

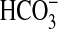
Abstract
In the medullary thick ascending limb, inhibiting the basolateral NHE1
Na+/H+ exchanger with nerve growth factor (NGF) induces
actin cytoskeleton remodeling that secondarily inhibits apical NHE3 and
transepithelial 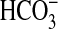 absorption. The
inhibition by NGF is mediated 50% through activation of extracellular
signal-regulated kinase (ERK). Here we examined the signaling pathway
responsible for the remainder of the NGF-induced inhibition. Inhibition of
absorption. The
inhibition by NGF is mediated 50% through activation of extracellular
signal-regulated kinase (ERK). Here we examined the signaling pathway
responsible for the remainder of the NGF-induced inhibition. Inhibition of
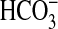 absorption was reduced 45% by the
phosphatidylinositol 3-kinase (PI3K) inhibitors wortmannin or LY294002 and 50%
by rapamycin, a specific inhibitor of mammalian target of rapamycin (mTOR), a
downstream effector of PI3K. The combination of a PI3K inhibitor plus
rapamycin did not cause a further reduction in the inhibition by NGF. In
contrast, the combination of a PI3K inhibitor plus the MEK/ERK inhibitor U0126
completely eliminated inhibition by NGF. Rapamycin decreased NGF-induced
inhibition of basolateral NHE1 by 45%. NGF induced a 2-fold increase in
phosphorylation of Akt, a PI3K target linked to mTOR activation, and a
2.2-fold increase in the activity of p70 S6 kinase, a downstream effector of
mTOR. p70 S6 kinase activation was blocked by wortmannin and rapamycin,
consistent with PI3K, mTOR, and p70 S6 kinase in a linear pathway.
Rapamycin-sensitive inhibition of NHE1 by NGF was associated with an increased
level of phosphorylated mTOR in the basolateral membrane domain. These
findings indicate that NGF inhibits
absorption was reduced 45% by the
phosphatidylinositol 3-kinase (PI3K) inhibitors wortmannin or LY294002 and 50%
by rapamycin, a specific inhibitor of mammalian target of rapamycin (mTOR), a
downstream effector of PI3K. The combination of a PI3K inhibitor plus
rapamycin did not cause a further reduction in the inhibition by NGF. In
contrast, the combination of a PI3K inhibitor plus the MEK/ERK inhibitor U0126
completely eliminated inhibition by NGF. Rapamycin decreased NGF-induced
inhibition of basolateral NHE1 by 45%. NGF induced a 2-fold increase in
phosphorylation of Akt, a PI3K target linked to mTOR activation, and a
2.2-fold increase in the activity of p70 S6 kinase, a downstream effector of
mTOR. p70 S6 kinase activation was blocked by wortmannin and rapamycin,
consistent with PI3K, mTOR, and p70 S6 kinase in a linear pathway.
Rapamycin-sensitive inhibition of NHE1 by NGF was associated with an increased
level of phosphorylated mTOR in the basolateral membrane domain. These
findings indicate that NGF inhibits
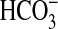 absorption in the medullary thick
ascending limb through the parallel activation of PI3K-mTOR and ERK signaling
pathways, which converge to inhibit NHE1. The results identify a role for mTOR
in the regulation of Na+/H+ exchange activity and
implicate NHE1 as a possible downstream effector contributing to mTOR's
effects on cell growth, proliferation, survival, and tumorigenesis.
absorption in the medullary thick
ascending limb through the parallel activation of PI3K-mTOR and ERK signaling
pathways, which converge to inhibit NHE1. The results identify a role for mTOR
in the regulation of Na+/H+ exchange activity and
implicate NHE1 as a possible downstream effector contributing to mTOR's
effects on cell growth, proliferation, survival, and tumorigenesis.
The Na+/H+ exchanger isoform NHE12 is expressed ubiquitously in the plasma membrane of nonpolarized cells and in the basolateral membrane of epithelial cells, where it plays essential roles in basic cell functions such as the maintenance of intracellular pH (pHi) and cell volume (1–3). NHE1 is involved in other important cellular processes, including proliferation, survival, adhesion, migration, and tumor formation (2–7). These specialized functions involve regulation of NHE1 by a variety of receptor-mediated signaling networks as well as physical interactions of NHE1 with the actin cytoskeleton (2, 3, 5). By comparison, the role of NHE1 in epithelial function remains poorly understood. In particular, the contributions of NHE1 to transcellular acid-base transport and the possible mechanisms involved are largely undefined.
The medullary thick ascending limb (MTAL) of the mammalian kidney
participates in acid-base regulation by reabsorbing most of the filtered
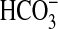 not reabsorbed by the proximal
tubule (8,
9). Absorption of
not reabsorbed by the proximal
tubule (8,
9). Absorption of
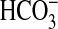 by the MTAL depends on
H+ secretion mediated by the apical membrane NHE3
Na+/H+ exchanger
(8,
10–13)
and basolateral
by the MTAL depends on
H+ secretion mediated by the apical membrane NHE3
Na+/H+ exchanger
(8,
10–13)
and basolateral 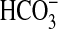 efflux, which
involves Cl-/
efflux, which
involves Cl-/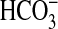 exchange
(14). The MTAL also expresses
basolateral NHE1, and we have recently identified a novel role for this
exchanger in transepithelial
exchange
(14). The MTAL also expresses
basolateral NHE1, and we have recently identified a novel role for this
exchanger in transepithelial 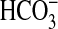 absorption. Inhibition of NHE1 with amiloride or nerve growth factor (NGF) or
by NHE1 knock-out results secondarily in inhibition of apical NHE3, thereby
decreasing
absorption. Inhibition of NHE1 with amiloride or nerve growth factor (NGF) or
by NHE1 knock-out results secondarily in inhibition of apical NHE3, thereby
decreasing 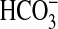 absorption
(15–17).
NHE1 modulates NHE3 activity by regulating the organization of the actin
cytoskeleton (18). The rate of
luminal H+ secretion and transepithelial
absorption
(15–17).
NHE1 modulates NHE3 activity by regulating the organization of the actin
cytoskeleton (18). The rate of
luminal H+ secretion and transepithelial
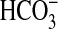 absorption in the MTAL thus
depends on a regulatory interaction between the basolateral and apical
membrane Na+/H+ exchangers, whereby basolateral NHE1
enhances the activity of apical NHE3
(15–18).
absorption in the MTAL thus
depends on a regulatory interaction between the basolateral and apical
membrane Na+/H+ exchangers, whereby basolateral NHE1
enhances the activity of apical NHE3
(15–18).
Based on the above findings, a key to understanding the role of NHE1 in
epithelial function lies in identifying cell signals that modify transcellular
acid transport through effects on basolateral NHE1 activity. Recently we
demonstrated that NGF inhibits 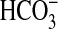 absorption in the MTAL by inhibiting NHE1 through activation of the
extracellular signal-regulated kinase (ERK) signaling pathway
(19). However, blocking ERK
activation eliminated only ∼50% of the NGF-induced transport regulation
(19). Thus, an additional
signaling pathway must play a role in mediating the inhibition of NHE1 by
NGF.
absorption in the MTAL by inhibiting NHE1 through activation of the
extracellular signal-regulated kinase (ERK) signaling pathway
(19). However, blocking ERK
activation eliminated only ∼50% of the NGF-induced transport regulation
(19). Thus, an additional
signaling pathway must play a role in mediating the inhibition of NHE1 by
NGF.
The present study was designed to identify the signaling pathway
responsible for the remainder of NGF-induced inhibition of NHE1 and
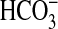 absorption in the MTAL. We show
that NGF decreases
absorption in the MTAL. We show
that NGF decreases 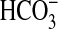 absorption
through a phosphatidylinositol 3-kinase (PI3K)-mammalian target of rapamycin
(mTOR)-dependent signaling pathway that functions in parallel with ERK to
inhibit NHE1. These studies identify a previously unrecognized role for mTOR
in regulating Na+/H+ exchange activity and the
absorptive function of renal tubules. The results implicate NHE1 as a possible
downstream effector contributing to mTOR's effects on cell growth,
proliferation, and tumorigenesis.
absorption
through a phosphatidylinositol 3-kinase (PI3K)-mammalian target of rapamycin
(mTOR)-dependent signaling pathway that functions in parallel with ERK to
inhibit NHE1. These studies identify a previously unrecognized role for mTOR
in regulating Na+/H+ exchange activity and the
absorptive function of renal tubules. The results implicate NHE1 as a possible
downstream effector contributing to mTOR's effects on cell growth,
proliferation, and tumorigenesis.
EXPERIMENTAL PROCEDURES
Tubule Perfusion and Measurement of Net
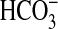 Absorption—MTALs from
male Sprague-Dawley rats (50–100 g body wt; Taconic, Germantown, NY)
were isolated and perfused in vitro as previously described
(16,
20). Tubules were dissected
from the inner stripe of the outer medulla at 10 °C in control bath
solution (see below), transferred to a bath chamber on the stage of an
inverted microscope, and mounted on concentric glass pipettes for perfusion at
37 °C. For
Absorption—MTALs from
male Sprague-Dawley rats (50–100 g body wt; Taconic, Germantown, NY)
were isolated and perfused in vitro as previously described
(16,
20). Tubules were dissected
from the inner stripe of the outer medulla at 10 °C in control bath
solution (see below), transferred to a bath chamber on the stage of an
inverted microscope, and mounted on concentric glass pipettes for perfusion at
37 °C. For 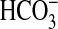 transport
experiments, the tubules were perfused and bathed in control solution that
contained 146 mm Na+, 4 mm K+, 122
mm Cl-, 25 mm
transport
experiments, the tubules were perfused and bathed in control solution that
contained 146 mm Na+, 4 mm K+, 122
mm Cl-, 25 mm
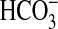 , 2.0 mm
Ca2+, 1.5 mm Mg2+, 2.0 mm
phosphate, 1.2 mm
, 2.0 mm
Ca2+, 1.5 mm Mg2+, 2.0 mm
phosphate, 1.2 mm 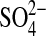 , 1.0
mm citrate, 2.0 mm lactate, and 5.5 mm
glucose (equilibrated with 95% O2-5% CO2, pH 7.45, at 37
°C). Bath solutions also contained 0.2% fatty acid-free bovine albumin.
Experimental agents were added to the bath solution as described under
“Results.” Solutions containing experimental agents were prepared
as described (19,
21). Equal concentrations of
vehicle were added to control solutions in all protocols.
, 1.0
mm citrate, 2.0 mm lactate, and 5.5 mm
glucose (equilibrated with 95% O2-5% CO2, pH 7.45, at 37
°C). Bath solutions also contained 0.2% fatty acid-free bovine albumin.
Experimental agents were added to the bath solution as described under
“Results.” Solutions containing experimental agents were prepared
as described (19,
21). Equal concentrations of
vehicle were added to control solutions in all protocols.
The protocol for study of transepithelial
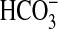 absorption was as described
(16,
20). Tubules were equilibrated
for 20–30 min at 37 °C in the initial perfusion and bath solutions,
and the luminal flow rate (normalized per unit tubule length) was adjusted to
1.5–1.9 nl/min/mm. One to three 10-min tubule fluid samples were then
collected for each period (initial, experimental, and recovery). The tubules
were allowed to re-equilibrate for 5–10 min after an experimental agent
was added to or removed from the bath solution. The absolute rate of
absorption was as described
(16,
20). Tubules were equilibrated
for 20–30 min at 37 °C in the initial perfusion and bath solutions,
and the luminal flow rate (normalized per unit tubule length) was adjusted to
1.5–1.9 nl/min/mm. One to three 10-min tubule fluid samples were then
collected for each period (initial, experimental, and recovery). The tubules
were allowed to re-equilibrate for 5–10 min after an experimental agent
was added to or removed from the bath solution. The absolute rate of
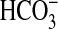 absorption
(
absorption
(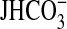 , pmol/min/mm) was calculated
from the luminal flow rate and the difference between total CO2
concentrations measured in perfused and collected fluids
(20). An average
, pmol/min/mm) was calculated
from the luminal flow rate and the difference between total CO2
concentrations measured in perfused and collected fluids
(20). An average
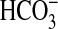 absorption rate was calculated for
each period studied in a given tubule. When repeat measurements were made at
the beginning and end of an experiment (initial and recovery periods), the
values were averaged. Single tubule values are presented in Figs.
1,
2,
3,
4 and
6. Mean values ± S.E.
(n = number of tubules) are presented under
“Results.”
absorption rate was calculated for
each period studied in a given tubule. When repeat measurements were made at
the beginning and end of an experiment (initial and recovery periods), the
values were averaged. Single tubule values are presented in Figs.
1,
2,
3,
4 and
6. Mean values ± S.E.
(n = number of tubules) are presented under
“Results.”
FIGURE 1.

Inhibition of 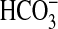 absorption by
NGF is reduced by PI3K inhibitors. Rat MTALs were isolated and
perfusedin vitroin control solution (A), bathed with 100
nm wortmannin or 20 μm LY294002 (B), or
bathed with a PI3K inhibitor plus the MEK/ERK inhibitor U0126 (15
μm) (C). NGF (0.7 nm) was then added to and
removed from the bath solution. Data points are average values for single
tubules. Lines connect paired measurements made in the same tubule.
p values are for paired t tests. NS, not
significant.
absorption by
NGF is reduced by PI3K inhibitors. Rat MTALs were isolated and
perfusedin vitroin control solution (A), bathed with 100
nm wortmannin or 20 μm LY294002 (B), or
bathed with a PI3K inhibitor plus the MEK/ERK inhibitor U0126 (15
μm) (C). NGF (0.7 nm) was then added to and
removed from the bath solution. Data points are average values for single
tubules. Lines connect paired measurements made in the same tubule.
p values are for paired t tests. NS, not
significant. 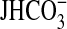 , absolute rate of
, absolute rate of
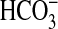 absorption. Mean values are given
under “Results.”
absorption. Mean values are given
under “Results.”
FIGURE 2.

Inhibition of 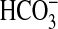 absorption by
NGF is reduced by rapamycin. MTALs were studied in control solution
(A), bathed with 20 nm rapamycin (Rap,
B), or bathed with rapamycin + 15 μm U0126
(C). NGF (0.7 nm) was then added to and removed from the
bath solution.
absorption by
NGF is reduced by rapamycin. MTALs were studied in control solution
(A), bathed with 20 nm rapamycin (Rap,
B), or bathed with rapamycin + 15 μm U0126
(C). NGF (0.7 nm) was then added to and removed from the
bath solution. 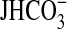 , data points,
lines, and p values are as in Fig.
1. Mean values are given under “Results.”
, data points,
lines, and p values are as in Fig.
1. Mean values are given under “Results.”
FIGURE 3.

Effect of PI3K inhibitors plus rapamycin on inhibition of
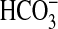 absorption by NGF. MTALs were
bathed with wortmannin (100 nm) or LY294002 (20 μm) +
rapamycin (Rap, 20 nm), and then NGF (0.7 nm)
was added to and removed from the bath solution.
absorption by NGF. MTALs were
bathed with wortmannin (100 nm) or LY294002 (20 μm) +
rapamycin (Rap, 20 nm), and then NGF (0.7 nm)
was added to and removed from the bath solution.
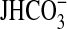 , data points, lines, and
p values are as in Fig.
1. Mean values are given under “Results.”
, data points, lines, and
p values are as in Fig.
1. Mean values are given under “Results.”
FIGURE 4.

Specificity of inhibitors in regulation of
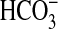 absorption. A, MTALs
were bathed with rapamycin (Rap, 20 nm), and then
angiotensin II (Ang II, 10 nm) was added to and removed
from the bath solution. B, MTALs were bathed with angiotensin II (10
nm), and then NGF (0.7 nm) was added to and removed from
the bath solution. C, MTALs were bathed with LY294002 (20
μm) + U0126 (15 μm), and then arginine vasopressin
(AVP, 0.1 nm) was added to and removed from the bath
solution.
absorption. A, MTALs
were bathed with rapamycin (Rap, 20 nm), and then
angiotensin II (Ang II, 10 nm) was added to and removed
from the bath solution. B, MTALs were bathed with angiotensin II (10
nm), and then NGF (0.7 nm) was added to and removed from
the bath solution. C, MTALs were bathed with LY294002 (20
μm) + U0126 (15 μm), and then arginine vasopressin
(AVP, 0.1 nm) was added to and removed from the bath
solution. 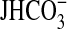 , data points, lines,
and p values are as in Fig.
1. Mean values are given under “Results.”
, data points, lines,
and p values are as in Fig.
1. Mean values are given under “Results.”
FIGURE 6.

Inhibition of 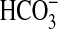 absorption by
bath amiloride is not affected by PI3K inhibitors or rapamycin. MTALs were
studied in control solution (A), bathed with 100 nm
wortmannin or 20 μm LY294002 (B), or bathed with 20
nm rapamycin (Rap, C). Amiloride (Amil,
10 μm) was then added to and removed from the bath solution.
absorption by
bath amiloride is not affected by PI3K inhibitors or rapamycin. MTALs were
studied in control solution (A), bathed with 100 nm
wortmannin or 20 μm LY294002 (B), or bathed with 20
nm rapamycin (Rap, C). Amiloride (Amil,
10 μm) was then added to and removed from the bath solution.
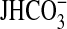 , data points, lines, and
p values are as in Fig.
1. Mean values are given under “Results.”
, data points, lines, and
p values are as in Fig.
1. Mean values are given under “Results.”
Measurement of Intracellular pH (pHi) and Basolateral
Na+/H+ Exchange
Activity—pHi was measured in isolated, perfused
MTALs by use of the pH-sensitive dye BCECF
(2′,7′-bis(2-carboxyethyl)-5(6)-carboxyfluorescein) as described
(16,
22). For
pHi experiments, tubules were perfused and bathed in
Na+-free HEPES-buffered solution that contained 145 mm
N-methyl-d-glucammonium, 4 mm K+,
147 mm Cl-, 2.0 mm Ca2+, 1.5
mm Mg2+, 1.0 mm phosphate, 1.0 mm
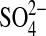 , 1.0 mm citrate, 2.0
mm lactate, 5.5 mm glucose, and 5 mm HEPES
(equilibrated with 100% O2; titrated to pH 7.4). Basolateral
Na+/H+ exchange activity was determined by measurement
of the initial rate of pHi increase after the addition of
145 mm Na+ to the bath solution (Na+ replaced
N-methyl-d-glucammonium), the intracellular buffering
power, and cell volume as described
(16,
19). Interruption of
pHi recovery at various points along the recovery curve
permits determination of the Na+/H+ exchange rate over a
range of pHi values, with appropriate corrections for a
variable background acid loading rate
(16,
22). The
Na+-dependent pHi recovery rate was inhibited
≥90% by bath ethylisopropyl amiloride (50 μm) under all
experimental conditions.
, 1.0 mm citrate, 2.0
mm lactate, 5.5 mm glucose, and 5 mm HEPES
(equilibrated with 100% O2; titrated to pH 7.4). Basolateral
Na+/H+ exchange activity was determined by measurement
of the initial rate of pHi increase after the addition of
145 mm Na+ to the bath solution (Na+ replaced
N-methyl-d-glucammonium), the intracellular buffering
power, and cell volume as described
(16,
19). Interruption of
pHi recovery at various points along the recovery curve
permits determination of the Na+/H+ exchange rate over a
range of pHi values, with appropriate corrections for a
variable background acid loading rate
(16,
22). The
Na+-dependent pHi recovery rate was inhibited
≥90% by bath ethylisopropyl amiloride (50 μm) under all
experimental conditions.
Inner Stripe Tissue Preparation and Immunoblotting—The inner
stripe tissue preparation used to study signaling proteins has been previously
described (19,
21,
23). In brief, thin strips of
tissue were microdissected at 10 °C from the inner stripe of the outer
medulla, the region of the kidney highly enriched in MTALs. The tissue strips
were then divided into four samples of equal amount and incubated in
vitro at 37 °C in the same solutions used for
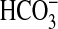 transport experiments
(19,
21,
23). The specific protocols
used for incubations are given under “Results” (Figs.
7 and
9D). After incubation,
the tissue was resuspended in ice-cold modified radioimmune precipitation
assay lysis buffer, pH 7.5, plus 1 mm NaF, 1 mm
Na3VO4, 1 mm phenylmethylsulfonyl fluoride,
and protease inhibitor mixture (1:400; Sigma), homogenized, and lysed for 4 h
at 4 °C. Lysates were cleared by centrifugation (4000 × g
for 10 min), and supernatants were separated into aliquots and stored at -80
°C. Samples of equal protein content were separated by SDS-PAGE on 8 or
10% gels and transferred to polyvinylidene difluoride membranes. Membranes
were blocked with Tris-buffered saline + 0.1% Tween 20 + 1–5% bovine
serum albumin at 4 °C and incubated overnight at 4 °C with
antiphospho-Akt-Ser-473 (1:1000) or anti-Akt (1:2500) antibodies (Cell
Signaling Technology), or with antiphospho-mTOR-Ser-2448 (1:500; BIOSOURCE) or
anti-mTOR (1:250; Santa Cruz Biotechnology) antibodies. After washing in
Tris-buffered saline + 0.1% Tween 20, horseradish peroxidase-conjugated goat
anti-rabbit secondary antibody was applied, and immunoreactive bands were
detected by chemiluminescence (Luminol Reagent, Santa Cruz Biotechnology).
Band intensities were quantified by densitometry.
transport experiments
(19,
21,
23). The specific protocols
used for incubations are given under “Results” (Figs.
7 and
9D). After incubation,
the tissue was resuspended in ice-cold modified radioimmune precipitation
assay lysis buffer, pH 7.5, plus 1 mm NaF, 1 mm
Na3VO4, 1 mm phenylmethylsulfonyl fluoride,
and protease inhibitor mixture (1:400; Sigma), homogenized, and lysed for 4 h
at 4 °C. Lysates were cleared by centrifugation (4000 × g
for 10 min), and supernatants were separated into aliquots and stored at -80
°C. Samples of equal protein content were separated by SDS-PAGE on 8 or
10% gels and transferred to polyvinylidene difluoride membranes. Membranes
were blocked with Tris-buffered saline + 0.1% Tween 20 + 1–5% bovine
serum albumin at 4 °C and incubated overnight at 4 °C with
antiphospho-Akt-Ser-473 (1:1000) or anti-Akt (1:2500) antibodies (Cell
Signaling Technology), or with antiphospho-mTOR-Ser-2448 (1:500; BIOSOURCE) or
anti-mTOR (1:250; Santa Cruz Biotechnology) antibodies. After washing in
Tris-buffered saline + 0.1% Tween 20, horseradish peroxidase-conjugated goat
anti-rabbit secondary antibody was applied, and immunoreactive bands were
detected by chemiluminescence (Luminol Reagent, Santa Cruz Biotechnology).
Band intensities were quantified by densitometry.
FIGURE 7.

NGF increases Akt phosphorylation. A, inner stripe tissue was incubated in vitro at 37 °C in the absence (Cont) and presence of 100 nm wortmannin (Wort) for 15 min, then treated with 0.7 nm NGF for 15 min. Cell lysates were immunoblotted with antiphospho-Akt-Ser-473 antibody (p-Akt) to analyze Akt phosphorylation and anti-Akt antibody for total Akt level. Data are representative of three independent experiments. B, phosphorylated Akt was analyzed by densitometry and presented as a percentage of the control level measured in the same experiment. Bars are means ± S.E. (n = 3). *, p < 0.05 versus Control.
FIGURE 9.

NGF increases p-mTOR labeling in the basolateral membrane domain. A–C, MTALs were incubated in vitro in control solution (A), 0.7 nm NGF (B), or 100 nm wortmannin + NGF (C) for 15 min, then fixed, permeabilized, and stained with antiphospho-mTOR-Ser-2448 antibody (p-mTOR). Tubules were analyzed by confocal immunofluorescence as described under “Experimental Procedures.” Images are z-axis sections (<0.4 μm) taken through a plane at the center of the tubule showing a cross-sectional view of cells in the lateral tubule walls (18). NGF increased p-mTOR labeling in the basolateral membrane domain (arrowheads). Quantification of membrane fluorescence intensity is given under “Results.” Images are representative of five independent experiments. Bar, 5 μm. D, inner stripe tissue was incubated in vitro at 37 °C in the absence (Cont) and presence of 0.7 nm NGF for 15 min. Cell lysates were immunoblotted with antiphospho-mTOR-Ser-2448 (p-mTOR) and anti-mTOR (mTOR) antibodies. Blots are representative of three separate experiments. Bars show densitometric analysis of mTOR phosphorylation, presented as a percentage of the control level measured in the same experiment. Data are means ± S.E. (n = 3). *, p < 0.05 versus control.
p70 S6 Kinase (S6K) Assay—Inner stripe tissue was incubated in vitro at 37 °C in the absence and presence of NGF and various inhibitors, and cell lysates were prepared as described above. The specific incubation conditions are indicated under “Results” (Fig. 8). For immunoprecipitation, equal amounts of sample protein (500 μg) were incubated for 2 h at 4 °C with 4 μg of rabbit anti-p70 S6K antibody (Upstate Biotechnology) coupled to protein A/G-agarose beads (Santa Cruz). The immune complexes were then washed 3 times in Triton X-100 lysis buffer and one time in assay dilution buffer (20 mm MOPS, pH 7.2, 25 mm sodium β-glycerophosphate, 5 mm EGTA, 1 mm Na3VO4, and 1 mm dithiothreitol). Kinase activity in immunoprecipitates was measured using a p70 S6K assay kit (Upstate). In brief, immunoprecipitates were incubated for 15 min at 30 °C in 50 μl final volume of assay dilution buffer containing 5 μm S6K substrate peptide 2 (KKRNRTLTK), inhibitor mixture containing protein kinase C and A and CdK inhibitors, magnesium acetate/ATP mixture, and 10 μCi of [γ-32P]ATP. Sample aliquots (25 μl) were then spotted onto P81 phosphocellulose paper. The paper was washed 5 times with 0.75% phosphoric acid and 1 time with acetone, and phosphorylated substrate was quantified by liquid scintillation counting. Results were corrected for background determined in each experiment by a negative control assay in which endogenous peptide substrate was omitted from the reaction mixture. Experimental values are presented as a percentage of the control value measured in the same experiment. Equal amounts of S6K in immunoprecipitates were verified within experiments by immunoblotting. We have demonstrated previously that changes in kinase activities measured in the inner stripe preparation reproduce accurately changes measured in the MTAL (19, 21, 23, 24).
FIGURE 8.

NGF increases S6K activity. Inner stripe tissue was incubated in vitro at 37 °C in the absence (Cont) and presence of 100 nm wortmannin (Wort), 20 nm rapamycin (Rap), or 15 μm U0126 for 15 min, then treated with 0.7 nm NGF for 15 min. S6K was immunoprecipitated, and in vitro kinase activity determined by phosphorylation of S6 peptide substrate (see “Experimental Procedures”). Phosphorylated products were spotted onto phosphocellulose paper and quantified by scintillation counting. Phosphotransferase activity of S6K is presented as a percentage of control activity measured in the same experiment. Bars are means ± S.E. for five independent experiments.
Confocal Immunofluorescence Microscopy—MTALs were studied by
confocal microscopy as previously described
(18). MTALs were
microdissected and mounted on Cell-Tak-coated coverslips at 10 °C. The
tubules were then incubated in the absence and presence of NGF for 15 min at
37 °C in a flowing bath using the same solutions as in
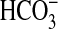 transport experiments. After
incubation, the tubules were washed with phosphate-buffered saline (PBS),
fixed with 4% paraformaldehyde in PBS for 15 min, and permeabilized with 0.3%
Triton X-100 in PBS for 15 min. The tubules were incubated in Image-iT FX
signal enhancer (Invitrogen) for 30 min at room temperature, washed, and
blocked in phosphate-buffered saline + 0.25% Tween 20 + 10% normal goat serum
for 1 h at room temperature. The tubules were then incubated overnight at 4
°C with a 1:50 dilution of antiphospho-mTOR-Ser-2448 antibody, washed, and
then incubated for 1 h at room temperature in Alexa 488-conjugated goat
anti-rabbit secondary antibody (1:100; Invitrogen). Fluorescence staining was
examined in the UTMB Optical Imaging Core using a Zeiss laser-scanning
confocal microscope (LSM 510 UV META) as described
(18). Tubules were imaged
longitudinally, and z-axis optical sections (<0.4 μm) were
obtained through a plane at the center of the tubule, which provides a
cross-sectional view of cells in the lateral tubule walls
(18). For individual
experiments, two to four tubules from the same kidney for each experimental
condition were fixed and stained identically and imaged in a single session at
identical settings of illumination, gain, and exposure time. Two-dimensional
image analysis was performed using MetaMorph software in which boxes (4.5
× 1.2 μm) were positioned on linear regions of basolateral and apical
membrane domains, and pixel intensity per unit area was determined for each
region. Two to four cells were analyzed in each tubule, and the values were
averaged. Fluorescence intensity for NGF-treated tubules was expressed as a
percentage of the control value measured in the same experiment.
transport experiments. After
incubation, the tubules were washed with phosphate-buffered saline (PBS),
fixed with 4% paraformaldehyde in PBS for 15 min, and permeabilized with 0.3%
Triton X-100 in PBS for 15 min. The tubules were incubated in Image-iT FX
signal enhancer (Invitrogen) for 30 min at room temperature, washed, and
blocked in phosphate-buffered saline + 0.25% Tween 20 + 10% normal goat serum
for 1 h at room temperature. The tubules were then incubated overnight at 4
°C with a 1:50 dilution of antiphospho-mTOR-Ser-2448 antibody, washed, and
then incubated for 1 h at room temperature in Alexa 488-conjugated goat
anti-rabbit secondary antibody (1:100; Invitrogen). Fluorescence staining was
examined in the UTMB Optical Imaging Core using a Zeiss laser-scanning
confocal microscope (LSM 510 UV META) as described
(18). Tubules were imaged
longitudinally, and z-axis optical sections (<0.4 μm) were
obtained through a plane at the center of the tubule, which provides a
cross-sectional view of cells in the lateral tubule walls
(18). For individual
experiments, two to four tubules from the same kidney for each experimental
condition were fixed and stained identically and imaged in a single session at
identical settings of illumination, gain, and exposure time. Two-dimensional
image analysis was performed using MetaMorph software in which boxes (4.5
× 1.2 μm) were positioned on linear regions of basolateral and apical
membrane domains, and pixel intensity per unit area was determined for each
region. Two to four cells were analyzed in each tubule, and the values were
averaged. Fluorescence intensity for NGF-treated tubules was expressed as a
percentage of the control value measured in the same experiment.
RESULTS
PI3K Inhibitors Reduce Inhibition of
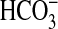 Absorption by NGF—Under
control conditions, adding 0.7 nm NGF to the bath decreased
Absorption by NGF—Under
control conditions, adding 0.7 nm NGF to the bath decreased
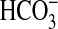 absorption in isolated MTALs by
39%, from 13.7 ± 0.6 to 8.4 ± 0.6 pmol/min/mm
(Fig. 1A). In MTALs
bathed with the PI3K inhibitor wortmannin (100 nm) or LY294002 (20
μm), NGF decreased
absorption in isolated MTALs by
39%, from 13.7 ± 0.6 to 8.4 ± 0.6 pmol/min/mm
(Fig. 1A). In MTALs
bathed with the PI3K inhibitor wortmannin (100 nm) or LY294002 (20
μm), NGF decreased 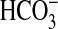 absorption only by 21%, from 13.5 ± 0.8 to 10.6 ± 0.7
pmol/min/mm (Fig. 1B).
The net decrease in
absorption only by 21%, from 13.5 ± 0.8 to 10.6 ± 0.7
pmol/min/mm (Fig. 1B).
The net decrease in 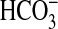 absorption
induced by NGF was reduced 45% by the PI3K inhibitors (5.3 ± 0.3
pmol/min/mm without inhibitors versus 2.9 ± 0.3 pmol/min/mm
with inhibitors; p < 0.001). In previous studies, inhibition of
absorption
induced by NGF was reduced 45% by the PI3K inhibitors (5.3 ± 0.3
pmol/min/mm without inhibitors versus 2.9 ± 0.3 pmol/min/mm
with inhibitors; p < 0.001). In previous studies, inhibition of
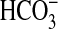 absorption by NGF was reduced
50–60% by inhibitors of ERK activation
(19). As shown in
Fig. 1C, the
combination of a PI3K inhibitor plus a MEK/ERK inhibitor (U0126) completely
eliminated the inhibition by NGF, indicating that the inhibitory effects of
PI3K and ERK are additive. These results support the view that NGF inhibits
absorption by NGF was reduced
50–60% by inhibitors of ERK activation
(19). As shown in
Fig. 1C, the
combination of a PI3K inhibitor plus a MEK/ERK inhibitor (U0126) completely
eliminated the inhibition by NGF, indicating that the inhibitory effects of
PI3K and ERK are additive. These results support the view that NGF inhibits
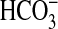 absorption through the parallel
activation of PI3K- and ERK-dependent signaling pathways.
absorption through the parallel
activation of PI3K- and ERK-dependent signaling pathways.
Rapamycin Reduces Inhibition of
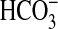 Absorption by NGF—An
important downstream effector of PI3K in growth factor signaling is mTOR,
which is specifically inhibited by the immunosuppressive drug rapamycin
(25). Similar to the preceding
results with PI3K inhibitors, NGF decreased
Absorption by NGF—An
important downstream effector of PI3K in growth factor signaling is mTOR,
which is specifically inhibited by the immunosuppressive drug rapamycin
(25). Similar to the preceding
results with PI3K inhibitors, NGF decreased
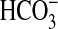 absorption by 38% under control
conditions (from 14.8 ± 0.4 to 9.2 ± 0.5 pmol/min/mm) but only
by 19% (from 14.3 ± 0.4 to 11.6 ± 0.6 pmol/min/mm) in MTALs
bathed with 20 nm rapamycin
(Fig. 2, A and
B). The net decrease in
absorption by 38% under control
conditions (from 14.8 ± 0.4 to 9.2 ± 0.5 pmol/min/mm) but only
by 19% (from 14.3 ± 0.4 to 11.6 ± 0.6 pmol/min/mm) in MTALs
bathed with 20 nm rapamycin
(Fig. 2, A and
B). The net decrease in
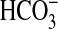 absorption induced by NGF was
reduced 50% by rapamycin (p < 0.001). The combination of rapamycin
plus U0126 again completely eliminated the inhibition by NGF
(Fig. 2C). These
results support a role for mTOR in mediating the inhibition of
absorption induced by NGF was
reduced 50% by rapamycin (p < 0.001). The combination of rapamycin
plus U0126 again completely eliminated the inhibition by NGF
(Fig. 2C). These
results support a role for mTOR in mediating the inhibition of
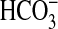 absorption by NGF and show that
inhibition via the rapamycin-sensitive pathway is additive to inhibition
mediated through ERK.
absorption by NGF and show that
inhibition via the rapamycin-sensitive pathway is additive to inhibition
mediated through ERK.
Effects of PI3K Inhibitors and Rapamycin Are Not
Additive—Because PI3K inhibitors and rapamycin reduced the
inhibition of 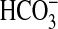 absorption by NGF by
a similar amount, further experiments were carried out to test whether these
agents block a common pathway. In MTALs bathed with a PI3K inhibitor plus
rapamycin, NGF decreased
absorption by NGF by
a similar amount, further experiments were carried out to test whether these
agents block a common pathway. In MTALs bathed with a PI3K inhibitor plus
rapamycin, NGF decreased 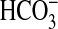 absorption by 22%, from 13.4 ± 0.3 to 10.4 ± 0.1 pmol/min/mm
(Fig. 3), a decrease similar to
that observed with either inhibitor alone (Figs.
1B and
2B). Thus, combining a
PI3K inhibitor with rapamycin does not cause a further reduction in the
inhibition by NGF, consistent with these agents blocking a common regulatory
pathway. These results support the view that PI3K and mTOR are components of a
common signaling pathway that inhibits
absorption by 22%, from 13.4 ± 0.3 to 10.4 ± 0.1 pmol/min/mm
(Fig. 3), a decrease similar to
that observed with either inhibitor alone (Figs.
1B and
2B). Thus, combining a
PI3K inhibitor with rapamycin does not cause a further reduction in the
inhibition by NGF, consistent with these agents blocking a common regulatory
pathway. These results support the view that PI3K and mTOR are components of a
common signaling pathway that inhibits
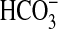 absorption.
absorption.
Specificity of Inhibitors in Regulation of
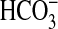 Absorption—To assess
the specificity of rapamycin actions on
Absorption—To assess
the specificity of rapamycin actions on
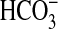 absorption, we examined factors
that inhibit
absorption, we examined factors
that inhibit 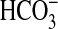 absorption through
signaling pathways not involving PI3K. Angiotensin II inhibits
absorption through
signaling pathways not involving PI3K. Angiotensin II inhibits
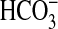 absorption in the MTAL through
cytochrome P450 (26). In MTALs
bathed with rapamycin, angiotensin II decreased
absorption in the MTAL through
cytochrome P450 (26). In MTALs
bathed with rapamycin, angiotensin II decreased
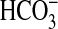 absorption by 32 ± 1%
(Fig. 4A), an effect
similar to that observed under identical conditions in the absence of the
inhibitor (26). In previous
studies we found that rapamycin also has no effect on inhibition of
absorption by 32 ± 1%
(Fig. 4A), an effect
similar to that observed under identical conditions in the absence of the
inhibitor (26). In previous
studies we found that rapamycin also has no effect on inhibition of
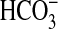 absorption by aldosterone, which
is mediated through ERK (24).
Thus, rapamycin selectively reduces inhibition of
absorption by aldosterone, which
is mediated through ERK (24).
Thus, rapamycin selectively reduces inhibition of
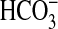 absorption by NGF that depends on
PI3K. Consistent with these findings, the inhibition of
absorption by NGF that depends on
PI3K. Consistent with these findings, the inhibition of
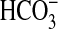 absorption by NGF is additive to
inhibition by angiotensin II (Fig.
4B) and aldosterone
(27), further confirming that
these factors act through distinct signaling pathways. Additional experiments
evaluated the specificity of the PI3K inhibitor + ERK inhibitor combination by
examining arginine vasopressin, which inhibits
absorption by NGF is additive to
inhibition by angiotensin II (Fig.
4B) and aldosterone
(27), further confirming that
these factors act through distinct signaling pathways. Additional experiments
evaluated the specificity of the PI3K inhibitor + ERK inhibitor combination by
examining arginine vasopressin, which inhibits
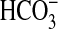 absorption via cAMP
(20). In MTALs bathed with
LY294002 plus U0126, vasopressin decreased
absorption via cAMP
(20). In MTALs bathed with
LY294002 plus U0126, vasopressin decreased
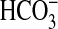 absorption by 45 ± 5%
(Fig. 4C), an effect
similar to that observed in the absence of the inhibitors
(20). Thus, the effect of the
PI3K-ERK inhibitor combination to eliminate inhibition of
absorption by 45 ± 5%
(Fig. 4C), an effect
similar to that observed in the absence of the inhibitors
(20). Thus, the effect of the
PI3K-ERK inhibitor combination to eliminate inhibition of
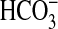 absorption is selective for NGF
(Fig. 1C) and is not
the result of nonspecific metabolic or cytotoxic effects on the tubule
cells.
absorption is selective for NGF
(Fig. 1C) and is not
the result of nonspecific metabolic or cytotoxic effects on the tubule
cells.
Rapamycin Reduces Inhibition of Basolateral
Na+/H+ Exchange by NGF—In
the MTAL, NGF decreases 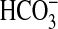 absorption
through primary inhibition of the basolateral NHE1
Na+/H+ exchanger
(16,
17). To determine whether the
PI3K-mTOR pathway is involved in mediating inhibition of basolateral
Na+/H+ exchange, we examined the effects of NGF in the
absence and presence of rapamycin. Under control conditions, NGF decreased
basolateral Na+/H+ exchange activity at all
pHi values studied (control versus NGF;
Fig. 5A). In MTALs
bathed with rapamycin, the inhibition by NGF was significantly reduced
(rapamycin + NGF, Fig.
5A). Overall, the net decrease in basolateral
Na+/H+ exchange activity induced by NGF was reduced 45%
by rapamycin (p < 0.05; Fig.
5B). Rapamycin alone did not affect basolateral
Na+/H+ exchange activity
(Fig. 5B). These
results demonstrate that NGF inhibits basolateral Na+/H+
exchange via a rapamycin-sensitive pathway and are consistent with mTOR acting
downstream of PI3K to mediate NGF-induced inhibition of NHE1 and
absorption
through primary inhibition of the basolateral NHE1
Na+/H+ exchanger
(16,
17). To determine whether the
PI3K-mTOR pathway is involved in mediating inhibition of basolateral
Na+/H+ exchange, we examined the effects of NGF in the
absence and presence of rapamycin. Under control conditions, NGF decreased
basolateral Na+/H+ exchange activity at all
pHi values studied (control versus NGF;
Fig. 5A). In MTALs
bathed with rapamycin, the inhibition by NGF was significantly reduced
(rapamycin + NGF, Fig.
5A). Overall, the net decrease in basolateral
Na+/H+ exchange activity induced by NGF was reduced 45%
by rapamycin (p < 0.05; Fig.
5B). Rapamycin alone did not affect basolateral
Na+/H+ exchange activity
(Fig. 5B). These
results demonstrate that NGF inhibits basolateral Na+/H+
exchange via a rapamycin-sensitive pathway and are consistent with mTOR acting
downstream of PI3K to mediate NGF-induced inhibition of NHE1 and
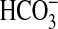 absorption.
absorption.
FIGURE 5.

Inhibition of basolateral Na+/H+ exchange by NGF is reduced by rapamycin. A, MTALs were studied under control conditions and with NGF (0.7 nm) or NGF + rapamycin (Rap, 20 nm) in the bath solution. Basolateral Na+/H+ exchange rates (JNa+/H+) were determined at various pHi values from initial rates of pHi increase after the addition of Na+ to the bath solution (see “Experimental Procedures”). Data points are from 11 control tubules, 11 tubules with NGF, and 7 tubules with rapamycin + NGF. B, mean basolateral Na+/H+ exchange rates for the three conditions in panel A plus additional data from seven tubules studied with rapamycin alone. *, p < 0.05 versus control or rapamycin; #, p < 0.05 versus NGF (analysis of variance).
PI3K Inhibitors and Rapamycin Do Not Affect Inhibition By Bath
Amiloride—Inhibiting basolateral Na+/H+
exchange decreases 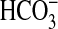 absorption in
the MTAL by inducing actin cytoskeleton remodeling that secondarily inhibits
apical Na+/H+ exchange
(18). Thus, an additional
mechanism through which PI3K-mTOR signaling could affect
absorption in
the MTAL by inducing actin cytoskeleton remodeling that secondarily inhibits
apical Na+/H+ exchange
(18). Thus, an additional
mechanism through which PI3K-mTOR signaling could affect
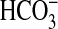 absorption is by modifying the
regulatory interaction between basolateral and apical
Na+/H+ exchangers. To test this, we took advantage of
our previous finding that the interaction between exchangers and the resulting
inhibition of
absorption is by modifying the
regulatory interaction between basolateral and apical
Na+/H+ exchangers. To test this, we took advantage of
our previous finding that the interaction between exchangers and the resulting
inhibition of 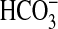 absorption can be
induced directly by inhibiting basolateral NHE1 with bath amiloride
(16–18).
Under control conditions, the addition of 10 μm amiloride to the
bath decreased
absorption can be
induced directly by inhibiting basolateral NHE1 with bath amiloride
(16–18).
Under control conditions, the addition of 10 μm amiloride to the
bath decreased 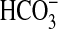 absorption by 31%,
from 14.1 ± 0.6 to 9.7 ± 0.7 pmol/min/mm
(Fig. 6A). This
inhibition was not affected by either PI3K inhibitors
(Fig. 6B) or rapamycin
(Fig. 6C). These
results indicate that PI3K and mTOR are not involved in mediating the
regulatory interaction between the basolateral NHE1 and apical NHE3
Na+/H+ exchangers. Taken together, our findings indicate
that the role of the PI3K-mTOR pathway in inhibition of
absorption by 31%,
from 14.1 ± 0.6 to 9.7 ± 0.7 pmol/min/mm
(Fig. 6A). This
inhibition was not affected by either PI3K inhibitors
(Fig. 6B) or rapamycin
(Fig. 6C). These
results indicate that PI3K and mTOR are not involved in mediating the
regulatory interaction between the basolateral NHE1 and apical NHE3
Na+/H+ exchangers. Taken together, our findings indicate
that the role of the PI3K-mTOR pathway in inhibition of
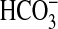 absorption is to mediate the
effect of NGF to decrease basolateral Na+/H+ exchange
activity.
absorption is to mediate the
effect of NGF to decrease basolateral Na+/H+ exchange
activity.
NGF Activates Akt and S6K—Further studies were carried out
to examine the effects of NGF on additional signaling components in the
PI3K-mTOR pathway. The serine/threonine kinase Akt is a downstream target of
PI3K that links PI3K to activation of mTOR
(25). To test whether NGF
activates Akt, inner stripe tissue was incubated in vitro in the
absence and presence of NGF for 15 min, and Akt phosphorylation was analyzed
by immunoblotting using antiphospho-Akt-Ser-473 antibody. As shown in
Fig. 7, NGF increased Akt
phosphorylation 2-fold without a change in total Akt expression. The
phosphorylation of Akt was blocked by wortmannin
(Fig. 7A), consistent
with a requirement for PI3K in the NGF-induced Akt activation. These results
are consistent with a role for Akt in mediating PI3K-dependent inhibition of
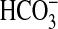 absorption in the MTAL.
absorption in the MTAL.
In contrast to Akt, which functions as an upstream activator of mTOR, S6K
is an important downstream effector of mTOR
(25). To test the effect of
NGF on S6K, inner stripe tissue was exposed to NGF in vitro for 15
min, and S6K activity was measured by immune complex assay. As shown in
Fig. 8, NGF increased S6K
activity 2.2-fold. This activation was blocked by wortmannin or rapamycin,
consistent with PI3K, mTOR, and S6K in a linear signaling pathway. In
contrast, S6K activation was not blocked by the MEK/ERK inhibitor U0126,
confirming further that activation of the PI3K-mTOR-S6K pathway by NGF occurs
independently of the activation of ERK. These results indicate that NGF
increases S6K activity via a PI3K- and mTOR-dependent pathway and suggest that
S6K may be a downstream effector of PI3K and mTOR in mediating NGF-induced
inhibition of NHE1 and 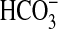 absorption
(see “Discussion”).
absorption
(see “Discussion”).
NGF Increases Phosphorylated mTOR Level in the Basolateral Membrane
Domain—The sensitivity to rapamycin (Figs.
2 and
5) suggests that the inhibition
of NHE1 and 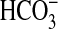 absorption by NGF
depends on activation of mTOR. To verify mTOR regulation, we examined the
effects of NGF on the level and subcellular location of phosphorylated mTOR.
Microdissected MTALs were incubated in vitro in the absence and
presence of NGF for 15 min, stained with antiphospho-mTOR-Ser-2448 antibody
(p-mTOR), and then analyzed by confocal immunofluorescence. Phosphorylation of
mTOR at Ser-2448 correlates with growth factor stimulation of the
PI3K-mTOR-S6K pathway (28). In
control tubules, staining for p-mTOR was seen in the cytoplasm and along the
plasma membranes (Fig.
9A). Stimulation with NGF induced a clear increase in
p-mTOR labeling along the basolateral membrane domain
(Fig. 9B).
Two-dimensional image analysis (see “Experimental Procedures”)
showed that NGF increased the intensity of p-mTOR staining in the basolateral
membrane 1.5 ± 0.1-fold (n = 5; p < 0.05) with no
effect on signal intensity in the apical membrane (1.1 ± 0.1-fold;
p = NS). The phosphorylation of mTOR was inhibited by wortmannin
(Fig. 9C). The effect
of NGF on mTOR phosphorylation was examined further by immunoblot analysis of
inner stripe tissue. As shown in Fig.
9D, NGF increased mTOR phosphorylation 1.3-fold without a
change in total mTOR level. These results confirm NGF-induced regulation of
mTOR in the MTAL and show that the PI3K-mTOR-dependent inhibition of
basolateral Na+/H+ exchange activity is associated with
an increased level of phosphorylated mTOR in the basolateral membrane
domain.
absorption by NGF
depends on activation of mTOR. To verify mTOR regulation, we examined the
effects of NGF on the level and subcellular location of phosphorylated mTOR.
Microdissected MTALs were incubated in vitro in the absence and
presence of NGF for 15 min, stained with antiphospho-mTOR-Ser-2448 antibody
(p-mTOR), and then analyzed by confocal immunofluorescence. Phosphorylation of
mTOR at Ser-2448 correlates with growth factor stimulation of the
PI3K-mTOR-S6K pathway (28). In
control tubules, staining for p-mTOR was seen in the cytoplasm and along the
plasma membranes (Fig.
9A). Stimulation with NGF induced a clear increase in
p-mTOR labeling along the basolateral membrane domain
(Fig. 9B).
Two-dimensional image analysis (see “Experimental Procedures”)
showed that NGF increased the intensity of p-mTOR staining in the basolateral
membrane 1.5 ± 0.1-fold (n = 5; p < 0.05) with no
effect on signal intensity in the apical membrane (1.1 ± 0.1-fold;
p = NS). The phosphorylation of mTOR was inhibited by wortmannin
(Fig. 9C). The effect
of NGF on mTOR phosphorylation was examined further by immunoblot analysis of
inner stripe tissue. As shown in Fig.
9D, NGF increased mTOR phosphorylation 1.3-fold without a
change in total mTOR level. These results confirm NGF-induced regulation of
mTOR in the MTAL and show that the PI3K-mTOR-dependent inhibition of
basolateral Na+/H+ exchange activity is associated with
an increased level of phosphorylated mTOR in the basolateral membrane
domain.
DISCUSSION
Previously we identified a novel role for the basolateral NHE1
Na+/H+ exchanger in transepithelial
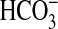 absorption in the renal MTAL
(15–17).
In the present study we examined the effects of NGF to identify signal
transduction pathways that regulate transcellular H+ secretion
through effects on NHE1 activity. The results reveal a new role for PI3K-mTOR
signaling in the acute regulation of NHE1 and provide the first evidence that
mTOR is involved in regulating the absorptive function of renal tubules.
absorption in the renal MTAL
(15–17).
In the present study we examined the effects of NGF to identify signal
transduction pathways that regulate transcellular H+ secretion
through effects on NHE1 activity. The results reveal a new role for PI3K-mTOR
signaling in the acute regulation of NHE1 and provide the first evidence that
mTOR is involved in regulating the absorptive function of renal tubules.
A model for NGF-induced regulation in the MTAL based on our current and
previous
(16–19)
findings is presented in Fig.
10. Binding of NGF to its basolateral cell surface receptor (TrkA)
induces parallel activation of PI3K and ERK signaling cascades. Activation of
PI3K leads to the downstream activation of Akt, mTOR, and S6K. The PI3K-mTOR
and ERK pathways converge to inhibit basolateral NHE1. This in turn induces
actin cytoskeleton remodeling that secondarily inhibits apical NHE3, resulting
in decreased luminal H+ secretion and transepithelial
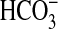 absorption
(16,
18). The PI3K-mTOR and ERK
pathways function independently to inhibit NHE1, with each pathway accounting
for ∼50% of the NGF-induced transport inhibition.
absorption
(16,
18). The PI3K-mTOR and ERK
pathways function independently to inhibit NHE1, with each pathway accounting
for ∼50% of the NGF-induced transport inhibition.
FIGURE 10.

Model of signaling pathways mediating inhibition of NHE1 and
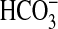 absorption by NGF in the MTAL.
NGF induces parallel activation of PI3K-mTOR and ERK signaling pathways, which
function independently to inhibit basolateral NHE1. Inhibition of NHE1 induces
actin cytoskeleton remodeling that secondarily inhibits apical NHE3, resulting
in decreased luminal H+ secretion and transepithelial
absorption by NGF in the MTAL.
NGF induces parallel activation of PI3K-mTOR and ERK signaling pathways, which
function independently to inhibit basolateral NHE1. Inhibition of NHE1 induces
actin cytoskeleton remodeling that secondarily inhibits apical NHE3, resulting
in decreased luminal H+ secretion and transepithelial
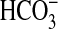 absorption. S6K is proposed to
function downstream of mTOR to inhibit NHE1. Regulatory steps linked by
arrows are not necessarily direct and may involve additional
signaling proteins.
absorption. S6K is proposed to
function downstream of mTOR to inhibit NHE1. Regulatory steps linked by
arrows are not necessarily direct and may involve additional
signaling proteins.
The role of NHE1 in processes such as proliferation and survival, adhesion
and migration, and the motile and invasive properties of tumor cells
(1–7)
has led to extensive investigation of cell signaling pathways that regulate
NHE1 activity. NHE1 is regulated by diverse stimuli acting through receptor
tyrosine kinases, G protein-coupled receptors, and integrin receptors
(2,
3). Activation of NHE1 in
response to growth factors and other stimuli involves phosphorylation of the
exchanger by various kinases, including the ERK-regulated kinase
p90RSK (29), the
RhoA target p160 Rho-associated kinase 1 (p160ROCK)
(30), and the serine/threonine
kinase Nck-interacting kinase (NIK)
(31). NHE1 also is modulated
through physical interactions with other regulatory proteins such as
Ca2+-calmodulin and calcineurin B homologous proteins (CHIPs)
(32–34).
A signaling molecule importantly involved in the control of cell growth,
proliferation, motility, and transformation is PI3K
(25,
35–37).
Although PI3K has been implicated in regulation of
Na+/H+ exchange activity in mammary epithelial cells and
breast tumor cells (38,
39), PI3K signaling generally
has not been ascribed a significant role in regulating NHE1 and its
physiological functions
(1–3,
5). In the present study we
demonstrate that PI3K plays a major role in NGF-induced inhibition of
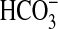 absorption, mediated through
inhibition of NHE1. Thus, PI3K signaling is importantly involved in regulating
the epithelial function of NHE1 to control transcellular H+
secretion in MTAL cells. Of significance, we found that PI3K signaling
inhibits NHE1 activity, contrary to the stimulation of NHE1 by other
growth-related signaling pathways
(1–3).
It remains to be determined whether PI3K inhibits NHE1 in other epithelial or
nonepithelial cells or if this represents a specialized response of the MTAL.
It is conceivable that PI3K could function in a negative feedback or
compensatory pathway that serves to modulate the stimulation of NHE1 by other
mitogenic pathways. Such a system would be analogous to the role of PI3K-Akt
signaling in innate immune regulation, where it serves as a negative regulator
of immune receptor signaling to limit the magnitude of proinflammatory
responses that can lead to organ damage
(40,
41). Consistent with this
possibility, inhibiting PI3K potentiated activation of
Na+/H+ exchange that was responsible for increased
motility and invasion of breast epithelial tumor cells during serum
deprivation (39). These
results support a role for PI3K in suppressing NHE1 stimulation that leads to
tumor cell transformation and metastasis.
absorption, mediated through
inhibition of NHE1. Thus, PI3K signaling is importantly involved in regulating
the epithelial function of NHE1 to control transcellular H+
secretion in MTAL cells. Of significance, we found that PI3K signaling
inhibits NHE1 activity, contrary to the stimulation of NHE1 by other
growth-related signaling pathways
(1–3).
It remains to be determined whether PI3K inhibits NHE1 in other epithelial or
nonepithelial cells or if this represents a specialized response of the MTAL.
It is conceivable that PI3K could function in a negative feedback or
compensatory pathway that serves to modulate the stimulation of NHE1 by other
mitogenic pathways. Such a system would be analogous to the role of PI3K-Akt
signaling in innate immune regulation, where it serves as a negative regulator
of immune receptor signaling to limit the magnitude of proinflammatory
responses that can lead to organ damage
(40,
41). Consistent with this
possibility, inhibiting PI3K potentiated activation of
Na+/H+ exchange that was responsible for increased
motility and invasion of breast epithelial tumor cells during serum
deprivation (39). These
results support a role for PI3K in suppressing NHE1 stimulation that leads to
tumor cell transformation and metastasis.
mTOR plays a central role in the control of cell growth, survival, and
proliferation, with abnormal elevation of PI3K-mTOR signaling a contributing
factor in tumorigenesis (25,
37,
42). To our knowledge, no
previous studies have reported a role for mTOR in the regulation of
Na+/H+ exchange activity. Results of the present study
indicate that mTOR functions downstream of PI3K to mediate NGF-induced
inhibition of NHE1 and 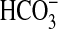 absorption.
This conclusion is supported by several lines of evidence. 1) Inhibition of
absorption.
This conclusion is supported by several lines of evidence. 1) Inhibition of
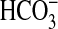 absorption by NGF was reduced by
wortmannin or LY294002, two chemically unrelated PI3K inhibitors with
different mechanisms of action shown previously to block PI3K-dependent
transport regulation in the MTAL
(21). Wortmannin was studied
at a concentration that selectively inhibits PI3K, with no significant effects
against mTOR, other PI3K-related kinases, and a broad range of protein kinases
(43–45).
2) NGF increased wortmannin-sensitive phosphorylation of the PI3K substrate
Akt, confirming PI3K activation. 3) Inhibition of NHE1 and
absorption by NGF was reduced by
wortmannin or LY294002, two chemically unrelated PI3K inhibitors with
different mechanisms of action shown previously to block PI3K-dependent
transport regulation in the MTAL
(21). Wortmannin was studied
at a concentration that selectively inhibits PI3K, with no significant effects
against mTOR, other PI3K-related kinases, and a broad range of protein kinases
(43–45).
2) NGF increased wortmannin-sensitive phosphorylation of the PI3K substrate
Akt, confirming PI3K activation. 3) Inhibition of NHE1 and
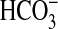 absorption were reduced by
rapamycin, a specific inhibitor of mTOR signaling (see below). 4) The effects
of PI3K inhibitors and rapamycin to reduce inhibition by NGF were similar in
magnitude and not additive, consistent with PI3K and mTOR as components of a
common inhibitory pathway. 5) Rapamycin selectively blocked PI3K-dependent
inhibition by NGF but had no effect on inhibition of
absorption were reduced by
rapamycin, a specific inhibitor of mTOR signaling (see below). 4) The effects
of PI3K inhibitors and rapamycin to reduce inhibition by NGF were similar in
magnitude and not additive, consistent with PI3K and mTOR as components of a
common inhibitory pathway. 5) Rapamycin selectively blocked PI3K-dependent
inhibition by NGF but had no effect on inhibition of
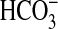 absorption by stimuli that act
through pathways not involving PI3K (angiotensin II and aldosterone). 6)
Inhibition of NHE1 by NGF was associated with an increased level of
phosphorylated mTOR in the basolateral membrane domain. 7) NGF-induced
stimulation of the mTOR target S6K was blocked by both PI3K inhibitors and
rapamycin. Taken together, these results support PI3K and mTOR as components
of a common signaling pathway that inhibits NHE1 and
absorption by stimuli that act
through pathways not involving PI3K (angiotensin II and aldosterone). 6)
Inhibition of NHE1 by NGF was associated with an increased level of
phosphorylated mTOR in the basolateral membrane domain. 7) NGF-induced
stimulation of the mTOR target S6K was blocked by both PI3K inhibitors and
rapamycin. Taken together, these results support PI3K and mTOR as components
of a common signaling pathway that inhibits NHE1 and
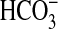 absorption. In mammalian cells,
the biochemical link between PI3K and mTOR involves Akt-induced
phosphorylation and inactivation of the protein tuberous sclerosis 2 (TSC2),
which negatively regulates mTOR through the small G protein Rheb
(25,
42,
46). Whether TSC2 and Rheb
play a role in mediating PI3K-mTOR-dependent regulation of NHE1 in the MTAL
remains to be determined. Inactivating mutations in the TSC2 gene lead to the
development of renal tumors that are sensitive to rapamycin, indicating an
important role for TSC suppression of mTOR signaling in renal cells
(25,
47,
48). The results of the
present study raise the possibility that mTOR-mediated regulation of NHE1
could play a role in mTOR-dependent renal cell proliferation and tumor
development.
absorption. In mammalian cells,
the biochemical link between PI3K and mTOR involves Akt-induced
phosphorylation and inactivation of the protein tuberous sclerosis 2 (TSC2),
which negatively regulates mTOR through the small G protein Rheb
(25,
42,
46). Whether TSC2 and Rheb
play a role in mediating PI3K-mTOR-dependent regulation of NHE1 in the MTAL
remains to be determined. Inactivating mutations in the TSC2 gene lead to the
development of renal tumors that are sensitive to rapamycin, indicating an
important role for TSC suppression of mTOR signaling in renal cells
(25,
47,
48). The results of the
present study raise the possibility that mTOR-mediated regulation of NHE1
could play a role in mTOR-dependent renal cell proliferation and tumor
development.
Rapamycin specifically inhibits mTOR by forming an inhibitory complex with
FKBP12, the intracellular rapamycin receptor. The rapamycin-FKBP12 complex
binds directly to mTOR, which destabilizes multiprotein mTOR signaling
complexes and inhibits the ability of mTOR to activate S6K and other
downstream signals (25,
46). The efficacy of rapamycin
in suppressing cell proliferation and the mammalian immune system has led to
its use in multiple clinical settings, including as an immunosuppressant in
kidney transplants, to prevent restinosis in cardiovascular stents, and as an
antitumor agent (25,
37,
42,
49,
50). Beneficial effects of
rapamycin to prevent progression of chronic kidney disease also have been
reported in experimental models
(51). Our study provides new
evidence that rapamycin at therapeutic concentrations influences the
regulation of NHE1 and its cell functions through inhibition of PI3K-mTOR
signaling. Rapamycin had no significant effect on NHE1 activity or
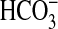 absorption under basal conditions
but impaired their regulation by NGF. In view of the defined roles of NHE1 in
cell growth, proliferation, and migration, it will be important in future
studies to determine whether the ability of rapamycin to modify NHE1
regulation via mTOR may contribute to the antiproliferative,
immunosuppressive, or antitumor properties of this drug. Our findings in the
MTAL also raise the possibility that rapamycin could impair the regulation of
renal tubule functions such as Na+ absorption and H+
secretion that depend on Na+/H+ exchange activity.
absorption under basal conditions
but impaired their regulation by NGF. In view of the defined roles of NHE1 in
cell growth, proliferation, and migration, it will be important in future
studies to determine whether the ability of rapamycin to modify NHE1
regulation via mTOR may contribute to the antiproliferative,
immunosuppressive, or antitumor properties of this drug. Our findings in the
MTAL also raise the possibility that rapamycin could impair the regulation of
renal tubule functions such as Na+ absorption and H+
secretion that depend on Na+/H+ exchange activity.
The two major downstream effectors of mTOR are S6K1, which enhances
translational efficiency, and eukaryotic initiation factor 4E-binding protein
(4E-BP1), a translational repressor protein
(25,
46). We propose that S6K1
functions downstream of PI3K-mTOR to mediate NGF-induced inhibition of NHE1 in
the MTAL based on the following observations. First, NGF increased S6K
activity under conditions similar to those used in
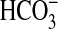 transport experiments. Second, the
stimulation of S6K was blocked by wortmannin and rapamycin but not by the
MEK/ERK inhibitor U0126. Thus, the inhibitor sensitivity of S6K activation
correlates directly with that observed for PI3K-dependent inhibition of
transport experiments. Second, the
stimulation of S6K was blocked by wortmannin and rapamycin but not by the
MEK/ERK inhibitor U0126. Thus, the inhibitor sensitivity of S6K activation
correlates directly with that observed for PI3K-dependent inhibition of
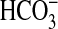 absorption. Third, the
PI3K-mTOR-dependent transport regulation occurs within a time frame (<15
min) that correlates with increased S6K activity but is unlikely to be
mediated through mTOR-dependent up-regulation of transcriptional factors and
protein synthesis. Fourth, S6K is a member of the AGC family of
serine/threonine kinases that includes several well defined regulators of
epithelial transport proteins, including protein kinases A, C, and G, serum-
and glucocorticoid-induced protein kinase, and p90 ribosomal S6 kinase
(52,
53). Our study is the first to
implicate a role for S6K in regulating Na+/H+ exchange
activity and the transport function of renal tubules. Because of the lack of
selective S6K inhibitors, we were unable to evaluate the functional
significance of S6K activation for Na+/H+ exchange
regulation in the perfused MTAL. Future studies using cell systems that enable
constitutive activation and/or knockdown of S6K will be required to establish
directly a role for S6K in NHE1 regulation. Additional mTOR-associated
proteins, such as protein phosphatase 2
(25,
54,
55), also could be involved in
mTOR regulation of NHE1 activity.
absorption. Third, the
PI3K-mTOR-dependent transport regulation occurs within a time frame (<15
min) that correlates with increased S6K activity but is unlikely to be
mediated through mTOR-dependent up-regulation of transcriptional factors and
protein synthesis. Fourth, S6K is a member of the AGC family of
serine/threonine kinases that includes several well defined regulators of
epithelial transport proteins, including protein kinases A, C, and G, serum-
and glucocorticoid-induced protein kinase, and p90 ribosomal S6 kinase
(52,
53). Our study is the first to
implicate a role for S6K in regulating Na+/H+ exchange
activity and the transport function of renal tubules. Because of the lack of
selective S6K inhibitors, we were unable to evaluate the functional
significance of S6K activation for Na+/H+ exchange
regulation in the perfused MTAL. Future studies using cell systems that enable
constitutive activation and/or knockdown of S6K will be required to establish
directly a role for S6K in NHE1 regulation. Additional mTOR-associated
proteins, such as protein phosphatase 2
(25,
54,
55), also could be involved in
mTOR regulation of NHE1 activity.
Regulatory interactions between the PI3K-mTOR and ERK signaling pathways
have been described in other systems. For example, ERK1/2 can induce
p90RSK-catalyzed phosphorylation and inactivation of TSC2,
resulting in increased mTOR signaling and S6K activation independent of PI3K
(46,
56,
57). Conversely, rapamycin has
been shown to diminish growth factor-induced ERK activation in some cells, an
effect that may involve mTOR modulating ERK phosphorylation through PP2A
(55). Several findings suggest
that these interactions are minimal or absent in the MTAL, at least with
respect to NGF-induced inhibition of NHE1 and
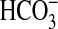 absorption. First, the effect of
ERK inhibitors to diminish inhibition by NGF is quantitatively similar in the
absence or presence of a functional PI3K-mTOR pathway (Figs.
1 and
2) (Ref.
19), arguing against a
contribution of mTOR to ERK activation. Second, PI3K inhibitors reduce
NGF-induced inhibition by an amount virtually identical to that observed with
rapamycin (Figs. 1,
2,
3), arguing against an effect
of ERK to activate mTOR-dependent regulation independently of PI3K. Third,
activation of S6K by NGF was similar in the absence and presence of a MEK/ERK
inhibitor (Fig. 8). Thus, our
results provide no evidence for significant cross-talk between the PI3K-mTOR
and ERK pathways and suggest that these pathways function independently to
inhibit NHE1 and
absorption. First, the effect of
ERK inhibitors to diminish inhibition by NGF is quantitatively similar in the
absence or presence of a functional PI3K-mTOR pathway (Figs.
1 and
2) (Ref.
19), arguing against a
contribution of mTOR to ERK activation. Second, PI3K inhibitors reduce
NGF-induced inhibition by an amount virtually identical to that observed with
rapamycin (Figs. 1,
2,
3), arguing against an effect
of ERK to activate mTOR-dependent regulation independently of PI3K. Third,
activation of S6K by NGF was similar in the absence and presence of a MEK/ERK
inhibitor (Fig. 8). Thus, our
results provide no evidence for significant cross-talk between the PI3K-mTOR
and ERK pathways and suggest that these pathways function independently to
inhibit NHE1 and 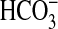 absorption.
Previous studies also have identified an internal feedback loop within the
PI3K-mTOR-S6K pathway involving phosphorylation of mTOR by S6K. In this
mechanism, S6K is primarily activated downstream of PI3K via the
Akt-TSC1/2-Rheb-mTOR pathway. Activated S6K in turn directly phosphorylates
mTOR at Ser-2448 (28,
58). It is presently unclear
whether this feedback loop functions as a positive or negative regulator of
mTOR signaling (28,
58). In the MTAL we found that
NGF activates S6K via a PI3K- and mTOR-dependent pathway and that NGF
stimulation results in increased Ser-2448 phosphorylation of mTOR in the
basolateral membrane domain, consistent with feedback regulation of mTOR
through S6K. Further studies will be required to confirm this and to evaluate
the possibility that phosphorylation of mTOR by S6K may be involved in
basolateral mTOR targeting and/or in mTOR-dependent regulation of NHE1
activity.
absorption.
Previous studies also have identified an internal feedback loop within the
PI3K-mTOR-S6K pathway involving phosphorylation of mTOR by S6K. In this
mechanism, S6K is primarily activated downstream of PI3K via the
Akt-TSC1/2-Rheb-mTOR pathway. Activated S6K in turn directly phosphorylates
mTOR at Ser-2448 (28,
58). It is presently unclear
whether this feedback loop functions as a positive or negative regulator of
mTOR signaling (28,
58). In the MTAL we found that
NGF activates S6K via a PI3K- and mTOR-dependent pathway and that NGF
stimulation results in increased Ser-2448 phosphorylation of mTOR in the
basolateral membrane domain, consistent with feedback regulation of mTOR
through S6K. Further studies will be required to confirm this and to evaluate
the possibility that phosphorylation of mTOR by S6K may be involved in
basolateral mTOR targeting and/or in mTOR-dependent regulation of NHE1
activity.
PI3K has been shown to play a role in acute regulation of the epithelial
Na+/H+ exchanger NHE3 by growth factors and other
stimuli (59,
60). In the MTAL,
hyposmolality increases NHE3 activity and
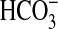 absorption through activation of
PI3K (13,
21). These effects are blocked
by rapamycin, indicating that mTOR functions downstream of PI3K to stimulate
NHE3 and
absorption through activation of
PI3K (13,
21). These effects are blocked
by rapamycin, indicating that mTOR functions downstream of PI3K to stimulate
NHE3 and 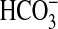 absorption in MTAL cells
(61). These findings can be
contrasted directly with the regulatory effects of NGF; activation of
PI3K-mTOR signaling by NGF results in inhibition of
absorption in MTAL cells
(61). These findings can be
contrasted directly with the regulatory effects of NGF; activation of
PI3K-mTOR signaling by NGF results in inhibition of
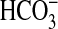 absorption through primary
inhibition of NHE1, with no direct coupling to NHE3
(Fig. 5) (Ref.
16). Thus, in the MTAL the
PI3K-mTOR pathway can be targeted specifically to inhibit basolateral NHE1 or
stimulate apical NHE3 depending on the physiological stimulus. The exact
mechanisms that target PI3K-mTOR signals to regulate different
Na+/H+ exchangers in different epithelial membrane
domains will be important to identify. The NGF-induced inhibition of NHE1 is
associated with an increase in phosphorylated mTOR in the basolateral
membrane. Thus, targeted activation of mTOR may be a component of PI3K-mTOR
signal specificity in MTAL cells.
absorption through primary
inhibition of NHE1, with no direct coupling to NHE3
(Fig. 5) (Ref.
16). Thus, in the MTAL the
PI3K-mTOR pathway can be targeted specifically to inhibit basolateral NHE1 or
stimulate apical NHE3 depending on the physiological stimulus. The exact
mechanisms that target PI3K-mTOR signals to regulate different
Na+/H+ exchangers in different epithelial membrane
domains will be important to identify. The NGF-induced inhibition of NHE1 is
associated with an increase in phosphorylated mTOR in the basolateral
membrane. Thus, targeted activation of mTOR may be a component of PI3K-mTOR
signal specificity in MTAL cells.
Neurotrophins and their receptors are highly expressed in the kidney, but
their roles in kidney function are not understood
(16). Our studies establish
directly that NGF can influence the transport function of renal tubules. The
change in 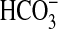 absorption induced by
NGF in the MTAL is comparable in magnitude to that observed with other
regulatory factors, including angiotensin II, aldosterone, chronic metabolic
acidosis and alkalosis, and dietary sodium intake
(8,
26,
27). The results of the
present study show that the inhibition of
absorption induced by
NGF in the MTAL is comparable in magnitude to that observed with other
regulatory factors, including angiotensin II, aldosterone, chronic metabolic
acidosis and alkalosis, and dietary sodium intake
(8,
26,
27). The results of the
present study show that the inhibition of
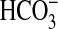 absorption by NGF is mediated
through a rapamycin-sensitive PI3K-mTOR pathway that is distinct from the
signaling pathways involved in inhibition by angiotensin II, aldosterone, and
vasopressin, thereby enabling NGF to regulate MTAL
absorption by NGF is mediated
through a rapamycin-sensitive PI3K-mTOR pathway that is distinct from the
signaling pathways involved in inhibition by angiotensin II, aldosterone, and
vasopressin, thereby enabling NGF to regulate MTAL
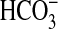 absorption in the presence of
these other hormones (Fig. 4)
(Refs. 20,
24,
26,
27, and
62). These findings support a
significant role for NGF in the physiological control of MTAL function.
absorption in the presence of
these other hormones (Fig. 4)
(Refs. 20,
24,
26,
27, and
62). These findings support a
significant role for NGF in the physiological control of MTAL function.
In summary, NGF inhibits NHE1 in the MTAL through the parallel activation
of PI3K-mTOR and ERK signaling pathways. The decrease in NHE1 activity results
secondarily in inhibition of apical NHE3 and transepithelial
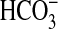 absorption
(16–18).
These studies provide the first evidence that mTOR regulates
Na+/H+ exchange activity and identify an important role
for PI3K-mTOR signaling in regulating the epithelial function of NHE1 to
enhance transcellular H+ secretion
(Fig. 10). Our findings raise
the possibility that NHE1 could function as a downstream target of mTOR that
plays a role in mediating mTOR's effects on cell growth, survival,
proliferation, and tumorigenesis.
absorption
(16–18).
These studies provide the first evidence that mTOR regulates
Na+/H+ exchange activity and identify an important role
for PI3K-mTOR signaling in regulating the epithelial function of NHE1 to
enhance transcellular H+ secretion
(Fig. 10). Our findings raise
the possibility that NHE1 could function as a downstream target of mTOR that
plays a role in mediating mTOR's effects on cell growth, survival,
proliferation, and tumorigenesis.
This work was supported, in whole or in part, by National Institutes of Health Grant R01 DK-038217. The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
Footnotes
The abbreviations used are: NHE, Na+/H+ exchanger;
pHi, intracellular pH; MTAL, medullary thick ascending
limb; NGF, nerve growth factor; ERK, extracellular signal-regulated kinase;
PI3K, phosphatidylinositol 3-kinase; mTOR, mammalian target of rapamycin; S6K,
p70 ribosomal S6 kinase; MEK, mitogen-activated protein kinase/ERK kinase;
TSC, tuberous sclerosis complex; 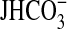 ,
absolute rate of
,
absolute rate of 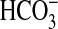 absorption; MOPS,
4-morpholinepropanesulfonic acid.
absorption; MOPS,
4-morpholinepropanesulfonic acid.
References
- 1.Counillon, L., and Pouyssegur, J. (2000) J. Biol. Chem. 275 1-4 [DOI] [PubMed] [Google Scholar]
- 2.Orlowski, J., and Grinstein, S. (1997) J. Biol. Chem. 272 22373-22376 [DOI] [PubMed] [Google Scholar]
- 3.Putney, L. K., Denker, S. P., and Barber, D. L. (2002) Annu. Rev. Pharmacol. Toxicol. 42 527-552 [DOI] [PubMed] [Google Scholar]
- 4.Stock, C., and Schwab, A. (2006) Acta Physiol. 187 149-157 [DOI] [PubMed] [Google Scholar]
- 5.Slepkov, E. R., Rainey, J. K., Sykes, B. D., and Fliegel, L. (2007) Biochem. J. 401 623-633 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lagana, A., Vadnais, J., Le, P. U., Nguyen, T. N., Laprade, R., Nabi, I. R., and Noel, J. (2000) J. Cell Sci. 113 3649-3662 [DOI] [PubMed] [Google Scholar]
- 7.Cardone, R. A., Casavola, V., and Reshkin, S. J. (2005) Nat. Rev. Cancer 5 786-795 [DOI] [PubMed] [Google Scholar]
- 8.Good, D. W. (1993) Semin. Nephrol. 13 225-235 [PubMed] [Google Scholar]
- 9.Alpern, R. J. (2000) in The Kidney (Brenner, B. M., ed) Vol. I, pp. 455-519, W. B. Saunders Co., Philadelphia, PA [Google Scholar]
- 10.Amemiya, M., Loffing, J., Lotscher, M., Kaissling, B., Alpern, R. J., and Moe, O. W. (1995) Kidney Int. 48 1206-1215 [DOI] [PubMed] [Google Scholar]
- 11.Good, D. W., and Watts, B. A., III (1996) Am. J. Physiol. 270 F691-F699 [DOI] [PubMed] [Google Scholar]
- 12.Biemesderfer, D., Rutherford, P. A., Nagy, T., Pizzonia, J. H., Abu-Alfa, A. K., and Aronson, P. S. (1997) Am. J. Physiol. 273 F289-F299 [DOI] [PubMed] [Google Scholar]
- 13.Watts, B. A., III, and Good, D. W. (1999) J. Clin. Investig. 104 1593-1602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Alper, S. L., Stuart-Tilley, A. K., Biemesderfer, D., Shmukler, B. E., and Brown, D. (1997) Am. J. Physiol. 273 F601-F614 [DOI] [PubMed] [Google Scholar]
- 15.Good, D. W., George, T., and Watts, B. A., III (1995) Proc. Natl. Acad. Sci. U. S. A. 92 12525-12529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Watts, B. A., III, George, T., and Good, D. W. (1999) J. Biol. Chem. 274 7841-7847 [DOI] [PubMed] [Google Scholar]
- 17.Good, D. W., Watts, B. A., III, George, T., Meyer, J., and Shull, G. E. (2004) Am. J. Physiol. 287 F1244-F1249 [DOI] [PubMed] [Google Scholar]
- 18.Watts, B. A., III, George, T., and Good, D. W. (2005) J. Biol. Chem. 280 11439-11447 [DOI] [PubMed] [Google Scholar]
- 19.Watts, B. A., III, and Good, D. W. (2002) Am. J. Physiol. 282 F1056-F1063 [DOI] [PubMed] [Google Scholar]
- 20.Good, D. W. (1990) J. Clin. Investig. 85 1006-1013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Good, D. W., Di Mari, J., and Watts, B. A., III (2000) Am. J. Physiol. 279 C1443-C1454 [DOI] [PubMed] [Google Scholar]
- 22.Watts, B. A., III, and Good, D. W. (1994) J. Biol. Chem. 269 20250-20255 [PubMed] [Google Scholar]
- 23.Watts, B. A., III, Di Mari, J. F., Davis, R. J., and Good, D. W. (1998) Am. J. Physiol. 275 F478-F486 [DOI] [PubMed] [Google Scholar]
- 24.Watts, B. A., III, George, T., and Good, D. W. (2005) Am. J. Physiol. 291 F1005-F1013 [DOI] [PubMed] [Google Scholar]
- 25.Fingar, D. C., and Blenis, J. (2004) Oncogene 23 3151-3171 [DOI] [PubMed] [Google Scholar]
- 26.Good, D. W., George, T., and Wang, D. W. (1999) Am. J. Physiol. 276 F726-F736 [DOI] [PubMed] [Google Scholar]
- 27.Good, D. W., George, T., and Watts, B. A., III (2006) Am. J. Physiol. 290 C757-C763 [DOI] [PubMed] [Google Scholar]
- 28.Holz, M. K., and Blenis, J. (2005) J. Biol. Chem. 280 26089-26093 [DOI] [PubMed] [Google Scholar]
- 29.Takahashi, E., Abe, J., Gallis, B., Aebersold, R., Springs, D. J., Krebs, E. G., and Berk, B. C. (1999) J. Biol. Chem. 274 20206-20214 [DOI] [PubMed] [Google Scholar]
- 30.Tominaga, T., Ishizaki, T., Narumiya, S., and Barber, D. L. (1998) EMBO J. 17 4712-4722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yan, W., Nehrke, K., Choi, J., and Barber, D. L. (2001) J. Biol. Chem. 276 31349-31356 [DOI] [PubMed] [Google Scholar]
- 32.Wakabayashi, S., Bertrand, B., Ikeda, T., Pouyssegur, J., and Shigekawa, M. (1994) J. Biol. Chem. 269 13710-13715 [PubMed] [Google Scholar]
- 33.Pang, T., Su, X., Wakabayashi, S., and Shigekawa, R. (2001) J. Biol. Chem. 276 17367-17372 [DOI] [PubMed] [Google Scholar]
- 34.Lin, X., and Barber, D. L. (1996) Proc. Natl. Acad. Sci. U. S. A. 93 12631-12636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rameh, L. E., and Cantley, L. C. (1999) J. Biol. Chem. 274 8347-8350 [DOI] [PubMed] [Google Scholar]
- 36.Cantley, L. C. (2002) Science 296 1655-1657 [DOI] [PubMed] [Google Scholar]
- 37.Wee, S., Lengauer, C., and Wiederschain, D. (2008) Curr. Opin. Oncol. 20 77-82 [DOI] [PubMed] [Google Scholar]
- 38.Ma, Y.-H., Reusch, H. P., Wilson, E., Escobedo, J. A., Fantl, W. J., Williams, L. T., and Ives, H. E. (1994) J. Biol. Chem. 269 30734-30739 [PubMed] [Google Scholar]
- 39.Reshkin, S. J., Bellizzi, A., Albarani, V., Guerra, L., Tommasino, M., Paradiso, A., and Casavola, V. (2000) J. Biol. Chem. 275 5361-5369 [DOI] [PubMed] [Google Scholar]
- 40.Fukao, T., and Koyasu, S. (2003) Trends Immunol. 24 358-363 [DOI] [PubMed] [Google Scholar]
- 41.Williams, D. L., Ozment-Skelton, T., and Li, C. (2006) Shock 25 432-439 [DOI] [PubMed] [Google Scholar]
- 42.Guertin, D. A., and Sabatini, D. M. (2007) Cancer Cell 12 9-22 [DOI] [PubMed] [Google Scholar]
- 43.Yano, H., Nakanishi, S., Kimura, K., Hanai, N., Saitoh, Y., Fukui, Y., Nonomura, Y., and Matsuda, Y. (1993) J. Biol. Chem. 268 25846-25856 [PubMed] [Google Scholar]
- 44.Brunn, G. J., Williams, J., Sabers, C., Wiederrecht, G., Lawrence, J. C., Jr., and Abraham, R. T. (1996) EMBO J. 15 5256-5267 [PMC free article] [PubMed] [Google Scholar]
- 45.Davies, S. P., Reddy, H., and Cohen, P. (2000) Biochem. J. 351 95-105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Avruch, J., Lin, Y., Long, X., Murthy, S., and Ortiz-Vega, S. (2005) Curr. Opin. Clin. Nutr. Metab. Care 8 67-72 [DOI] [PubMed] [Google Scholar]
- 47.Kenerson, H. L., Aicher, L. D., True, L. D., and Yeung, R. S. (2002) Cancer Res. 62 5645-5650 [PubMed] [Google Scholar]
- 48.Manning, B. D., Logsdon, M. N., Lipovsky, A. I., Abbott, D., Kwiatkowski, D. J., and Cantley, L. C. (2005) Genes Dev. 19 1773-1778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Woods, T. C., and Marks, A. R. (2004) Annu. Rev. Med. 55 169-178 [DOI] [PubMed] [Google Scholar]
- 50.Buhaescu, I., Izzedine, H., and Covic, A. (2006) Ther. Drug Monit. 28 577-584 [DOI] [PubMed] [Google Scholar]
- 51.Liu, Y. (2006) Kidney Int. 69 1925-1927 [DOI] [PubMed] [Google Scholar]
- 52.Volarevic, S., and Thomas, G. (2001) Prog. Nucleic Acids Res. Mol. Biol. 65 101-127 [DOI] [PubMed] [Google Scholar]
- 53.Mora, A., Komander, D., van Aalten, D. M. F., and Alessi, D. R. (2004) Semin. Cell Dev. Biol. 15 161-170 [DOI] [PubMed] [Google Scholar]
- 54.Peterson, R. T., Desai, B. N., Hardwick, J. S., and Schreiber, S. L. (1999) Proc. Natl. Acad. Sci. U. S. A. 96 4438-4442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Harwood, F. C., Shu, L., and Houghton, P. J. (2008) J. Biol. Chem. 283 2575-2585 [DOI] [PubMed] [Google Scholar]
- 56.Eguchi, S., Iwasaki, H., Ueno, H., Frank, G. D., Motley, E. D., Eguchi, K., Marumo, F., Hirata, Y., and Inagami, T. (1999) J. Biol. Chem. 274 36843-36851 [DOI] [PubMed] [Google Scholar]
- 57.Tee, A. R., Anjum, R., and Blenis, J. (2003) J. Biol. Chem. 278 37288-37296 [DOI] [PubMed] [Google Scholar]
- 58.Chiang, G. G., and Abraham, R. T. (2005) J. Biol. Chem. 280 25485-25490 [DOI] [PubMed] [Google Scholar]
- 59.Kurashima, K., Szabo, E. Z., Lukacs, G., Orlowski, J., and Grinstein, S. (1998) J. Biol. Chem. 273 20828-20836 [DOI] [PubMed] [Google Scholar]
- 60.Donowitz, M., and Li, X. (2007) Physiol. Rev. 87 825-872 [DOI] [PubMed] [Google Scholar]
- 61.Watts, B. A., III, George, T., and Good, D. W. (2006) J. Am. Soc. Nephrol. 17 14A (abstr.) [Google Scholar]
- 62.Good, D. W. (1998) Am. J. Physiol. 274 C931-C939 [DOI] [PubMed] [Google Scholar]