

Abstract
The conversion of a substituted dioxinone to a pyrone was used in an improved synthesis of the AB spiroketal subunit of the spongistatins. This transformation occurred via a hetero-Diels-Alder reaction of an acyl ketene with butyl vinyl ether. A double diastereoselective Mukaiyama aldol reaction is used to provide the hetero-Diels-Alder precursor.
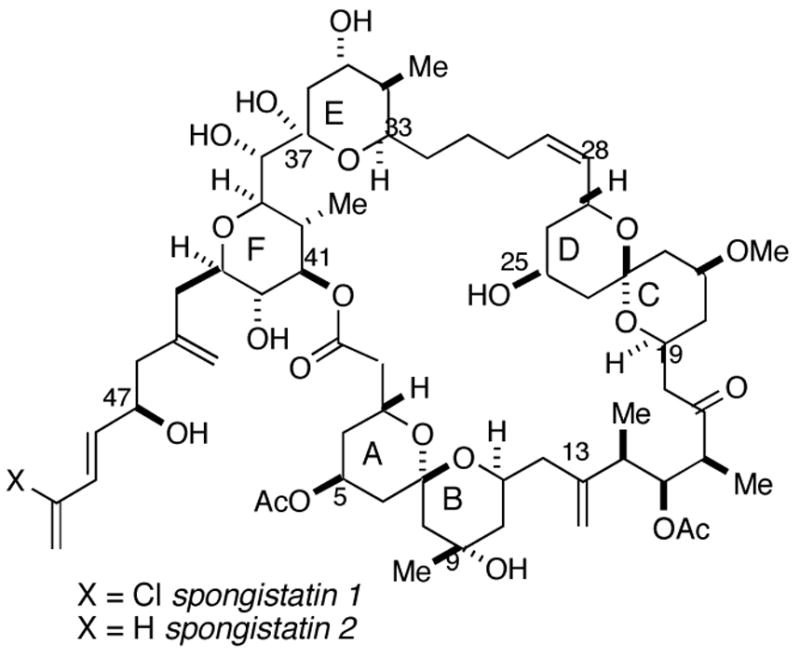
The spongistatins (Figure 1) are potent antitumor agents1 initially isolated in 1993 from marine sponges of the genus Spongia.2 The potenial clinical importance of the spongistatins has created substantial interest in their synthesis, and several total syntheses have been accomplished. 3 The total synthesis of spongistatins 1 and 2 (1, 2) from our laboratory was based on the assembly of three fragments including the C1–13 AB spiroketal 3 (Figure 1).4
Figure 1.
The Spongistatins
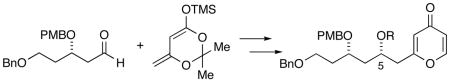
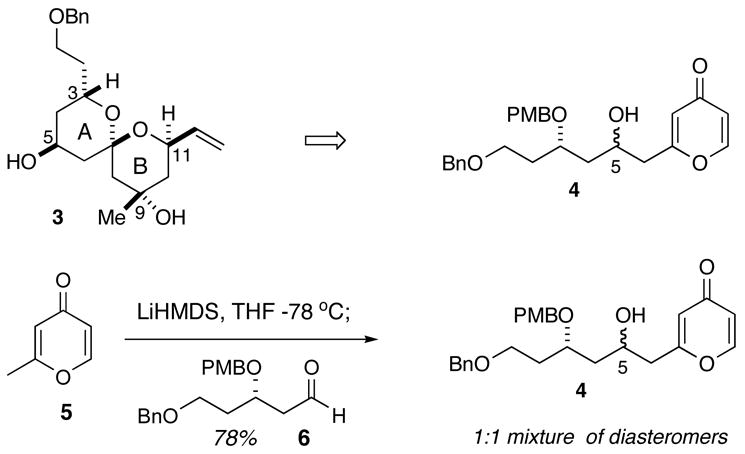
Our original synthesis of the AB spiroketal (Scheme 1) centered on the preparation of pyrone 4 through addition of a metallated pyrone to an aldehyde. As shown in Scheme 1, deprotonation of pyrone 5 with LiHMDS followed by addition of aldehyde 6 led to the generation of a 1:1 mixture of diastereomers. All attempts to improve the diastereoselection of the addition by changing the counterion, solvents and additives were unsatisfactory.5 Additionally, attempts to prepare the enol silyl ether of pyrone 5 to investigate Mukaiyama type additions to aldehyde 6 were thwarted since all silylating conditions led to C-silylation of pyrone 5. The lack of stereocontrol at C5 required a four-step sequence to correct and could not be accomplished until after the spiroketalization event, thus requiring that both diastereomers be carried forward several steps prior to convergence. An improvement in the preparation of the pyrone precursor to the spiroketal which overcomes this stereochemical shortcoming is reported herein.
Scheme 1.
Addition of Metallated Pyrones to Aldehydes
Retrosynthetically, the AB spiroketal subunit was envisioned to come from an acid-catalyzed cyclization6 of pyrone 9 (Scheme 2), which would be formed through a hetero-Diels-Alder cycloaddition-elimination sequence from dioxinone 10. The dioxinone would derive from a selective Mukaiyama aldol addition of silyl dienolate 11 to aldehyde 6.
Scheme 2.

Hetero-Diels-Alder Cycloaddition Strategy
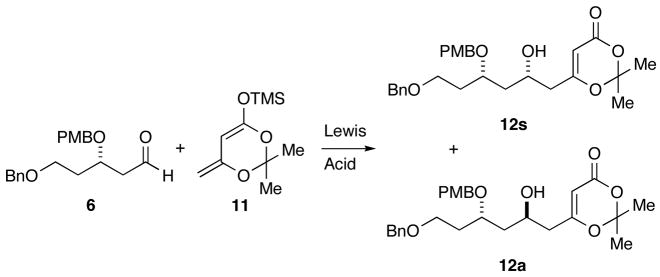
To improve the synthesis of the AB spiroketal, a general method to selectively access the Mukaiyama aldol adduct of silyl dienolate 11 with aldehyde 6 was needed. First attempts to achieve this goal focused on the use of different Lewis acids to take advantage of the potential directing ability of the β-alkoxyaldehyde. The use of a common Lewis acid, BF3-OEt2 (Table 1, entry 1), proved nonselective, albeit high yielding. We next investigated the use of stronger chelating Lewis acids, such asMe2AlCl (entry 2), which has been used in similar casesto achieve rigid chelating transition states with β-alkoxyaldehydes and addition of silyl enol ethers leading to high anti selectivity.7 Unfortunately, only a slight preference (1.3:1) in favor of the anti diastereomer was observed. The use of titanium Lewis acids (entries 3–7) slightly improved the anti selectivity, but the best case with titanium(IV) dichlorodiisopropoxide (entry 6) provided only a 3.3:1 preference for the anti diastereomer.
Table 1.
Use of Lewis Acids in Addition to Aldehyde
 | ||||
|---|---|---|---|---|
| Lewis acid | ligand | Yield (%) | Selectivity (12a:12s) | |
| 1 | BF3·OEt2 | - | 90 | 1:1 |
| 2 | Me2AlCl | - | 45 | 1.3:1 |
| 3 | TiCl4 | - | 61 | 1.5:1 |
| 4 | Ti(Oi-Pr)4 | - | 0 | - |
| 5 | Ti(Oi-Pr)Cl3 | - | 82 | 2.0:1 |
| 6 | Ti(Oi-Pr)2Cl2 | - | 89 | 3.3:1 |
| 7 | Ti(Oi-Pr)3Cl | - | 21 | 2.4:1 |
| 8 | Ti(Oi-Pr)4 | S-BINOL | 28 | 1:1 |
| 9 | Ti(Oi-Pr)2Cl2 | (S,S)-(+)-TADDOL | 64 | 1:5 |
| 10 | Ti(Oi-Pr)2Cl2 | (R,R)-(−)-TADDOL | 60 | 1:2.1 |
| 11 | Ti(Oi-Pr)4 | (−)-13 | 81 | 8.6:1 |
| 12 | Ti(Oi-Pr)4 | (+)-13 | 95 | 1:10 |
Chiral ligands were then explored in an attempt to use reagent control to influence the diastereoselectivity of the reaction. BINOL in conjunction with Ti(i-PrO)4 has been reported to result in good selectivity in Mukaiyama aldol additions,8 but when applied to the case at hand (entry 8), low yield and poor selectivity were observed. TADDOL has also been shown to provide high levels of reagent conrol in Lewis acid catalyzed aldol additions.9 Exposure of dienolate 11 and aldehyde 6 to titanium(IV) dichlorodiisopropoxide and S,S-TADDOL (entry 9) delivered a 5:1 mixture favoring the anti diastereomer. Unfortunately, R,R-TADDOL gave only a 2:1 preference for the syn diastereomer (entry 10).
Satisfactory diastereoselection was ultimately obtained taking advantage of Carreira’s catalyst,10 which has been reported to effect enantioselective Mukiayama aldol additions of silyl dienolate 11 with a variety of achiral aldehydes. Use of the Carreira protocol with dienolate 11 and aldehyde 6, allowed access to either the syn diastereomer 12s or anti diastereomer 12a in high yield and selectivity with low catalyst loadings. Using the (+)-enantiomer of 13 (entry 12) led to formation of a 10:1 mixture of aldol adducts favoring the desired syn diastereomer 12s (95% yield), while (−)-13 produced the anti diastereomer 12a (entry 11) as the major product (8.6:1 dr, 81% yield).
Having accomplished an efficient synthesis of the desired dioxinone 12s, its conversion to the required pyrone was investigated. An extension of the method reported by Coleman and Grant11 to include more complex substrates, by performing a hetero-Diels-Alder reaction of an acyl ketene with butyl vinyl ether, was envisioned for conversion of dioxinone 12s to the desired pyrone. To this end, the C5 hydroxyl was readily protected as a benzyloxymethyl ether to provide hetero-Diels-Alder precursor 14 (Scheme 3). Heating dioxinone 14 in toluene in the presence of butyl vinyl ether led to formation of butyl acetal 16 presumably through a hetero-Diels-Alder reaction of intermediate acyl ketene 15 with butyl vinyl ether. Immediate exposure of the butyl acetal to p-TsOH in THF led to rapid elimination of butanol to produce pyrone 17 (65% over two steps).12
Scheme 3.

Hetero-Diels-Alder Cycloaddition
Efficient conversion of the dioxinone 14 to the butyl acetal 16 required that all materials be rigorously dried to avoid trapping of the acyl ketene intermediate by advantitious water. Failure to scrupulously dry the dioxinone, the solvent or butyl vinyl ether led to formation of β-keto acid 19 (Scheme 4, path a). Additionally, the choice of protecting group on the C5 hydroxyl group was critical. Early attempts to effect the hetero-Diels–Alder reaction using triethylsilyl13 and t -butyl dimethylsilyl ethers at C5 resulted in formation of β-keto lactone 20 presumably via interception of the acyl ketene by the C5 ether oxygen followed by loss of the labile silicon protecting group (Scheme 4, path b).14 Incorporating the more robust benzyloxymethyl group precluded formation of the β-keto lactone 20.
Scheme 4.

Side reactions of acyl ketene intermediate
Taking advantage of the chemistry previously employed in the total synthesis of spongistatin, the p-methoxybenzyl group was removed by the action of DDQ to provide the free alcohol at C3 (Scheme 5). Exposure of the hydroxypyrone to catalytic trifluoroacetic acid in benzene provided spiroenone 21 in 64% yield after recycle. The minor diastereomer (<10% which had been introduced in the Mukaiyama aldol addition) could be readily removed after the spiroketalization. Treatment of the spiroenone 21 with the vinyl cuprate reagent formed from vinylmagnesium bromide and tetrakis[copper(I) iodide-tributylphosphine] led to the formation of alkene 22 as the major diastereomer (5:1 dr). The C9 tertiary carbinol was introduced by addition of methylmagnesium iodide to the C9 ketone. Cleavage of the benzyloxymethyl ether occurred upon treatment with acid to provide the desired diol 3. Comparison of spectral data confirmed the interception of the previously synthesized fragment utilized in the reported total synthesis of spongistatin.4
Scheme 5.

Completion of AB Spiroketal
In summary, an improved synthesis of the AB spiroketal subunit of spongistatin has been developed. This synthesis takes advantage of a double diastereoselective Mukaiyama aldol reaction and subsequent hetero-Diels-Alder cycloaddition to contstruct a pyrone precursor for a spiroketalization reaction.
Supplementary Material
Copies of 1H NMR spectra and experimental procedures. This material is available via the Internet at http://pubs.acs.org.
Figure 2.
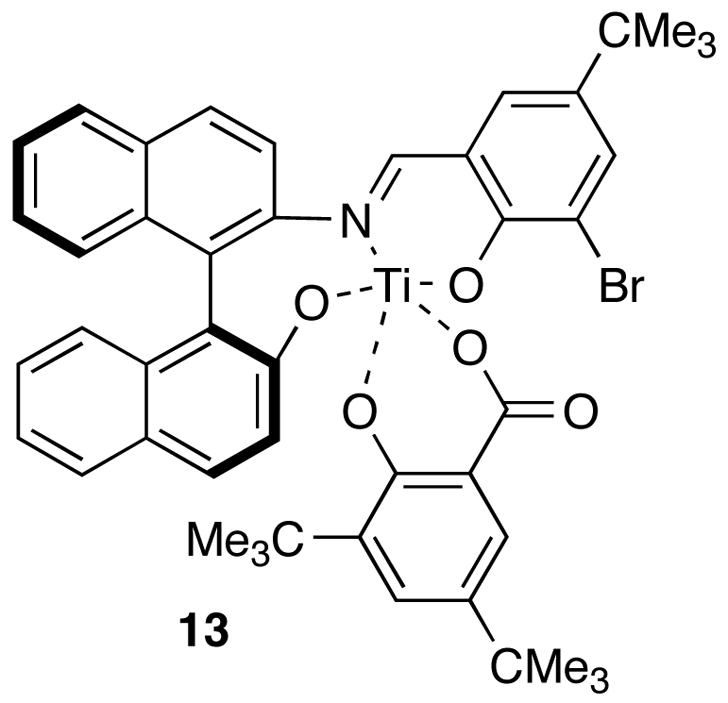
Carreira’s Catalyst
Acknowledgments
Special thanks to Professor Xumu Zhang at Pennsylvania State University for providing an enantiopure precursor to Carreira’s catalyst. Financial support from the National Cancer Institute (CA63572) is gratefully acknowledged.
References
- 1.(a) Bai R, Cichacz ZA, Herald CL, Pettit GR, Hamel E. Mol Pharmacol. 1993;44:757–66. [PubMed] [Google Scholar]; (b) Bai R, Taylor GF, Cichacz ZA, Herald CL, Kepler JA, Pettit GR, Hamel E. Biochemistry. 1995;34:9714–21. doi: 10.1021/bi00030a009. [DOI] [PubMed] [Google Scholar]
- 2.(a) Pettit GR, Herald CL, Cichacz ZA, Gao F, Schmidt JM, Byd MR, Christie ND, Boettner FE. J Chem Soc, Chem Commun. 1993:1805–7. [Google Scholar]; (b) Pettit GR, Cichacz ZA, Herald CL, Gao F, Boyd MR, Schmidt JM, Hamel E, Bai R. J Chem Soc, Chem Commun. 1994:1605–6. [Google Scholar]; (c) Pettit GR, Cichacz ZA, Gao F, Herald CL, Boyd MR. J Chem Soc, Chem Commun. 1993:1166–8. [Google Scholar]; (d) Pettit GR, Chicacz ZA, Gao F, Herald CL, Boyd MR, Schmidt JM, Hooper JNA. J Org Chem. 1993;58:1302–4. [Google Scholar]
- 3.Yeung KS, Paterson I. Chem Rev. 2005;105:4237–4313. doi: 10.1021/cr040614c.For other recent synthetic efforts see: Smith AB, III, Sfouggatakis C, Gotchev DB, Shirakami S, Bauer D, Zhu W, Doughty VA. Org Lett. 2004;6:3637–40. doi: 10.1021/ol048418f. and refernces therein.
- 4.(a) Crimmins MT, Katz JD, Washburn DG, Allwein SP, McAtee LF. J Am Chem Soc. 2002;124:5661–3. doi: 10.1021/ja0262683. [DOI] [PubMed] [Google Scholar]; Crimmins MT, Katz JD, McAtee LC, Tabet EA, Kirincich SJ. Org Lett. 2001;3:949–952. doi: 10.1021/ol015652m. [DOI] [PubMed] [Google Scholar]; (b) Crimmins MT, Katz JD. Org Lett. 2000;2:957–60. doi: 10.1021/ol005605e. [DOI] [PubMed] [Google Scholar]; (c) Crimmins MT, Washburn DG. Tetrahedron Lett. 1998;39:7487–90. [Google Scholar]
- 5.Crimmins MT, Washburn DG, Katz JD, Zawacki FJ. Tetrahedron Lett. 1998;39:3439–42. [Google Scholar]
- 6.Crimmins MT, O’Mahony R. J Org Chem. 1990;55:5894–900. [Google Scholar]
- 7.Evans DA, Allison BD, Yang MG, Masse CE. J Am Chem Soc. 2001;123:10840–52. doi: 10.1021/ja011337j. [DOI] [PubMed] [Google Scholar]
- 8.(a) De Rosa M, Soriente A, Scettri A. Tetrahedron: Asymmetry. 2000;11:3187–95. [Google Scholar]; (b) De Rosa M, Acocella MR, Soriente A, Scettri A. Tetrahedron: Asymmetry. 2001;12:1529–31. [Google Scholar]
- 9.(a) Seebach D, Beck AK, Heckel A. Angew Chem, Int Ed. 2001;40:92–138. [PubMed] [Google Scholar]; (b) Szlosek M, Jullian JC, Hocquemiller R, Figadere B. Heterocycles. 2000;52:1005–13. [Google Scholar]
- 10.Singer RA, Carreira EM. J Am Chem Soc. 1995;117:12360–61. [Google Scholar]
- 11.Coleman RS, Grant EB. Tetrahedron Lett. 1990;31:3677–80. see also Sato M, Ogasawara H, Kato K, Sakai M, Kato T. Chem Pharm Bull. 1983;31:4300–05.
- 12.Zawacki FJ, Crimmins MT. Tetrahedron Lett. 1996;37:6499–6502. [Google Scholar]
- 13.Dr. Jason D. Katz thesis, University of North Carolina
- 14.Sato M, Sunami S, Sugita Y, Kaneko C. Heterocycles. 1995;41:1435–44. [Google Scholar]; Sato M, Sunami S, Sugita Y, Kaneko C. Chem Pharm Bull. 1994;42:839–45. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Copies of 1H NMR spectra and experimental procedures. This material is available via the Internet at http://pubs.acs.org.