

Abstract
Background
Major depressive disorder (MDD) shows increased coronary artery disease (CAD) risk of unknown mechanism(s). MDD is more common in women than men; CAD diagnosis can be difficult in women. Elevations of the inflammatory markers C-reactive protein (CRP) and serum amyloid A (SAA) predict increased CAD risk in populations; few data on these markers exist in MDD, particularly in remitted patients.
Methods
We measured fasting am serum CRP (high sensitivity, CRPhs) and SAA in 18 unmedicated, remitted women with MDD (mean age 41± (SD)12, body mass index (BMI) 25.2 ± 4.1 kg/m2) and 18 BMI-matched healthy control subjects (age 36 ± 10, BMI 25.3 ± 3.8 kg/m2) on 2 separate occasions, ≥ 6 days apart.
Results
Repeat SAA and CRPhs measurements strongly correlated across study days (SAA: r = .83, p< .001; CRPhs: r = .94, p < .001). Both SAA (5.30 ± 3.39 vs. 2.84 ± 1.87 mg/L, p < .005) and CRPhs (3.23 ± 3.17 vs. 1.12 ± 1.45 mg/L; p < .01) were significantly elevated in MDD women versus controls.
Conclusions
Elevated SAA and CRPhs in remitted, unmedicated women with MDD indicate a pro-inflammatory state unrelated to current depressive symptoms or pharmacotherapy. These findings suggest that inflammatory mechanisms may in part underlie findings of increased CAD risk in MDD.
Numerous studies have now shown that depressive disorders are associated with an increased risk of coronary artery disease (CAD), independent of smoking, hypertension, and other potentially confounding risk factors for poor health (Lett et al 2004; Wulsin 2004). However, information is limited regarding biologic mechanisms or risk factors accounting for this association between major depressive disorder (MDD) and CAD. Such information would be important for screening patients with MDD for increased CAD risk, in order that intervention strategies to mitigate this risk could be considered. Such strategies are routinely employed in clinical medicine, including screening patients for evidence of modifiable risk factors for CAD. For example, hypertension, frank or incipient diabetes, and dyslipidemia can be treated with antihypertensives, oral hypoglycemics and/or insulin sensitizers, and statins, respectively; in addition, all of these risk factors can be modified by lifestyle changes such as dietary modifications and regular exercise.
Major depressive disorder is twice as common in women as in men (Kessler et al 1994) Coronary artery disease, although more common in men, can be more difficult to diagnose in women (Mosca et al 1997). Thus, biological markers of CAD risk may be particularly valuable in women with MDD for identifying those at increased risk and in whom closer follow-up or more aggressive preventive measures are warranted.
Elevated serum levels of inflammatory markers have been shown to predict CAD outcomes (Choudhury and Leyva 1999; Delanghe et al 2002; Morrow and Ridker 2000; Morrow et al 2000). These elevations are believed to reflect the important role of chronic inflammation in the progression of atherosclerotic lesions (Murtagh and Anderson 2004).
Acute phase proteins (APP) are important inflammatory mediators of the innate immune system, and are implicated as both risk markers and pathogenetic factors in CAD (Kisilevsky and Tam 2002; Rattazzi et al 2003) Although APPs were initially identified because of their role in acute inflammatory responses to microbial agents or products, they are often persistently elevated above baseline levels during chronic inflammatory processes, unrelated to infections (Chait et al 2005).
C-reactive protein (CRP) and serum amyloid A (SAA) are the earliest and most robustly-responding of the APPs, making them the most sensitive known markers of acute phase responses to inflammatory stimuli (Gabay and Kushner 1999). During acute responses to infectious agents, levels of CRP and SAA can be elevated a thousand-fold over baseline (Gabay and Kushner 1999) However, low-grade elevations in basal levels of these mediators, assessed in the absence of an acute infection or other inflammatory stimulus, are predictive of CAD outcomes in multiple studies (Choudhury and Leyva 1999; Delanghe et al 2002; Johnson et al 2004; Morrow and Ridker 2000; Morrow et al 2000) These predictive effects are believed to reflect active participation of these mediators in chronic inflammatory processes contributing to the progression of atherosclerotic lesions (Murtagh and Anderson 2004). Hence, CRP has many pro-inflammatory effects and is a sensitive marker of ongoing inflammation (Chait et al 2005; Pepys and Hirschfield 2003) SAA is an apolipoprotein that, when incorporated into high-density lipoproteins (HDL), renders them atherogenic rather than atheroprotective (Chait et al 2005; Kisilevsky and Tam 2002).
Recent studies have shown elevation of CRP (Lanquillon et al 2000; Miller et al 2002; Penninx et al 2003) in association with current depressive symptoms. However, it is not clear whether such changes also occur in individuals with MDD who are symptomatically recovered from depressive episodes for extended periods. Identification of a sustained, state-independent low-grade pro-inflammatory state in symptomatically remitted MDD patients would have important pathophysiologic implications for the increased CAD risk in MDD and suggest treatments that otherwise might not be considered, such as agents with anti-inflammatory actions. Indeed, there is evidence that several of the classes of agents known to reduce CAD risk, including aspirin, statins, and the insulin-sensitizing thiazolidinedione drugs, may act in part by reducing inflammatory responses that contribute to atherosclerotic plaque development (Jain and Ridker 2005; Plutzky 2003).
In this study, we have examined serum levels of SAA and CRP, in a rigorously selected group of women with a lifetime MDD diagnosis, studied while in clinical remission and not taking medications, and a group of healthy women matched by BMI who served as control subjects. Matching by BMI was considered to be critical for this study given the tendency of SAA and CRP to vary with BMI in previous studies (Tracy 2001), particularly in women (Johnson et al 2004; Thorand et al 2006) We hypothesized that serum SAA and CRP might be elevated to clinically important levels in our remitted, unmedicated female patients with MDD, and as such would provide evidence for a sustained mechanism contributing to increased CAD risk in MDD.
Methods and Materials
Participants
Eighteen women (mean age 41 ± (SD) 12 years; mean body mass index [BMI] 25.2 ± 4.1 kg/m2) with MDD in remission after at least 2 prior episodes, and off psychotropic medications for ≥ 3 months, and 18 individually BMI-matched (within ± 2 kg/m2) healthy control subjects (HCS; age 36 ± 10 years; mean BMI 25.3 ± 3.8 kg/m2) were included in the study. Two of the MDD patients and 4 control subjects identified themselves as African-American; the remaining subjects were self-identified as Caucasian, of whom 1 patient and 3 controls were self-identified as Hispanic. Patients and controls were recruited from the greater Washington, DC metropolitan area through newspaper and radio advertisements; respondents passing a telephone screen for eligibility were evaluated in the National Institute of Mental Health (NIMH) outpatient clinic at the National Institutes of Health (NIH) Clinical Center in Bethesda, Maryland. All subjects gave written, informed consent for participation in the study, which was conducted under protocols approved by the NIMH Institutional Review Board, after having the study explained to them in detail and having an opportunity to ask questions of the investigator.
Diagnosis was based on the Structured Clinical Interview for DSM-IV–Nonpatient Version (SCID-I-NP) with Past History of MDD addendum. Remission was defined by a period of at least 3 months prior to enrollment during which the patient did not take an antidepressant agent and had 24-item Hamilton Depression Rating Scale (HDRS) scores < 8. HCS had no personal (by SCID-I-NP) or family (first-degree relatives) history of psychiatric disorders. SCID and Hamilton interviews were conducted by mental health professionals (a mental health nurse and a psychologist) with extensive clinical and research experience who participated in an ongoing program of rater quality assurance and inter-rater reliability monitoring under the NIMH Mood and Anxiety Disorders Program. All participants were free of potentially confounding illnesses on the basis of history and results of physical examination, electrocardiography, and routine laboratory tests, including liver and kidney function tests, hematologic profile, thyroid function tests, urinalysis, and toxicology. Pregnant and nursing women were excluded. All subjects were nonsmokers, and none of the subjects was taking medication for treatment or prevention of a medical condition. Premenopausal women were studied during the follicular phase of the menstrual cycle, rigorously determined as previously described (Neumeister et al 2004).
Patients were studied on two separate occasions, at least six days apart. The median interval between sampling days was 15 days (interquartile interval, 10–25 days). Blood samples were obtained between 0800–0900 hours after an overnight fast. No subject showed evidence of infection or any inflammatory condition, including fever or symptoms of malaise, at the time of blood sampling.
Laboratory Measures
Serum SAA and CRP (high sensitivity, CRPhs) were measured by immunonephelometry using a BN II automated analyzer (Dade Behring, Newark, Denmark). Detection limits (DL) are .72 mg/L for SAA and .16 mg/L for CRPhs Inter-assay coefficients of variation (CVs) were 7.5% at a mean of 13.6 mg/L (n = 22) for SAA, and 5.3% at a mean of 14.2 mg/L (n = 17) and 2.7% at a mean of 53.1 mg/L (n = 6) for CRPhs. Specimens were coded so that laboratory personnel were blinded to subjects’ diagnosis and other identifying characteristics, but paired samples from a given subject were analyzed in the same batch. Results below the DL were assigned the value of the DL for computational purposes. This occurred in 4 of the CRPhs measurements, all in HCS. All values for SAA were above the DL.
Subjects also underwent 24-hour urine collections for urinary free cortisol (UFC) determination prior to both study days. Urinary free cortisol was measured by chemiluminescence immunoassay on an automated analyzer (Nichols Advantage, San Juan Capistrano, California).
Statistical Analyses
Pearson correlations were first calculated for SAA and CRPhs across the two test dates for all participants. Once found to be highly reproducible across the two test dates (see Results), means of the values from these dates were used in subsequent analyses. Discrepancies between SAA and CRPhs values on different test days were taken to represent day-to-day variation and were not repeated.
To improve suitability for parametric statistics, SAA and CRPhs data were log transformed. Results obtained with log-transformed data were similar to those obtained using raw data (see Results). Because subjects were individually BMI-matched, comparisons of SAA and CRPhs between diagnostic groups were performed with paired t-tests. We also conducted chi-square tests of the frequency of subjects in intermediate and high cardiac risk categories defined in previous studies, using cutoffs of 4 and 8 mg/L for SAA (Johnson et al 2004) and 1 and 3 mg/L for CRP (Willerson and Ridker 2004) for intermediate and high risk, respectively.
The relationship of SAA and CRPhs to each other, and that of each analyte with age, were assessed with Pearson correlations. All tests were evaluated at p < .05, two-tailed. All means in figures are those of raw values. The SPSS version 13.0.1 software package (SPSS Inc., Chicago, Illinois) was used for all analyses.
Results
The mean age between subject groups did not differ significantly (paired |t| = 1.37, df = 17, p = .19). The differences in ethnic composition of the 2 groups did not differ significantly. The mean 24-item HDRS score was .76 ± .96 (mean ± SD; range: 0–4) in the MDD women, and .39 ± .5 (range: 0–2) in controls.
Within-subject correlations for SAA and CRPhs levels between the two sampling dates were robust (SAA: r = .83, p < .001; CRPhs: r = .94, p < .001). In addition, the absolute values of these measures did not differ across the 2 determinations by paired t-tests (SAA: t = 1.12, df = 48, p = .27; CRP: t = .77, df = 48, p = .44). Based on these findings, subsequent analyses of SAA and CRPhs were conducted on the mean value for each subject across the two determinations.
Fasting morning SAA levels in the MDD women were significantly elevated compared with HCS (mean ± SD: 5.30 ± 3.39 mg/L vs. 2.84 ± 1.87 mg/L; |t| = 3.27; df = 17; p < .005) (Figure 1). This difference remained significant using the log-transformed values (|t| = 3.52; df = 17; p < .005). Using cutoffs of 4 and 8 mg/L for intermediate and high risk, respectively, for future CAD (Johnson et al 2004), 7/18 MDD patients versus 1/18 HCS had values of at least intermediate risk, and 4/18 MDD patients versus 1/18 HCS had values corresponding to high cardiac risk. These differences were significant by chi-square test (χ2 = 9.82, df = 2, p < .01).
Figure 1.
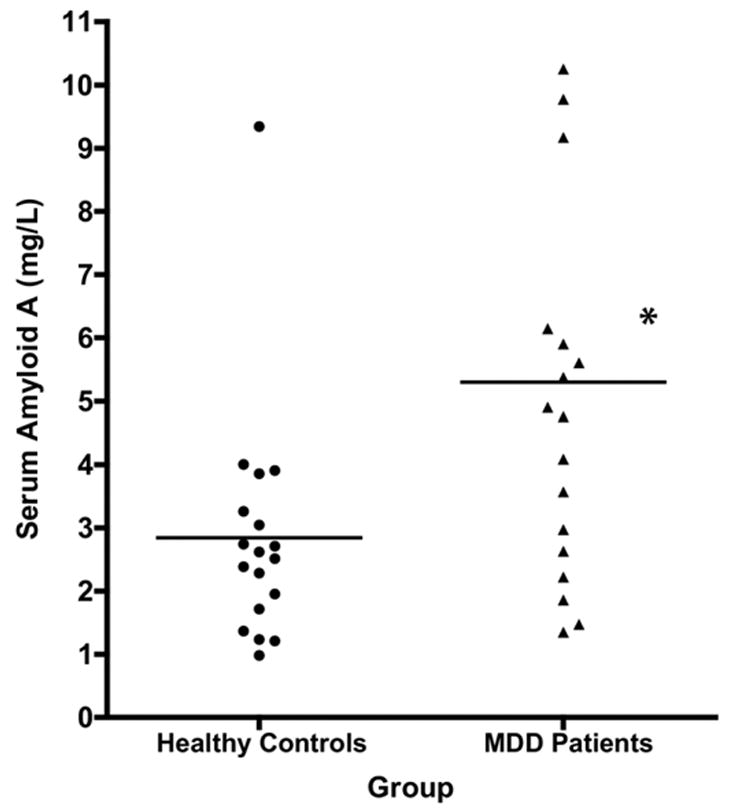
Serum amyloid A (SAA) levels in serum of remitted, unmedicated women with major depressive disorder (MDD) versus body mass index (BMI)-matched female healthy control subjects (HCS). Points represent the mean of two fasting am SAA levels obtained on two different dates, at least six days apart, in 18 MDD women and 18 HCS. Horizontal bars represent group means. * p < .005 versus controls by paired t-test.
Serum CRPhs was also significantly elevated in the MDD women (3.23 ± 3.17 mg/L) versus HCS (1.12 ± 1.45 mg/L) (|t| = 2.90; df = 17; p < .01) (Figure 2). This difference remained significant using the log-transformed values (|t| = 2.94; df = 17; p < .01). Using cutoffs of 1 and 3 mg/L for intermediate and high CAD risk (Willerson and Ridker 2004), respectively, 4/18 MDD patients versus 3/18 HCS had values of at least intermediate risk, while 8/18 versus 2/18 HCS showed values corresponding to high cardiac risk, respectively. These differences were significant by chi-square test (χ2 = 6.32, p < .05). No systematic differences in patient characteristics could be identified between those with SAA or CRPhs values in the higher versus lower risk categories.
Figure 2.
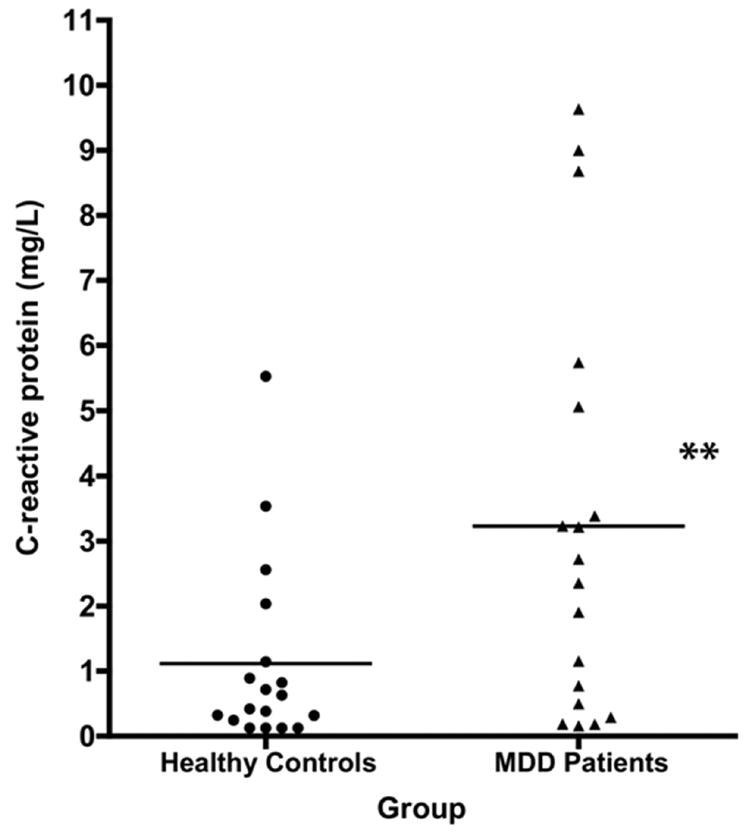
Serum C-reactive protein (CRP) levels in remitted, unmedicated women with major depressive disorder (MDD) versus body mass index (BMI)-matched female healthy control subjects (HCS). Points represent the mean of two fasting am CRP levels obtained on two different dates, at least six days apart, in 18 MDD women and 18 HCS. Horizontal bars represent group means. ** p < 01 versus controls by paired t-test.
Differences between subject groups in SAA and CRPhs levels were not attributable to racial or ethnic differences between pairs of subjects.
There was a nonsignificant positive relationship between SAA and CRPhs in the MDD patients (r = .38, p = .12), which was less robust in the HCS (r = .29, p > .2).
Neither SAA nor CRPhs was correlated with age either overall or by diagnostic group (all r values < .1, p > .2). Also, ethnic differences between subject pairs did not influence the group differences in SAA and CRPhs levels (data not shown).
Mean 24-hour UFC excretion did not differ between the remitted unmedicated MDD patients (mean 45.0 ± 16.0 μg/d) and the healthy controls (mean 40.7 ± 19.9 μg/d; paired |t| = .65, p > .2). Neither SAA (r = .02, p > .2) nor CRPhs (r = .14, p > .2) was significantly correlated with mean 24-hour UFC.
Discussion
Serum SAA was increased significantly and, on average, by over 86% in the group of remitted, unmedicated MDD women versus BMI-matched control subjects. Serum CRPhs was increased significantly and by an average of almost three-fold in these patients. The proportion of women at high risk for future CAD, on the basis of established cut-off values for SAA and CRP, was 4 times higher in MDD women than in controls. These differences were based on two separate determinations showing high test-retest reliability, indicating their stability over time. Measurements were done with an automated system that minimizes human error, reducing intra- and interassay variability, and were done in a blinded fashion.
These findings are unlikely to represent artifacts of BMI or phase of the menstrual cycle, and cannot be attributed to a transient effect of a current depressive episode. Given that the patients had been off psychotropic medications for at least three months prior to study, it is also difficult to attribute these findings to an effect of antidepressant medications, although a long-term effect of such medications cannot be ruled out. The MDD patients were slightly, but not significantly older on average than the control subjects; moreover, there was no significant correlation between age and either SAA or CRPhs, analyzing either in the combined sample or by study group. Thus, these elevated levels of SAA and CRPhs are also unlikely to be secondary to an age effect. As expected given their clinically remitted status, hypercortisolism was not present in the patients compared with controls, and there was no evidence of a significant association between 24 hour urinary free cortisol and serum levels of either SAA or CRPhs.
Elevations of CRP and, in one study, SAA, have been previously reported in actively depressed individuals (Lanquillon et al 2000; Miller et al 2002; Panagiotakos et al 2004; Penninx et al 2003). In some of these studies (Lanquillon et al 2000; Miller et al 2002), diagnosis was made using structured interviews; others (Panagiotakos et al 2004; Penninx et al 2003) utilized self-report measures of depressive symptoms. These studies did not utilize individually BMI-matched controls, as was done in the present study. The degree of elevation of CRP versus control levels varied in these previous studies from 40% (Lanquillon et al 2000; Miller et al 2002) to about 2.8-fold (Lanquillon et al 2000; Miller et al 2002); in the one prior study of SAA (Panagiotakos et al 2004; Penninx et al 2003), a population-based survey, women with severe depression (defined by a score of > 70 on the Zung Depression Rating Scale) showed 37% higher SAA levels than women who were classified as “normal” based on a Zung score of < 50. Thus, the degree of elevation in CRP and SAA in the present study of remitted, unmedicated women with MDD was comparable to or greater than those reported in previous studies of actively depressed individuals.
Our findings indicate that elevation of CRP is not confined to the depressive state, and indicate that SAA is also increased during sustained remission of MDD. These data suggest the presence of a persistent pro-inflammatory state in women who have clinically recovered from depressive episodes and have discontinued antidepressant medications for several months or more.
Our results do not allow conclusions as to whether elevated SAA and CRP in unmedicated remitted patients is a consequence of prior depressive episodes, or represents a shared vulnerability to CAD and MDD. Studies of individuals at high risk for development of MDD, but who have not yet manifested the depressive phenotype, would help to address this question.
The elevation of both SAA and CRP, the two most robustly-responding APPs, in female MDD patients may indicate that a common pro-inflammatory stimulus is influencing these two mediators in MDD. One candidate for such a factor is interleukin-6 (IL-6), a systemically secreted, pro-inflammatory cytokine. We (Alesci et al 2005) and others (Brambilla and Maggioni 1998; Maes et al 1995, 1997; Miller et al 2002; Motivala et al 2005; Penninx et al 2003) have reported elevated IL-6 during active depressive states or in early remission of MDD. IL-6 stimulates the production of both SAA (Uhlar and Whitehead 1999) and CRP (Pepys and Hirschfield 2003); thus, elevation of APPs may represent a sustained consequence of IL-6 activation associated with depressive episodes.
CRP and SAA are not simply surrogate biochemical markers of CAD risk, but play important roles during inflammatory responses Although both likely confer survival value by their roles in the response to acute infection or tissue injury, more sustained, low-grade elevations in these mediators are proposed to participate in the chronic inflammatory process leading to progression and/or rupture of atherosclerotic lesions (Kisilevsky and Tam 2002; Pepys and Hirschfield 2003) Both CRP and SAA are found in the walls of arterial plaques and inflamed endothelial linings. Moreover, SAA functions as an apolipoprotein for HDLs that modulates “reverse” cholesterol transport, with the net effect of reducing cholesterol retrieval by HDL from most tissue sites other than macrophages (Kisilevsky and Tam 2002). Thus, these mediators may directly participate in the pathogenic process of CAD.
The elevation of both SAA and CRP reported here is of interest given possible differential tissue origins of these mediators. Thus, CRP derives mainly from liver, while recent data suggest a greater derivation of SAA in humans from visceral fat than from liver (Poitou et al 2005; Sjoholm et al 2004; Yang et al 2004) Thus, secretion of inflammatory mediators by visceral fat may play a role in the CAD risk associated with MDD in addition to increased CRP production by the liver in response to pro-inflammatory signals. The relatively low concordance rate between elevated SAA and CRP in our study subjects may reflect their distinct origin and suggests the need for further studies of both of these mediators in MDD.
Our findings support the premise that MDD is associated with clinically significant systemic manifestations, even following remission of depressive symptoms. The presence of a persistent pro-inflammatory state may contribute to the increased CAD risk associated with MDD (Lett et al 2004; Wulsin 2004) We thus propose that further studies be conducted to: 1) confirm these findings in larger and more diverse samples of patients with MDD; 2) examine whether our observations extend to men as well as women with MDD; 3) explore their biological determinants and relationship to other known CAD risk factors; and 4) examine their clinical implications for CAD risk in both cross-sectional and longitudinal studies Should these findings be confirmed and found to have value in predicting increased CAD risk, MDD patients manifesting pro-inflammatory risk markers may be candidates for preventive therapies such as drugs with anti-inflammatory actions including aspirin statins or other agents (Jain and Ridker 2005; Plutzky 2003) to reduce their likelihood of developing clinically significant CAD.
Acknowledgments
We wish to gratefully acknowledge the clinical support of Marilla Geraci, RN, Tracy Waldeck, Ph.D., and the entire clinical staff of the NIMH Mood and Anxiety Disorders Program.
Footnotes
Author contributions – study design: AN, PWG, SA, WCD, HKM, DSC; subject recruitment, assessment, and clinical care: AN, OB; assay development and conduct: GC, RC; data analysis: MAK, DAL, RD; manuscript writing: MAK, SA, GC, DAL, PWG, AN.
Drs. Neumeister and Gold contributed equally to the study.
Funding for this study was provided by the Division of Intramural Research Programs, National Institute of Mental Health (NIMH) and the Department of Laboratory Medicine, Clinical Center, National Institutes of Health.
References
- Alesci S, Martinez PE, Kelkar S, Ilias I, Ronsaville DS, Listwak SJ, et al. Major Depression is associated with significant diurnal elevations in plasma IL-6 levels, a shift of its circadian rhythm, and loss of physiologic complexity in its secretion: Clinical implications. J Clin Endocrinol Metab. 2005;90:2522–2530. doi: 10.1210/jc.2004-1667. [DOI] [PubMed] [Google Scholar]
- Brambilla F, Maggioni M. Blood levels of cytokines in elderly patients with major depressive disorder. Acta Psychiatr Scand. 1998;97:309–313. doi: 10.1111/j.1600-0447.1998.tb10005.x. [DOI] [PubMed] [Google Scholar]
- Chait A, Han CY, Oram JF, Heinecke JW. Thematic review series: The immune system and atherogenesis. Lipoprotein-associated inflammatory proteins: markers or mediators of cardiovascular disease? J Lipid Res. 2005;46:389–403. doi: 10.1194/jlr.R400017-JLR200. [DOI] [PubMed] [Google Scholar]
- Choudhury RP, Leyva F. C-Reactive protein, serum amyloid A protein, and coronary events. Circulation. 1999;100:e65–66. doi: 10.1161/01.cir.100.15.e65. [DOI] [PubMed] [Google Scholar]
- Delanghe JR, Langlois MR, De Bacquer D, Mak R, Capel P, Van Renterghem L, De Backer G. Discriminative value of serum amyloid A and other acute-phase proteins for coronary heart disease. Atherosclerosis. 2002;160:471–476. doi: 10.1016/s0021-9150(01)00607-4. [DOI] [PubMed] [Google Scholar]
- Gabay C, Kushner I. Acute-phase proteins and other systemic responses to inflammation. N Engl J Med. 1999;340:448–454. doi: 10.1056/NEJM199902113400607. [DOI] [PubMed] [Google Scholar]
- Jain MK, Ridker PM. Anti-inflammatory effects of statins: clinical evidence and basic mechanisms. Nat Rev Drug Discov. 2005;4:977–987. doi: 10.1038/nrd1901. [DOI] [PubMed] [Google Scholar]
- Johnson BD, Kip KE, Marroquin OC, Ridker PM, Kelsey SF, Shaw LJ, et al. Serum amyloid A as a predictor of coronary artery disease and cardiovascular outcome in women: the National Heart, Lung, and Blood Institute-Sponsored Women’s Ischemia Syndrome Evaluation (WISE) Circulation. 2004;109:726–732. doi: 10.1161/01.CIR.0000115516.54550.B1. [DOI] [PubMed] [Google Scholar]
- Kessler RC, McGonagle KA, Zhao S, Nelson CB, Hughes M, Eshleman S, et al. Lifetime and 12-month prevalence of DSM-III-R psychiatric disorders in the United States. Results from the National Comorbidity Survey. Arch Gen Psychiatry. 1994;51:8–19. doi: 10.1001/archpsyc.1994.03950010008002. [DOI] [PubMed] [Google Scholar]
- Kisilevsky R, Tam SP. Acute phase serum amyloid A, cholesterol metabolism, and cardiovascular disease. Pediatr Pathol Mol Med. 2002;21:291–305. doi: 10.1080/02770930290056523. [DOI] [PubMed] [Google Scholar]
- Lanquillon S, Krieg J-C, Bening-Abu-Shach U, Vedder H. Cytokine production and treatment response in major depressive disorder. Neuropsychopharmacology. 2000;22:370–379. doi: 10.1016/S0893-133X(99)00134-7. [DOI] [PubMed] [Google Scholar]
- Lett HS, Blumenthal JA, Babyak MA, Sherwood A, Strauman T, Robins C, Newman MF. Depression as a risk factor for coronary artery disease: evidence, mechanisms, and treatment. Psychosom Med. 2004;66:305–315. doi: 10.1097/01.psy.0000126207.43307.c0. [DOI] [PubMed] [Google Scholar]
- Maes M, Bosmans E, De Jongh R, Kenis G, Vandoolaeghe E, Neels H. Increased serum IL-6 and IL-1 receptor antagonist concentrations in major depression and treatment resistant depression. Cytokine. 1997;9:853–858. doi: 10.1006/cyto.1997.0238. [DOI] [PubMed] [Google Scholar]
- Maes M, Meltzer HY, Bosmans E, Bergmans R, Vandoolaeghe E, Ranjan R, Desnyder R. Increased plasma concentrations of interleukin-6, soluble interleukin-6, soluble interleukin-2 and transferrin receptor in major depression. J Affect Disord. 1995;34:301–309. doi: 10.1016/0165-0327(95)00028-l. [DOI] [PubMed] [Google Scholar]
- Miller GE, Stetler CA, Carney RM, Freedland KE, Banks WA. Clinical depression and inflammatory risk markers for coronary heart disease. Am J Cardiol. 2002;90:1279–1283. doi: 10.1016/s0002-9149(02)02863-1. [DOI] [PubMed] [Google Scholar]
- Morrow DA, Ridker PM. C-reactive protein, inflammation, and coronary risk. Med Clin North Am. 2000;84:149–161. doi: 10.1016/s0025-7125(05)70211-x. [DOI] [PubMed] [Google Scholar]
- Morrow DA, Rifai N, Antman EM, Weiner DL, McCabe CH, Cannon CP, Braunwald E. Serum amyloid A predicts early mortality in acute coronary syndromes: A TIMI 11A substudy. J Am Coll Cardiol. 2000;35:358–362. doi: 10.1016/s0735-1097(99)00574-4. [DOI] [PubMed] [Google Scholar]
- Mosca L, Manson JE, Sutherland SE, Langer RD, Manolio T, Barrett-Connor E. Cardiovascular Disease in Women : A Statement for Healthcare Professionals From the American Heart Association. Circulation. 1997;96:2468–2482. doi: 10.1161/01.cir.96.7.2468. [DOI] [PubMed] [Google Scholar]
- Motivala SJ, Sarfatti A, Olmos L, Irwin MR. Inflammatory markers and sleep disturbance in major depression. Psychosom Med. 2005;67:187–194. doi: 10.1097/01.psy.0000149259.72488.09. [DOI] [PubMed] [Google Scholar]
- Murtagh BM, Anderson HV. Inflammation and atherosclerosis in acute coronary syndromes. J Invasive Cardiol. 2004;16:377–384. [PubMed] [Google Scholar]
- Neumeister A, Nugent AC, Waldeck T, Geraci M, Schwarz M, Bonne O, et al. Neural and behavioral responses to tryptophan depletion in unmedicated patients with remitted Major Depressive Disorder and controls. Arch Gen Psychiatry. 2004;61:765–773. doi: 10.1001/archpsyc.61.8.765. [DOI] [PubMed] [Google Scholar]
- Panagiotakos DB, Pitsavos C, Chrysohoou C, Tsetsekou E, Papageorgiou C, Christodoulou G, Stefanadis C. Inflammation, coagulation, and depressive symptomatology in cardiovascular disease-free people; the ATTICA study. Eur Heart J. 2004;25:492–499. doi: 10.1016/j.ehj.2004.01.018. [DOI] [PubMed] [Google Scholar]
- Penninx BWJH, Kritchevsky SB, Yaffe K, Newman AB, Simonsick EM, Rubin S, et al. Inflammatory markers and depressed mood in older persons: results from the health, aging, and body composition study. Biol Psychiatry. 2003;54:566–572. doi: 10.1016/s0006-3223(02)01811-5. [DOI] [PubMed] [Google Scholar]
- Pepys MB, Hirschfield GM. C-reactive protein: a critical update. J Clin Invest. 2003;111:1805–1812. doi: 10.1172/JCI18921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plutzky J. Peroxisome proliferator-activated receptors as therapeutic targets in inflammation. J Am Coll Cardiol. 2003;42:1764–1766. doi: 10.1016/j.jacc.2003.08.017. [DOI] [PubMed] [Google Scholar]
- Poitou C, Viguerie N, Cancello R, De Matteis R, Cinti S, Stich V, et al. Serum amyloid A: production by human white adipocyte and regulation by obesity and nutrition. Diabetologia. 2005;48:519–528. doi: 10.1007/s00125-004-1654-6. [DOI] [PubMed] [Google Scholar]
- Rattazzi M, Puato M, Faggin E, Bertipaglia B, Zambon A, Pauletto P. C-reactive protein and interleukin-6 in vascular disease: culprits or passive bystanders? J Hypertens. 2003;21:1787–1803. doi: 10.1097/00004872-200310000-00002. [DOI] [PubMed] [Google Scholar]
- Sjoholm K, Palming J, Olofsson LE, Gummesson A, Svensson PA, Lystig TC, et al. A microarray search for genes predominantly expressed in human omental adipocytes: Adipose tissue as a major production site of Serum Amyloid A. J Clin Endocrinol Metab. 2004;90:2233–2239. doi: 10.1210/jc.2004-1830. [DOI] [PubMed] [Google Scholar]
- Thorand B, Baumert J, Doring A, Herder C, Kolb H, Rathmann W, et al. Sex differences in the relation of body composition to markers of inflammation. Atherosclerosis. 2006;184:216–224. doi: 10.1016/j.atherosclerosis.2005.04.011. [DOI] [PubMed] [Google Scholar]
- Tracy RP. Is visceral adiposity the “enemy within”? Arterioscler Thromb Vasc Biol. 2001;21:881–883. doi: 10.1161/01.atv.21.6.881. [DOI] [PubMed] [Google Scholar]
- Uhlar CM, Whitehead AS. Serum amyloid A, the major vertebrate acute-phase reactant. Eur J Biochem. 1999;265:501–523. doi: 10.1046/j.1432-1327.1999.00657.x. [DOI] [PubMed] [Google Scholar]
- Willerson JT, Ridker PM. Inflammation as a cardiovascular risk factor. Circulation. 2004;109(suppl II):II-2–II-10. doi: 10.1161/01.CIR.0000129535.04194.38. [DOI] [PubMed] [Google Scholar]
- Wulsin LR. Is depression a major risk factor for coronary disease? A systematic review of the epidemiologic evidence. Harv Rev Psychiatry. 2004;12:79–93. doi: 10.1080/10673220490447191. [DOI] [PubMed] [Google Scholar]
- Yang R-Z, Lee M-J, Hu H, Duan L, Pollin T, Nicklas B, et al. Acute-phase protein serum amyloid A (SAA) is a pro-inflammatory adipocytokine in humans. Annual Meeting, American Diabetes Association. 2004:A12. [Google Scholar]