated that the presence of His103 appears to increase the polarity within a specific sub-domain of the active site, in the vicinity of the ‘signature residue’ Glu124. This would promote greater access of water molecules to the active site, favoring the hydrolytic pathway leading to ADPR at the expense of the cyclization reaction and cADPR production.
Although the active site of S. mansoni NACE and S. japonicum NACE contain a His instead of the Trp residue that is found in all other ADP-ribosyl cyclase family members (Figure 2), this change does not affect the overall catalytic activity of the enzyme but only influences the cyclase efficiency of the enzyme. This leads to the question of why WT SmNACE is as efficient as the other ADP-ribosyl cyclases in the catalytic cleavage of the nicotinamide-ribose bond of NAD+ which is the most energy-demanding step of the kinetic mechanism of these enzymes. Our modeling studies indicate that the active site Trp103, found in all other CD38 family members and in H103W SmNACE, partners with the other Trp found in the active site (Trp165 in SmNACE), to efficiently engage in stacking interactions (most probably edge-to-face type) with the nicotinamide moiety of the substrate. This stacking interaction can be mimicked by a neutral His103 residue found in WT SmNACE (Figure 7). Such an aromatic/hydrophobic environment – which also includes Leu123 of the signature region – and the absence of solvent in the vicinity of the positively charged pyridinium moiety in NAD+ was previously shown to significantly lower the barrier of dissociation of the nicotinamide-ribose bond (48-50). Moreover, our docking experiments predict that uncharged His103 and Trp103, via its 5-membered pyrrole ring, are both placed above the reacting ribose and thus could also stabilize – via σ-donor (His103) (51) or cation-π (Trp103) (52, 53) type interactions – the oxocarbenium ion-like transition state occurring en route to the ADP-ribosyl reaction intermediate (Figure 1B) (3) and, as above, promote the cleavage of the scissile bond (Figure 7). Therefore we can infer that a neutral imidazole side-chain is needed for catalysis and that protonation of His103 should result in a drastic decrease of activity in SmNACE. In agreement with this hypothesis, we have found experimentally in the WT enzyme a group that titrates with a pKa of ~ 6.0 (Figure 4), i.e. a value close to the one predicted for His103 (Table 2). This group was not detected in the H103W mutant but interestingly another ionizable group important for catalysis with a pKa of 5.4 was found. We have tentatively attributed this group to Glu202, the catalytic residue essential for activity in the ADP-ribosyl cyclase enzyme family (9, 13, 54). According to the prevalent reaction mechanism for this family of enzymes (3) and in analogy with reactions catalyzed by glycosidases (55) this acid residue, when ionized, is involved in the stabilization of the transition state and of the reaction intermediate (Figure 1B). As discussed earlier, because of the presence of His103, the residue Glu202 is more acidic in the WT SmNACE (Table 2) than in the H103W mutant and thus could not be detected within the pH range investigated where the active site His103 titrates first.
FIGURE 7.
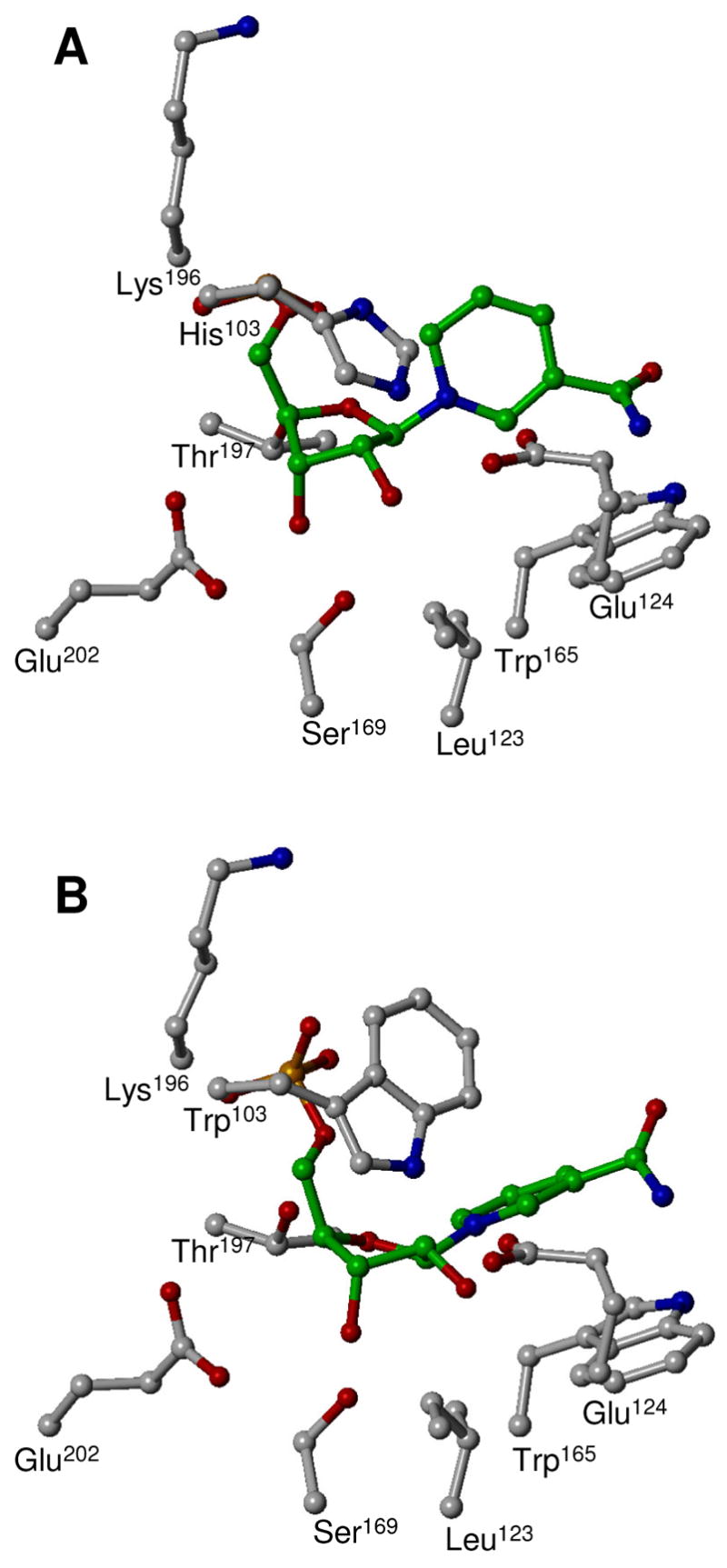
Interaction of the NMN+ moiety of NAD+ docked into the active site of WT (A) and H103W mutant (B) of SmNACE. Heavy atoms of NMN+ and side chains of the enzyme key residues are depicted using ball-and-stick representation. Carbon atoms of NMN+ are colored in green, all the other atoms are colored by atom type as defined in Figure 5. The rendering was performed using SYBYL 7.1. The ‘catalytic’ Glu202 is believed to be involved in the stabilization of the putative oxocarbenium ion transition state formed during the nicotinamide-ribosyl bond cleavage and in the stabilization of the ADP-ribosyl reaction intermediate, see (3). The ‘signature’ Glu124 is taking part in the partitioning of the intermediate between cyclization and hydrolysis.
We previously showed that in dramatic contrast with NAD+, the cyclization reaction pathway is favored in the catalytic transformation of the alternate substrate NGD+ by SmNACE (16). As shown here, the H103W mutation does not significantly alter this reaction which overwhelmingly yields the cyclized reaction product, cGDPR. This suggests that the interaction of NAD+ and NGD+, and their cyclic derivatives cADPR and cGDPR, with the active site of SmNACE is different enough to affect the outcome of the catalytic reaction. Docking of cADPR in the active site to model the ADP-ribosyl intermediate poised for the intramolecular cyclization reaction (Figure 1B), suggests that the SmNACE residue 103 directly interacts with the adenine moiety of NAD+ (Figure 6B). In contrast, this residue does not seem to be a key determinant for the positioning of the guanine moiety in cGDPR, in either the WT or H103W mutant SmNACE (Figure 6C). To explain the NAD+ cyclization vs. hydrolysis selectivity by SmNACE, we suggest that replacement of His103 by a Trp residue might allow a better π-stacking of the adenine ring in the H103W mutant (Figure 6B), providing an orientation within the ADP-ribosyl intermediate that favors the intramolecular cyclization reaction between adenine-N1 and ribose-C1″. The absence of such interactions in the transformation of NGD+ is consistent with our experimental results indicating only minor changes in the proportions of cyclic and solvolytic products upon H103W mutation (for a comparison with known adenine and guanine binding sites in proteins, see Supporting Information).
In conclusion, it is of interest to understand why the active site of S. mansoni NACE contains a His residue at position 103 instead of the tryptophan residue that is found in all other ADP-ribosyl cyclase family members. We suggest that SmNACE has evolved to be an efficient NAD+glycohydrolase and a very poor ADP-ribosyl cyclase. This implies two separate conditions: i) that the first step of the reaction mechanism, which generates an ADP-ribosyl intermediate from NAD+ (Figure 1), is – as demonstrated in this work – equally efficient for the enzymes of the ADP-ribosyl cyclase family regardless of whether the active site residue at position 103 in SmNACE (or the analogous position in CD38 or Aplysia cyclase) is a Trp or a His, and ii) that the specificity of these enzymes in terms of hydrolysis vs. cyclization must depend on the outcome of competing reaction pathways during the second part of the kinetic mechanism. Why this particular “mutation” was selected for during the evolution of the schistosomes is unknown. However, the fact that S. mansoni NACE, which is a GPI-anchored ecto-enzyme preferentially expressed at the surface (tegument) of adult worms (16), is lacking the “hallmark” enzyme activity of all other cyclases, suggests that there might be some functional advantage to the parasite to express this “defective” form of the enzyme. Regardless, the data presented here clearly demonstrate that SmNACE can be converted from an enzyme that only generates ADPR into one that can produce both cADPR and ADPR by simply changing the active site His103 “back” to the canonical tryptophan. In this respect residue-103 which shifts the product selectivity in SmNACE without affecting its catalytic activity fulfills the criteria of a “plasticity residue” in a promiscuous (or multifunctional) enzyme (56).
Supplementary Material
Details about the docking protocol of ligands into the active site of WT and H103W mutant; conformational analysis of cADPR and cGDPR (Figure S1); comparison of adenine and guanine binding mode into SmNACE obtained by docking with experimental data available in the Protein Data Bank. This material is available free of charge via the Internet at http://pubs.acs.org.
Acknowledgments
We thank Dr. G. Marcou (UMR 7175 CNRS-ULP) and Prof. A. Bernardi (University of Milano, Italy) for performing calculations using Macromodel and Prof. N.J. Oppenheimer (UCSF) for his generous gift of araF-NAD+.
Footnotes
Support for this work was provided by the CNRS, MERNT and ANR Grant NT05-1-41905 (FS and DR) and NIH AI-057996 (FL).
Abbreviations: SmNACE, Schistosoma mansoni NAD(P)+ catabolizing enzyme; ADPR, ADP-ribose; cADPR, cyclic ADP-ribose; cGDPR, cyclic GDP-ribose; araF-NAD+, nicotinamide 2′-deoxy-2′-fluoroarabinoside adenine dinucleotide; RMS, root mean square; WT, wild type.
References
- 1.Howard M, Grimaldi JC, Bazan JF, Lund FE, Santos-Argumedo L, Parkhouse RM, Walseth TF, Lee HC. Formation and hydrolysis of cyclic ADP-ribose catalyzed by lymphocyte antigen CD38. Science. 1993;262:1056–1059. doi: 10.1126/science.8235624. [DOI] [PubMed] [Google Scholar]
- 2.Lee HC. Enzymatic functions and structures of CD38 and homologs. Chem Immunol. 2000;75:39–59. doi: 10.1159/000058774. [DOI] [PubMed] [Google Scholar]
- 3.Schuber F, Lund FE. Structure and enzymology of ADP-ribosyl cyclases: Conserved enzymes that produce multiple calcium mobilizing metabolites. Curr Mol Med. 2004;4:249–261. doi: 10.2174/1566524043360708. [DOI] [PubMed] [Google Scholar]
- 4.Lee HC. Nicotinic acid adenine dinucleotide phosphate (NAADP)-mediated calcium signaling. J Biol Chem. 2005;280:33693–33696. doi: 10.1074/jbc.R500012200. [DOI] [PubMed] [Google Scholar]
- 5.Hirata Y, Kimura N, Sato K, Ohsugi Y, Takasawa S, Okamoto H, Ishikawa J, Kaisho T, Ishihara K, Hirano T. ADP ribosyl cyclase activity of a novel bone marrow stromal cell surface molecule, BST-1. FEBS Lett. 1994;356:244–248. doi: 10.1016/0014-5793(94)01279-2. [DOI] [PubMed] [Google Scholar]
- 6.Lee HC, Aarhus R. ADP-ribosyl cyclase: an enzyme that cyclizes NAD+ into a calcium-mobilizing metabolite. Cell Regul. 1991;2:203–209. doi: 10.1091/mbc.2.3.203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cakir-Kiefer C, Muller-Steffner H, Schuber F. Unifying mechanism for Aplysia ADP-ribosyl cyclase and CD38/NAD+glycohydrolases. Biochem J. 2000;349:203–210. doi: 10.1042/0264-6021:3490203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Muller-Steffner HM, Augustin A, Schuber F. Mechanism of cyclization of pyridine nucleotides by bovine spleen NAD+glycohydrolase. J Biol Chem. 1996;271:23967–23972. doi: 10.1074/jbc.271.39.23967. [DOI] [PubMed] [Google Scholar]
- 9.Sauve AA, Deng HT, Angeletti RH, Schramm VL. A covalent intermediate in CD38 is responsible for ADP-ribosylation and cyclization reactions. J Am Chem Soc. 2000;122:7855–7859. [Google Scholar]
- 10.Prasad GS, McRee DE, Stura EA, Levitt DG, Lee HC, Stout CD. Crystal structure of Aplysia ADP ribosyl cyclase, a homologue of the bifunctional ectozyme CD38. Nature Struct Biol. 1996;3:957–964. doi: 10.1038/nsb1196-957. [DOI] [PubMed] [Google Scholar]
- 11.Yamamoto-Katayama S, Ariyoshi M, Ishihara K, Hirano T, Jingami H, Morikawa K. Crystallographic studies on human BST-1/CD157 with ADP- ribosyl cyclase and NAD glycohydrolase activities. J Mol Biol. 2002;316:711–723. doi: 10.1006/jmbi.2001.5386. [DOI] [PubMed] [Google Scholar]
- 12.Liu Q, Kriksunov IA, Graeff R, Munshi C, Lee HC, Hao Q. Crystal structure of human CD38 extracellular domain. Structure. 2005;13:1331–1339. doi: 10.1016/j.str.2005.05.012. [DOI] [PubMed] [Google Scholar]
- 13.Munshi C, Aarhus R, Graeff R, Walseth TF, Levitt D, Lee HC. Identification of the enzymatic active site of CD38 by site-directed mutagenesis. J Biol Chem. 2000;275:21566–21571. doi: 10.1074/jbc.M909365199. [DOI] [PubMed] [Google Scholar]
- 14.Graeff R, Munshi C, Aarhus R, Johns M, Lee HC. A single residue at the active site of CD38 determines its NAD cyclizing and hydrolyzing activities. J Biol Chem. 2001;276:12169–12173. doi: 10.1074/jbc.M011299200. [DOI] [PubMed] [Google Scholar]
- 15.Lund FE, Muller-Steffner HM, Yu N, Stout CD, Schuber F, Howard MC. CD38 signaling in B lymphocytes is controlled by its ectodomain but occurs independently of enzymatically generated ADP-Ribose or cyclic ADP-Ribose. J Immunol. 1999;162:2693–2702. [PubMed] [Google Scholar]
- 16.Goodrich SP, Muller-Steffner H, Osman A, Moutin M-J, Kusser K, Roberts A, Woodland DL, Randall TD, Kellenberger E, LoVerde PT, Schuber F, Lund FE. Production of calcium-mobilizing metabolites by a novel member of the ADP-ribosyl cyclase family expressed in Schistosoma mansoni. Biochemistry. 2005;44:11082–11097. doi: 10.1021/bi050704r. [DOI] [PubMed] [Google Scholar]
- 17.Graeff RM, Walseth TF, Fryxell K, Branton WD, Lee HC. Enzymatic synthesis and characterizations of cyclic GDP-ribose. A procedure for distinguishing enzymes with ADP-ribosyl cyclase activity. J Biol Chem. 1994;269:30260–30267. [PubMed] [Google Scholar]
- 18.Hu W, Yan Q, Shen DK, Liu F, Zhu ZD, Song HD, Xu XR, Wang ZJ, Rong YP, Zeng LC, Wu J, Zhang X, Wang JJ, Xu XN, Wang SY, Fu G, Zhang XL, Wang ZQ, Brindley PJ, McManus DP, Xue CL, Feng Z, Chen Z, Han ZG. Evolutionary and biomedical implications of a Schistosoma japonicum complementary DNA resource. Nat Genet. 2003;35:139–147. doi: 10.1038/ng1236. [DOI] [PubMed] [Google Scholar]
- 19.Clare JJ, Romanos MA, Rayment FB, Rowedder JE, Smith MA, Payne MM, Sreekrishna K, Henwood CA. Production of mouse epidermal growth factor in yeast: high-level secretion using Pichia pastoris strains containing multiple gene copies. Gene. 1991;105:205–212. doi: 10.1016/0378-1119(91)90152-2. [DOI] [PubMed] [Google Scholar]
- 20.Morrissey JH. Silver stain for proteins in polyacrylamide gels: a modified procedure with enhanced uniform sensitivity. Anal Biochem. 1981;117:307–310. doi: 10.1016/0003-2697(81)90783-1. [DOI] [PubMed] [Google Scholar]
- 21.Muller CD, Tarnus C, Schuber F. Preparation of analogues of NAD+ as substrates for a sensitive fluorimetric assay of nucleotide pyrophosphatase. Biochem J. 1984;223:715–721. doi: 10.1042/bj2230715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Orsi BA, Tipton KF. Kinetic analysis of progress curves. Methods Enzymol. 1979;63:159–183. doi: 10.1016/0076-6879(79)63010-0. [DOI] [PubMed] [Google Scholar]
- 23.Meyer-Almes FJ, Auer M. Enzyme inhibition assays using fluorescence correlation spectroscopy: A new algorithm for the derivation of k(cat)/K(M) and K(i) values at substrate concentrations much lower than the Michaelis constant. Biochemistry. 2000;39:13261–13268. doi: 10.1021/bi000057y. [DOI] [PubMed] [Google Scholar]
- 24.Tipton AK, Dixon HBF. Effect of pH on enzymes. Methods Enzymol. 1979;63:183–234. doi: 10.1016/0076-6879(79)63011-2. [DOI] [PubMed] [Google Scholar]
- 25.Berthelier V, Tixier JM, Muller-Steffner H, Schuber F, Deterre P. Human CD38 is an authentic NAD(P)+ glycohydrolase. Biochem J. 1998;330:1383–1390. doi: 10.1042/bj3301383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Muller-Steffner HM, Malver O, Hosie L, Oppenheimer NJ, Schuber F. Slow-binding inhibition of NAD+glycohydrolase by arabino analogues of beta-NAD+ J Biol Chem. 1992;267:9606–9611. [PubMed] [Google Scholar]
- 27.Sali A, Blundell TL. Comparative protein modelling by satisfaction of spatial restraints. J Mol Biol. 1993;234:779–815. doi: 10.1006/jmbi.1993.1626. [DOI] [PubMed] [Google Scholar]
- 28.Case DA, Cheatham TE, 3rd, Darden T, Gohlke H, Luo R, Merz KM, Jr, Onufriev A, Simmerling C, Wang B, Woods RJ. The Amber biomolecular simulation programs. J Comput Chem. 2005;26:1668–1688. doi: 10.1002/jcc.20290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dolinsky TJ, Nielsen JE, McCammon JA, Baker NA. PDB2PQR: an automated pipeline for the setup of Poisson-Boltzmann electrostatics calculations. Nucleic Acids Res. 2004;32:W665–667. doi: 10.1093/nar/gkh381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yang AS, Gunner MR, Sampogna R, Sharp K, Honig B. On the calculation of pKas in proteins. Proteins. 1993;15:252–265. doi: 10.1002/prot.340150304. [DOI] [PubMed] [Google Scholar]
- 31.Bashford D, Karplus M. pKa’s of ionizable groups in proteins: atomic detail from a continuum electrostatic model. Biochemistry. 1990;29:10219–10225. doi: 10.1021/bi00496a010. [DOI] [PubMed] [Google Scholar]
- 32.Lee HC, Aarhus R, Levitt D. The crystal structure of cyclic ADP-ribose [letter] Nature Struct Biol. 1994;1:143–144. doi: 10.1038/nsb0394-143. [DOI] [PubMed] [Google Scholar]
- 33.Lee HC. Cyclic ADP-ribose and NAADP. In: Lee HC, editor. Cyclic ADP-ribose and NAADP Structures, metabolism and functions. Kluwer Academic Publishers; Boston: 2002. pp. 1–21. [Google Scholar]
- 34.Verdonk ML, Cole JC, Hartshorn MJ, Murray CW, Taylor RD. Improved protein-ligand docking using GOLD. Proteins. 2003;52:609–623. doi: 10.1002/prot.10465. [DOI] [PubMed] [Google Scholar]
- 35.Lund FE, Moutin MJ, Muller-Steffner H, Schuber F. ADP-ribosyl cyclase and GDP-ribosyl cyclase activities are not always equivalent: Impact on the study of the physiological role of cyclic ADP-ribose. Anal Biochem. 2005;346:336–338. doi: 10.1016/j.ab.2005.08.016. [DOI] [PubMed] [Google Scholar]
- 36.Muller-Steffner H, Muzard M, Oppenheimer N, Schuber F. Mechanistic implications of cyclic ADP-ribose hydrolysis and methanolysis catalyzed by calf spleen NAD+glycohydrolase. Biochem Biophys Res Commun. 1994;204:1279–1285. doi: 10.1006/bbrc.1994.2601. [DOI] [PubMed] [Google Scholar]
- 37.Sauve AA, Munshi C, Lee HC, Schramm VL. The reaction mechanism for CD38. A single intermediate is responsible for cyclization, hydrolysis, and base-exchange chemistries. Biochemistry. 1998;37:13239–13249. doi: 10.1021/bi981248s. [DOI] [PubMed] [Google Scholar]
- 38.Morrison JF, Walsh CT. The behavior and significance of slow-binding enzyme inhibitors. Adv Enzymol Relat Areas Mol Biol. 1988;61:201–301. doi: 10.1002/9780470123072.ch5. [DOI] [PubMed] [Google Scholar]
- 39.Thompson ST, Stellwagen E. Binding of Cibacron blue F3GA to proteins containing the dinucleotide fold. Proc Natl Acad Sci USA. 1976;73:361–365. doi: 10.1073/pnas.73.2.361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Biellmann JF, Samama JP, Brändén CI, Eklund H. X-ray studies of the binding of Cibacron blue F3GA to liver alcohol dehydrogenase. Eur J Biochem. 1979;102:107–110. doi: 10.1111/j.1432-1033.1979.tb06268.x. [DOI] [PubMed] [Google Scholar]
- 41.Prestera T, Prochaska HJ, Talalay P. Inhibition of NAD(P)H:(quinone-acceptor) oxidoreductase by cibacron blue and related anthraquinone dyes: a structure-activity study. Biochemistry. 1992;31:824–833. doi: 10.1021/bi00118a027. [DOI] [PubMed] [Google Scholar]
- 42.Thompson ST, Cass KH, Stellwagen E. Blue dextran-sepharose: an affinity column for the dinucleotide fold in proteins. Proc Natl Acad Sci USA. 1975;72:669–672. doi: 10.1073/pnas.72.2.669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Denizli A, Piskin E. Dye-ligand affinity systems. J Biochem Biophys Meth. 2001;49:391–416. doi: 10.1016/s0165-022x(01)00209-3. [DOI] [PubMed] [Google Scholar]
- 44.Schuber F, Pascal M. Interaction of Blue Dextran and Cibacron Blue F3GA with calf spleen NAD+-glycohydrolase. Biochimie. 1977;59:735–737. doi: 10.1016/s0300-9084(77)80254-x. [DOI] [PubMed] [Google Scholar]
- 45.del Carmen Fernandez-Alonso M, Canada FJ, Jimenez-Barbero J, Cuevas G. Molecular recognition of saccharides by proteins. Insights on the origin of the carbohydrate-aromatic interactions. J Am Chem Soc. 2005;127:7379–7386. doi: 10.1021/ja051020+. [DOI] [PubMed] [Google Scholar]
- 46.Love ML, Szebenyi DME, Kriksunov IA, Thiel DJ, Munshi C, Graeff R, Lee HC, Hao Q. ADP-ribosyl cyclase: Crystal structures reveal a covalent intermediate. Structure. 2004;12:477–486. doi: 10.1016/j.str.2004.02.006. [DOI] [PubMed] [Google Scholar]
- 47.Lund FE, Muller-Steffner H, Romero-Ramirez H, Moreno-Garcia ME, Partida-Sanchez S, Makris M, Oppenheimer NJ, Santos-Argumedo L, Schuber F. CD38 induces apoptosis of a murine pro-B leukemic cell line by a tyrosine kinase-dependent but ADP-ribosyl cyclase- and NAD glycohydrolase-independent mechanism. Int Immunol. 2006;18:1029–1042. doi: 10.1093/intimm/dxl037. [DOI] [PubMed] [Google Scholar]
- 48.Oppenheimer NJ. NAD hydrolysis: Chemical and enzymatic mechanisms. Mol Cell Biochem. 1994;138:245–251. doi: 10.1007/BF00928468. [DOI] [PubMed] [Google Scholar]
- 49.Buckley N, Handlon AL, Maltby D, Burlingame AL, Oppenheimer NJ. Reactions of charged substrates. 2 Gas-phase dissociation of 2′-substituted nicotinamide arabinosides. J Org Chem. 1994;59:3609–3615. [Google Scholar]
- 50.Buckley N, Oppenheimer NJ. Reactions of Charged Substrates. 5 The Solvolysis and Sodium Azide Substitution Reactions of Benzylpyridinium Ions in Deuterium Oxide. J Org Chem. 1996;61:7360–7372. doi: 10.1021/jo960729j. [DOI] [PubMed] [Google Scholar]
- 51.Hu J, Barbour LJ, Ferdani R, Gokel GW. Sodium cation complexation behavior of the heteroaromatic sidechains of histidine and tryptophan. Chem Commun (Camb) 2002:1810–1811. doi: 10.1039/b204321c. [DOI] [PubMed] [Google Scholar]
- 52.Zacharias N, Dougherty DA. Cation-pi interactions in ligand recognition and catalysis. Trends Pharmacol Sci. 2002;23:281–287. doi: 10.1016/s0165-6147(02)02027-8. [DOI] [PubMed] [Google Scholar]
- 53.Hu J, Barbour LJ, Gokel GW. The indole side chain of tryptophan as a versatile pi-donor. J Am Chem Soc. 2002;124:10940–10941. doi: 10.1021/ja020586k. [DOI] [PubMed] [Google Scholar]
- 54.Munshi C, Thiel DJ, Mathews II, Aarhus R, Walseth TF, Lee HC. Characterization of the active site of ADP-ribosyl cyclase. J Biol Chem. 1999;274:30770–30777. doi: 10.1074/jbc.274.43.30770. [DOI] [PubMed] [Google Scholar]
- 55.Zechel DL, Withers SG. Dissection of nucleophilic and acid-base catalysis in glycosidases. Curr Opin Chem Biol. 2001;5:643–649. doi: 10.1016/s1367-5931(01)00260-5. [DOI] [PubMed] [Google Scholar]
- 56.Yoshikuni Y, Ferrin TE, Keasling JD. Designed divergent evolution of enzyme function. Nature. 2006;440:1078–1082. doi: 10.1038/nature04607. [DOI] [PubMed] [Google Scholar]
- 57.Tarnus C, Muller HM, Schuber F. Chemical evidence in favour of a stabilized oxocarbonium-ion intermediate in the NAD+glycohydrolase catalyzed reactions. Bioorg Chem. 1988;16:38–51. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Details about the docking protocol of ligands into the active site of WT and H103W mutant; conformational analysis of cADPR and cGDPR (Figure S1); comparison of adenine and guanine binding mode into SmNACE obtained by docking with experimental data available in the Protein Data Bank. This material is available free of charge via the Internet at http://pubs.acs.org.