

Abstract
The selectivity filter of K+ channels comprises four contiguous ion binding sites, S1 through S4. Structural and functional data indicate that the filter contains on average two K+ ions at any given time and that these ions reside primarily in two configurations, namely to sites S1 and S3 or to sites S2 and S4. Maximum ion flux through the channel is expected to occur when the energy difference between these two binding configurations is zero. In this study, we have used protein semisynthesis to selectively perturb site 1 within the filter of the KcsA channel through use of an amide-to-ester substitution. The modification alters K+ conduction properties. The structure of the selectivity filter is largely unperturbed by the modification, despite the loss of an ordered water molecule normally located just behind the filter. Introduction of the ester moiety was found to alter the distribution of K+, Rb+ and Cs+ within the filter, with the most dramatic change found for Rb+. The redistribution of ions is associated with the appearance of a partially hydrated ion just external to the filter, at a position where no ion is observed in the wild type channel. The appearance of this new ion-binding site creates a change in the distance between a pair of K+ ions some fraction of the time, apparently leading to a reduction in the ion conduction rate. Importantly, this finding suggests that the selectivity filter of a potassium channel is optimized both in terms of absolute ion occupancy and in terms of the separation in distance between the conducting ions.
Potassium channels are tetrameric proteins that conduct K+ across cellular membranes, selectively and at very high rates.1 All K+ channels contain a highly conserved stretch of amino acids, the signature sequence (T/S)XG(Y/F)G, that lines the narrowest part of the ion conduction pathway, referred to as the selectivity filter (Figure 1A). The selectivity filter contains four sequential K+ binding sites, S1 to S4, each of which is formed by eight oxygen atoms, which surround K+ similar to an inner shell of water molecules when the ion is hydrated.2, 3 This form of molecular mimicry is at the heart of selective ion conduction by the channel; binding of K+, but not other ions such as Na+, to the protein offsets the energetic cost of dehydration. High-resolution structural studies indicate that, in the conducting state, the selectivity filter contains two K+ ions at any given time and that the occupancy of each of the four binding sites is around one half.4, 5 Based on several lines of evidence,4, 5 two discrete binding configurations are thought to dominate: in the (S1,S3) configuration K+ ions occupy positions S1 and S3 with water molecules at the other sites, while in the (S2,S4) configuration K+ ions occupy positions S2 and S4 with, again, intervening water molecules (Figure 1B). Ion movement between these conducting states is likely concerted, that is to say the two ions in the filter are kinetically coupled. Simulations predict that maximal ion flux through the filter is achieved when the two states are energetically equivalent. This effect can be understood at an intuitive level by realizing that the energy difference between the two configurations represents an energy barrier that must be overcome when the (S1,S3) and (S2,S4) configurations interconvert during the throughput cycle.
Figure 1.

The selectivity filter of K+ channels. (A) The KcsA channel is depicted in ribbon representation. Only two opposite subunits of the tetrameric protein are shown. The selectivity filter region (residues 75–79) is colored red. K+ ions bound to the selectivity filter are depicted as green spheres. (B) Close-up view of the selectivity filter. Two of the subunits are shown in stick representation. The K+ binding sites in the selectivity filter are labeled. The arrow indicates the peptide bond that was replaced by an ester bond.
The above model of ion conduction predicts that perturbation of ion binding at one site in the selectivity filter should be felt at the coupled site. This is, in fact, observed for the T75C mutant of the bacterial potassium channel, KcsA, in which a reduction of ion occupancy at site 4 is accompanied by a diminution of ion occupancy at site 2.6 That study exploited that fact that four of the eight oxygen atoms in site 4 are contributed by the side-chain of Thr75, which is part of the signature sequence, and is accessible to modification using conventional mutagenesis. Extending this type of structure-activity analysis to positions S1-S3 is non-trivial since they are constructed exclusively from backbone amide carbonyl oxygen atoms (Figure 1B). Techniques of chemical synthesis, rather than conventional mutagenesis, are better suited to problems of this type.7, 8 For example, amide-to-ester substitutions have been widely used by chemists as a way of engineering the backbone of proteins.9–12 This isosteric substitution alters the hydrogen bonding characteristics of the backbone and, of particular relevance to the present study, reduces the electronegativity of the carbonyl oxygen by roughly one half as compared to a typical amide.13 Thus, incorporation of an ester linkage within the selectivity filter of a channel would be expected to alter K+ binding and hence conduction.
The nonsense suppression approach has previously been used to incorporate backbone esters into the filter of the inward rectifier K+ channel, Kir2.1.14 The tyrosine residue in the signature sequence was replaced with α-hydroxytyrosine. This substitution is expected to perturb both site 1 and site 2 in filter and indeed was found to reduce the conductance rate and mean open time of the channel. This functional study did not, however, examine the effects of the ester substitution on the structure or ion occupancy of the filter. Thus, the origins of the observed changes in channel conduction could not be determined. In the present study we have used protein semisynthesis to perturb selectively position S1 within the filter of the KcsA channel, also through use of an amide-to-ester substitution. The consequences of this modification on the structure and function of the channel have been investigated using electrophysiology and x-ray crystallography. Overall, these studies reveal that subtle alterations in the electronics and hydrogen bonding characteristics of the selectivity filter can lead to significant changes in the binding and conduction of ions.
Materials and Methods
Chemical synthesis of depsi-peptides
Synthetic C-peptides corresponded either to residues 69–122 (with substitutions S69C and A98G) or residues 70–122 (with the substitution V70C) of KcsA and in both cases contained an ester linkage between residues Y78 and G79. The depsi-peptides were assembled by manual solid phase peptide synthesis using a slightly modified version of the in situ neutralization/HBTU (2-(1H-Benzotriazol-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate) activation protocol for Boc chemistry, as previously described.15 The ester linkage was incorporated as follows: Glycolic acid (2.2 mmoles) dissolved in 4 mLs of DMF:DCM (1:1) was activated with DIC (diisopropyl carbodiimide) (2.0 mmoles) in the presence of HOBT (N-hydroxybenzotriazole) (2.4 mmoles) at 0 °C for 15 min. The mixture was then added to the resin in the presence of 0.8 mmoles NEM (N-ethyl morpholine) and coupled for 10 min at RT. The resin was then treated with a solution consisting of ethanolamine (16.3 mmoles) and H2O (5% v/v) in DMF for 30 min. This procedure resulted in removal of the formyl protecting groups on the Trp residues and also eliminated any instances of double coupling of glycolic acid through hydrolysis of the resulting ester bond. (In initial work, generation of the ester side-product was found to be a serious problem). Boc-Tyr(2,6-dichloro-Z)-OH was then coupled to the alcohol using first DIC and then PyAOP ((7-azabenzotriazol-1-yloxy)tripyrrolidinophosphonium hexafluorophosphate) as the activating agents in a double coupling procedure. Further chain assembly was carried out as previously described.16 Protocols for cleavage of the peptide from the resin and side chain deprotection using anhydrous HF and purification of the peptide using RP-HPLC have been previously described and were used without modification.16
Purification of the N-peptide α-thioester
A dual fusion strategy was used for the expression of the N-peptides.16 The constructs consisted of KcsA residues 1–68 or 1–69 sandwiched between Glutathione-S-transferase (GST) at the N-terminus and the gyrA intein-chitin binding domain at the C-terminus. Bacterial expression and initial isolation of the fusion protein from inclusion bodies were carried out as previously described.16 Minor modifications to our published cleavage and purification protocol were made in order to improve the final yield of protein, most notably the order of removal of the GST and intein tags was reversed. In addition, a tricholoroacetic acid precipitation step (10% at 0 °C) was introduced following removal of the GST tag. Washing of the resulting precipitate with ice cold acetone was found to efficiently remove Triton X-100 detergent allowing superior protein recovery from the subsequent RP-HPLC purification step.
Semisynthesis of KcsAester
Two versions of KcsAester were prepared and involved the ligation of complementary N- and C-peptides using previously optimized conditions.16 Semisynthetic KcsAester analogues were folded and purified essentially as described,17 except that the purification steps were carried out at 4 °C and the concentration of KCl in the buffers was increased to 0.3 M from 0.15 M.
Electrophysiology
KcsAester and the semisynthetic KcsA (also truncated at residue 122 and containing a A98G mutation, referred to as wild-type throughout) were reconstituted into lipid vesicles composed of 1-palmitoyl-2-oleoyl-phosphatidylethanolamine (POPE, 7.5 mg/ml) and 1-palmitoyl-2-oleoyl-phosphatidylglycerol (POPG, 2.5 mg/ml).4 For measurements of single channel activity, the lipid vesicles were fused with planar lipid bilayers composed of POPE (15 mg/ml) and POPG (5 mg/ml) painted over a 50 μm hole in overhead transparency film separating the internal and external solutions.18 The recording solutions used consisted of 10 mM succinate, pH 4.0, 150 mM KCl on the internal side and 10 mM HEPES, pH 7.0, 150 mM KCl on the external side. At least two conductance states were observed for KcsAester and the wild type control. Data reported are for the larger conductance state.
Crystallization of the KcsAester
The KcsA-IgG was purified from mouse hybridoma cell culture supernatant and the Fab fragment was obtained by papain proteolysis as previously described.3 A complex of KcsAester with Fab was formed using published protocols3 and crystallized in the presence of 300 mM KCl at 20 °C using the sitting drop method. For each drop, concentrated KcsAester-Fab complex (7–15 mg/mL) was mixed with an equal volume of reservoir (20–25% PEG400, 50 mM magnesium acetate, pH 5.5–7.5). Cryo protection was achieved by increasing the PEG concentration in the reservoir to 40%. All crystals were flash frozen in liquid nitrogen cooled liquid propane for data collection. For crystallization in the presence of RbCl, the KcsAester-Fab complex was prepared in buffers containing KCl and then dialyzed against a buffer containing 300 mM RbCl prior to crystallization. Crystals of the KcsAester-Fab complex in the presence of CsCl were obtained as follows; crystals of the KcsAester-Fab complex grown in the presence of 300 mM KCl were washed 2× in a similar solution containing 300 mM CsCl, incubated overnight in the presence of 0.3M CsCl and then cryoprotected by increasing the PEG concentration in the reservoir to 40%.
Crystallographic Analysis
Data were collected at beam line X25 of the National Synchrotron Light Source, Brookhaven National Laboratory. The data were processed and scaled using Denzo and Scalepack from the HKL program suite.19 The structures were solved by molecular replacement using the published KcsA-Fab structure (PDB code: 1K4C). The model obtained after molecular replacement was modified to incorporate the amino acid substitutions that are present in the ester mutant. The model was then refined by cycles of manual rebuilding using the program O20 and refinement using CNS.21 One dimensional electron density profiles were obtained as follows. For the purposes of comparing the electron density in the ester mutant to the wild type protein, the data sets for the ester mutant were scaled to 2.25 Å for the K+ data set, 2.4 Å for the Rb+ data set and 2.75 Å for the Cs+ data set using the corresponding wild type data set as the reference set. An Fo−Fc omit map was then calculated using the refined model with ions and the selectivity filter residues removed as previously described.45 The one dimensional electron density profile was obtained by sampling the omit map along the central axis of the selectivity filter using Mapman.22 Coordinates of the KcsAester-Fab complexes have been deposited in the protein data bank.
Results
Semisynthesis of an ester analogue of KcsA
The goal of this study is to determine the effects on structure and function of perturbing a single K+ binding site within the selectivity filter of KcsA using an amide-to-ester substitution. Of the four backbone amides in the filter, only that between Tyr78 and Gly79 contributes to a single ion binding site, namely site 1 (Figure 1B). Hence, the peptide bond between Tyr78 and Gly79 was selected for replacement with an ester bond. We have previously reported the semisynthesis of a truncated but functional form of KcsA.16, 17 These protocols provide chemical access to the selectivity filter of KcsA and were used to replace the amide bond between Tyr78 and Gly79 with an ester bond. Briefly, the synthesis employed expressed protein ligation to assemble the target molecule from a recombinant N-terminal fragment (residues 1–68) and a synthetic C-terminal fragment (residues 69–122) containing the desired S1 ester linkage (Figure 2A). The ligation product was folded to the tetrameric state using lipid vesicles (Figure 2B).
Figure 2.

Semisynthesis of KcsAester. (A) SDS-PAGE gel (12%) of the expressed protein ligation reaction at 0 min. (lane 1) and 24 h (lane 2) showing the C-peptide (C), N-peptide (N) and the ligation product (L). (B) SDS-PAGE gel (12%) of the folding reaction showing the ligation product before (lane 1) and after the addition of lipid vesicles (lane 2). (C) SDS-PAGE gel (12% at 4 °C) of wild type KcsA (lane 1) and purified KcsAester (lane 2). For panels B and C; T, tetramer; M, monomer. (D) Electrospray MS of purified KcsAester; Inset, reconstructed spectrum (expected mass, 13059.0 Da).
The presence of a His6 tag on the N-terminus enabled initial purification of the folded and unfolded protein from the lipids using metal affinity chromatography. The folded tetrameric protein was then separated from the unfolded molecules and unreacted peptides using gel filtration chromatography. ES-MS of the purified protein indicated the expected mass for KcsA-Ψ[COO]-Gly79 (Figure 2D).
Unlike wild type KcsA, which migrates as a native tetramer on SDS-PAGE over a range of temperatures,23 the KcsA-Ψ[COO]-Gly79 analogue (hereafter referred to as KcsAester) migrates predominantly as a monomer at RT (data not shown) and as a mixture of monomeric and tetrameric states at 4 °C (Figure 2C). This finding reflects lower stability of KcsAester compared to the wild type channel. Similar behavior has been observed for a number of KcsA mutants with substitutions around the selectivity filter.16, 24
Single channel activity of KcsAester
We reconstituted the purified folded KcsAester protein into planar lipid bilayers for measurement of electrical activity. Single channel activity for the KcsAester protein was readily detected. Representative single channel traces of the ester and wild type proteins are shown (Figure 3A). The S1-ester substitution in the KcsA filter lowers the single channel conductance: current-voltage curves show that the zero-voltage conductance of KcsAester is approximately 50% that of the wild type channel. Notably, the shape of the current-voltage curve is strongly affected by the presence of the ester (Figure 3B). At large positive (> 100 mV) and large negative (< −100 mV) membrane voltages current through the wild type channel is nearly voltage independent, whereas the ester mutant exhibits a nearly linear dependence on voltage.
Figure 3.
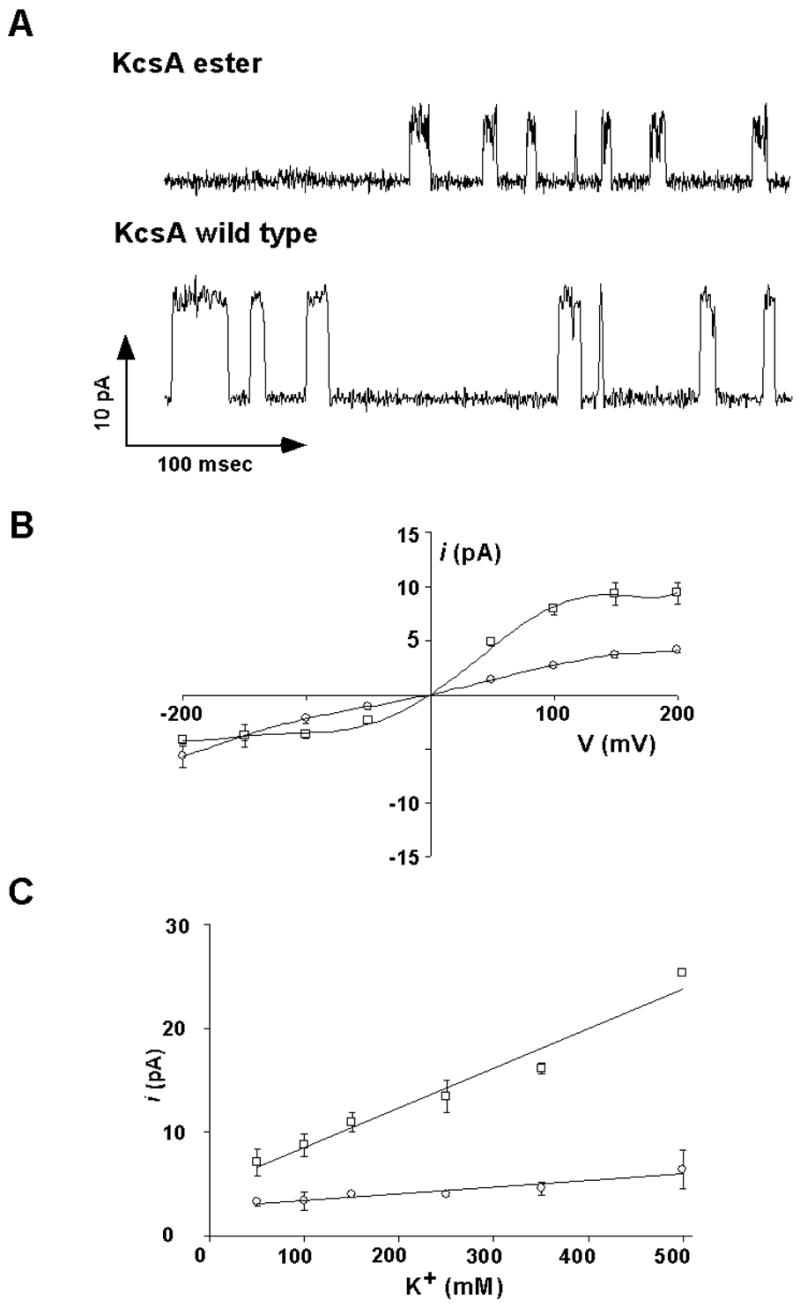
Electrophysiological analysis of KcsAester. (A) Representative single channel traces of the KcsAester or wild type KcsA channels recorded at +200 mV in 10 mM succinate/150 mM KCl (pH 4.0) inside and 10 mM HEPES/150 mM KCl (pH 7.0) outside. (B) Single channel current as a function of the membrane voltage in the above solutions for wild type KcsA (□) and KcsAester (○). Solid lines have no theoretical meaning. C) Single channel current at +200 mV as a function of the K+ ion concentration for wild type KcsA (□) and KcsAester (○). Solid lines have no theoretical meaning.
The single channel current at +200 mV was measured as a function of K+ concentration in the KcsAester analogue and wild type KcsA (Figure 3C). The wild type channel current increases in a nearly linear fashion over the range of 50–500 mM KCl, with no indication of leveling off. This linear regime is a feature of many K+ channels18 and is expected if the two K+ binding configurations, (1,3) and (2,4), are energetically balanced and if the rates of transition between the configurations is rapid compared to the rate of entry of a third ion from solution (which must occur in association with the exit of an ion from the opposite side of the filter).4 In contrast, the current through the KcsAester channel is smaller and increases only slightly over the same K+ concentration range. Indeed, the K+ current nearly levels off with a maximum value that is much lower than for the wild type channel. This “saturation” is reminiscent of the Rb+ conduction-concentration curve of the wild type channel4 and of the K+ conduction-concentration curve of the T75C mutant of KcsA.6 In both these cases, the observed effects on conduction were attributed in part to loss of the energetic equivalency of (1,3) and (2,4) configurations.4,6 Of note, the Rb+ conduction properties of the KcsAester channel were also measured (data not shown) and were found to be very similar to those of the wild-type channel.
Crystallization of the KcsAester channel
To characterize the effects of the amide-to-ester replacement on ion binding to the selectivity filter we determined the crystal structure of the KcsAester channel in the presence of K+, Rb+ and Cs+. The semisynthetic protein used in these crystallographic studies differed slightly from that used in the preceding functional studies. Firstly, the ligation site employed in the semisynthesis was moved to between S69 and V70, with these two residues being replaced with Ala and Cys, respectively. This adjustment significantly improved the yield of the ligation product, providing sufficient material for crystallographic analysis. Secondly, we reversed the A98G mutation in the inner helix, which permits electrophysiological measurements on the truncated protein.17 This change afforded protein crystals that diffracted to higher resolution. Crystals of the KcsAester channel were grown as a complex with an antibody Fab fragment as previously described.3
Structure of the KcsAester channel
The structures of the KcsAester analogue in the presence of K+, Rb+ and Cs+ were solved by molecular replacement using the wild type KcsA-Fab structure (pdb code, 1K4C) as a search model. The structures containing K+ and Rb+ were both refined to 2.25 Å resolution, while the Cs+ containing structure was refined to 2.75 Å resolution. Data collection and structure refinement statistics are given in Table 1. Overall, the KcsAester structure is very similar to that of the wild type channel determined at 2.0 Å resolution.3 The root mean square deviation (rmsd) between the KcsAester and wild type KcsA structures in the presence of K+ is 0.60 Å over the backbone and 0.64 Å for all non hydrogen atoms. An electron density map calculated using Fourier coefficients (2Fo−Fc), for the selectivity filter of the KcsAester analogue in the presence of K+ (Figure 4A), and the refined model (Figure 4B), show that the selectivity filter region of KcsAester is nearly identical to that of the wild type channel.
Table 1.
Crystallographic data collection and refinement statistics
| KCl | RbCl | CsCl | |
|---|---|---|---|
| Resolution (Å) | 2.25 | 2.25 | 2.75 |
| Rsyma | 0.073(0.51) | 0.079(0.462) | 0.087(0.434) |
| Completeness(%) | 98.1(96.3) | 98.9(99.9) | 97.0(98.5) |
| Refinement statistics | |||
| Rfree/Rwork(%)b | 24.2/23.3 | 25.1/23.3 | 27.1/23.9 |
| Mean B-factor (Å2) | 58.9 | 52.2 | 57.9 |
| Root mean square difference | |||
| Bond lengths (Å) | 0.013 | 0.014 | 0.008 |
| Bond angles (°) | 1.6 | 1.7 | 1.4 |
Rsym = Σ |Ii − <Ii>|/ΣIi where <Ii> is the average intensity of symmetry-equivalent reflections.
R = Σ |Fo − Fc|/ΣFo, 5% of the reflections that were excluded from the refinement were used in the Rfree calculation.
Numbers in brackets are statistics for the last resolution shell.
Figure 4.
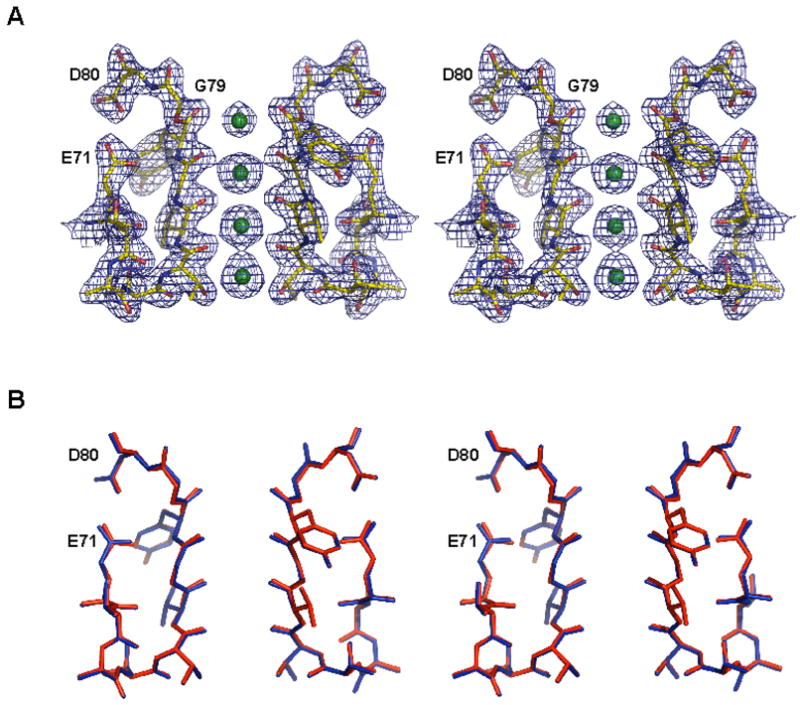
Structure of the selectivity filter of KcsAester. (A) Stereo view of the electron density of the selectivity filter of KcsAester. The 2Fo−Fc electron density map contoured at 2.0σ is shown with residues 71–80 shown as sticks and K+ ions in the selectivity filter depicted as green spheres. (B) Stereo view of the superimposition of residues 71–80 of the KcsAester (red) and the wild type KcsA (blue).
Amide-to-ester substitutions have been widely used in protein engineering,9–12 however, there has only been one high-resolution structure of an ester-containing protein reported to date, a synthetic analogue of a small domain from a serine proteinase inhibitor.25 In this case, the ester bond was found to be trans-planar. Based on analysis of the Fo − Fc electron density omit map (with residues 77 to 80 omitted), the ester linkage between residues 78 and 79 is also in the trans conformation (Figure 5A), although we cannot rule out minor deviations from planarity. Also of note was the electron density corresponding to the ligation site (Figure 5B). In the Fo−Fc electron density omit map (with residues 68 to 71 omitted) of this region, the peptide backbone is clearly defined and confirms that an amide bond has indeed been formed between Ala69 and Cys70 and that the L-stereoisomer of Ala69 is present.
Figure 5.
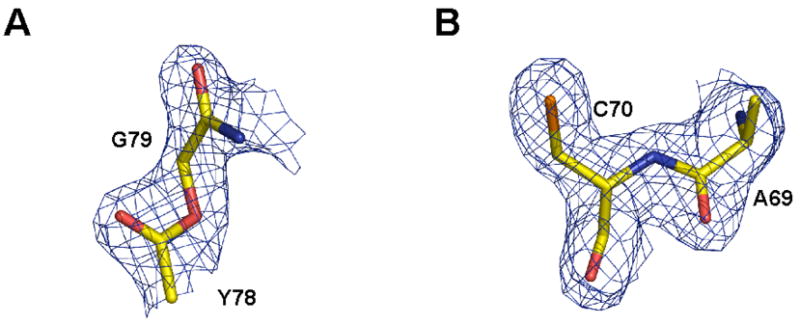
A) Fo−Fc omit map (residues 77–80 omitted) contoured at 3.0σ showing the ester bond introduced between Y78 and G79 B) Fo−Fc omit map (residues 68–71 omitted) contoured at 3.0σ showing the peptide bond formed between A69 and C70 by the expressed protein ligation reaction.
Replacement of an amide bond with an ester results in the removal of a hydrogen bond donor. In the wild type channel, the G79 amide donates a hydrogen bond to a highly ordered water molecule located in the protein core surrounding the selectivity filter (Figure 6A, B). This buried water molecule mediates a hydrogen bond network that may play a role in stabilizing the filter (Figure 6A, B). Thus, we were keen to see the effect of the ester modification on this region of the protein. We did not observe electron density corresponding to this buried water molecule in any of the three KcsAester structures. Electron density is shown for the KcsAester analogue in the presence of K+ (Figure 6C). The absence of defined density for the water molecule in our structures indicates that the water molecule is no longer present or that it is disordered. Despite the apparent absence of the water molecule, we see that the positions of protein atoms remain essentially unchanged (Figures 4B).
Figure 6.
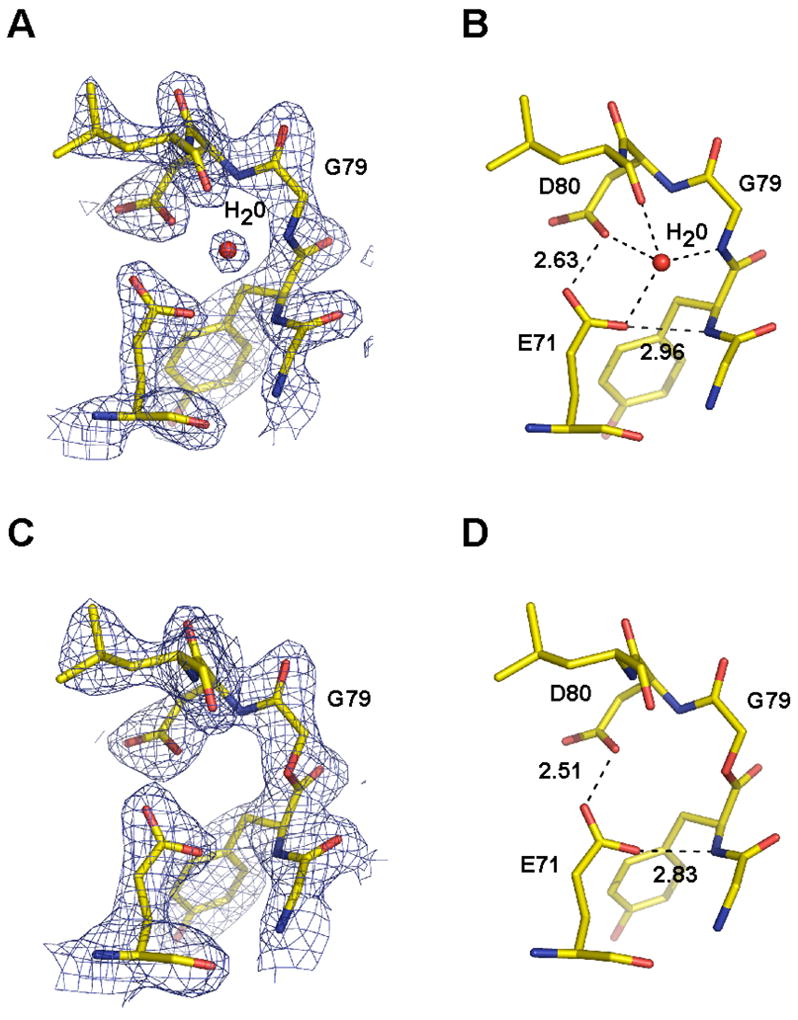
A) 2Fo−Fc map contoured at 2.0σ showing the buried water molecule present behind the selectivity filter of KcsA. B) Hydrogen bond interactions formed by the water molecule with the surrounding protein residues in wild type KcsA are indicated. C) 2Fo−Fc map contoured at 2.0σ of KcsAester showing the lack of a buried water molecule. D) Hydrogen bond interactions formed behind the selectivity filter in KcsAester are indicated.
Of particular interest are the positions of Glu71 and Asp80. In the wild type KcsA structure, the side-chains of these residues are within hydrogen bonding distance of the ordered water molecule, but are also close enough to form a direct carboxyl-carboxylate hydrogen bond (Figure 6B). Mutagenesis studies suggest that the Glu71-Asp80 interaction is important for the stability of the native KcsA tetramer.24 The KcsAester tetramer is destabilized relative to the wild type, as evidenced by the dissociation of subunits into monomers on SDS PAGE, raising the possibility that the carboxyl-carboxylate interaction is disrupted. This is clearly not the case. In fact, the carboxyl-carboxylate hydrogen bond is slightly shorter in the KcsAester structures compared to the wild type KcsA structure (Figure 6B, 6D): 2.63 Å in wild type KcsA3 vs. 2.51 Å (K+) and 2.55 Å (Rb+) in the KcsAester structures. The Cs+ KcsAester structure was not included in this comparison because of its lower resolution.
Distribution of ions in the KcsAester filter
The amide-to-ester substitution has significant effects on the distribution of K+, Rb+ and Cs+ ions in the selectivity filter. Electron density corresponding to K+ ions bound in the selectivity filter is shown for wild type KcsA (Figure 7A) and for KcsAester (Figure 7B). A smaller peak of electron density is observed at site 1 of the ester filter relative to that of the wild type, suggesting lower K+ occupancy at this site. In contrast, the electron density at sites 2–4 is not significantly altered. The magnitude of peaks can be appreciated in one-dimensional electron density profiles, which suggest that K+ occupancy at site 1 has been reduced by ~50% in the ester channel (Figure 7C).
Figure 7.
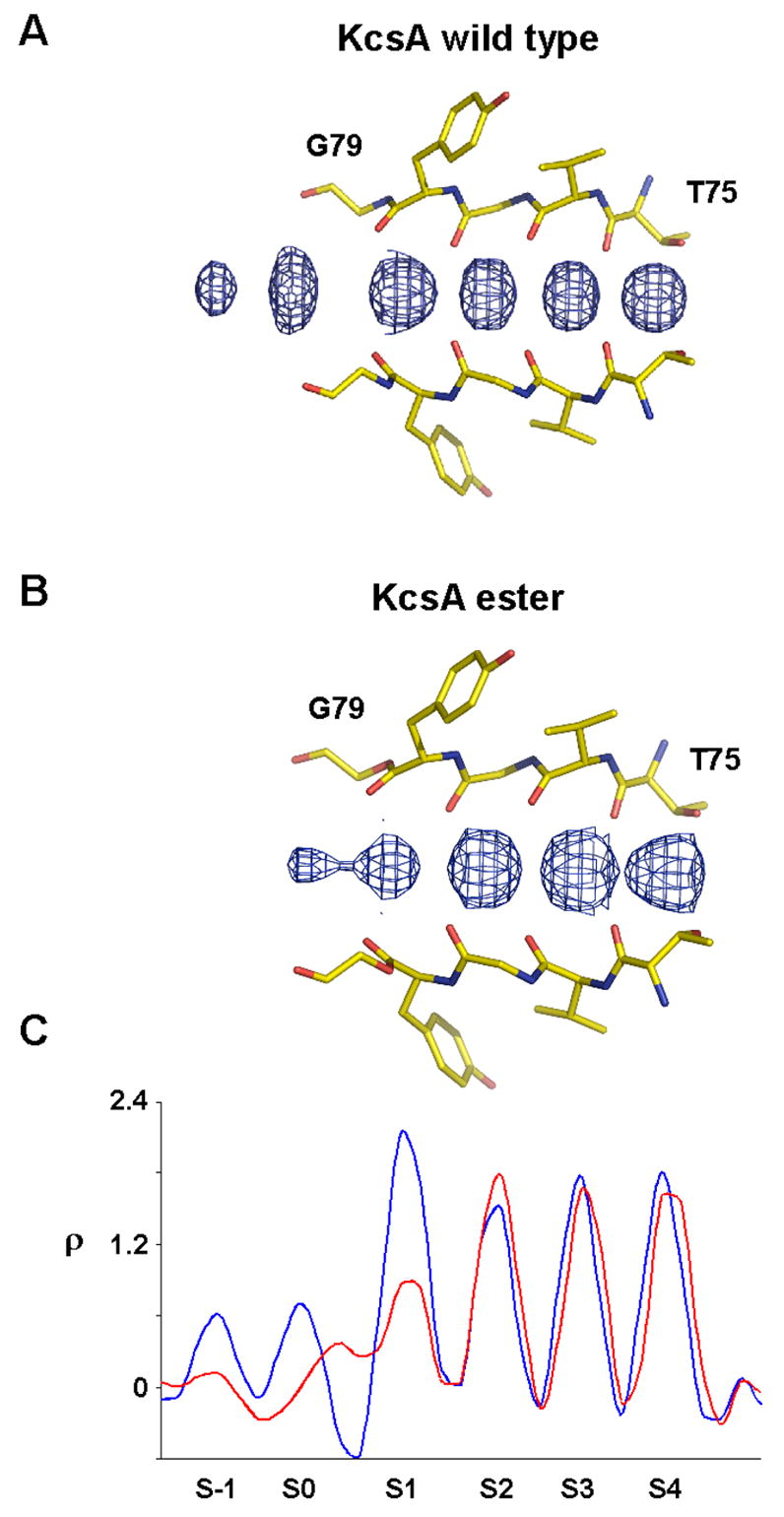
K+ binding to the selectivity filter of KcsAester. Electron density along the central axis of the selectivity filter of the wild type KcsA (A) and KcsAester (B) in a Fo−Fc omit map (residues 75–79 and K+ ions omitted) contoured at 3.0σ. (C) One dimensional plot of the electron density sampled axis of the selectivity filter for wild type KcsA (blue) and KcsAester (red).
In addition to differences in the distribution of ions within the filter, important information is also provided by ester-induced changes in the electron density for ions just outside the filter, at its entryway. In the K+ complex of the wild type channel, two electron density peaks at the external entryway, labeled S-1 and S0 in Figure 7A,C, were explained on the basis of (S1,S3) and (S2,S4) configurations within the filter. Specifically, a K+ ion at the external entryway was proposed to reside at S-1 when two ions inside the filter reside at the S1 and S3 positions ((S1,S3) configuration) and at S0 when two filter ions reside at the S2 and S4 positions ((S2,S4) configuration). In the ester mutant two ions at the entryway are not clearly present. Rather, one peak of density ‘outside’ the filter is near to S1, as if the location of the S1 ion is split between its normal (S1) position and a new (S0.5) position, which is shifted slightly toward the external solution (Figure 7C). Thus, it appears as if the S1 ion of the wild type channel is distributed over S1 and S0.5 positions in KcsAester (see discussion).
The ester substitution also changes the distribution of Rb+ and Cs+ ions in the selectivity filter (Figure 8). The consequences are most dramatic for Rb+. In the wild type channel, Rb+ ions are observed at sites 1, 3 and 4, but essentially not at all at site 2.4, 5 In the case of the KcsAester channel, we see strong electron density peaks at sites 2, 3 and 4, but, conversely, very little density at site 1 (Figure 8A). This change in Rb+ occupancy is clearly seen in the one dimensional electron density profiles of the wild type and KcsAester channel structures (Figure 8B). In addition to the changes within the filter, we also see the appearance of new electron density in the entryway to the pore of the KcsAester channel. This density is quite broad but has a strong peak at a position between sites S0 and S1. The amide-to-ester substitution appears to have only a small effect on the binding of Cs+ ions in the selectivity filter (Figure 8A, B). Careful comparison with the wild type channel shows that for Cs+ the three peaks of electron density are very slightly shifted toward the intracellular side in the KcsAester channel structure and the first peak is reduced and somewhat broadened (Figure 8A, B).
Figure 8.
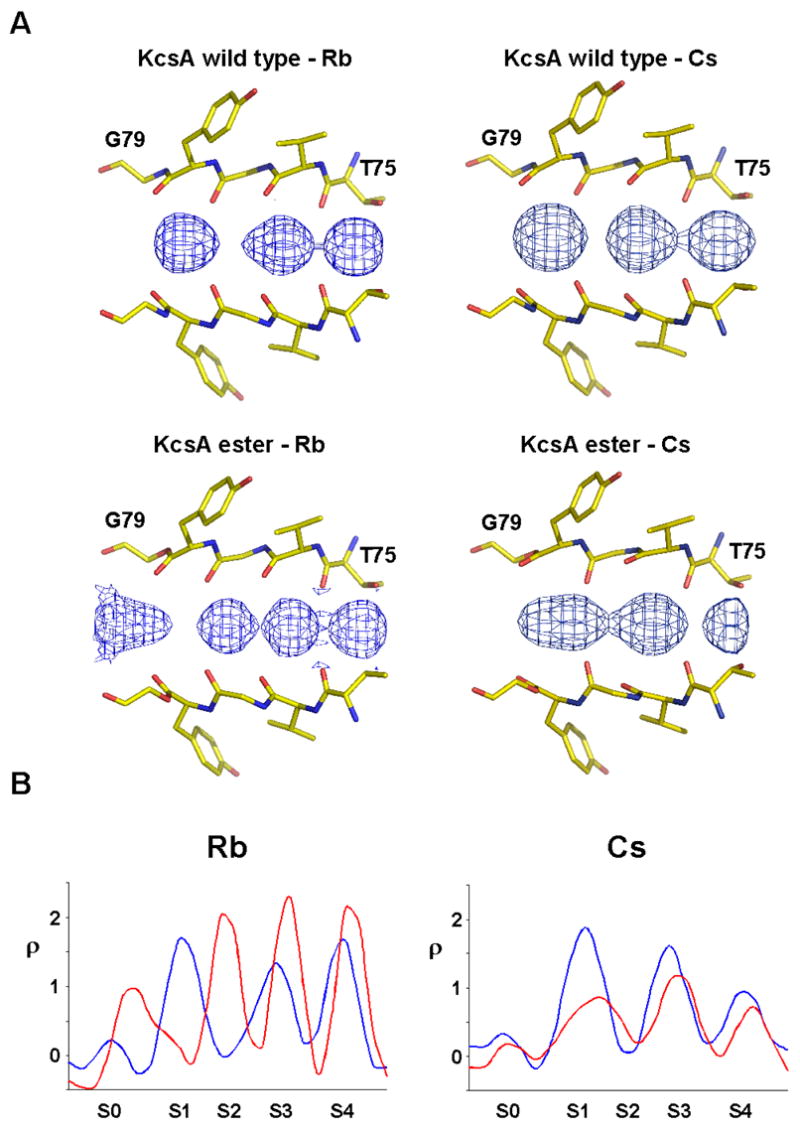
Rb+ and Cs+ binding to the selectivity filter of KcsAester. A) Electron density along the central axis of the selectivity filter of wild type KcsA and KcsAester in a Fo−Fc omit map (residues 75–79 and ions omitted) contoured at 3.0σ is shown. B) One dimensional plot of the electron density sampled along the central axis of the selectivity filter for wild type KcsA (blue) and KcsAester (red).
Discussion
The conduction properties of potassium ion channels are exceptionally sensitive to mutations within the selectivity filter,26 reflecting the fact that the steric and electronic properties of this region of the channel are critical to ion conduction. The goal of the present study is to evaluate the structural and functional consequences of an amide to ester substitution. This form of mutation is much more subtle than any protein alteration achievable through amino acid substitution and allows us to examine the role of main chain oxygen atoms in the K+ selectivity filter.
Esters are excellent peptide bond isosteres since they display a strong preference for the trans conformation, although the rotational barrier for an ester bond is somewhat smaller than for an amide (10–15 kcal mol−1 vs. 18–21 kcal mol−1).27 An amide-to-ester substitution has two consequences for hydrogen bonding; a hydrogen bond donor is deleted by replacing the amide NH with an ester oxygen and a hydrogen bond acceptor is weakened by replacing the amide carbonyl with an ester carbonyl.10 We observe that the KcsAester tetramer is less stable than the wild type KcsA tetramer (Figure 2C). It is tempting to attribute this reduction in stability solely to changes in hydrogen bonding, particularly since we see the complete disappearance of electron density corresponding to a buried water molecule that normally accepts a hydrogen bond from the G79 amide (Figure 6). Such an interpretation is, however, over simplistic since it does not take into account possible effects of the amide-to-ester substitution on solvation, van der Waals interactions, conformational entropy and electrostatics. While some of these effects might be small, especially the van der Waals and entropic contributions,10 the change in electrostatics caused by amide-to-ester substitution is likely to be a contributing factor to the lower stability of KcsAester, particularly since K+ binding to the selectivity filter is significantly altered (Figure 7).
An unexpected consequence of the amide-to-ester replacement is a reduction in the length of the Glu71-Asp80 carboxyl-carboxylate hydrogen bond (Figure 6B, D). In the wild type channel, the length of this carboxyl-carboxylate hydrogen bond is in the upper range of that typically seen for carboxyl-carboxylates (average ~ 2.5 Å), whereas in the ester analogue the bond length is closer to the expected distance.28 It is interesting that shortening of the carboxyl-carboxylate hydrogen bond should accompany the loss of the ordered water molecule in the protein core next to the filter. One interpretation of this is that the hydrogen bonds from the buried water molecule compensate for, and perhaps lead to, a weaker carboxyl-carboxylate hydrogen bond. With this in mind, we note that the δ-oxygen of Asp80 appears to hydrogen bond to both the Glu71 carboxyl group and the buried water in the structure of the wild type channel (Figure 6B). The absence of the water in the KcsAester structure suggests that this oxygen no longer acts as a bifurcate hydrogen bond acceptor, possibly leading to a stronger carboxyl-carboxylate hydrogen bond (Figure 6D). It is also conceivable that the ionization properties of the carboxyl-carboxylate pair are altered by the absence of the buried water molecule. One possibility is that in the wild type channel there is exchange between a carboxyl-carboxylate-water configuration and a carboxylate-carboxylate-hydroxonium configuration. In other words, the proton might actually be shared between the two carboxylates and the water. Intuitively, this would lead to a net lengthening of the carboxyl-carboxylate bond for electrostatic reasons. Additional analysis, perhaps employing quantum mechanical calculations, will be required to explore this idea further.
The conductance of KcsAester channel is lower than the wild type channel as indicated by single channel measurements in planar lipid bilayers (Figure 3A). At positive voltages, the K+ current through the KcsAester channel is approximately 50% that of the wild type (Figure 3B). The amide-to-ester substitution also leads to a pronounced saturation in the K+ conductance-concentration curve (Figure 3C). Such a behavior has been observed previously in two contexts: Rb+ conduction through wild type KcsA 4 and K+ conduction through the T75C mutant of KcsA.6 In these cases, the effect was explained in terms of a loss in energetic equivalency of the two ion binding configurations. The amide-to-ester substitution does not alter the backbone structure of the filter (Figure 4B); the backbone rmsd for the wild type KcsA and KcsAester structures from residue 75 to residue 80 is 0.15 Å. Thus, the change in K+ conductivity must be related to the altered electronic properties of the filter resulting from the reduced electronegativity of the ester carbonyl oxygen.13
Crystallographic analysis shows that the ion occupancy of KcsAester is altered compared to the wild type channel: K+ ion electron density at S1 is reduced by about 50% (Figure 7). This reduction is qualitatively consistent with the diminished electronegativity of the S1 binding site due to the smaller dipole moment of an ester compared to an amide (1.7 D vs. 3.5 D).13 The reduction appears to come about through a shift of the outermost K+ ion (S1 in wild type) to the S0.5 position some fraction of the time (in other words, in some fraction of channels in the crystal) (Figure 7C). This shift implies that in KcsAester a partially hydrated K+ ion at S0.5 is almost equally stable as a dehydrated K+ ion at S1. It is likely relevant that the dipole moment of a water molecule (1.8 D) is close to that of an ester.13 So we think that the amide-to-ester conversion at site 1 of the channel tends to give the K+ ion to the partially hydrated S0.5 position. This interpretation of the K+ ion electron density in KcsAester implies the existence of three prevalent ion configurations ((S0.5, S3), (S1, S3) and (S2, S4)) instead of the two ((S1, S3) and (S2, S4)) that dominate in wild type KcsA. Altered K+ conduction in KcsAester could therefore be described not simply as a result of a shift in energy equality of (S1, S3) and (S2, S4) configurations, but as the creation of a (S0.5, S3) configuration that replaces the (S1, S3) configuration some fraction of the time.
What consequence would such a change have on ion conduction? Rates are not predictable from structure alone, however, it is apparent that instead of having a conduction cycle in which the filter fluctuates between (S1,S3) and (S2,S4) configurations it will sometimes fluctuate between (S0.5,S3) and (S2,S4) configurations. As long as the throughput rate is influenced by the transitions between configurations of paired ions in the filter (which is thought to be the case4) then the creation of a new configuration is expected, except under very special circumstances, to alter the conduction rate. Why in mechanistic terms might the rate be reduced? One likely possibility is that the (S0.5,S3) configuration has an ‘unnatural’ spacing between the pair of ions (i.e. center-to-center spacing between the pair of ions is 9.2 Å for the (S0.5,S3) configuration compared to 6.6 Å and 6.5 Å for the (S1,S3) and (S2,S4) configurations, respectively). Among other effects, a change in distance between the ions will influence the electrostatic interaction between them. The observed effects of the ester substitution on the ion distributions in the filter and on conduction rates leads us to propose that equal spacing of ions in the selectivity filter is an important aspect of high conduction rates through K+ channels.
The occupancy of Rb+ and Cs+ ions is also altered in the KcsAester channel (Figure 7). The effect is most dramatic for Rb+, in which case we see strong peaks of ion electron density at sites 2, 3 and 4 in the KcsAester channel, as compared to sites 1, 3 and 4 in the wild type channel. We also observe new Rb+ ion electron density centered between site S0 and S1 in the KcsAester structure, analogous to the situation with K+. Indeed, the K+ and Rb+ electron density profiles are quite similar in the KcsAester channel. It is likely that creation of a S0.5 Rb+ binding site in the KcsAester channel is at least partly responsible for the similarities in Rb+ and K+ conduction. A change in ion occupancy is also observed for Cs+, though the effects are less obvious than for K+ and Rb+. It may be that Cs+, having the largest radius and lowest charge density, may be less sensitive to perturbations of binding site electronegativity.
In summary, we have analyzed the functional and structural consequences of an amide-to-ester substitution at the outermost position of the K+ selectivity filter. The perturbation causes a water molecule that forms a hydrogen bond with the amide nitrogen in the wild type channel to disappear. The protein atoms maintain their wild type positions and thus the main effect on ions in the selectivity filter should be mediated by the reduction of the dipole moment of the carbonyls that form the K+ binding site. The conductance of the channel is reduced, as is the occupancy of the binding site. The reduction of the occupancy comes about because the K+ ion tends to shift to a position in which it can become partially hydrated. This change in ion configuration is expected to effect the normal through-put cycle of K+ conduction.
Acknowledgments
We thank Seok-Yong Lee for help with crystallographic data collection and analysis and Dr. Eric Gouaux for providing computer access to FIV. This work was supported by grants from the NIH to T. W. M. (EB001991, GM55843) and R. M. (GM43949). R. M. is an Investigator in the HHMI.
References
- 1.MacKinnon R. Angew Chem Int Ed Engl. 2004;43:4265–77. doi: 10.1002/anie.200400662. [DOI] [PubMed] [Google Scholar]
- 2.Doyle DA, Morais Cabral J, Pfuetzner RA, Kuo A, Gulbis JM, Cohen SL, Chait BT, MacKinnon R. Science. 1998;280:69–77. doi: 10.1126/science.280.5360.69. [DOI] [PubMed] [Google Scholar]
- 3.Zhou Y, Morais-Cabral JH, Kaufman A, MacKinnon R. Nature. 2001;414:43–8. doi: 10.1038/35102009. [DOI] [PubMed] [Google Scholar]
- 4.Morais-Cabral JH, Zhou Y, MacKinnon R. Nature. 2001;414:37–42. doi: 10.1038/35102000. [DOI] [PubMed] [Google Scholar]
- 5.Zhou Y, MacKinnon R. J Mol Biol. 2003;333:965–75. doi: 10.1016/j.jmb.2003.09.022. [DOI] [PubMed] [Google Scholar]
- 6.Zhou M, MacKinnon R. J Mol Biol. 2004;338:839–46. doi: 10.1016/j.jmb.2004.03.020. [DOI] [PubMed] [Google Scholar]
- 7.Muir TW. Annu Rev Biochem. 2003;72:249–89. doi: 10.1146/annurev.biochem.72.121801.161900. [DOI] [PubMed] [Google Scholar]
- 8.Wang L, Schultz PG. Angew Chem Int Ed Engl. 2004;44:34–66. doi: 10.1002/anie.200460627. [DOI] [PubMed] [Google Scholar]
- 9.Lu W, Qasim MA, Laskowski M, Jr, Kent SB. Biochemistry. 1997;36:673–9. doi: 10.1021/bi9625612. [DOI] [PubMed] [Google Scholar]
- 10.Koh JT, Cornish VW, Schultz PG. Biochemistry. 1997;36:11314–22. doi: 10.1021/bi9707685. [DOI] [PubMed] [Google Scholar]
- 11.England PM, Zhang Y, Dougherty DA, Lester HA. Cell. 1999;96:89–98. doi: 10.1016/s0092-8674(00)80962-9. [DOI] [PubMed] [Google Scholar]
- 12.Deechongkit S, Nguyen H, Powers ET, Dawson PE, Gruebele M, Kelly JW. Nature. 2004;430:101–5. doi: 10.1038/nature02611. [DOI] [PubMed] [Google Scholar]
- 13.Lide DR. Handbook of Chemistry and Physics. 1990 [Google Scholar]
- 14.Lu T, Ting AY, Mainland J, Jan LY, Schultz PG, Yang J. Nat Neurosci. 2001;4:239–46. doi: 10.1038/85080. [DOI] [PubMed] [Google Scholar]
- 15.Schnolzer M, Alewood P, Jones A, Alewood D, Kent SB. Int J Pept Protein Res. 1992;40:180–93. doi: 10.1111/j.1399-3011.1992.tb00291.x. [DOI] [PubMed] [Google Scholar]
- 16.Valiyaveetil FI, MacKinnon R, Muir TW. J Am Chem Soc. 2002;124:9113–20. doi: 10.1021/ja0266722. [DOI] [PubMed] [Google Scholar]
- 17.Valiyaveetil FI, Sekedat M, Muir TW, MacKinnon R. Angew Chem Int Ed Engl. 2004;43:2504–7. doi: 10.1002/anie.200453849. [DOI] [PubMed] [Google Scholar]
- 18.LeMasurier M, Heginbotham L, Miller C. J Gen Physiol. 2001;118:303–14. doi: 10.1085/jgp.118.3.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Otwinowski Z, Minor W. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 20.Jones TA, Zou JY, Cowan SW, Kjeldgaard Acta Crystallogr A. 1991;47:110–9. doi: 10.1107/s0108767390010224. [DOI] [PubMed] [Google Scholar]
- 21.Brunger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, Read RJ, Rice LM, Simonson T, Warren GL. Acta Crystallogr D Biol Crystallogr. 1998;54:905–21. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 22.Kleywegt GJ, Jones TA. Acta Crystallogr D Biol Crystallogr. 1996;52:826–8. doi: 10.1107/S0907444995014983. [DOI] [PubMed] [Google Scholar]
- 23.Cortes DM, Perozo E. Biochemistry. 1997;36:10343–52. doi: 10.1021/bi971018y. [DOI] [PubMed] [Google Scholar]
- 24.Choi H, Heginbotham L. Biophys J. 2004;86:2137–44. doi: 10.1016/S0006-3495(04)74273-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bateman KS, Huang K, Anderson S, Lu W, Qasim MA, Laskowski M, Jr, James MN. J Mol Biol. 2001;305:839–49. doi: 10.1006/jmbi.2000.4343. [DOI] [PubMed] [Google Scholar]
- 26.Heginbotham L, Lu Z, Abramson T, MacKinnon R. Biophys J. 1994;66:1061–7. doi: 10.1016/S0006-3495(94)80887-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wiberg KB, Laidig KE. J Am Chem Soc. 1987;109:5935–5943. [Google Scholar]
- 28.Speakman JC. Struct Bonding (Berlin) 1972;12:141–199. [Google Scholar]