

Abstract
In the airways, adenine nucleotides support a complex signaling network mediating host defenses. Released by the epithelium into the airway surface liquid (ASL) layer, they regulate mucus clearance through P2 (ATP) receptors, and following surface metabolism through P1 (adenosine; Ado) receptors. The complexity of ASL nucleotide regulation provides an ideal subject for biochemical network modeling. A mathematical model was developed to integrate nucleotide release, the ectoenzymes supporting the dephosphorylation of ATP into Ado, Ado deamination into inosine (Ino), and nucleoside uptake. The model also includes ecto-adenylate kinase activity and feed-forward inhibition of Ado production by ATP and ADP. The parameters were optimized by fitting the model to experimental data for the steady-state and transient concentration profiles generated by adding ATP to polarized primary cultures of human bronchial epithelial (HBE) cells. The model captures major aspects of ATP and Ado regulation, including their >4-fold increase in concentration induced by mechanical stress mimicking normal breathing. The model also confirmed the independence of steady-state nucleotide concentrations on the ASL volume, an important regulator of airway clearance. An interactive approach between simulations and assays revealed that feed-forward inhibition is mediated by selective inhibition of ecto-5′-nucleotidase. Importantly, the model identifies ecto-adenylate kinase as a key regulator of ASL ATP and proposes novel strategies for the treatment of airway diseases characterized by impaired nucleotide-mediated clearance. These new insights into the biochemical processes supporting ASL nucleotide regulation illustrate the potential of this mathematical model for fundamental and clinical research.
Mucociliary clearance (MCC)4 constitutes the first line of defense against airway infection (1). Inhaled pathogens are trapped by a mucus layer, positioned above ciliated epithelia by a periciliary (PCL) layer. Together, mucus and PCL form the ASL layer, which is continuously transported cephalad by coordinated ciliary beating. Through their interactions with P2 receptors, adenine nucleotides regulate all major epithelial functions supporting MCC, including Cl-/liquid secretion via the Ca2+-activated Cl- channel and the cystic fibrosis transmembrane regulator (CFTR) (2), mucin secretion (3, 4), and ciliary beat activity (5, 6). Nucleotides are released by the epithelium into the ASL layer under basal conditions (7, 8), and their release rate increases in response to mechanical stress imparted by tidal breathing (8-12). Nucleotide-mediated epithelial responses are modified by surface enzymes (ectonucleotidases) that convert a fraction of the ATP into Ado (13-15). This nucleoside activates airway epithelial signaling pathways through P1 receptors, typically the A2B receptor, which stimulates ciliary beat activity (16) and Cl-/liquid secretion by CFTR (17). In healthy airways, the purinergic regulation of MCC is mediated by both P1 and P2 receptor-mediated responses (10, 18). In chronic lung diseases, the accelerated metabolism of ATP (14) can lead to severe complications related to airway obstruction and inflammation (2, 19). The cystic fibrosis (CF) patients are particularly vulnerable to infection because of the sole dependence of MCC on P2 receptor-mediated signaling, reflecting the absence of the final effector for P1 signaling, i.e. functional CFTR. This information led to the current interest in aerosolized nucleotide analogs for the improvement of MCC (19).
Despite the importance of purinergic signaling for airway defenses, it has not been possible, until recently, to develop a mathematical model of ASL nucleotide regulation because of the absence of quantitative information regarding ASL nucleotide and Ado levels, the rates of nucleotide release and Ado uptake, and the kinetic properties of the ectonucleotidases. In the past decade, extensive work has been conducted to characterize ATP release from the apical surface of HBE cells (7, 8, 10-12, 20, 21). Members of the three ecto-ATPase families (alkaline phosphatases, ecto-nucleoside triphosphate diphosphohydrolases (E-NTPDases), and nucleotide pyrophosphatase/phosphodiesterases (E-NPPs)) (22, 23) have been identified on HBE cells (13-15, 24-26), along with ecto-AK (2ADP ↔ ATP + AMP) (27, 28). Also, Ado levels are regulated on HBEs by a balance between the production rate (AMP → Ado) by ecto-5′-NT and NSAP (13) and the elimination rate by Ado deaminase 1 (ADA1; Ado → Ino) and cellular uptake by concentrative nucleoside transporter 3 (CNT3) (29).
Based on these findings, we developed a mathematical model of ASL nucleotide regulation. The kinetic coefficients were optimized by fitting the output of the model to experimental values of ASL nucleotide and Ado concentrations, nucleotide release rates (12, 20), ATP metabolism (13, 26, 28), and Ado uptake (29). The capacity of the model to reproduce the impact of physical parameters (ASL volume and mechanical stress) and functional interactions between network components was verified by biochemical assays on primary cultures of HBE cells. This study provides the first mathematical approach to the investigation of an important defense mechanism in human airways.
EXPERIMENTAL PROCEDURES
Cell Culture—Model validation was conducted on HBE cultures maintained at air-liquid interface, as described previously (30). The epithelial cells were obtained from healthy donors at the time of transplantation by the Tissue Culture Core of the Cystic Fibrosis Center (University of North Carolina) under the auspices of protocols approved by the Institutional Board of Review. They were isolated by protease digestion, seeded on 12-mm permeable supports (0.45 μm pores, T-Clear; Costar) coated with human type IV collagen, and kept in Ham's F-12-based medium supplemented with 5 μg·ml-1 transferrin, 30 nm triiodothyronine, 1 μm hydrocortisone, and 3.75 μg·ml-1 endothelial cell growth substance. The confluent cultures were maintained at air-liquid interface with medium on the basolateral side. After 4 weeks, they were composed of ciliated and secretory cells, with transepithelial electrical resistance ≥300 ohms/cm2.
Cyclic Compressive Stress (CCS)—The HBE cultures were subjected to 1 h of CCS mimicking normal tidal breathing (20 cm of H2O; 20 cycles/min) using a system enclosed in an incubator (37 °C; 5% CO2) (10). Two pieces of tubing, each containing a high speed solenoid valve, were inserted into a conical silicon plug fitted to the top of the T-Clear, one connected to a pressurized chamber and the other connected to the incubator atmosphere. A microprocessor regulated the amplitude and frequency of the chamber pressure applied to the culture by controlling the open/closed state of the solenoid valves (10).
Biochemical Measurements of Steady-state Nucleotide Concentrations—The steady-state ASL ATP concentration was measured on HBE cultures by luminometry using soluble luciferase or a chimeric Staphylococcus aureus protein A-luciferase attached to the apical surface via keratan sulfate-specific antibodies (20). Steady-state concentrations of ATP, ADP, AMP, and Ado were determined on HBE cultures by ethenyl derivatization and fluorescence high pressure liquid chromatography (HPLC) (12).
Transient Nucleotide Concentration Profiles and Ectonucleotidase Activities—HBE cultures were preincubated 30 min (37 °C; 5% CO2) in Krebs buffer (pH 7.5) (0.35 ml; bilateral). Then 0.1-1000 μm ATP was added to the apical surface to determine the transient concentration profile and initial rate of ATP hydrolysis by HPLC analysis of buffer samples collected over 60 min (14). The contribution of each ectoenzyme was calculated from paired assays conducted on the same culture wells before and after pretreatment with selective noncompetitive inhibitor(s). The kinetic parameters of NSAP for ATP and ADP were determined as described previously for AMP (13), but in the presence of 0.5 mm Ap5A to inhibit ecto-AK (ATP + AMP ↔ 2ADP) (28). NSAP activity was calculated from the difference in linear rate of substrate hydrolysis measured before and after exposure to 10 mm levamisole, at each substrate concentration (0.1-1000 μm). Michaelis constants (KM) and maximal velocities (Vmax) were calculated as shown previously (13).
Kinetic Model of Airway ATP/Ado Regulation—This section
outlines the theory and methodology used to build the mathematical model of
airway nucleotide regulation on the apical surface of human airway epithelia.
A biochemical network was derived using quantitative data gathered from the
literature on nucleotide release
(12,
20) and metabolism
(13-15,
24,
26) by HBE cultures
(Fig. 1). Because UTP levels
are about 15% that of ATP (25)
in the ASL layer, the influence of uridine nucleotides on ATP regulation was
considered negligible. Therefore, the reactions governing ATP
dephosphorylation to Ado and Ado deamination are as shown in Reactions
1-5,
FIGURE 1.

Diagram of adenine nucleotides transport and metabolism on human bronchial epithelia. Released by the epithelium, ATP is sequentially dephosphorylated into Ado by E-NPPs, ecto-AK, NTPDase 1, NTPDase 3, NSAP, and ecto-5′-NT. Ado is converted into Ino by ADA1, both returning to the cytosol via CNT3. The model also includes the feed-forward inhibition of Ado production by ATP and ADP.
Each of the above reactions was assumed to follow Michaelis-Menten kinetics
of the form shown in Equation
1,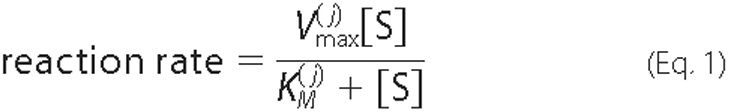
where [S] is the substrate concentration, and
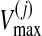 and
and
 are the maximum velocity and Michaelis
constant. The superscript j is used to index the multiple enzymes
involved in the pathway. In contrast, the reversible ecto-AK Reaction
6,
are the maximum velocity and Michaelis
constant. The superscript j is used to index the multiple enzymes
involved in the pathway. In contrast, the reversible ecto-AK Reaction
6,
which involves two binding sites, has been shown to follow a Bi Bi
mechanism (32) as shown in
Equation 2,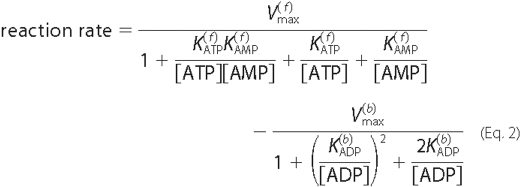
This expression is a generalization of Michaelis-Menten kinetics that
relies on the same underlying assumptions. The model also incorporates the
feed-forward inhibition of AMP hydrolysis by ATP and ADP. Experimental
evidence indicates that ATP and ADP competitively bind to enzymes that
catalyze AMP dephosphorylation
(13,
33). Equation 3 was used to
model this effect,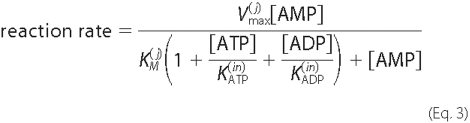
Although it is possible that the inhibition constants
 and
and
 may differ for
ecto-5′-NT and NSAP, these values were assumed identical for simplicity.
The remaining elements that were incorporated into the model are the rates of
ATP release and Ado/Ino uptake. In accordance with experimental observations
(12), the basal rate of ATP
release (JATP) was considered constant. Because Ado and
Ino are taken back into the cell through CNT3, and therefore compete for
access to the transporter, this effect is modeled using the following rates
for Ado and Ino uptake as shown in Equations 4 and
5,
may differ for
ecto-5′-NT and NSAP, these values were assumed identical for simplicity.
The remaining elements that were incorporated into the model are the rates of
ATP release and Ado/Ino uptake. In accordance with experimental observations
(12), the basal rate of ATP
release (JATP) was considered constant. Because Ado and
Ino are taken back into the cell through CNT3, and therefore compete for
access to the transporter, this effect is modeled using the following rates
for Ado and Ino uptake as shown in Equations 4 and
5,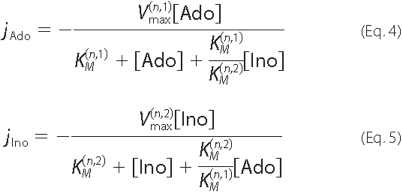
which allows for the possibility of different Vmax and KM values for the two molecules.
One of the functions of nucleotide release is to stimulate liquid
secretion, which in turn may affect ASL nucleotide concentrations. Under
static conditions, the basal rate of release does not maintain ATP at
sufficiently high levels to activate P2 receptors
(12). However, Ado levels
generated from ATP metabolism under static conditions stimulate liquid
secretion into the ASL layer via the A2B receptor-CFTR pathway
(17). Therefore, the model
equations were derived for the general condition of a time-dependent ASL
volume. To illustrate how the ASL volume enters the model, consider a simple
case in which ATP is released into the ASL at a constant rate of
JATP molecules/s, and then hydrolyzed by enzyme
E. The surface area is denoted by A, and
[E]T represents the surface density of E,
expressed in mol·cm-2.If V indicates ASL volume,
the equation governing ATP concentration is shown in Equation
6,
where
(kcatA)/(V)[E]T
corresponds to maximum velocity (Vmax) and the last term
models changes in ATP concentration because of the changing ASL volume. The
considerations discussed above led to the formulation of five equations to
describe the regulation of adenine nucleotides in the ASL layer (Equations
7-11). All model simulations were performed in MatLab (Mathworks, Natick, MA)
using the ordinary differential equation solver of 15
s.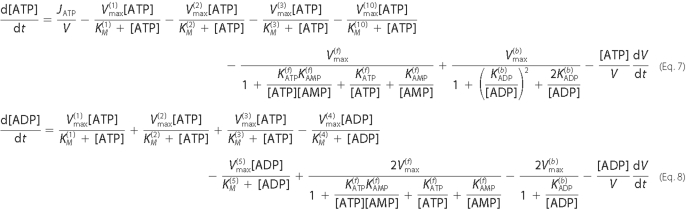
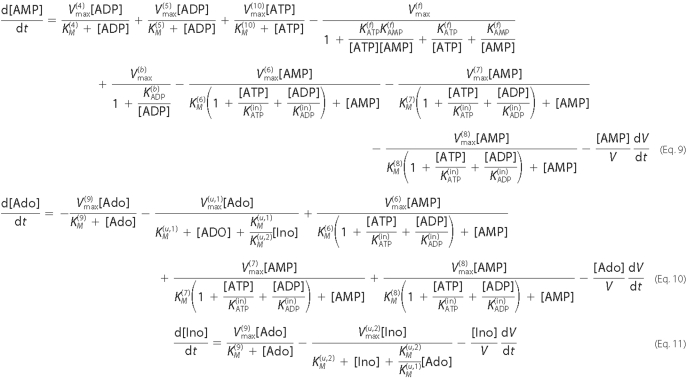
Linear Model for the Steady-state Nucleotide
Concentrations—The biochemical data showed that, under
physiological conditions, steady-state nucleotide concentrations are orders of
magnitude lower than the KM values of the enzymatic
reactions (12). Hence, to a
good approximation, the Michaelis-Menten reaction rates can be simplified
(linearized) as shown in Equation
12,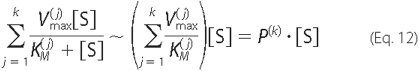
where the parameter P(K) denotes the sum of
the ratios of  to
to
 for all enzymes catalyzing the same
reaction. The expressions for Ado and Ino uptake can be simplified in a
similar way. The reversible ecto-AK reaction (Reaction 6) is a second order
reaction requiring two substrates and therefore does not contribute
significantly to the regulation of ASL nucleotide concentrations under
steady-state conditions. Also, steady-state ATP and ADP levels are 1000-fold
lower than their Ki values for the feed-forward inhibition
reaction (13) and therefore
insufficient to inhibit AMP. Therefore, these equations were excluded from the
linear model. Using these simplifications, Equations 7-11 can be approximated
under steady-state conditions by the following set of linear Equations
13-17,
for all enzymes catalyzing the same
reaction. The expressions for Ado and Ino uptake can be simplified in a
similar way. The reversible ecto-AK reaction (Reaction 6) is a second order
reaction requiring two substrates and therefore does not contribute
significantly to the regulation of ASL nucleotide concentrations under
steady-state conditions. Also, steady-state ATP and ADP levels are 1000-fold
lower than their Ki values for the feed-forward inhibition
reaction (13) and therefore
insufficient to inhibit AMP. Therefore, these equations were excluded from the
linear model. Using these simplifications, Equations 7-11 can be approximated
under steady-state conditions by the following set of linear Equations
13-17,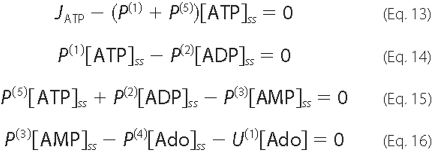
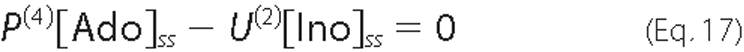
where U(1) and U(2) result from linearizing the expressions for Ado (Equation 4) and Ino (Equation 5) uptake, respectively.
Statistical Analysis—All biochemical assays were conducted on HBE cultures from ≥5 subjects (±S.E.), and the mean experimental value was considered nonsignificantly different from the corresponding simulated value when <15% different.
RESULTS
Generation of the Experimental Data Set for Model Parameter Optimization—The strength of this model resides in the fact that all biochemical data were acquired from a single experimental system, polarized primary cultures of HBE cells maintained under air-liquid interface conditions and assayed in a physiological buffered solution (pH 7.5). Such consistency allowed us to combine data gathered over a decade into one experimental data set (Table 1, column A).
TABLE 1.
Kinetic parameters of ASL nucleotide/nucleoside release, uptake, and metabolism measured on primary cultures of HBE cultures or estimated by the model equations without/with ADP and AMP release
Unknown experimental values are labeled Unk. The units for Vmax or J/V are nmol·min−1·ml−1, and the units for KM are μm.
1 REF indicates references for the experimental data from our previous studies.

The first kinetic parameters of ATP and ADP hydrolysis measured on HBE
cultures represent the sum of all apical ecto-ATPase and ecto-ADPase
activities (26). More
recently, we determined the kinetic properties of ecto-AK
( ,
,
 ,
,
 ,
,
 , and
, and
 ;
Table 1, column A)
(28) and E-NPPs
(
;
Table 1, column A)
(28) and E-NPPs
( and
and
 ;
Table 1, column A)
(24) on these cells. Based on
this information, the kinetic measurements of ATP and ADP hydrolysis were
repeated in the presence of the ecto-AK inhibitor, Ap5A
(28), to eliminate the
influence of the nonlinear reaction (ATP + AMP ↔ 2ADP). Under these
conditions, the ecto-ATPase and ecto-ADPase activities described by
;
Table 1, column A)
(24) on these cells. Based on
this information, the kinetic measurements of ATP and ADP hydrolysis were
repeated in the presence of the ecto-AK inhibitor, Ap5A
(28), to eliminate the
influence of the nonlinear reaction (ATP + AMP ↔ 2ADP). Under these
conditions, the ecto-ATPase and ecto-ADPase activities described by
 (Table 1, column A) combined
the activities of two enzyme families: E-NTPDases and alkaline phosphatases.
We also recently showed that HBE cells express NTPDase 1 and NTPDase 3
(15). It is important to note
that the KM corresponds to the substrate affinity of an
enzyme, which is independent of cell type or expression level, whereas
Vmax constitutes a quantitative measure of the expression
level of an enzyme (33).
Because specific inhibitors of NTPDase 1 are currently unavailable, NTPDase 1
and NTPDase 3 were assigned to the parameter sets with KM
values approximating (±10%) their KM
(Table 1, column A), as
reported for purified human NTPDase 1 (ATP, 10 μm; ADP, 20
μm) (34) and
NTPDase 3 (ATP, 98 μm; ADP, 128 μm)
(35).
(Table 1, column A) combined
the activities of two enzyme families: E-NTPDases and alkaline phosphatases.
We also recently showed that HBE cells express NTPDase 1 and NTPDase 3
(15). It is important to note
that the KM corresponds to the substrate affinity of an
enzyme, which is independent of cell type or expression level, whereas
Vmax constitutes a quantitative measure of the expression
level of an enzyme (33).
Because specific inhibitors of NTPDase 1 are currently unavailable, NTPDase 1
and NTPDase 3 were assigned to the parameter sets with KM
values approximating (±10%) their KM
(Table 1, column A), as
reported for purified human NTPDase 1 (ATP, 10 μm; ADP, 20
μm) (34) and
NTPDase 3 (ATP, 98 μm; ADP, 128 μm)
(35).
We recently demonstrated that NSAP is the alkaline phosphatase isoform
functionally expressed by human airway epithelia
(13). Clinical assays,
generally performed in basic solutions (pH 9-10), depict NSAP as a low
affinity enzyme (KM >400 μm)
(23). However, NSAP has been
shown to exhibit both low (lowNSAP; KM >400
μm) and high (highNSAP; KM = 12-20
μm) affinities for adenine nucleotides at pH 7.5
(36-38).
We reported that airway NSAP hydrolyzes AMP with KM values
of 36 μm (highNSAP,  ;
Table 1, column A) and 717
μm (lowNSAP,
;
Table 1, column A) and 717
μm (lowNSAP,  ;
Table 1, column A) at pH 7.5
(13). Because the kinetic
parameters of airway NSAP for ATP and ADP were unavailable, we conducted these
experiments, as described previously
(13) but in the presence of
the ecto-AK inhibitor, Ap5A
(28). Two kinetic parameter
sets were identified for ATP and ADP hydrolysis
(Table 2), with
KM values within the ranges reported for lowNSAP and
highNSAP activities
(36-38).
These values also approximated the KM values of the high
affinity and low affinity ecto-ATPase
(
;
Table 1, column A) at pH 7.5
(13). Because the kinetic
parameters of airway NSAP for ATP and ADP were unavailable, we conducted these
experiments, as described previously
(13) but in the presence of
the ecto-AK inhibitor, Ap5A
(28). Two kinetic parameter
sets were identified for ATP and ADP hydrolysis
(Table 2), with
KM values within the ranges reported for lowNSAP and
highNSAP activities
(36-38).
These values also approximated the KM values of the high
affinity and low affinity ecto-ATPase
( and
and
 ) and ecto-ADPase
(
) and ecto-ADPase
( and
and
 ) activities
(Table 1, column A). Because
NSAP, NTPDase 1, and NTPDase 3 all support the dephosphorylation of ATP and
ADP, enzyme activities with similar KM values were
combined and designated as the high affinity (NTPDase 1/highNSAP) and the low
affinity (NTPDase 3/lowNSAP) enzymes (Table
1, column A).
) activities
(Table 1, column A). Because
NSAP, NTPDase 1, and NTPDase 3 all support the dephosphorylation of ATP and
ADP, enzyme activities with similar KM values were
combined and designated as the high affinity (NTPDase 1/highNSAP) and the low
affinity (NTPDase 3/lowNSAP) enzymes (Table
1, column A).
TABLE 2.
Kinetic parameters of ATP and ADP dephosphorylation by NSAP on primary cultures of HBE cells
Michaelis constants (Km; μm) and maximal velocities (Vmax: nmol·min−1·ml−1) were calculated from Woolf-Augustinson Hoftsee plots.
| Reactions | Enzyme identity | Parameters | Experimental data |
|---|---|---|---|
| ATP → ADP + Pi | HighNSAP | Vmax, Km | 1.0, 17 |
| LowNSAP | Vmax, Km | 14.1, 451 | |
| ADP → AMP + Pi | HighNSAP | Vmax, Km | 0.5, 11 |
| LowNSAP | Vmax, Km | 5.9, 117 |
The list of experimental data were completed by adding the kinetic parameters of ADA1 (29), the cellular uptake of Ado by CNT3 (29), the basal rate of ATP release (12), and the feed-forward inhibition of Ado production by ATP and ADP (13). Altogether, these assays and studies provided the experimental data set that was used to develop the mathematical model of nucleotide regulation on human airway epithelia (Table 1, column A).
Model Validation with Experimental Data—The model was tested
by comparing simulated and previously reported experimental measurements of
transient (26) and
steady-state (12) nucleotide
and nucleoside concentrations on the apical surface of HBE cells. We started
our investigations using the multicomponent model
(Fig. 1) described by Equations
7-11. Experimental values were available for all parameters from earlier
studies (Table 1), except the
KM of ecto-AK for AMP and the KM and
Vmax values of CNT3 for Ino. When used as free parameters
to fit the data, considerable discrepancies were found between the simulated
and experimental transient profiles of ATP hydrolysis
(Fig. 2A) and
steady-state concentrations (Fig.
2B). The performance of the model was considerably
improved by using the experimental data set as initial guesses in the
nonlinear least square fitting routine in MatLab (Mathworks, Natick, MA).
Using this approach, the model closely reproduced the transient and
steady-state profiles (Fig. 2, C
and D). Among the 30 estimated parameters, only 6 were
>2-fold different from the experimental values
(Table 1, column B) as follows:
high affinity ecto-ATPase (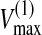 ),
E-NPPs (
),
E-NPPs ( and
and
 ), ecto-AK
(
), ecto-AK
( ) and the feed-forward
inhibition constants (
) and the feed-forward
inhibition constants ( and
and 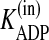 ). However, a
12-fold discrepancy persisted between measured and predicted ATP release
rates. Similar experimental values were reported by different research groups
using various techniques (10,
12,
20), ruling out experimental
error as the source of discrepancy. On the other hand, if the rate of ATP
release was held fixed at the experimental value and the other parameters free
to be determined by data fitting, the model reproduced the transient behavior
of the system (Fig.
2E) but not the steady-state profile
(Fig. 2F).
). However, a
12-fold discrepancy persisted between measured and predicted ATP release
rates. Similar experimental values were reported by different research groups
using various techniques (10,
12,
20), ruling out experimental
error as the source of discrepancy. On the other hand, if the rate of ATP
release was held fixed at the experimental value and the other parameters free
to be determined by data fitting, the model reproduced the transient behavior
of the system (Fig.
2E) but not the steady-state profile
(Fig. 2F).
FIGURE 2.

Validation of the transient and steady-state concentration profiles generated by the nonlinear model without data fitting (A and B) or by data fitting using the MatLab nonlinear least square program without (C and D) or with (E and F) JATP fixed at the experimental value. A, C, and E, simulation (lines) and experimental data (symbols; n = 5; S.E. <10% of mean) for nucleotide/nucleoside concentrations measured by HPLC of buffer samples collected from HBE cultures after the addition of 100 μm ATP (n = 6). B, D, and F, steady-state concentrations obtained by model simulation (empty bars) and experimental data (filled bars) from previous publications by our group (n = 5; ±S.E.) (12, 26).
Steady-state Analysis with a Linear Model—In an attempt to resolve the differences between experimental and predicted steady-state ATP concentrations, we developed a linear model to describe the system near steady state, as described by Equations 13-17. The model was analyzed using two strategies. First, the experimental steady-state concentrations were used as inputs, and the model was solved for the parameter values. Because the steady-state Ino level cannot be measured by fluorescent HPLC, it was considered an unknown value in this procedure. In this case, solving the linear equations generated a negative Ino concentration, which is biologically impossible. Second, the experimental data set (Table 1, column A) was used as input parameters, and the model was solved for the steady-state concentration profile. This approach generated a good approximation of the experimental ATP concentration, but it resulted in estimated ADP, AMP, and Ado levels >10-fold lower than the experimental values (Table 3). Altogether, these results suggested that the model was incompatible with the experimental data and was therefore missing key components.
TABLE 3.
Steady-state concentrations (nm) calculated from the linear model without or with parameters of ADP and AMP release
Unk indicates unknown.
| ATP | ADP | AMP | Ado | Ino | |
|---|---|---|---|---|---|
| Experimental data | 2.2 | 38.7 | 70.2 | 168 | Unk |
| Model optimization (JATP) | 3.9 | 2.8 | 4.6 | 15.3 | 6.7 |
| Model optimization (JATP, JADP, JAMP) | 4.0 | 38.1 | 70.3 | 231.7 | 102.0 |
Our current knowledge on nucleotide release is limited to ATP because the inhibitor mixture designed to prevent nucleotide metabolism (20) interferes with fluorescence HPLC. However, emerging studies suggest that exocytosis mediates the release of ATP, ADP, and AMP from mammalian cells (39-41), including polarized epithelia (4, 7, 42). We recently demonstrated that human airway epithelia co-release ATP and mucin by Ca2+-regulated exocytosis (42). During their maturation, secretory vesicles accumulate ATP- and UDP-sugars in concentrations ≥50-fold above cytosolic levels, a fraction of which is converted to ADP and UDP, and eventually to AMP and UMP (40). As a result, vesicle fusion to the apical membrane is expected to release ATP, ADP, and AMP into the ASL layer. These observations motivated the addition of terms describing ADP and AMP release to the mathematical model. Constant fluxes for ADP (JADP) and AMP (JADP) were added to the linear equations (Equations 13-17), and the values were adjusted so the model would generate steady-state concentrations closest to the experimental values. Table 3 shows that adding ADP and AMP release to the model eliminated the discrepancies between the experimental and predicted steady-state concentration profiles.
Reanalysis of the Nonlinear Model—The multienzyme model with the nonlinear equations (Equations 7-11) was updated to include terms describing the release of ADP (JADP/V in Equation 8) and AMP (JATP/V in Equation 9). The model was then refitted using the experimental data set (Table 1, column A) and the parameters of JADP and JAMP (Table 1, column C). Under these conditions, the simulations closely reproduced the experimental transient (Fig. 3A) and steady-state (Fig. 3B) concentration profiles, and eliminated the 12-fold difference between measured and estimated JATP. Finally, the predicted Vmax and KM values (Table 1, column C) were in closest agreement with the experimental data (Table 1, column A). Collectively, these simulations and experiments suggest that the model captures all major elements of nucleotide regulation on human airway epithelia.
FIGURE 3.

Validation of the transient and steady-state concentration profiles generated by the model equations, including basal rates of ADP and AMP release. A, simulated (lines) and experimental (symbols; n = 5; S.E. <10% of mean) transient nucleotide/nucleoside concentration profiles generated by the addition of 100 μm ATP on HBE cultures. B, simulated (empty bars) and experimental (filled bars) steady-state concentration profiles from previous publications by our group (n = 5; ±S.E.) (12, 26).
Impact of ASL Volume on Nucleotide Regulation—The model identified an important property of airway nucleotide regulation regarding ASL volume, which has been identified as a key determinant of mucus clearance (1, 2). Because the rate at which ATP release raises ATP concentration (JATP) and the maximum reaction velocity (Vmax = (kcatA)/(V)[E]T), both scale inversely with ASL volume in Equation 6, the model predicts that steady-state concentrations are independent of ASL volume. Following a simulated 2-fold increase in ASL volume, which acutely dilutes ATP and ADP, nucleotide concentrations are predicted to rapidly return to steady-state values (Fig. 4A). These simulation data were validated experimentally on HBE cultures using two luminometry assays, soluble luciferase and S. aureus protein A-luciferase attached to the epithelial surface. Fig. 4B shows that steady-state ATP and ADP levels, measured 60 min after the addition of 10-500 μl of phosphate-buffered saline, were unaffected by ASL volume changes. This result provides new insights into the biochemical processes supporting MCC. For instance, the airways of CF patients exhibit an ∼70% reduction in ASL volume, which plays a critical role in airway obstruction (1, 2). Under steady-state conditions, the model predicts that ASL dehydration in CF does not concentrate, i.e. increase the availability of ATP and Ado for P2 and P1 receptor-mediated mucus clearance as a compensatory mechanism to restore ASL volume.
FIGURE 4.

Modeling the impact of ASL volume and mechanical stress mimicking tidal breathing on ASL nucleotide regulation. A, model simulation of the impact of increasing ASL volume by 2-fold (20 μl) on steady-state ATP/ADP concentrations. B, model validation by biochemical measurements of steady-state ATP concentration using soluble luciferase (solid black box) or cell-attached S. aureus protein A-luciferase (open box) on HBE cultures after the addition of 1-500 μl of phosphate-buffered saline (n = 5; ±S.E.). C, impact of CCS (solid red box) on the steady-state concentration profile. HBE cultures were exposed 1 h to static conditions (control) or CCS mimicking tidal breathing (20 cm H2O; 20 cycles/min) (10). Then ASL nucleotide concentrations were measured by HPLC (n = 7; ±S.E.). Model simulations conducted to reproduce the impact of CCS on the steady-state concentration profile by increasing the release rates of ATP (JATP; black box), ATP and ADP (JATP, ADP; striped box) or ATP, ADP and AMP (JATP, ADP, AMP; open box) by 9.3-fold (10). Values expressed as fold increase from static exposure.
Model Adaptation to Mechanical Stress—Normal tidal breathing applies compressive and shear stresses on the airway walls that stimulate epithelial ATP release and ATP-mediated MCC (8, 10). This physiological response is particularly relevant to airway homeostasis as CCS mimicking tidal breathing was shown to restore ASL volume and mucus transport on the dehydrated airways of CF patients (10). Because the mathematical model was developed using experimental data acquired on static cultures, we tested its capacity to predict the ASL nucleotide and nucleoside concentrations maintained by mechanical stress mimicking tidal breathing. We recently demonstrated that CCS increases the rate of ATP release by 9.3-fold on HBE cultures (10). In this study, we conducted experiments to test the impact of CCS on the entire steady-state nucleotide concentration profile. Fig. 4C shows that CCS raised ATP, ADP, AMP, and Ado concentrations by 4-8-fold within the ASL layer. Model simulations were conducted to determine whether this response to mechanical stress was best fitted by an increase in the rate of ATP release or by concomitant increases in the release of several nucleotides. The equations solved with a 9.3-fold higher rate of ATP release (JATP; Table 1, column C) reproduced the effect of CCS on ATP but not on ADP or AMP (Fig. 4C). This simulation suggested that ATP release and metabolism were unable to account for the impact of CCS on ADP and AMP levels. Simultaneous 9.3-fold increases in JATP and JADP generated ATP and ADP levels within experimental range, whereas AMP levels remained >2-fold lower. The impact of CCS on the ASL concentration profile was best fitted by a model simulation in which JATP, JADP, and JAMP were all 9.3-fold higher than base line. Altogether, these data are consistent with the general proposal that mechanical stress induces the release of all three nucleotides through exocytosis in polarized epithelia (39-42). Furthermore, they demonstrate the ability of the mathematical model to predict the complex behavior of ASL nucleotide regulation under static and dynamic conditions.
Novel Approach to Biochemical Network Analysis—Although
several ectonucleotidases have been characterized on the apical surface of HBE
cells
(13-15,
24-29),
the key enzyme(s) regulating nucleotide-mediated MCC remain unidentified
because of the complexity of the multienzyme network. The mathematical model
allowed us, for the first time, to determine the contribution of each enzyme
and enzyme group to the regulation of both physiological and pathological ASL
nucleotide concentrations. Several airway ectonucleotidases have been shown to
hydrolyze ATP, ADP, and/or AMP as follows: NTPDase 1, NTPDase 3, NSAP, E-NPPs,
and ecto-AK (14,
15,
28). Based on their kinetic
properties, we hypothesized that specific enzymes regulate ASL nucleotide
levels encountered under physiological conditions (<0.1 μm)
(8,
12,
20), whereas others regulate
higher ASL concentrations reached during tissue injury (0.1-1
μm)
(43-46)
or aerosolization (1-100 μm) for diagnostic
(47,
48) or therapeutic
(49-52)
purposes. The biochemical network was first simplified by classifying the
enzymes into the following two groups: 1) low affinity high capacity
(KM ≥ 50 μm; NTPDase 3, lowNSAP), and 2)
high affinity low capacity (KM < 50 μm;
NTPDase 1, E-NPPs, highNSAP, and ecto-AK). Model simulations were conducted by
blocking the Vmax of each enzyme
(Table 1, column C) to
reproduce the effects of selective inhibitors on HBE cultures. One of them,
levamisole, inhibits both the high and low affinity activities of NSAP
(13), which could introduce
validation errors into group comparisons. This possibility was addressed by
comparing the impact of eliminating highNSAP, lowNSAP, or high/low NSAP
activities on ASL nucleotide regulation within the two concentration ranges of
interest: aerosolized/pathological (10-100 μm) and physiological
(0.1-1 μm). For Vmax values corresponding to
the total maximum activities of two enzymes, NTPDase 1/highNSAP
( and
and
 ) and NTPDase 3/lowNSAP
(
) and NTPDase 3/lowNSAP
( ), NSAP activity was blocked by
reducing the estimated parameter (Table
1, column C) proportionally by its experimental contribution
(Table 2) to total enzyme
activity (Table 1, column A).
Fig. 5A shows that
blocking highNSAP reduced the elimination rate of 100 μm ATP by
<10%. In contrast, blocking lowNSAP or high/lowNSAP reproduced the effects
of 10 mm levamisole on the HBE cultures and increased the half-life
(t½) of 100 μm ATP by 1.6-fold.
Conversely, the metabolism of 1 μm ATP was not affected by
blocking lowNSAP, whereas blocking highNSAP or high/lowNSAP mimicked the
effects of levamisole with a 1.9-fold increase in the
t½ of ATP (Fig.
5B). These data indicate that lowNSAP and highNSAP
activities exhibit negligible overlap, regulating excess and physiological ATP
concentrations selectively. Consequently, model simulations in which both NSAP
activities are blocked are expected to reproduce the effects of levamisole
within the concentration range of interest.
), NSAP activity was blocked by
reducing the estimated parameter (Table
1, column C) proportionally by its experimental contribution
(Table 2) to total enzyme
activity (Table 1, column A).
Fig. 5A shows that
blocking highNSAP reduced the elimination rate of 100 μm ATP by
<10%. In contrast, blocking lowNSAP or high/lowNSAP reproduced the effects
of 10 mm levamisole on the HBE cultures and increased the half-life
(t½) of 100 μm ATP by 1.6-fold.
Conversely, the metabolism of 1 μm ATP was not affected by
blocking lowNSAP, whereas blocking highNSAP or high/lowNSAP mimicked the
effects of levamisole with a 1.9-fold increase in the
t½ of ATP (Fig.
5B). These data indicate that lowNSAP and highNSAP
activities exhibit negligible overlap, regulating excess and physiological ATP
concentrations selectively. Consequently, model simulations in which both NSAP
activities are blocked are expected to reproduce the effects of levamisole
within the concentration range of interest.
FIGURE 5.

Contribution of the low affinity and high affinity enzyme groups to ASL nucleotide regulation. A and B, simulations comparing hydrolysis rates of 100 μm ATP (A) or 1 μm ATP (B) under control conditions (solid black line) or after blocking highNSAP (solid red line), lowNSAP (solid blue line), or both (high/lowNSAP) (dashed blue line). Validation conducted on HBE cultures for control conditions (•) and high/lowNSAP inhibition by 10 mm levamisole (open blue box). C and D, simulated metabolic profile for 100 μm ATP before (solid line) and after (dashed line) blocking the low affinity (LA; lowNSAP and NTPDase 3) (C) or high affinity (HA; NTPDase 1, highNSAP, E-NPPs, and ecto-AK) enzymes (D). Validation for the control (C and D, filled symbols) and low affinity block (C; open symbols) using an inhibitor mixture (10 mm levamisole + 10 μm NF-279) (n = 5; S.E. <10% of mean). E, simulated half-life (t½) of 0.1-100 μm ATP generated by blocking the high affinity (solid black box) or low affinity (striped box) group, and experimental data (solid red box) for the low affinity block as above (n = 5; ±S.E.). F, simulated impact of blocking the high affinity (solid black box) or low affinity (striped box) group on the steady-state profile. Validation (solid red box) was by comparing ASL nucleotide levels before/after incubating HBE cultures with the low affinity inhibitor mixture (n = 5; ±S.E.).
We first acquired general information on the biochemical network by comparing the relative contribution of the low affinity and high affinity enzyme groups to the regulation of aerosolized/pathological nucleotide concentrations. Model simulations in which the kinetic parameters of the low affinity enzymes (high/lowNSAP and NTPDase 3) were blocked predicted a 4-fold decrease in elimination rate of 100 μm ATP and significant delays in the accumulation of ADP, AMP, and Ado (Fig. 5C). Comparable profiles were obtained experimentally using HBE cultures assayed with 100 μm ATP in the absence (control) or presence of an inhibitor mixture specific for the low affinity enzymes (Fig. 5C) as follows: 10 mm levamisole (NSAP) (13) + 10 μm NF-279 (NTPDase 3) (53). In contrast, model simulation in which all the high affinity enzymes (high/lowNSAP, NTPDase 1, E-NPPs, and ecto-AK) were blocked yielded minor modifications in the transient profile, with a 1.5-fold decrease in elimination rate of 100 μm ATP (Fig. 5D). Although experimental validation of the high affinity block awaits the identification of noncompetitive inhibitors for NTPDase 1 and E-NPPs, these data suggest that high ATP levels (>10 μm) are regulated mainly by the low affinity enzymes on airway epithelia.
Comparative analyses conducted over a broad range of ATP concentrations predicted a shift in the relative contribution of the high and low affinity enzyme groups. To carefully scrutinize the involvement of each group, the two NSAP activities were analyzed independently. Model simulations in which the high affinity enzymes (highNSAP, NTPDase 1, E-NPPs, and ecto-AK) or the low affinity enzymes (lowNSAP and NTPDase 3) were blocked raised the t½ of 100 μm ATP by 1.2- and 2.3-fold, respectively (Fig. 5E). Although the t½ of 10 μm ATP would be equally affected by either group, the high affinity group is predicted to dominate at lower ATP concentrations. These simulations were reproduced experimentally on HBE cultures with a low affinity inhibitor mixture (10 mm levamisole + 10 μm NF-279) (Fig. 5E). The model suggests that the higher contribution of the low affinity group measured experimentally for 0.1 μm ATP reflects the inhibitory effect of levamisole on highNSAP. Together, simulated and experimental results support a major role for high affinity enzymes in the regulation of ASL ATP within the effective concentration range of P2Y2 receptors (0.01-1 μm) (16, 17).
Recent studies showed that ASL nucleotide metabolism is accelerated in chronic lung diseases, including CF, chronic obstructive pulmonary disease, and primary ciliary dyskinesia (14), which could compromise nucleotide-mediated MCC (19, 52). Consequently, activation of P2Y2 receptors by aerosolized nucleotides is being tested as a therapy for CF (52, 54). As an alternative strategy to raise steady-state ASL ATP concentration, we conducted simulations to test the potential of an aerosolized inhibitor mixture against specific enzymes. As expected from their kinetic properties, model simulations in which the low affinity enzymes (lowNSAP and NTPDase 3) were blocked predicted a <2-fold increase in steady-state nucleotide concentrations (Fig. 5F). This simulation was reproduced experimentally by assays conducted on HBE cultures exposed to the low affinity inhibitor mixture (10 mm levamisole + 10 μm NF-279) (Fig. 5F). Because levamisole does not discriminate between the lowNSAP and highNSAP activities, these assays demonstrate that NSAP does not contribute significantly to the regulation of steady-state ATP concentrations (i.e. <0.1 μm) on airway epithelial surfaces. To our surprise, the model predicts that the steady-state nucleotide levels are sensitive to the high affinity enzyme group with respect to ADP but not ATP (Fig. 5F), which suggested the influence of a nonlinear component within the biochemical network.
The properties of the reversible ecto-AK reaction suggest that the net directionality depends on the relative substrate concentrations (Fig. 6A). Micromolar ATP, encountered during injury or aerosolization, would drive the forward reaction, whereas steady-state conditions would favor the reverse reaction because of the relatively high ADP concentration (Table 3). Therefore, simulations and assays were designed to test whether ecto-AK influences the ATP/ADP concentration ratio on airway epithelial surfaces in these two situations (Fig. 6A). Model simulations predicted that blocking ecto-AK would not affect the metabolism of 100 μm ATP (Fig. 6B) or the steady-state concentration profile (Fig. 6E). These simulated data were reproduced experimentally for the transient (Fig. 6B) and steady-state (Fig. 6E) concentration profiles by HBE cultures exposed to the vehicle control or buffer containing the ecto-AK inhibitor, 0.5 mm Ap5A (28). We next tested the hypothesis that ecto-AK activity may be significant within a functional unit addressing a specific nucleotide concentration range. Fig. 6C shows that blocking ecto-AK is not expected to influence the metabolic profile of 100 μm ATP regulated by the low affinity enzymes, i.e. determined in the absence of the other high affinity enzymes. In contrast, model simulations and experimental data suggest that ecto-AK contributes to the regulation of ASL nucleotides under conditions where the high affinity enzyme activities predominate. First, the model predicts that blocking ecto-AK would not affect the elimination rate of 100 μm ATP by a high affinity biochemical network, i.e. determined in the absence of the low affinity enzymes (Fig. 6D). However, when ADP concentration rises above that of ATP (∼20 min), the absence of ecto-AK would be associated with higher rates of ADP elimination and Ado accumulation (Fig. 6D). Similar results were obtained experimentally with HBE cultures using the low affinity inhibitor mixture (10 mm levamisole and 10 μm NF-279) with/without 0.5 mm Ap5A (Fig. 6D). These data were also consistent with the behavior of ecto-AK reported on HBE cultures exposed to an ATP concentration (1 μm) (28) predicted to be regulated predominantly by high affinity enzymes (Fig. 5E). The reverse ecto-AK reaction would compete with the high affinity ADP-hydrolyzing enzymes to trap adenine nucleotides into a trans-phosphorylation cycle (28). These findings suggest that airway ecto-AK regulates ASL nucleotide levels within a high affinity biochemical network exposed to a high ADP/ATP concentration ratio.
FIGURE 6.

Role of ecto-AK in ASL nucleotide regulation. A, reaction directionality during nucleotide aerosolization (>10 μm ATP) and under steady-state conditions (1-10 nm ATP) with ADP/ATP >20 (Table 3). B-D, simulations (lines) of the impact of blocking ecto-AK on the metabolism of 100 μm ATP (B) in the presence of all enzymes and after blocking (C) the high affinity (NTPDase 1, highNSAP, and E-NPPs) or (D) low affinity (LA; lowNSAP and NTPDase 3) enzymes. B, validation using HBEs pretreated with the vehicle (closed symbols) or the ecto-AK inhibitor (0.5 mm Ap5A; open symbols). D, validation on HBEs pretreated with the low affinity inhibitor mixture (10 mm levamisole + 10 μm NF-279) without (closed symbols) or with (open symbols) 0.5 mm Ap5A (n = 6; S.E. < 10% of mean). E, impact of blocking ecto-AK on the steady-state profile by stimulation (solid black box) or exposing HBEs to 0.5 mm Ap5A (solid red box) (n = 5; ±S.E.). Data were compared with the simulated impact of blocking the other high affinity enzymes (open box).
Based on this information, we revisited the impact of ecto-AK on ASL nucleotide regulation at steady-state conditions, which is expected to favor the reverse ecto-AK reaction (2ADP → ATP + AMP) (Fig. 6A) because of the high ADP/ATP ratio (∼20; see Table 3). A model simulation with a high affinity inhibitor mixture, excluding the ecto-AK inhibitor, resulted in a 6-fold increase in steady-state ATP concentration (Fig. 6E), compared with 2-fold for a complete high affinity block (Fig. 5F). The physiological relevance of this information is illustrated by the fact that a comparable increase in ASL ATP level was generated by CCS mimicking normal tidal breathing (Fig. 4C), which restored normal ASL height and mucus transport on the dehydrated airway epithelial surfaces of CF patients (10). The mathematical model of ASL nucleotide regulation therefore identified the key enzymes regulating ASL ATP and proposes a novel approach for the treatment of airway obstruction in CF, an aerosolized inhibitor mixture against high affinity NTPDase 1 and E-NPPs.
Adenosine Regulation in Airway Defenses—The model was instrumental to our investigation of airway Ado regulation, supplementing the biochemical assays with simulated information that would not be available experimentally. First, we modeled the relative contribution of the two ectonucleotidases supporting the conversion of AMP to Ado on airway epithelial surfaces, NSAP and ecto-5′-NT (13). Model simulations were run by blocking the parameters of the following: 1) ecto-5′-NT; 2) high/lowNSAP toward AMP [NSAP (AMP)], or 3) high/lowNSAP toward ATP, ADP, and AMP [NSAP (all)]. These simulations were verified by biochemical assays conducted on HBE cultures before and after the inhibition of ecto-5′-NT (10 mm concanavalin A) (13, 55) or NSAP (10 mm levamisole) (13, 56). Although blocking all parameters of NSAP would mimic the effects of levamisole, we tested whether the concomitant reductions in ecto-ATPase and ecto-ADPase activities would influence the feed-forward inhibition of Ado accumulation (Equation 3) (13). Hence, all simulations and assays were initiated with ATP to probe the response of the entire biochemical network.
Comparative analysis conducted over a range of ATP concentrations revealed a shift in the relative contribution of ecto-5′-NT and NSAP (Fig. 7, A-C). Both simulated and experimental data showed that ecto-5′-NT dominates at physiological ATP levels (0.1-1.0 μm), as indicated by larger reductions in the accumulation rate of Ado (Fig. 7, A and B). In contrast, Ado production from 10 μm ATP would be equally reduced by blocking ecto-5′-NT or NSAP (AMP) (Fig. 7C). Interestingly, an intermediate response was predicted by blocking NSAP (all) (Fig. 7C) or measured experimentally with levamisole (Fig. 7C). These data suggest that levamisole increases the availability of ATP and ADP for the feed-forward inhibition of Ado production by the remaining enzyme, ecto-5′-NT.
FIGURE 7.

Relative contribution of ecto-5′NT and NSAP to Ado
regulation. A-C, simulated (lines) and experimental
(symbols) time courses of Ado accumulation from 0.1 to 10
μm ATP under control conditions and after blocking
ecto-5′-NT (solid red line) or NSAP (solid blue line)
activities. For NSAP, simulations were run by blocking the parameters
regulating AMP (NSAP (AMP)) (dashed blue line) or all nucleotides
(NSAP (all)) (solid blue line). Experimental data using HBE cultures
were incubated with 0.1-10 μm ATP, without/with an inhibitor of
ecto-5′-NT (10 mm concanavalin A) (open red
triangle), or NSAP (all) (10 mm levamisole) (open blue
box) (n = 6; S.E. < 10% of mean). D, simulated
(solid black box) and experimental (solid red box) delay to
the accumulation of 0.3 μm Ado from 1 to 1000 μm
ATP. Simulations also compared the impact of blocking ecto-5′-NT with
(striped box) or without (open box) feed-forward inhibition
(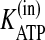 ,
,
 block;
Table 1, column C). E,
simulated (solid black box) and experimental (solid blue
box; n = 5, p < 0.05) impact of blocking NSAP (all)
on the delay to reach 0.3 μm Ado (fold from controls in
D). Simulated impact of blocking both NSAP (all) and feed-forward
inhibition (striped black box) is shown. F, simulated impact
of blocking NSAP (all) (solid black box) or ecto-5′-NT
(open box) on the steady-state profile. Experimental block of NSAP
(all) (solid blue box) or ecto-5′-NT (solid red box)
was obtained by measuring nucleotide levels before/after incubating cultures
with the inhibitors (n = 5, p < 0.05).
block;
Table 1, column C). E,
simulated (solid black box) and experimental (solid blue
box; n = 5, p < 0.05) impact of blocking NSAP (all)
on the delay to reach 0.3 μm Ado (fold from controls in
D). Simulated impact of blocking both NSAP (all) and feed-forward
inhibition (striped black box) is shown. F, simulated impact
of blocking NSAP (all) (solid black box) or ecto-5′-NT
(open box) on the steady-state profile. Experimental block of NSAP
(all) (solid blue box) or ecto-5′-NT (solid red box)
was obtained by measuring nucleotide levels before/after incubating cultures
with the inhibitors (n = 5, p < 0.05).
Model simulations were designed to evaluate the physiological relevance of feed-forward inhibition for airway homeostasis by calculating the time required for 1-1000 μm ATP to generate an Ado concentration equivalent to the EC50 of A2B receptors (0.3 μm) (16, 17). Both simulated and biochemical data demonstrated that the delay to reach 0.3 μm Ado is influenced by the ASL ATP concentration in a biphasic manner (Fig. 7D). A relatively long delay would be expected for concentrations (i.e. 1 μm ATP) below the catalytic capacity of most airway ectonucleotidases (Table 1, column A). In the 10-100 μm ATP range, both enzymes efficiently converted AMP to Ado, whereas longer delays are required for ATP levels challenging their Vmax.
The mathematical model was able to identify the enzyme targeted by
feed-forward inhibition using simulations that cannot be reproduced
experimentally: by blocking the kinetic constants
( and
and
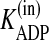 ;
Table 1, column C). We compared
the impact of blocking each AMP-hydrolyzing enzyme on the ATP-mediated
regulation of Ado production in the presence or absence of feed-forward
inhibition. Model simulations in which the Vmax of
ecto-5′-NT activity was blocked resulted in slightly longer Ado delays
(< 30%) at all ATP concentrations, which were partially reduced to control
values by blocking the inhibition constants
(Fig. 7D). These
simulations predict a weak control of feed-forward inhibition over the
remaining enzyme, NSAP, which may be ascribed to its capacity to eliminate ATP
and ADP in close vicinity to the catalytic site.
;
Table 1, column C). We compared
the impact of blocking each AMP-hydrolyzing enzyme on the ATP-mediated
regulation of Ado production in the presence or absence of feed-forward
inhibition. Model simulations in which the Vmax of
ecto-5′-NT activity was blocked resulted in slightly longer Ado delays
(< 30%) at all ATP concentrations, which were partially reduced to control
values by blocking the inhibition constants
(Fig. 7D). These
simulations predict a weak control of feed-forward inhibition over the
remaining enzyme, NSAP, which may be ascribed to its capacity to eliminate ATP
and ADP in close vicinity to the catalytic site.
The reciprocal simulation exposed a strong relationship between ASL ATP concentration and Ado production by ecto-5′-NT. The time required for Ado to reach the EC50 of A2B receptors (0.3 μm) (16, 17) increased by up to 6-fold when ecto-5′-NT dominated, i.e. in the absence of NSAP, with a regulatory threshold of 1 μm ATP (Fig. 7E). These simulations were reproduced by HBE cultures pretreated with the NSAP inhibitor (10 mm levamisole). The model predicts that this relationship would be completely abrogated by simultaneously blocking NSAP (all) and the feed-forward inhibition (Fig. 7E). The mathematical model therefore provided new insights into the biochemical processes regulating ASL Ado, which would not have been available otherwise. The simulations predict that feed-forward inhibition targets specifically Ado production by ecto-5′-NT on HBE surfaces. However, whereas the sensitivity of ecto-5′-NT activity to ≥1 μm ATP is consistent with the properties of the vascular model of nucleotide regulation (57-59), airway epithelia seldom experience such high ATP concentrations, except locally during injury or aerosolization.
Finally, we determined the contribution of NSAP and ecto-5′-NT to the regulation of the steady-state nucleotide concentrations. The model predicts that NSAP inhibition would raise ATP, ADP, and AMP concentrations by 1.4-, 2.5-, and 2.8-fold, respectively (Fig. 7F). This inverse relationship between NSAP activity and the degree of phosphorylation of the substrate was closely reproduced experimentally by levamisole on HBE cultures (Fig. 7F) and is consistent with the properties of the purified enzyme: ATP < ADP < AMP (60, 61). In contrast, model simulations in which ecto-5′-NT activity was eliminated only increased AMP concentration, in agreement with the substrate specificity of the enzyme (56). These results were reproduced by HBE cultures exposed to the selective inhibitor, concanavalin A (Fig. 7F). Altogether, these data demonstrate that the model captures the contributions of ecto-5′-NT and NSAP to the regulation of steady-state nucleotide concentrations. On the other hand, inhibiting ecto-5′-NT or NSAP did not influence the steady-state concentration of Ado, which suggests that one AMP-hydrolyzing enzyme may be sufficient under conditions where AMP concentration is >100-fold lower than their KM values.
DISCUSSION
This study presents the first mathematical model of extracellular nucleotide and nucleoside regulation for the respiratory system. Based on experimental data reported in the literature (12, 13, 20, 26, 28, 29), and acquired in this study, for polarized HBE cultures, Equations 7-11 were designed to incorporate all known factors regulating ASL nucleotides and Ado as follows: epithelial ATP release, the reactions mediating the surface conversion of ATP into ADP, AMP, and Ado, the surface elimination of Ado by deamination and cellular uptake, the trans-phosphorylating ecto-AK activity, and the feed-forward inhibition of Ado production by ATP and ADP. The simulations closely reproduced the transient and steady-state concentration profiles generated on the apical surface following the addition of ATP. However, regardless of the fitting strategy, a 12-fold difference persisted between the simulated and experimental rates of ATP release, suggesting the model was missing key elements.
The current information on the mechanisms of nucleotide release is derived from measurements of ATP release (62, 63). Yet recent breakthroughs in our understanding of exocytosis support the notion that nucleotides, in addition to ATP, are also released via this route by a variety of cell types (4, 39-41), including HBE cells (42). During their maturation, the vesicles accumulate ATP, ADP, and AMP, which are released during exocytosis (39-41). Importantly, the addition of terms describing ADP and AMP release provided the best fit of the experimental data, as well as the transient and steady-state concentration profiles. Furthermore, it resolved the discrepancy between simulated and experimental rates of ATP release.
The resulting model was validated by testing the impact of mechanical stress on ASL nucleotide regulation. Airway epithelia, in vivo, are subjected to mechanical forces imparted by tidal breathing, which are known to stimulate ATP release (8, 10). We recently showed that CCS mimicking normal tidal breathing causes a 9.3-fold increase in the rate of ATP release from HBE cultures (10). In this study, we showed that CCS raises the ASL concentrations of ATP, ADP, AMP, and Ado by 4-7-fold. Model simulations in which JATP was increased 9.3-fold reproduced the effects of CCS on ATP but not on the other nucleotides. The model therefore predicted that mechanical stress-induced ATP release and surface metabolism are unable to account for the effects of CCS on the ASL nucleotide composition. In fact, the best fit was obtained by raising simultaneously JATP, JADP, and JAMP by 9.3-fold. These results support the importance of including parameters for the release of all three nucleotides into the mathematical model of ASL nucleotide regulation. Furthermore, they illustrate the capacity of the model to predict the impact of physiological phenomena such as breathing on ASL nucleotide regulation.
Multiple ATP-hydrolyzing enzymes compete for the same pool of nucleotides within the ASL layer. The development of a mathematical model incorporating all the ectonucleotidases provided, for the first time, an invaluable tool to assess their individual contribution within the multienzyme system. The complex biochemical network was simplified by sorting the enzymes into kinetically distinct groups as follows: low affinity high capacity (KM > 50 μm; lowNSAP and NTPDase 3) and high affinity low capacity (KM < 50 μm; highNSAP, E-NTPDase 1, E-NPPs and ecto-AK) (26, 64). Model simulations accurately predicted the predominant role of the low affinity group in the regulation of high ATP concentrations (>1 μm) (43-46), as demonstrated on HBE cultures using selective inhibitors of NSAP (10 mm levamisole) (13) and NTPDase 3 (10 μm NF-279) (53). Therapies inhibiting these ectoenzymes could potentially accentuate airway epithelial apoptosis (65, 66) and inflammatory responses (45) because of increased ATP-mediated activation of P2X7 receptors, which are up-regulated in lung diseases (67). In addition, these enzymes are likely responsible for the rapid elimination of aerosolized nucleotides (1-100 μm) used to diagnose asthma and chronic obstructive pulmonary disease (ATP and AMP) (47, 48), for the treatment of CF and primary ciliary dyskinesia (UTP) (49-52) and in animal studies (48, 68, 69).
The model predicts that high affinity enzymes play a major role in the regulation of ATP within the physiological concentration range encountered on airway epithelial surfaces (8, 12, 20). The recent report that CCS-mediated increase in ASL ATP level restores ASL volume and mucus transport on dehydrated HBE cultures from CF patients (10) motivated the search for an inhibitor mixture against the high affinity enzymes to enhance P2Y2 receptor-mediated mucus clearance. However, the model predicts that blocking all four enzymes would raise ADP concentrations, instead of ATP. An explanation was provided by separating the high affinity enzymes into linear and nonlinear components. Inhibition of the enzymes supporting the linear reactions (ATP → ADP → AMP; NTPDase 1, E-NPPs and highNSAP) is expected to raise the concentrations of all the released nucleotides. In contrast, the directionality of the reversible ecto-AK reaction (ATP + AMP ↔ 2ADP) would be influenced by the ASL nucleotide composition. Model simulations and biochemical assays suggest that ecto-AK does not influence the elimination of high ATP concentrations (>10 μm). However, blocking ecto-AK significantly modified the transient metabolic profile generated by the high affinity enzyme group once ADP level exceeded that of ATP by >3-fold, as depicted by enhanced ADP, but increased Ado, accumulation. This behavior is consistent with the substrate requirements of the reversible ecto-AK reaction. The forward reaction (ATP + AMP → 2ADP) is limited on airway epithelia by the high ATP/AMP ratio maintained through efficient conversion of AMP to Ado. In contrast, the reverse reaction (2ADP → ATP + AMP) is favored once ADP concentration exceeds that of [ATP + AMP]. However, the ATP generated by ecto-AK would be rapidly dephosphorylated by the high affinity ecto-ATPases (NTPDase 1, E-NPPs, and highNSAP). Consequently, these data suggest that removing the strong competitive effect of the linear high affinity enzymes may allow ecto-AK to raise ATP concentrations within a nucleotide mixture dominated by ADP.
Based on this analysis of the high affinity group, we evaluated the role of ecto-AK under steady-state conditions, in which ASL exhibits an ADP/ATP concentration ratio of ∼20 (Table 3). The model predicts that a high affinity inhibitor mixture, excluding the ecto-AK inhibitor (Ap5A), would raise the ASL ATP concentration by ∼6-fold. The therapeutic potential of this approach is illustrated by the fact that CCS mimicking normal breathing produced a similar increase in ASL ATP level (Fig. 4C), which restored mucus transport on the airway epithelial surfaces of CF patients (10). The physiological importance of ecto-AK for the regulation of steady-state nucleotide levels was also demonstrated in human hepatocytes, where it regulates P2Y13 (ADP) receptor-mediated high density lipoprotein endocytosis (70). The ecto-AK inhibitor stimulated high density lipoprotein endocytosis in the absence of exogenous substrates, supporting a significant role in cholesterol transport under steady-state conditions. Altogether, the model and experimental data suggest that aerosolization of an inhibitor mixture against the high affinity enzymes (excluding Ap5A) may stimulate airway clearance in CF patients. The fact that blocking NSAP did not affect the steady-state ATP level suggests that levamisole may be omitted. Further refinement of the high affinity enzyme inhibitor mixture for the treatment of obstructive lung diseases awaits the discovery of specific noncompetitive inhibitors for NTPDase 1 and the E-NPPs.
The regulation of ASL nucleotides through cell surface metabolism leads to
the accumulation of another important signaling molecule for airway defenses,
i.e. Ado. We previously demonstrated that two ectoenzymes contribute
to the production of Ado on airway epithelial surfaces, ecto-5′-NT and
NSAP (13). Although their
characterization was conducted using AMP as initial substrate
(13), this study documented
the impact of the entire biochemical network on ASL Ado production using ATP
as initial substrate. Based on model simulations and biochemical assays, we
identified a negative relationship between ATP ≥1 μm and the
time required for Ado concentration to reach the EC50 of
A2B receptors for CFTR activation (0.03 μm)
(16,
17). Model simulations, in
which the kinetic constants
 and
and
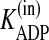 were blocked, suggest
that this relationship is caused by feed-forward inhibition of ATP on
ecto-5′-NT, but not on NSAP. These data are in agreement with the
reported inhibition of purified ecto-5′-NT by ATP and ADP
(71,
72). Although the inhibitory
mechanism has not been identified, the insensitivity of NSAP may be partially
because of its capacity to eliminate ATP and ADP. Altogether, these results
emphasize the critical importance of studying the properties of
ectonucleotidases using experimental designs accounting for the entire
biochemical network, as the suppression of ecto-5′-NT activity by ATP
would have been missed by protocols isolating the AMP → Ado reaction.
were blocked, suggest
that this relationship is caused by feed-forward inhibition of ATP on
ecto-5′-NT, but not on NSAP. These data are in agreement with the
reported inhibition of purified ecto-5′-NT by ATP and ADP
(71,
72). Although the inhibitory
mechanism has not been identified, the insensitivity of NSAP may be partially
because of its capacity to eliminate ATP and ADP. Altogether, these results
emphasize the critical importance of studying the properties of
ectonucleotidases using experimental designs accounting for the entire
biochemical network, as the suppression of ecto-5′-NT activity by ATP
would have been missed by protocols isolating the AMP → Ado reaction.
In human airways, feed-forward inhibition is not expected to play a significant role in the regulation of purinergic signaling, as epithelial surfaces are not normally exposed to >1 μm ATP (73-75). However, this regulatory mechanism may become relevant during injury-induced ATP release to prevent A2B receptor desensitization (76) and/or excessive inflammatory responses caused by chronically high Ado concentrations (77). But even under these circumstances, NSAP activity would be expected to compensate for the reduction in ecto-5′-NT activity caused by excess ATP. Consequently, this regulatory mechanism may be more relevant for purinergic signaling in tissues expressing ecto-5′-NT, with little or no NSAP. For instance, feed-forward inhibition is not detected on vascular smooth muscle cells (78), which express NSAP (79). In contrast, endothelial surfaces do not express NSAP (80) and exhibit an inhibitory effect of ADP on ecto-5′-NT activity (59, 81). Because ADP activates platelets and Ado suppresses their activation (31, 82), feed-forward inhibition was proposed to regulate thrombus formation during endothelial repair. A mathematical model of extracellular nucleotide metabolism on endothelial cells predicts an inhibition of Ado production by 1-10 μm ADP (57-59). Because activated platelets release >1 mm ADP on endothelial surfaces (31), feed-forward inhibition would create a time gap between ADP- and Ado-mediated responses, allowing sufficient time to consolidate the thrombus while minimizing spread beyond the site of damage. Consequently, two factors determine the importance of feed-forward inhibition for purinergic signaling, the ecto-5′-NT/NSAP activity ratio and local nucleotide concentrations.
In conclusion, this study provides a robust model for the regulation of two essential signaling molecules for airway defenses, ATP and Ado. The equations capture the complex behavior of the linear and nonlinear reactions and the dynamic interplay between the ectoenzymes. Furthermore, the model reproduces ASL nucleotide regulation under both static conditions and mechanical stress mimicking normal breathing. Finally, therapeutic applications are proposed based on an interactive approach between simulations and biochemical assays. We are currently developing a model for the regulation of the ASL volume by P2Y2 and A2B receptor-mediated ion channel activities. In the near future, the integration of these two models should provide insightful information on the interactions between the biochemical and functional networks of purinergic signaling on airway epithelia under normal and pathological conditions, as well as novel guidelines for the development of efficient treatments of chronic obstructive lung diseases.
Acknowledgments
We thank Catja van Heusden for technical assistance.
This work was supported, in whole or in part, by National Institutes of Health Grants PPG-P01-HL034332, P30 DK065988, SCOR 1-P50-HL060280, SCCOR 1-P50-HL084934, and R01-GM079271. This work was also supported by Cystic Fibrosis Foundation Grants BUTTON06G0, OKADA06I0, and PICHER07I0. The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
Footnotes
The abbreviations used are: MCC, mucociliary clearance; ADA1, adenosine deaminase 1; Ado, adenosine; ASL, airway surface liquid; CFTR, cystic fibrosis transmembrane regulator; CCS, cyclic compressive stress; CF, cystic fibrosis; CNT3, concentrative nucleoside transporter 3; ecto-5′-NT, ecto-5′-nucleotidase; ecto-AK, ecto-adenylate kinase; E-NTPDases, ecto-nucleoside triphosphate diphosphohydrolases; E-NPPs, ecto-nucleotide pyrophosphatase/phosphodiesterases; HDL, high density lipoprotein; highNSAP, high affinity NSAP activity; HPLC, high pressure liquid chromatography; Ino, inosine; jAdo, rate of Ado uptake; jIno, rate of Ino uptake; JADP, rate of ADP release; JAMP, rate of AMP release; JATP, rate of ATP release; lowNSAP, low affinity NSAP activity; NTPDase 1, nucleoside triphosphate diphosphohydrolase 1; NTPDase 3, nucleoside triphosphate diphosphohydrolase 3; NSAP, nonspecific alkaline phosphatase; PCL, periciliary liquid layer; HBE, human bronchial epithelial cell; Ap5A, P1,P5-di(adenosine 5′)-pentaphosphate.
References
- 1.Boucher, R. C. (2003) Pfluegers Arch. 445 495-498 [DOI] [PubMed] [Google Scholar]
- 2.Boucher, R. C. (2007) J. Int. Med. 261 5-16 [DOI] [PubMed] [Google Scholar]
- 3.Davis, C. (2002) Norvatis Found. Symp. 248 113-125 [PubMed] [Google Scholar]
- 4.Williams, O. W., Sharafkhaneh, A., Kim, V., Dickey, B. F., and Evans, C. M. (2006) Am. J. Respir. Cell Mol. Biol. 34 527-536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Donaldson, S. H., and Boucher, R. C. (1998) in The P2 Nucleotide Receptors (Turner, J. T., Weisman, G. A., and Fedan, J. S., eds) pp. 413-424, Humana Press Inc., Totowa, NJ
- 6.Marcet, B., and Boeynaems, J. M. (2006) Pharmacol. Ther. 112 719-732 [DOI] [PubMed] [Google Scholar]
- 7.Lazarowski, E. R., Boucher, R. C., and Harden, T. K. (2003) Mol. Pharmacol. 64 785-795 [DOI] [PubMed] [Google Scholar]
- 8.Tarran, R., Button, B., and Boucher, R. C. (2006) Annu. Rev. Physiol. 68 543-561 [DOI] [PubMed] [Google Scholar]
- 9.Kunzelmann, K., Scheidt, K., Scharf, B., Ousingsawat, J., Schreiber, R., Wainwright, B., and McMorran, B. (2006) FASEB J. 20 545-546 [DOI] [PubMed] [Google Scholar]
- 10.Button, B., Picher, M., and Boucher, R. C. (2007) J. Physiol. (Lond.) 580 577-592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Guyot, A., and Hanrahan, J. W. (2002) J. Physiol. (Lond.) 545 199-206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lazarowski, E. R., Tarran, R., Grubb, B. R., van Heusden, C. A., Okada, S., and Boucher, R. C. (2004) J. Biol. Chem. 279 36855-36864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Picher, M., Burch, L. H., Hirsh, A. J., Spychala, J., and Boucher, R. C. (2003) J. Biol. Chem. 278 13468-13479 [DOI] [PubMed] [Google Scholar]
- 14.Picher, M., Burch, L. H., and Boucher, R. C. (2004) J. Biol. Chem. 279 20234-20241 [DOI] [PubMed] [Google Scholar]
- 15.Picher, M., and Burch, L. (2006) Purin. Signal 2 399-408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Morse, D. M., Smullen, J. L., and Davis, C. W. (2001) Am. J. Physiol. 280 C1485-C1497 [DOI] [PubMed] [Google Scholar]
- 17.Huang, P., Lazarowski, E. R., Tarran, R., Milgram, S. L., Boucher, R. C., and Stutts, M. J. (2001) Proc. Natl. Acad. Sci. U. S. A. 98 14120-14125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tarran, R., Button, B., Picher, M., Paradiso, A. M., Ribeiro, C. M. P., Lazarowski, E. R., Zhang, L., Collins, P. L., Pickles, R. J., Fredburg, J. J., and Boucher, R. C. (2005) J. Biol. Chem. 280 35751-35759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Boucher, R. C. (2007) Annu. Rev. Med. 58 157-170 [DOI] [PubMed] [Google Scholar]
- 20.Okada, S. F., Nicholas, R. A., Kreda, S. M., Lazarowski, E. R., and Boucher, R. C. (2006) J. Biol. Chem. 281 22992-23002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Takemura, H., Takamura, Y., Isono, K., Tamaoki, J., Nagai, A., and Kawahara, K. (2003) Jpn. J. Physiol. 53 319-326 [DOI] [PubMed] [Google Scholar]
- 22.Zimmermann, H. (2000) Naunyn-Schmiedeberg's Arch. Pharmacol. 362 299-309 [DOI] [PubMed] [Google Scholar]
- 23.Millán, J. (2006) Purin. Signal 2 335-341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Picher, M., and Boucher, R. C. (2000) Am. J. Respir. 23 255-261 [DOI] [PubMed] [Google Scholar]
- 25.Donaldson, S. H., Lazarowski, E. R., Picher, M., Knowles, M. R., Stutts, M. J., and Boucher, R. C. (2000) Mol. Med. 6 969-982 [PMC free article] [PubMed] [Google Scholar]
- 26.Picher, M., and Boucher, R. C. (2001) Drug Dev. Res. 52 66-75 [Google Scholar]
- 27.Donaldson, S. H., Picher, M., and Boucher, R. C. (2002) Am. J. Respir. Cell Mol. Biol. 26 209-215 [DOI] [PubMed] [Google Scholar]
- 28.Picher, M., and Boucher, R. C. (2003) J. Biol. Chem. 278 11256-11264 [DOI] [PubMed] [Google Scholar]
- 29.Hirsh, A. J., Stonebraker, J., van Heusden, C. A., Lazarowski, E. R., Boucher, R. C., and Picher, M. (2007) Biochemistry 46 10373-10383 [DOI] [PubMed] [Google Scholar]
- 30.Fulcher, M. L., Gabriel, S., Burns, K. A., Yankaskas, J. R., and Randell, S. H. (2005) Methods Mol. Med. 107 183-206 [DOI] [PubMed] [Google Scholar]
- 31.Kahner, B. N., Shankar, H., Murugappan, S., Prasad, G. L., and Kunapuli, S. P. (2006) J. Thromb. Haemost. 4 2317-2326 [DOI] [PubMed] [Google Scholar]
- 32.Valero, E., Varon, R., and Garcia-Carmona, F. (2006) FEBS Lett. 273 3598-3613 [DOI] [PubMed] [Google Scholar]
- 33.Dixon, M., and Webb, E. C. (eds) (1979) Enzymes, 2nd Ed., 950 pp., Academic Press, New York
- 34.Christoforidis, S., Papamarcaki, T., Galaris, D., Kellner, R., and Tsolas, O. (1995) Eur. J. Biochem. 234 66-74 [DOI] [PubMed] [Google Scholar]
- 35.Smith, T. M., and Kirley, T. L. (1998) Biochim. Biophys. Acta 1386 65-78 [DOI] [PubMed] [Google Scholar]
- 36.Seargeant, L. E., and Stinson, R. A. (1979) Can. J. Biochem. 57 2000-2007 [PubMed] [Google Scholar]
- 37.Pizauro, J. M., Ciancaglini, P., and Leone, F. A. (1993) Biochim. Biophys. Acta 1202 22-28 [DOI] [PubMed] [Google Scholar]
- 38.Demenis, M. A., Furriel, R. P. M., and Leone, F. A. (2003) Biochim. Biophys. Acta 1646 216-225 [DOI] [PubMed] [Google Scholar]
- 39.Knight, G. E., Bodin, P., De Groat, W. C., and Burnstock, G. (2002) Am. J. Physiol. 282 F281-F288 [DOI] [PubMed] [Google Scholar]
- 40.Hirschberg, C. B., Robbins, P. W., and Abeijon, C. (1998) Annu. Rev. Biochem. 67 49-69 [DOI] [PubMed] [Google Scholar]
- 41.Bodin, P., and Burnstock, G. (2001) J. Cardiovasc. Pharmacol. 38 900-908 [DOI] [PubMed] [Google Scholar]
- 42.Kreda, S. M., Okada, S. F., van Heusden, C. A., O'Neal, W., Gabriel, S., Abdullah, L., Davis, C. W., Boucher, R. C., and Lazarowski, E. R. (2007) J. Physiol. (Lond.) 584 245-259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Adinolfi, E., Pizzirani, C., Idzko, M., Panther, E., Norgauer, J., Di Virgilio, F., and Ferrari, D. (2005) Purin. Signal 1 219-227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Schwiebert, E. M., and Zsembery, A. (2003) Biochim. Biophys. Acta 1615 7-32 [DOI] [PubMed] [Google Scholar]
- 45.Ferrari, D., Pizzirani, C., Adinolfi, E., Lemoli, R. M., Curti, A., Idzko, M., Panther, E., and Di Virgilio, F. (2006) J. Immunol. 176 3877-3883 [DOI] [PubMed] [Google Scholar]
- 46.Eltzschig, H., Weissmüller, T., Mager, A., and Eckle, T. (2006) Methods Mol. Biol. 341 73-87 [DOI] [PubMed] [Google Scholar]
- 47.Spicuzza, L., and Polosa, R. (2003) Curr. Opin. Allergy Clin. Immunol. 3 65-69 [DOI] [PubMed] [Google Scholar]
- 48.Basoglu, O. K., Pelleg, A., Essilfie-Quaye, S., Brindicci, C., Barnes, P. J., and Kharitonov, S. A. (2005) Chest 128 1905-1909 [DOI] [PubMed] [Google Scholar]
- 49.Bennett, W. D., Olivier, K. N., Zeman, K. L., Hohneker, K. W., Boucher, R. C., and Knowles, M. R. (1996) Am. J. Respir. Crit. Care Med. 153 1796-1801 [DOI] [PubMed] [Google Scholar]
- 50.Noone, P. G., Bennett, W. D., Regnis, J. A., Zeman, K. L., Carson, J. L., King, M., Boucher, R. C., and Knowles, M. R. (1999) Am. J. Respir. Crit. Care Med. 160 144-149 [DOI] [PubMed] [Google Scholar]
- 51.Yerxa, B. R., Sabater, J. R., Davis, C. W., Stutts, M. J., Lang-Furr, M., Picher, M., Jones, A. C., Cowlen, M., Dougherty, R., Boyer, J., Abraham, W. M., and Boucher, R. C. (2002) J. Pharmacol. Exp. Ther. 302 871-880 [DOI] [PubMed] [Google Scholar]
- 52.Shaver, S. R., Rideout, J. L., Perdergast, W., Douglass, J. G., Brown, E. G., Boyer, J. L., Patel, R. I., Redick, C. C., Jones, A. C., Picher, M., and Yerxa, B. (2005) Purin. Signal 1 183-191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Munkonda, M. N., Kauffenstein, G., Kukulski, F., Levesque, S. A., Legendre, C., Pelletier, J., Lavoie, E. G., Lecka, J., and Sevigny, J. (2007) Biochem. Pharmacol. 74 1524-1534 [DOI] [PubMed] [Google Scholar]
- 54.Deterding, R., Retsch-Bogart, G., Milgram, L., Gibson, R., Daines, C., Zeitlin, P. L., Milla, C., Marshall, B., Lavange, L., Engels, J., Mathews, D., Gorden, J., Schaberg, A., and Bonnie, W. (2005) Pediatr. Pulmonol. 39 339-348 [DOI] [PubMed] [Google Scholar]
- 55.Sunderman, F. W. (1990) Ann. Clin. Lab. Sci. 20 123-139 [PubMed] [Google Scholar]
- 56.Van Belle, H. (1976) Clin. Chem. 22 972-976 [PubMed] [Google Scholar]
- 57.Slakey, L. L., Cosimini, K., Earls, J. P., Thomas, C., and Gordon, J. L. (1986) J. Biol. Chem. 261 15505-15507 [PubMed] [Google Scholar]
- 58.Pearson, J. D., Carleton, J. S., and Gordon, J. L. (1980) Biochem. J. 190 421-429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Gordon, E. L., Pearson, J. D., and Slakey, L. L. (1986) J. Biol. Chem. 261 15496-15507 [PubMed] [Google Scholar]
- 60.Ikehara, Y., Mansho, K., Takahashi, K., and Kato, K. (1978) J. Biochem. (Tokyo) 83 1471-1483 [DOI] [PubMed] [Google Scholar]
- 61.Say, J. C., Ciuffi, K., Furriel, R. P., Ciancaglini, P., and Leone, F. A. (1991) Biochim. Biophys. Acta 1074 256-262 [DOI] [PubMed] [Google Scholar]
- 62.Burnstock, G. (2006) Novartis Found. Symp. 276 26-48 [PubMed] [Google Scholar]
- 63.Pankratov, Y., Lalo, U., Verkhratsky, A., and North, R. A. (2006) Pfluegers Arch. 452 589-597 [DOI] [PubMed] [Google Scholar]
- 64.Kukulski, F., Levesque, S. A., Lavoie, E. G., Lecka, J., Bigonnesse, F., Knowles, A. F., Robson, S., Kirley, T. L., and Sevigny, J. (2005) Purin. Signal 1 193-204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Gayle, S., and Burnstock, G. (2005) Cell Tissue Res. 319 27-36 [DOI] [PubMed] [Google Scholar]
- 66.Kim, C. H., Kim, S. S., Choi, J. Y., Shin, J. H., Kim, J. Y., Namkung, W., Lee, J. G., Lee, M. G., and Yoon, J. H. (2004) Am. J. Physiol. 287 L835-L842 [DOI] [PubMed] [Google Scholar]
- 67.Taylor, A. L., Schwiebert, L. M., Smith, J. J., King, C., Jones, J. R., Sorscher, E. J., and Schwiebert, E. M. (1999) J. Clin. Investig. 104 875-884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Evans, C. M., Williams, O. W., Tuvim, M. J., Nigam, R., Mixides, G. P., Blackburn, M. R., DeMayo, F. J., Burns, A. R., Smith, C., Reynolds, S. D., Stripp, B. R., and Dickey, B. F. (2004) Am. J. Respir. Cell Mol. Biol. 31 382-394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Vaughan, R. P., Szewczyk, M. T., Lanosa, M. J., DeSesa, C. R., Gianutsos, G., and Morris, J. B. (2006) Toxicol. Sci. 93 411-421 [DOI] [PubMed] [Google Scholar]
- 70.Fabre, A. C., Vantourout, P., Champagne, E., Tercé, F., Rolland, C., Perret, B., Collet, X., Barbaras, R., and Martinez, L. O. (2006) Cell. Mol. Life Sci. 63 2829-2837 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Le Hir, M., Gandhi, R., and Dubach, U. C. (1989) Enzyme 41 87-93 [DOI] [PubMed] [Google Scholar]
- 72.Stefanovic, V., Savic, V., Vlahovic, P., Ardaillou, N., and Ardaillou, R. (1988) Renal Physiol. Biochem. 11 89-102 [DOI] [PubMed] [Google Scholar]
- 73.Mitchell, C. H., Carre, D. A., McGlinn, A. M., Stone, R. A., and Civan, M. M. (1998) Proc. Natl. Acad. Sci. U. S. A. 95 7174-7178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Hazama, A., Hayashi, S., and Okada, Y. (1998) Pfluegers Archiv. Eur. J. Physiol. 437 31-35 [DOI] [PubMed] [Google Scholar]
- 75.Hazama, A., Shimizu, T., Ando-Akatsuka, Y., Hayashi, S., Tanaka, S., Maeno, E., and Okada, Y. (1999) J. Gen. Physiol. 114 525-533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Matharu, A.-L., Mundell, S. J., Benovic, J. L., and Kelly, E. (2001) J. Biol. Chem. 276 30199-30207 [DOI] [PubMed] [Google Scholar]
- 77.Mohsenin, A., and Blackburn, M. (2006) Curr. Opin. Pulm. Med. 12 54-59 [DOI] [PubMed] [Google Scholar]
- 78.Gordon, E. L., Pearson, J. D., Dickinson, E. S., Moreau, D., and Slakey, L. L. (1989) J. Biol. Chem. 264 18986-18995 [PubMed] [Google Scholar]
- 79.Fadini, G. P., Pauletto, P., Avogaro, A., and Rattazzi, M. (2007) Atherosclerosis 193 241-244 [DOI] [PubMed] [Google Scholar]
- 80.Fleetwood, G., Coade, S. B., Gordon, J. L., and Pearson, J. D. (1989) Am. J. Physiol. 256 H1565-H1572 [DOI] [PubMed] [Google Scholar]
- 81.Meghji, P., Pearson, J. D., and Slakey, L. L. (1995) Biochem. J. 308 725-731 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Hourani, S. M. (1996) J. Auton. Pharmacol. 16 349-352 [DOI] [PubMed] [Google Scholar]