

Abstract
The enantioselective synthesis of FD-891 has been achieved with a longest linear sequence of 21 steps. The synthetic strategy involves the use of aldol additions of a chlorotitanium enolate of N-acylthiazolidinthiones as the key reaction to establish 6 of the 10 stereogenic centers. A key cross-metathesis and a late stage Julia olefination serve to assemble three key subunits.
FD-891, a 16-membered macrolide isolated from the fermentation broth of Streptomyces graminofaciens A-8890, has been shown to have cytotoxic activity in vitro against several tumor cell lines.1 The activity is reportedly similar to that of concanamycin A, a specific inhibitor of vascular-type H+-ATPase.2 Recently, concanamycin A has been shown to specifically inhibit perforin-dependent cytotoxic T lymphocyte (CTL)-mediated cytotoxicity, but not affect Fas ligand (FasL)-dependent CTL-mediated cytotoxicity; these two cytotoxic pathways play an essential role in the maintenance of tissue homeostasis.3 Conversely, FD-891 was found to potently prevent both perforin and FasL-dependent CTL-mediated killing pathways, but did not inhibit vacuolar acidification.4
The relative and absolute configuration of FD-891 was elucidated after extensive spectroscopic analysis and partial degradation.5 The structure was subsequently revised based on x-ray analysis of partial structures.6 While substantial synthetic accomplishments have been made on related plecomacrolides,7 to date only three reports of synthesis of fragments of FD-891 have appeared.8
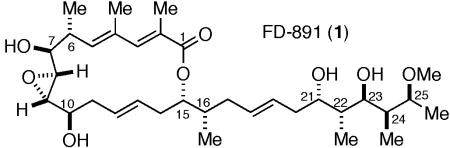
Herein we report the first total synthesis of FD-891. A convergent strategy exploiting the assembly of three subunits 2, 3, and 4 of similar complexity was anticipated for the synthesis of FD-891 (Scheme 1). Fragments 2 and 3 were envisioned to undergo selective cross-metathesis leading to a subsequent lactonization to give the macrocyclic core. The late-stage installation of the C19–C25 fragment 4 would be accomplished via a Julia olefination. The three key fragments would derive from synthons 5 and 6 accessible through the application of the versatile aldol additions of chlorotitanium enolates of N-acylthiazolidinethiones recently advanced in our laboratory.9
Scheme 1.
Retrosynthetic analysis of FD-891.
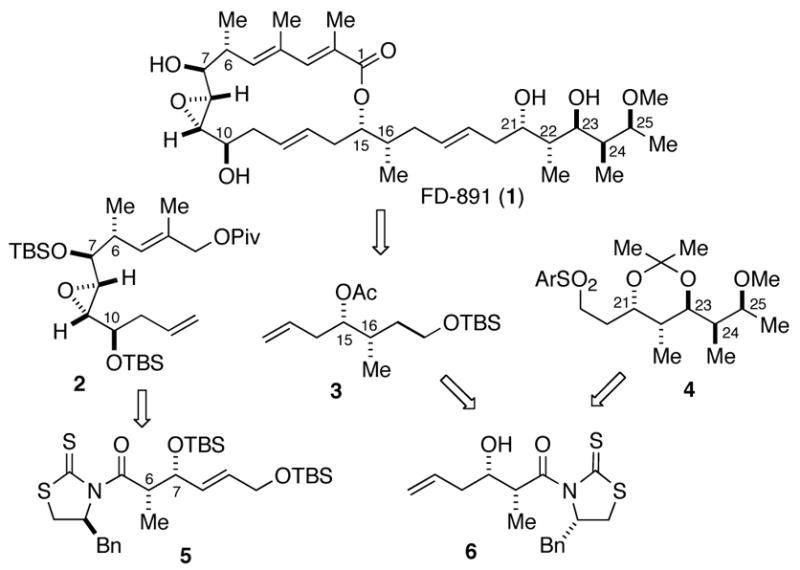
The synthesis of the C3–C12 unit 2 began with addition of known aldehyde 710 to the chlorotitanium enolate of thioimide 8 to deliver the protected Evans syn adduct 5 after silylation of the alcohol (Scheme 2). Reductive removal of the auxiliary directly gave aldehyde 9, which was rapidly transformed to pivaloate 10 in three steps. Selective removal of the primary silyl ether11 followed by Sharpless epoxidation12 gave epoxide 11 in 72% yield. Dess-Martin13 oxidation of alcohol 11, and subsequent chelate-controlled allylation of the resultant aldehyde14 delivered the expected epoxyalcohol as single detectable diastereomer (dr >20:1). Protection of the alcohol as its TBS ether produced the required C3–C12 unit 2.
Scheme 2.
Synthesis of epoxide 2.
Conditions: (a) TiCl4, (−)-sparteine, NMP, CH2Cl2, −78 to −40 °C, 78% (dr >20:1); (b) TBSOTf, 2,6-lutidine, CH2Cl2, −78 °C, 92%; (c) i-Bu2AlH, CH2Cl2, −78 °C, 74%; (d) Ph3P=C(CH3)CO2Et, toluene, 80 °C, 92%; (e) i-Bu2AlH, THF, −78 °C, 98%; (f) pyridine, PivCl, CH2Cl2, 99%; (g) NH4F, MeOH, 85%; (h) (+)-DET, (i-PrO)4Ti, t-BuOOH, molecular sieves 4Å, CH2Cl2, −23 °C, 72% (dr 15:1); (i) Dess-Martin periodinane, NaHCO3, CH2Cl2, 97%; (j) MgBr2(Et2O), CH2=CHCH2SiMe3, CH2Cl2, −20 to −10 °C, 70% (dr >20:1); (k) TBSCl, imid., DMAP, CH2Cl2, 99%.
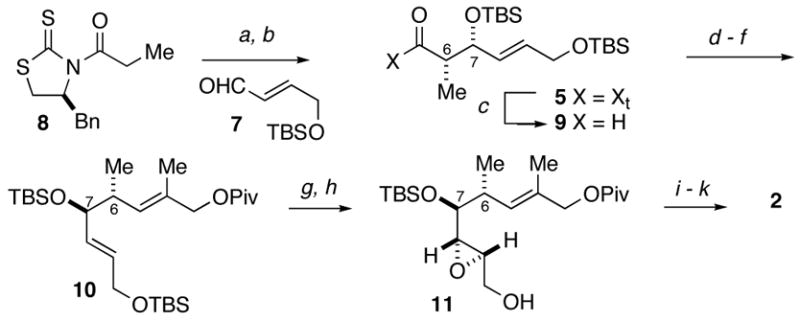
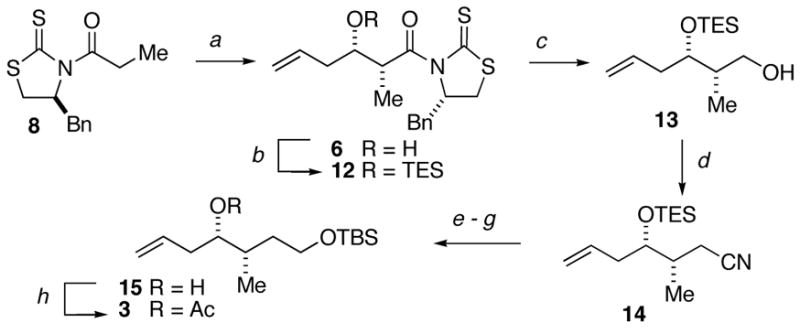
The synthesis of fragment 3 commenced with an aldol addition between 3-butenal15 and the enolate of thiazolidinethione 8 under conditions9a to give the non-Evans syn aldol adduct 6 in 73% yield (>15:1 dr) (Scheme 3). Protection of the alcohol delivered silyl ether 12 whereupon reduction of the N-acylthiomide gave alcohol 13. Homologation of alcohol 13 was accomplished by displacement of the hydroxyl with cyanide under Mitsunobu conditions16 to provide nitrile 14. Two-stage reduction gave the corresponding diol, which underwent selective protection to provide alcohol 15. Protection of the secondary alcohol as its acetate gave the C13–C18 fragment 3 in 98% yield.
Scheme 3.
Synthesis of the C13–C18 fragment 3.
Conditions: (a) TiCl4, (−)-sparteine, 3-butenal, CH2Cl2, 0 °C, 73%; (b) TESOTf, 2,6-lutidine, CH2Cl2, −78 °C, 96%; (c) LiBH4, Et2O, MeOH, 0°C to rt, 98%; (d) acetone cyanohydrin, DEAD, Ph3P, toluene, 70 °C,90%; (e) i-Bu2AlH, toluene, −78 °C, 88%; (f) NaBH4, CH2Cl2, MeOH, then HCl, 84%; (g) TBSCl, imidazole, CH2Cl2, 92%; (h) Ac2O, DMAP, Et3N, CH2Cl2, 0 to 25 °C, 98%.
The synthesis of sulfone 4 began with the thioimide 12 also used in the synthesis of the C13–C18 fragment. The aldehyde obtained by the direct reduction of thioimide 12 was subjected to a second aldol iteration to provide the aldol adduct 16 in 87% yield. The methyl ketone 17 was obtained by transacylation of the auxiliary to provide the corresponding Weinreb amide17 followed by protection of the alcohol and addition of methylmagnesium chloride. Chelation-controlled18 reduction of the ketone provided 75% of the alcohol 18 along with 15% of the C25 isomer, which could be recycled by oxidation-reduction. Methylation of the C25 hydroxyl gave the corresponding C25 methyl ether. Acid catalyzed deprotection of the MOM and TES groups followed by exposure of the diol to dimethoxypropane and p-TsOH provided acetonide 19. Ozonolysis with reductive work-up followed by a Mitsunobu reaction gave the desired sulfide, which was oxidized to sulfone 4.
With the three key fragments in hand, their assembly was undertaken. A cross-metathesis19 between terminal alkenes 2 and 3 was performed with the Grubbs catalyst (Scheme 5). The nature of the protecting group on the C15 alcohol had a profound influence on the selectivity of the cross metathesis. The best E:Z ratio was obtained with the C15 acetate compared to other esters or the free hydroxyl. The desired E olefin 20 was obtained in 68% yield along with 10% of the Z-isomer.
Scheme 5.
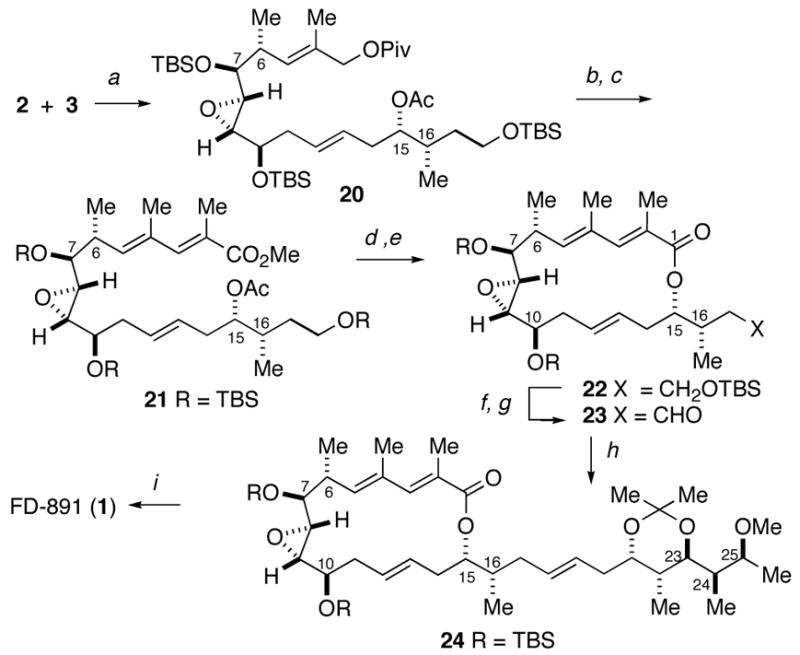
Completion of FD-891.
Conditions: (a) Cl2(Cy3P)(IMes)Ru=CHPh, CH2Cl2, 40 °C, 68% + 10% Z-isomer; (b) i-Bu2AlH, CH2Cl2, −78 °C, 85%; (c) MnO2, CH2Cl2, 40 °C, then BuLi, Ph3P(O)C(CH3)CO2Me, THF, 0 to 25 °C, 66% for 2 steps; (d) TMSOK, THF; (e) Cl2C6H3COCl, Et3N, THF; then DMAP, PhCH3, 61% for 2 steps; (f) PPTS, MeOH, 90%; (g) Dess-Martin periodinane, NaHCO3, CH2Cl2, 82%; (h) Sulfone 4, KHMDS, THF, −78 °C, then aldehyde 23, 80%; (i) H2SiF6 20% in H2O, CH3CN, 90%.
The completion of the macrolactone required the extension at C3 to the dienoate. To this end, reductive removal of the pivaloate preceded oxidation of the allylic alcohol with manganese dioxide, and Horner-Wadsworth-Emmons olefination to deliver the dienoate 21. Hydrolysis of ester 21 followed by Yamaguchi macrolactonization20 gave the desired macrocycle 22. Selective deprotection of the primary silyl ether and oxidation of the resultant alcohol provided aldehyde 23 in 70% yield, ready to be coupled with the C19–C26 fragment 4 via a Julia reaction.
Julia olefination21 between aldehyde 23 and sulfone 4 provided exclusively the E-olefin 24 (Scheme 5). Global deprotection by the action of H2SiF622 gave FD-891 in 90% yield. The spectral data of synthetic FD-891 were consistent in all respects with those reported for the natural product.1,5,6
In conclusion, we have completed the first total synthesis of the macrolide FD-891 in 21 steps (longest linear sequence). The versatile aldol reaction of N-acylthiazolidinethione 8 was used to create eight of the twelve stereocenters with the same enantiomer of the chiral auxiliary.
Supplementary Material
Experimental procedures, as well as 1H and 13C NMR spectra for all new compounds, and synthetic FD-891. This material is available free of charge via the Internet at htpp://pubs.acs.org.
Scheme 4.
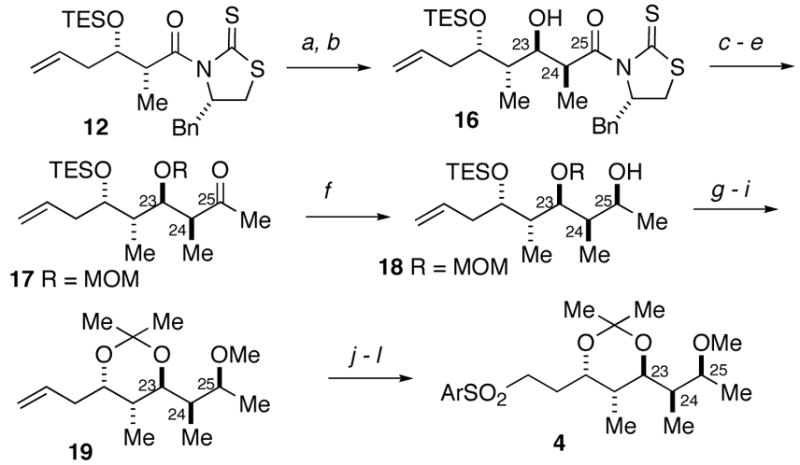
Synthesis of sulfone 4.
Conditions: (a) i-Bu2AlH, CH2Cl2, −78 °C, 84%; (b) Thioimide 8, TiCl4, (−)-sparteine, NMP, CH2Cl2, −78 to 0 °C, then add aldehyde, 87% (dr >20:1); (c) CH3N(OCH3)H·HCl, imidazole, CH2Cl2, 78%; (d) MOMCl, i-Pr2NEt, DMF, 50 °C, 87%; (e) MeMgCl, Et2O, 0 °C, 97%; (f) NaBH4, CeCl3·7(H2O), MeOH, 0 °C, 75% 18 + 15% C25 isomer which was recycled; (g) NaH, MeI, THF, 0 °C to rt, 81%; (h) HCl (conc.), MeOH, 78%; (i) 2,2-dimethoxypropane, p-TSA, 88 %; (j) O3, CH2Cl2, MeOH, −78 °C, then NaBH4 –78 to 25 °C, 81%; (k) DIAD, 2−mercaptobenzothiazole, PPh3, CH2Cl2, 93%; (l) H2O2 30%,(NH4)6Mo7O2 4·4H2O, EtOH, 89%.
Acknowledgments
This work was supported by a research grant from The National Cancer Institute (CA63572). We are grateful to professor T. Eguchi for providing authentic 1H and 13C spectra of the natural product.
References
- 1.(a) Seki-Asano M, Okazaki T, Yamagishi M, Sakai N, Hanada K, Mizoue K. J Antibiot. 1994;47:1226. doi: 10.7164/antibiotics.47.1226. [DOI] [PubMed] [Google Scholar]; (b) Seki-Asano M, Tsuchida Y, Hanada K, Mizoue K. J Antibiot. 1994;47:1234. doi: 10.7164/antibiotics.47.1234. [DOI] [PubMed] [Google Scholar]
- 2.(a) Muroi M, Shiragami N, Nagao K, Yamasaki M, Takatsuki A. Cell Struct Funct. 1993;18:139. doi: 10.1247/csf.18.139. [DOI] [PubMed] [Google Scholar]; (b) Drose S, Bindseil KU, Bowman EJ, Siebers A, Zeeck A, Altendork K. Biochemistry. 1993;32:3902. doi: 10.1021/bi00066a008. [DOI] [PubMed] [Google Scholar]
- 3.Kataoka T, Shinohara N, Takayama H, Takaku K, Kondo S, Yonehara S, Nagai K. J Immunol. 1996;156:3678. [PubMed] [Google Scholar]
- 4.Kataoka T, Yamada A, Bando M, Honna T, Mizoue K, Nagai K. Immunology. 2000;100:170. doi: 10.1046/j.1365-2567.2000.00039.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Egushi T, Kobayashi K, Uekusa H, Ohashi Y, Mizoue K, Matsushima Y, Kakinuma K. Org Lett. 2002;20:3383. doi: 10.1021/ol026518k. [DOI] [PubMed] [Google Scholar]
- 6.Egushi T, Kamamoto K, Mizoue K, Kakinuma K. J Antibiot. 2004;57:156. doi: 10.7164/antibiotics.57.156. [DOI] [PubMed] [Google Scholar]
- 7.Review: Dai M, Guan YC, Jin J. Curr Med Chem. 2005;21:1947. doi: 10.2174/0929867054546591.
- 8.(a) Murga J, Garcia-Fortanet J, Carda M, Marco JA. Synlett. 2004:2830. [Google Scholar]; Murga J, Garcia-Fortanet J, Carda M, Marco JA. Tetrahedron Lett. 2004;45:7499. [Google Scholar]; (c) Hang SS, Xu J, Loh TP. Tetrahedron Letters. 2003;27:4997. [Google Scholar]
- 9.(a) Crimmins MT, King BW, Tabet EA, Chaudhary K. J Org Chem. 2001;66:894. doi: 10.1021/jo001387r. [DOI] [PubMed] [Google Scholar]; (b) Crimmins MT, Christie HS, Chaudhary K, Long A. J Am Chem Soc. 2005;127:13810. doi: 10.1021/ja0549289. [DOI] [PubMed] [Google Scholar]
- 10.Roush WR, Koyama K. Tetrahedron Lett. 1992;33:6227. [Google Scholar]
- 11.Schinzer D, Bohm OM, Altmann KH, Wartmann M. Synlett. 2004:1375. [Google Scholar]
- 12.Gao Y, Hanson RM, Klunder JM, Ko YS, Masamune H, Sharpless BK. J Am Chem Soc. 1987;109:5765. [Google Scholar]
- 13.Dess DB, Martin JC. J Am Chem Soc. 1991;113:7277. [Google Scholar]
- 14.(a) Keck GE, Abbott DE. Tetrahedron Lett. 1984;25:1883. [Google Scholar]; (b) Howe GP, Wang S, Procter G. Tetrahedron Lett. 1987;28:2629. [Google Scholar]
- 15.Crimmins MT, Choy AL. J Am Chem Soc. 1999;121:5653. [Google Scholar]
- 16.Andrus MB, Hicken EJ, Meredith EL, Simmons BL, Cannon JF. Org Lett. 2003;21:3859. doi: 10.1021/ol035400g. [DOI] [PubMed] [Google Scholar]
- 17.Basha A, Lipton M, Weinreb SM. Tetrahedron Letter. 1997:4171. [Google Scholar]; Levin JI, Turos E, Weinreb SM. Synth Commun. 1982;12:989. [Google Scholar]
- 18.Ancerewicz J, Vogel P. Helv Chim Acta. 1996;79:1393. [Google Scholar]
- 19.Chatterjee AK, Choi TL, Sanders DP, Grubbs RH. J Am Chem Soc. 2003;125:11360. doi: 10.1021/ja0214882. [DOI] [PubMed] [Google Scholar]
- 20.Inanada J, Hirata K, Saeki H, Katsuki T, Yamaguchi M. Bull Chem Soc Jpn. 1979;52:1989. [Google Scholar]
- 21.Blakemore PR. J Chem Soc, Perkin Trans I. 2002:2563. [Google Scholar]
- 22.Pilcher AS, Hill DK, Shimshock SJ, Waltermire RE, DeShong P. J Org Chem. 1992;57:2492. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Experimental procedures, as well as 1H and 13C NMR spectra for all new compounds, and synthetic FD-891. This material is available free of charge via the Internet at htpp://pubs.acs.org.