

Abstract
Enzyme structures solved with and without bound substrate often show that substrate-induced conformational changes bring catalytic residues into alignment, alter the local environment, and position the substrate for catalysis. Although the structural data are compelling, the role of conformational changes in enzyme specificity has been controversial in that specificity is a kinetic property that is not easy to predict based upon structure alone. Recent studies on DNA polymerization have illuminated the role of substrate-induced conformational changes in enzyme specificity by showing that the rate at which the enzyme opens to release the bound substrate is a key kinetic parameter. The slow release of a correct substrate commits it to the forward reaction so that specificity is determined solely by the rate of substrate binding, including the isomerization step, and not by the slower rate of the chemical reaction. In contrast, fast dissociation of an incorrect substrate favors release rather than reaction. Thus, the conformational change acts as a molecular switch to select the right substrate and to recognize and disfavor the reaction of an incorrect substrate. A conformational switch may also favor release rather than reverse reaction of the product.
The role of induced fit in enzyme specificity has been controversial. On the one hand, it is apparent that catalysis is facilitated by the rapid binding of a substrate to an open form of the enzyme, whereas the chemical reaction is accelerated most efficiently by the precise alignment of amino acids surrounding the substrate and by the altered reaction environment in the closed state. At this level, it is apparent that changes in enzyme structure may simply solve the disparate demands for an open site to allow fast binding and a closed site to afford fast catalysis. It has been argued that one- and two-step binding reactions would lead to the same end point with the same free energy change, and therefore, induced fit can do nothing to improve catalytic efficiency or specificity (1). However, these arguments fail to consider the rates at which the enzyme closes and opens following the initial substrate binding.
A general mechanism, including isomerization after an initial weak substrate binding, can be described by the following reaction sequence (Scheme 1).
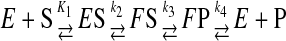 |
SCHEME 1 |
The substrate first binds to the open form of the enzyme (E) and then induces a change in structure to the closed form (FS). The relative values for the forward and reverse rates of the conformational change (k2 and k–2) and the rate of the chemical reaction (k3) provide the key to understanding the role of the conformational change in enzyme specificity. Surprisingly, the rate of the reverse conformational change (k–2) is an important specificity determinant (2).
The term induced fit was first proposed to explain why hexokinase had such low ATPase activity. The simple logic said that when ATP bound in the absence of glucose, the ATP must be protected from water, whereas the binding of glucose must induce a change in structure to make the ATP accessible (3). This basic concept was confirmed by the solution of the crystal structure of hexokinase (4). Decades later, the solution of the structure of the ribosome has revealed a “steric switch mechanism” that protects the peptidyl-tRNA from hydrolysis, whereas the binding of a cognate aa-tRNA2 induces a change in structure to allow reaction (5). Steps involved in delivering the aa-tRNA to the ribosome are complex, involving EF-Tu and the hydrolysis of GTP, but nonetheless reveal some common underlying themes (6).
Structures provide clues as to how a desired reaction can be favored by the alignment of catalytic residues. However, the manner in which changes in enzyme structure influence specificity remains obscure, bearing in mind that enzyme specificity is a purely kinetic property with less than obvious structural origins.
DNA polymerases offer a unique model for accessing specificity because the alternate substrates are well defined, and the discrimination between correct and incorrect substrates can be quite high. Analysis of DNA polymerases has led to the suggestion that the binding of the correct base (as defined by base pairing with the template strand) induces a change in structure to facilitate catalysis, whereas the binding of a mismatched base may somehow fail to induce the same organization of catalytic residues (7, 8). Early work on DNA polymerases focused on deducing whether the conformational change may limit and thereby regulate the rate of incorporation (7). Subsequent structural studies demonstrated a large change in the “fingers” domain after binding the correct dNTP, supporting the notion of an induced fit (9). Models were developed suggesting that each step along the pathway (ground state binding, conformational change, and chemistry) contributed a checkpoint such that the net fidelity was the product of the contributions at each step (10), and arguments focused on whether the conformational change or the chemistry step was rate-limiting (11, 12).
We now know that for the past two decades we have been asking the wrong question. To define the role of the conformational change in enzyme specificity, we need to compare the rate of the reverse of the conformational change with the rate of the chemistry step, not the rate of the forward conformational change step. The analysis leading to this unexpected conclusion is summarized below.
To examine the kinetics of the conformational change, a fluorescent label was placed on the fingers domain of T7 DNA polymerase at a position that would be sensitive to changes in protein structure in forming the closed complex (2). This allowed the rate and equilibrium constants governing substrate binding and the conformational change to be measured, and the results were combined with conventional measurement of the rate of the chemistry step by rapid quench-flow methods using radiolabeled DNA. These studies led to the following pathway for correct nucleotide binding and incorporation (Scheme 2),
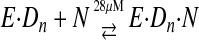 |
SCHEME 2 |
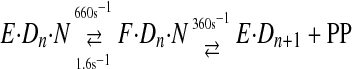 |
where E·Dn represents the enzyme·DNA complex with a primer strand n residues in length, N represents the incoming dNTP, and F·Dn·N represents the closed state of the enzyme. The rates of substrate binding and release (k1 and k–1) are not known but can be approximated based upon conservative estimates of diffusion-controlled binding (k1 ≥ 100 μm–1 s–1) and the measured dissociation constant of 28 μm (k–1 ≥ 2800 s–1). Pyrophosphate release appears to be fast, so this simple three-step mechanism is sufficient to account for processive synthesis (13).
Steady-state analysis of two competing substrates (or two competing enzymes) shows that specificity is a linear function of kcat/Km. Therefore, to understand the role of conformational changes, we need only to define the effect of the substrate-induced conformational changes on the value of kcat/Km. The simple math is reproduced here because the results are quite compelling. The specificity constant for this pathway is defined by Equation 1.
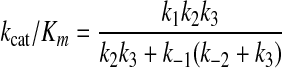 |
(Eq. 1) |
Because the rate of chemistry (k3) is much greater than the reverse of the conformational change step (k–2), this reduces to Equation 2.
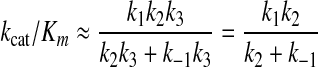 |
(Eq. 2) |
This leads to the surprising conclusion that the rate of chemistry does not enter into the definition of kcat/Km even though it is slower than the conformational change step! This is because the reverse of the conformational change step is so slow relative to chemistry that the substrate is committed to go forward after the conformational change. The equation defining kcat/Km can be further reduced based upon the rapid equilibrium binding of substrate in the collision complex by the comparison indicating that k–1 ≫ k2 to yield Equation 3.
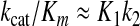 |
(Eq. 3) |
Thus, kcat/Km is defined by the product of the binding constant (K1) for the substrate in the collision complex (ES) and the rate of the conformational change (k2). It is important to note, however, that kcat is not equal to k2 and Km is not equal to 1/K1; additional terms in both kcat and Km cancel in the ratio kcat/Km.
Although it is often taught that kcat/Km is the ratio of kcat and Km, this view often leads to inaccurate conclusions, especially when the assumption is made that kcat measures the rate of chemistry and Km is equal to the ground state dissociation constant for substrate (1). Rather, the specificity constant is best understood as the second-order rate constant for substrate binding times the probability that, once bound, the substrate continues forward to form product. According to our model for correct dNTP incorporation, the second-order rate constant for substrate binding is K1k2, and the probability that the bound substrate continues forward is near unity. This result also brings forth the cautionary note that analysis of the separate contributions of kcat and Km to discrimination can be misleading (14). As seen here, kcat contains terms (notably k3) that do not contribute to the specificity constant for the correct base.
Analysis of the kinetics of the conformational change following the binding of a mismatch yielded more surprises. First, the binding of a mismatch caused an increase in fluorescence, opposite to that seen following the binding of a correct base. This implies that there are three distinct structural states, open (defined by the structure in the absence of bound nucleotide), closed (defined by the structure seen in the ternary enzyme·DNA·dNTP complex), and a unique mismatch recognition state that has not yet been solved crystallographically. Moreover, the kinetics of the fluorescence change after mismatch binding were unusual, suggesting multiple conformational states formed with unfavorable equilibrium constants. The data were fitted to a minimal model (Scheme 3).
 |
SCHEME 3 |
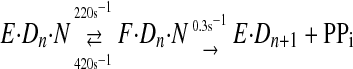 |
Kinetic constants governing misincorporation are quite different from those leading to incorporation of a correct base. The reverse rate of the conformational change is greater than the forward rate, so the isomerization is unfavorable (K2 = 0.5), and the rate of the chemical reaction is reduced ∼1000-fold from that seen with the correct substrate. In this case, simplification of the equation for the specificity constant yields Equation 4.
 |
(Eq. 4) |
The rate of incorporation is so slow relative to the reverse of the conformational change step that the steps equilibrate leading up to incorporation, and therefore, the specificity constant is the product of the two equilibrium constants (binding and isomerization) and the rate of the chemistry step.
The most important conclusion of this analysis is that binding of a mismatched nucleotide leads to a state from which dissociation of the bound nucleotide is favored while the incorporation rate is reduced. The data suggest that the enzyme has evolved to recognize a mismatch and to use binding energy to actively misalign catalytic residues and prevent formation of the tight binding state seen with a correct substrate. Thus, the conformational change acts as a switch to recognize a correct substrate and hold it tightly while aligning catalytic residues or, alternatively, to recognize an incorrect substrate and promote dissociation while misaligning catalytic residues.
The high fidelity of T7 DNA polymerase is due to the kinetic partitioning of the conformational intermediate formed after nucleotide binding. The kinetic partitioning of the conformational isomer (F·Dn·N) is defined by P = k3/(k–2 + k3). A correct base induces a state in which 99.5% of the bound substrates continue in the forward reaction, whereas a mismatch induces a state in which only 0.07% react (see Table 1). The kinetic partitioning of the conformational intermediate is also illustrated in the free energy profile in Fig. 1A. The arrows show how the correct substrate partitions to the forward reaction while the bound mismatch partitions toward release according to the relative heights of the barriers.
TABLE 1.
Kinetic partitioning of conformational intermediates for three enzymes
Shown is a summary of elementary rate constants governing specificity for T7 DNA polymerase (DNAP) (2), protein synthesis by the ribosome (19), and DHFR (20). Kinetic constants are defined in Scheme 1. For DHFR, the initial binding constant (K1) and the isomerization rate (k2) have not been resolved, so the observed second-order rate constant for substrate binding defines the product, K1k2. The kinetic partitioning of the conformational intermediate (FS) is calculated as P = k3/(k3 + k–2).
| Enzyme and substrate | K1 | k2 | k-2 | k3 | P |
|---|---|---|---|---|---|
| μ%m% | s-1 | s-1 | s-1 | ||
| T7 DNAP | |||||
| Correct | 28 | 660 | 1.6 | 360 | 99.5 |
| Mismatch | 200 | 220 | 420 | 0.3 | 0.07 |
| Ribosome | |||||
| Cognate | 0.6a | 190 | 0.23 | 260 | 99.9 |
| Non-cognate | 0.6 | 190 | 80 | 0.4 | 0.5 |
| DHFR | |||||
| H2Fb | K1k2 = 40 μ%m%-1 s-1 | 40 | 950 | 95.9 | |
| H4F | K1k2 = 5 μ%m%-1 s-1 | 200 | 0.6 | 0.3 | |
The initial binding of the aa-tRNA · EF-Tu · GTP complex to the ribosome is not a rapid equilibrium, so kcat/Km ∼ k1k2/(k-1 + k2) (19).
DHFR substrate, dihydrofolate (H2F), and product, tetrahydrofolate (H4F).
FIGURE 1.

Free energy profiles for the T7 DNA polymerase. A, conventional free energy diagram for correct (green; dCTP) and mismatched (red; dGTP) nucleotide incorporation reactions. The free energy was calculated as ΔG‡ = RT(ln(κT/h) – ln(kobs)) kcal/mol using rate constants from Scheme 1. κ is the Boltzmann constant, T is 293 K, h is Planck's constant, and kobs is the first-order rate constant. The nucleotide concentration was set equal to 100 μm. B, proposed three-dimensional free energy diagram taking conclusions from our fluorescence studies into account. The diagram includes the alignment of active-site residues as a third axis. This figure was reproduced from Ref. 2 with permission.
It has been argued that fidelity of a polymerase is a function of sequential kinetic checkpoints, the first during ground state binding, the second during the conformational change step, and the third attributable to the rate of chemistry (10). The proposal that the net selectivity is simply the product of the selectivity values at the three checkpoints is not valid, at least for the high fidelity T7 DNA polymerase. In this case, the net discrimination (D) is the ratio of kcat/Km values for correct and incorrect substrates and is given by Equation 5.
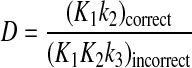 |
(Eq. 5) |
In this case, only the ratio of the ground state binding constants (K1) for correct and incorrect substrates enters into the value for the net discrimination.
It remains to be established whether other DNA polymerases follow this same pattern; as of this date, no studies have been reported in which the reverse rate (k–2) has been measured for any of the other thoroughly studied polymerases. For the past two decades, we and others have been focusing on whether the conformational change or chemistry was rate-limiting (comparing k2 and k3, respectively). However, we should have been comparing the rate of the reverse of the conformation change with the rate of the chemical reaction (k–2 and k3, respectively).
In relating these kinetic measurements to structure, one is left with the challenge of predicting how the rate of the conformational change can be influenced by the structure of the enzyme and the nature of the interactions with the weakly bound substrate. Certainly, the macroscopic parameters that we measure as K1k2 must be the composite of many multiple equilibria contributing to the ground state binding leading to a small fraction of enzyme that achieves a critical state whereby enough contacts are made to recognize the correct substrate and trigger the larger change in structure. There may be smaller structural changes to bring catalytic residues into precise alignment for catalysis, and recent work has suggested that coupled motions at higher frequency ultimately bring the reactants to the nearest alignment needed to reach the transition state (8, 15). These motions would be a component of the net observed rate constant for the chemistry step (k3). As enzymes evolve, kcat is an important parameter especially for a DNA polymerase where the demands cannot be met by expressing more enzyme. However, once a threshold is reached where k3 is greater than k–2, further enhancements in k3 do not improve specificity; they only increase kcat.
We proposed that the correct and incorrect substrates induce distinctly different structural states based upon the observation that the binding of a mismatch induced a change in fluorescence opposite in sign to that observed after binding a correct base (2). An incorrect base reacts at a rate 1000-fold slower than a correct base pair even though the chemical reaction centers for correct and incorrect base pairs are identical. Therefore, these data imply that the binding of a mismatch leads to misalignment of catalytic residues, suggesting a three-dimensional free energy plot shown in Fig. 1B. In this figure, the free energy at each of the minima and at each saddle point is defined by our measurements, but the scale of the structural alignment/misalignment is unspecified. Other investigators have proposed a three-dimensional free energy plot, but these theoretical plots expand upon the microheterogeneity within the saddle point in the transition state from the tightly bound substrate to product (8, 15) and therefore address the fine points of the transition state, but do not address the kinetic partitioning of the conformational intermediate as it relates to specificity.
Most theoretical analysis has focused on the proficiency (16) of enzyme catalysis by attempting to compute how the enzyme stabilizes the transition state and facilitates the chemical reaction at the active site (8, 15). Perhaps an even bigger challenge would be to compute how the weak binding of the substrate can then trigger a large conformational change leading to tighter binding and the alignment of catalytic residues. Some computational approaches may be capable to analyzing such largescale and relatively slow movements (17).
Analysis of enzyme families has led to the division of enzyme active sites into catalytic and specificity domains (18). Although this distinction is logical from a purely structural and genetic perspective, it is only when residues in the specificity domain bind and properly align the substrate, in concert with the catalytic residues, that such binding can be translated into catalytic efficiency and specificity. Nonetheless, it is conceivable that the specificity domains establish specificity by binding the substrates tightly (reducing k–2), thereby committing them to the forward reaction.
Selectivity of the cognate aa-tRNA during protein synthesis is also dominated by the attenuation of the substrate dissociation rate for the correct (cognate) substrate (19). The aa-tRNA, in a complex with EF-Tu·GTP, binds weakly to the ribosome. Codon recognition leads to activation of GTP hydrolysis, which is the rate-limiting step leading to accommodation of the aa-tRNA into the A-site of the ribosome and rapid peptide bond formation. Cognate and near-cognate aa-tRNA molecules are distinguished kinetically by the forward versus reverse partitioning of an intermediate formed after a conformational change (see Table 1). For a cognate aa-tRNA, 99.9% of the intermediates proceed forward (260 s–1 forward versus 0.23 s–1 reverse), whereas for a non-cognate aa-tRNA, 99.5% dissociate (0.4 s–1 forward versus 80 s–1 reverse). Although the subsequent reaction sequence is complex with multiple points where changes in conformation are responsible for rapid incorporation, it is interesting to note that the major point of discrimination occurs through the kinetic partitioning of the conformational isomer formed after the initial substrate binding.
DHFR has been shown to undergo conformational changes upon substrate binding, but none of these appear to be ratelimiting (20). Under these conditions, one can understand the second-order rate constant for substrate binding as the product of a weak binding constant for the initial collision complex and the rate of isomerization, kon = K1k2 (Scheme 1). Accordingly, the kinetic partitioning of the conformational isomer is defined by the relative values for k3 and k–2 (Table 1).
For the substrate dihydrofolate, the observed substrate release rate is 40 s–1, whereas the rate of the chemistry step is 950 s–1. Given the relative magnitudes of k–2 and k3, the specificity constant for dihydrofolate is determined solely by the rate of binding, kon = K1k2 = 40 μm–1 s–1. Although the kinetics of an incorrect substrate have not been defined, one can examine the rates observed for the product of the reaction, tetrahydrofolate. Here, considered as a reaction defined in the forward direction with tetrahydrofolate as the substrate, k–2 = 200 s–1, whereas k3 = 0.6 s–1, so clearly, the kinetic partitioning favors release rather than chemistry. Once again, the decision to do chemistry or release the bound substrate appears to be made during the conformational change.
Recent studies on DHFR have focused on coupled motions occurring on a faster time scale and thought to be critical for facilitating catalysis (21). Moreover, it has been shown that the motions occurring during or after the chemical reaction bring the enzyme from a “closed” state, with the substrates bound, to a so-called “occluded” state, with the products bound (22). This structural transition, apparently coupled to or following catalysis, also changes the kinetic partitioning from favoring reaction of the bound substrate to favoring release of the bound product. It is particularly interesting to note that enzymes may have evolved not only to use the conformational changes to favor the tight binding and alignment of the desired substrate, but also to use conformational changes to promote the release rather than reverse reaction of the product of the reaction.
The initial formulation of the concept of an induced fit was understandably superficial, that an undesired side reaction can be precluded, whereas the desired reaction can be facilitated by structural rearrangements dependent upon substrate binding. From this beginning, it is now apparent that the essential role of conformational changes in alignment of catalytic residues and changing the environment at the active site to promote catalysis seems to be nearly universal. Moreover, it now appears that the converse may also be true in suggesting that the misalignment of catalytic residues might be used to slow the reaction of an undesired substrate (or product of the reaction) while promoting its dissociation.
Theoretical arguments against the ability of an induced fit mechanism to alter specificity have each been predicated by assumptions that, in retrospect, appear to be circular. For example, Fersht (1) argued against an induced fit mechanism for specificity based upon the assumption that the isomerization reaction was fast and not rate-limiting and that the structure of the closed state was identical for all substrates. The pathway does not matter if we consider only equilibrium end points, and therefore, it is obvious that there is no difference between pathways with or without a conformational change if all steps reach equilibrium. In this sense, the assumption of rapid equilibria and the conclusion that the pathway does not matter are circularly connected. Herschlag (23) argued on theoretical grounds that the conformational change would need to be rate-limiting to affect specificity, and our early work (7) on DNA polymerases suggested that this was the case. Post and Ray (24) introduced the concept that the structure of the enzyme·substrate complex in the transition state may differ between correct and incorrect substrates, thus allowing for a specificity difference due to the induced fit even if the conformational change was part of a rapid equilibrium binding. Our most recent data suggest that the conformational change is not rate-limiting but still dominates specificity and that correct and incorrect substrates induce different conformational states.
In the examples described here, either the chemistry step is largely irreversible (k3 ≫ k–3) or the product release is fast (k4 ≫ k3), so the rates of k–3 and k4 do not influence specificity, although they can in the most general case. For example, it has been noted that the human mitochondrial DNA polymerase has a novel mechanism of discrimination against azidothymidine and 8-oxo-dGTP, whereby pyrophosphate release is slow, allowing the chemical reaction to come to equilibrium at the active site (13, 25). The reverse reaction increases the concentration of the ES complex, thereby increasing the probability of substrate release and decreasing kcat/Km. Perhaps an important lesson here is that, in the most general sense, specificity can be influenced by any step in the pathway, and all kinetically significant intermediates need to be measured to derive an accurate description.
In summary, we have now shown the importance of the substrate dissociation rate in governing selectivity, so the conformational change step need not be rate-limiting to dominate the specificity constant. Indeed, even if chemistry is rate-limiting in the steady state, it will have no influence on specificity as long as the rate of chemistry is greater than the rate at which the substrate dissociates. In addition, we have provided evidence that the conformational states differ after the binding of a correct versus an incorrect substrate (2).
In the final analysis, theories based upon simplifying assumptions are likely to be misleading. Rather, it appears as though enzymes have evolved to take advantage of any trick possible to improve specificity and efficiency at physiological substrate concentrations. A substrate can be favored by binding tighter in the collision complex, by inducing a faster rate of the conformational change, and by achieving a closed state with a lower substrate release rate and an optimal alignment of catalytic residues. In addition, reducing k–3 and increasing k4 (Scheme 1) promote product release. In contrast, an incorrect substrate may be disfavored by weaker binding to the collision complex and isomerization to a state that promotes rapid release while misaligning catalytic residues. Moreover, increasing k–3 and decreasing k4 can also decrease kcat/Km. There is no justification for assuming that an enzyme is somehow precluded from taking advantage of any of these structural and kinetic maneuvers to improve specificity.
The isomerization following substrate binding appears to operate as a molecular switch by sensing whether to hold on to a substrate tightly and organize residues to promote catalysis or to favor release of an undesired substrate while attenuating the rate of its reaction. Coupled motions of a smaller magnitude and on a faster time scale may then further facilitate the chemical reaction. The dynamic flexibility of enzymes is clearly an important component leading to their remarkable specificity and efficiency.
Supplementary Material
This work was supported, in whole or in part, by National Institutes of Health Grant R01 GM071404. This work was also supported by Welch Foundation Grant F-1604. This minireview will be reprinted in the 2008 Minireview Compendium, which will be available in January, 2009.
Footnotes
The abbreviations used are: aa-tRNA, aminoacyl-tRNA; EF-Tu, elongation factor Tu; dNTP, deoxynucleoside triphosphate; DHFR, dihydrofolate reductase.
References
- 1.Fersht, A. R. (1999) Enzyme Structure and Mechanism, 3rd Ed., W. H. Freeman, New York
- 2.Tsai, Y. C., and Johnson, K. A. (2006) Biochemistry 45 9675–9687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Koshland, D. E. (1959) in The Enzymes (Boyer, P. D., Lardy, H., and Myrback, K., eds) Vol. 1, 2nd Ed., pp. 305–346, Academic Press, New York [Google Scholar]
- 4.Bennett, W. S., Jr., and Steitz, T. A. (1978) Proc. Natl. Acad. Sci. U. S. A. 75 4848–4852 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schmeing, T. M., Huang, K. S., Strobel, S. A., and Steitz, T. A. (2005) Nature 438 520–524 [DOI] [PubMed] [Google Scholar]
- 6.Rodnina, M. V., Gromadski, K. B., Kothe, U., and Wieden, H. J. (2005) FEBS Lett. 579 938–942 [DOI] [PubMed] [Google Scholar]
- 7.Johnson, K. A. (1993) Annu. Rev. Biochem. 62 685–713 [DOI] [PubMed] [Google Scholar]
- 8.Xiang, Y., Goodman, M. F., Beard, W. A., Wilson, S. H., and Warshel, A. (2008) Proteins 70 231–247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Doublie, S., Tabor, S., Long, A. M., Richardson, C. C., and Ellenberger, T. (1998) Nature 391 251–258 [DOI] [PubMed] [Google Scholar]
- 10.Joyce, C. M., and Benkovic, S. J. (2004) Biochemistry 43 14317–14324 [DOI] [PubMed] [Google Scholar]
- 11.Showalter, A. K., and Tsai, M. D. (2002) Biochemistry 41 10571–10576 [DOI] [PubMed] [Google Scholar]
- 12.Rothwell, P. J., Mitaksov, V., and Waksman, G. (2005) Mol. Cell 19 345–355 [DOI] [PubMed] [Google Scholar]
- 13.Hanes, J. W., and Johnson, K. A. (2007) Nucleic Acids Res. 35 6973–6983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Echols, H., and Goodman, M. F. (1991) Annu. Rev. Biochem. 60 477–511 [DOI] [PubMed] [Google Scholar]
- 15.Benkovic, S. J., Hammes, G. G., Hammes-Schiffer, S., Wong, K. F., Selzer, T., Swanwick, R. S., Shrimpton, P. J., Allemann, R. K., Rajagopalan, P. T., Lutz, S., Agarwal, P. K., and Billeter, S. R. (2008) Biochemistry 47 3317–3321 [DOI] [PubMed] [Google Scholar]
- 16.Miller, B. G., and Wolfenden, R. (2002) Annu. Rev. Biochem. 71 847–885 [DOI] [PubMed] [Google Scholar]
- 17.Elber, R. (2005) Curr. Opin. Struct. Biol. 15 151–156 [DOI] [PubMed] [Google Scholar]
- 18.Gerlt, J. A., and Babbitt, P. C. (2001) Annu. Rev. Biochem. 70 209–246 [DOI] [PubMed] [Google Scholar]
- 19.Wilden, B., Savelsbergh, A., Rodnina, M. V., and Wintermeyer, W. (2006) Proc. Natl. Acad. Sci. U. S. A. 103 13670–13675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fierke, C. A., Johnson, K. A., and Benkovic, S. J. (1987) Biochemistry 26 4085–4092 [DOI] [PubMed] [Google Scholar]
- 21.Venkitakrishnan, R. P., Zaborowski, E., McElheny, D., Benkovic, S. J., Dyson, H. J., and Wright, P. E. (2004) Biochemistry 43 16046–16055 [DOI] [PubMed] [Google Scholar]
- 22.Hammes-Schiffer, S., Benkovic, S. J., Wong, K. F., Selzer, T., Swanwick, R. S., Shrimpton, P. J., Allemann, R. K., Rajagopalan, P. T., Lutz, S., Agarwal, P. K., and Billeter, S. R. (2006) Annu. Rev. Biochem. 75 519–541 [DOI] [PubMed] [Google Scholar]
- 23.Herschlag, D. (1988) Bioorg. Chem. 16 62–96 [Google Scholar]
- 24.Post, C. B., and Ray, W. J., Jr. (1995) Biochemistry 34 15881–15885 [DOI] [PubMed] [Google Scholar]
- 25.Hanes, J. W., Thal, D. M., and Johnson, K. A. (2006) J. Biol. Chem. 281 36241–36248 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.