

Abstract
Nuclear/steroid hormone receptors (NHRs) function as ligand-dependent transcriptional regulators of diverse sets of genes involved in development and homeostasis. Mutations to the androgen receptor (AR), a member of NHRs, have been linked to the failure of hormonal therapy in the treatment of prostate cancer (PCa). It has been proposed that AR mutations such as Thr877 → Ala, Trp741 → Leu and Trp741 → Cys that cause anti-androgens such as flutamide and bicalutamide to function as agonists are likely associated with anti-androgen withdrawal syndrome in PCa therapy. The recently solved crystal structure of bicalutamide was used to design analogs that would complement the Trp741 → Leu mutation effectively restoring antagonist action of the ligand. Three out of the six designed analogs showed potent antagonistic activity in all three mutations as well as wild-type, suggesting that these analogs may be considered “pan-antagonists” of AR.
Prostate cancer (PCa) remains the second leading cause of cancer death in men. As prostate tissue is dependent on androgens for growth, anti-androgens used alone or in conjunction with inhibitors of testosterone biosynthesis have been used in the treatment of PCa, however, often cancer cells escape such androgen blockade therapies. Androgen receptor (AR) mutations have been identified as one mechanism leading to anti-androgen resistance and often lead to a clinical phenomenon known as anti-androgen withdrawal syndrome wherein anti-androgen resistant patients show symptomatic improvement after cessation of anti-androgen treatment.1 It has been proposed that anti-androgen withdrawal syndrome is likely associated with AR mutations such as Thr877 → Ala, Trp741 → Leu and Trp741 → Cys which cause the antagonists flutamide and bicalutamide (Bic) to act as agonists.2,3 Anti-androgens are presumed to apply a selective pressure on cancer cell growth such that 31% of metastasies arising with flutamide treatment have been observed to possess the identical Thr877 → Ala mutation.4 As part of our program to rescue nuclear receptor mutations through ligand design, we describe the development of AR “pan-antagonists” that function with wild-type (AR(wt)) and mutant ARs associated with anti-androgen resistance.
The family of nuclear/steroid hormone receptors (NHRs) are ligand-dependent transcriptional regulators for diverse sets of genes involved in development and homeostasis. In the prototypical model for NHRs, ligand binding induces a conformational change in the ligand-binding domain that reveals a co-activator dimerization surface on the receptor composed of helicies 3, 5 and 12. As the ligand-binding site is adjacent to helix 12 (H12), NHR antagonists have commonly been designed by appending molecular extensions to the core structure of NHR agonists that interfere with the placement of H12, thereby disrupting co-activator recruitment.5
Recently, the structure of Bic with the Bic-resistant mutant AR(W741L), was solved in the receptor’s agonist conformation.6 It has also been shown that sequences that compete for AR’s co-activator binding site have been identified in both the N-terminal and C-terminal domains of the receptor and are believed to play a role in the transactivation function of AR. Therefore the structure of AR in its antagonist-bound form remains largely unknown.7
In the Bic/AR(W741L) co-crystal structure, the 4-fluorophenyl sulfone group of Bic is situated between residues of H12 and the side chain of Leu741 suggesting that in wild-type receptor, the larger Trp741 side chain would require Bic to push against H12.6 Based on the assumption that Bic functions similar to other NHR antagonists by blocking H12 from its agonist conformation, we modeled the antagonist conformation of Bic in AR(wt) by deleting H12 from an AR(W741L) site-model and changing the Leu741 residue back to Trp. Given the general lack of mechanistic and structural details of the antagonist form of the receptor, this model seemed a reasonable albeit crude model of the antagonist-bound form of AR. Molecular dynamics simulations of Bic into this site-model suggested that in the absence of the Tryp741 → Leu mutant, Bic prefers to bind in a manner that places the 4-fluorophenyl ring in the space otherwise occupied by H12 in its active conformation (Figure 1a).
Figure 1.
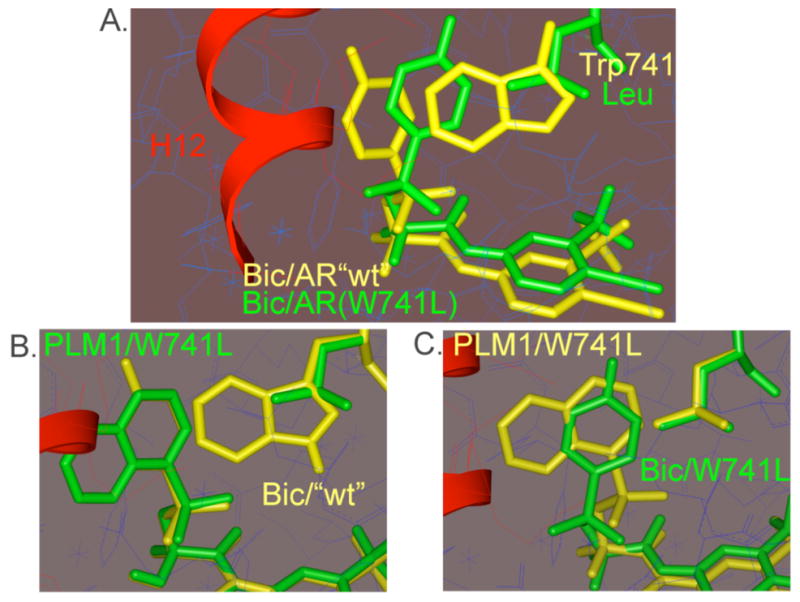
Comparison of modeled structures, a. Bic/AR(W741L) from x-ray (green) with Bic/AR(wt)-H12 (yellow); b. PLM1/AR(W741L)-H12 (green) with Bic/AR(wt)-H12 (yellow); c. PLM1/AR(W741L)-H12, conformation 2 (yellow) with Bic/AR(W741L) (green). Agonist position of H12 from x-ray shown in red.
Based on this proposed model for Bic antagonism, we hypothesized that derivatives with an expanded aryl sulfone core, such as PLM1, would similarly interfere with H12’s ability to adopt an agonist conformation but would be unable to be switched to an agonist by a simple missense mutation of Trp741 (Figure 1b, 1c). Simulations suggest that the lowest energy conformation of PLM1 in the AR(W741L)-H12 resembled the Bic/AR(wt)-H12 antagonist conformation (Figure 1b), whereas energetically accessible conformations of PLM1, which place the proximal aryl ring in the pocket created by the Trp741 → Leu mutation, still extend into the space occupied by H12 (Figure 1c). We synthesized PLM1 as well as a series of expanded aryl sulfone analogs that were conceived based upon the same design principle (Figure 2). Control compounds C1-C3 were also made. These analogs can be accommodated within the full AR(W741L) site model without any apparent clash with H12.
Figure 2.
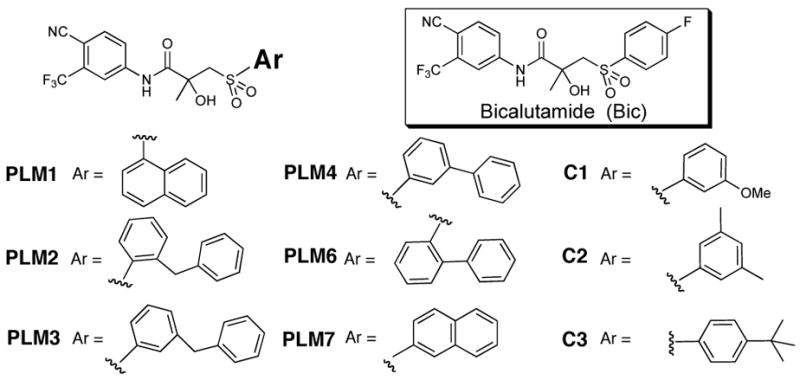
Structure of bicalutamide and analogs.
The analogs were evaluated alone and in competition with dihydrotestosterone (DHT) by cellular reporter gene assays using CV-1 cells transiently transfected with either wild-type or mutant ARs (pSG5AR) and an AR responsive luciferase reporter gene (ARE-luc). With AR(wt), all of the analogs have no agonist activity and in competition with 3 nM DHT, all of the ligands but PLM7 and C3 antagonize AR(wt) by greater than 50% at 25 uM (Figure 3a). In contrast, with the Bic-resistant mutant AR(W741L), Bic, C1 and C2 are strong agonists and C3 has intermediate activity, however, PLM1, PLM2, PLM6 and PLM7 are not agonists having activities at 10 μM comparable to Bic in AR(wt). PLM3 and PLM4 have weak to partial agonist activity (27% and 40%) (Figure 3b). Whereas Bic and the control compounds become agonists with AR(W741L), half of the designed analogs (3 of 6) retain potent antagonist activity with this mutant associated with anti-androgen resistance. PLM3 no longer acts as an antagonist whereas PLM4 appears to be a weak antagonist/partial agonist.
Figure 3.
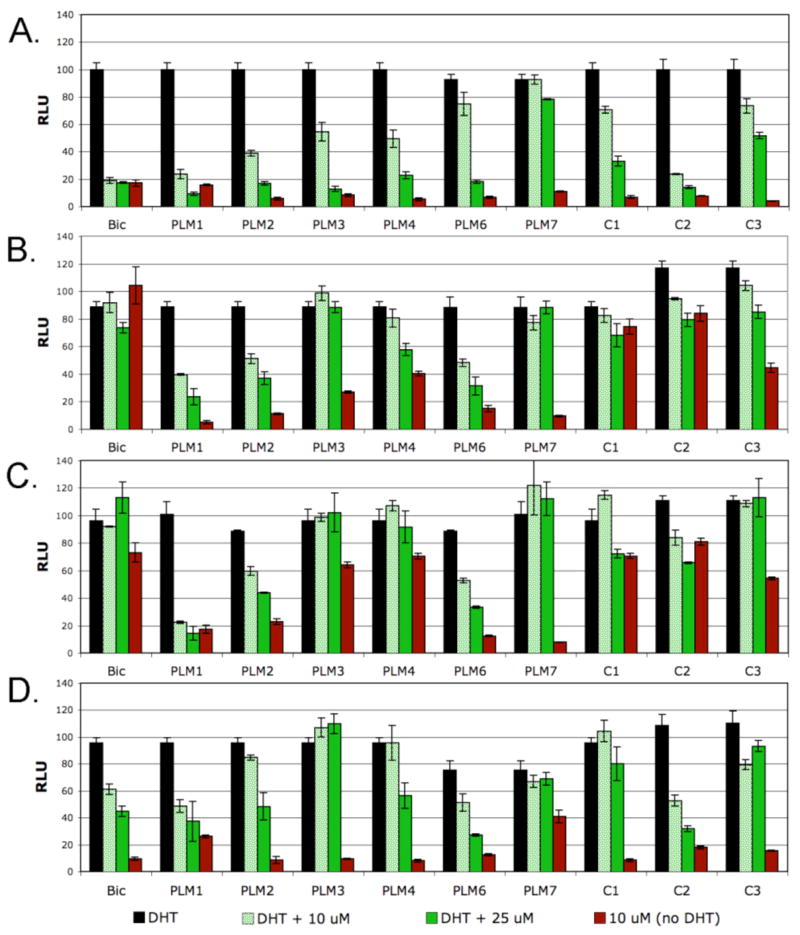
Cellular reporter gene activities of analogs alone and in competition with DHT; a. AR(wt) ±3 nM DHT; b. AR(W741L) ±250 nM DHT; c. AR(W741C) ±200 nM DHT and d. AR(T877A) ±10 nM DHT. RLU = relative light units ± SEM.
With AR(W741C), controls C1-C3 are all agonists with activities similar to those observed with AR(W741L). PLM1, PLM2 and PLM6 remain non-agonists and potent antagonists, however, the meta-substituted aryl sulfones, PLM3 and PLM4, now become potent agonists suggesting that these modifications are complemented by the larger binding pocket created by the Cys mutant compared to the Leu mutant (Figure 3c). Additionally, PLM1, PLM2 and PLM6 remain non-agonists/antagonists with the flutamide resistant mutant AR(T877A) (Figure 3d). Collectively these results suggest that these analogs have the unique property of acting as “pan-antagonists” being potent antagonists of AR(wt), AR(W741L), AR(W741C) and AR(T877A) (Table 1).
Table 1.
Cellular activities and competitive binding constants (μM).
| AR(wt) | AR(W741L) | AR(W741L) | AR(T877A) | ||||
|---|---|---|---|---|---|---|---|
| ligand | IC50 | Ki | IC50 | Ki | IC50 | Ki | IC50 |
| PLM1 | 3.8 | 0.5 | 9.7 | 2.1 | 3.3 | 5.9 | 6.0 |
| PLM2 | 12.5 | 6.3 | 23.0 | 2.9 | 11.6 | 2.8 | 16.5 |
| PLM6 | 21.4 | 2.1 | 7.9 | n.d. | 7.4 | n.d. | 5.4 |
To further demonstrate that the cellular reporter gene assays are reflective of inhibitory activities in prostate cell lines, cell growth inhibition studies were conducted in LNCaP cells which contain the AR(T877A) mutant. Consistent with reporter gene assays, PLM1 is able to repress androgen (R1881) induced cell growth to levels below media control similar to Bic (Figure 4).
Figure 4.
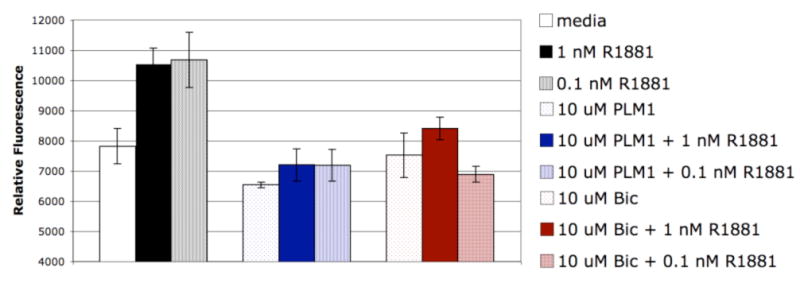
LNCaP cell proliferation (CyQuant) after 8 day ligand treatment ± R1881. Relative Fluorescence ± SEM. (R1881 ± ligand; P < 0.05, one-way ANOVA)
These studies demonstrate that structure-based design can identify design strategies that can be used to efficiently engineer second-generation anti-androgens that have the potential to circumvent anti-androgen resistance by complementing receptor mutations at the molecular level. Because in principle there is a limited subset of mutations that can cause an antagonist to function as an agonist, it is conceivable that such approaches may ultimately lead to the development of anti-androgens that resist mutations leading to anti-androgen withdrawal syndrome.
Supplementary Material
Complete experimental procedures, characterization and assay data. This material is available free of charge via the Internet at http://pubs.acs.org
Acknowledgments
We thank Professor Robert Sikes for helpful discussions and advice. This work was supported by the National Institutes of Health R01 DK054257.
References
- 1.a Navarro D, Luzardo OP, Fernandez L, Chesa N, Diaz-Chico BN. J Steroid Biochem Mol Biol. 2002;81:1091–201. doi: 10.1016/s0960-0760(02)00064-x. [DOI] [PubMed] [Google Scholar]; b Ko YJ, Balk SP. Curr Pharma Biotechnol. 2004;5:459–470. doi: 10.2174/1389201043376616. [DOI] [PubMed] [Google Scholar]
- 2.Linja MJ, Visakoipi T. J Steroid Biochem Mol Biol. 2004;92:255–264. doi: 10.1016/j.jsbmb.2004.10.012. [DOI] [PubMed] [Google Scholar]
- 3.Buchanan G, Greenberg NM, Scher HI, Harris JM, Marshall VR, Tilley WD. Clinical Cancer Res. 2001;7:1273–1281. [PubMed] [Google Scholar]
- 4.Taplin ME, Bubley GJ, Ko YJ, Small EJ, Upton M, Rajeshkumar B, Balk SP. Cancer Res. 1999;59:2511–2515. [PubMed] [Google Scholar]
- 5.a Weatherman RV, Fletterick RJ, Scanlan TS. Annu Rev Biochem. 1999;68:559–581. doi: 10.1146/annurev.biochem.68.1.559. [DOI] [PubMed] [Google Scholar]; b Hashimoto Y, Miyachi H. Bioorg Med Chem. 2005;13:5080–5093. doi: 10.1016/j.bmc.2005.03.027. [DOI] [PubMed] [Google Scholar]
- 6.Bohl CE, Gao WQ, Miller DD, Bell CE, Dalton JT. Proc Natl Acad Sci USA. 2005;102:6201–6206. doi: 10.1073/pnas.0500381102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gao W, Bohl CE, Dalton JT. Chem Rev. 2005;105:3352–3370. doi: 10.1021/cr020456u. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Complete experimental procedures, characterization and assay data. This material is available free of charge via the Internet at http://pubs.acs.org