

Abstract
A substantial body of evidence implicates TGFβ as a tumor promoter in epithelial cells that have become resistant to its tumor suppressor activity. To better understand early, genome-wide TGFβ responses in cells resistant to growth inhibition by TGFβ, we used microarray analysis in a well-defined cell culture system of sensitive and resistant intestinal epithelial cells. TGFβ-regulated gene expression in TGFβ-growth-sensitive, nontransformed rat intestinal epithelial cells (RIE-1) was compared to expression in TGFβ-growth-resistant RIE cells stably transformed by oncogenic Ras(12V). Treatment of RIE-1 cells with 2 ng/ml TGFβ1 for 1 hour increased the expression of eight gene sequences by 2.6-fold or more, whereas eight were down regulated 2.6-fold. In RIE-Ras(12V) cells, 42 gene sequences were upregulated and only 3 were down-regulated. Comparison of RIE and RIE-Ras(12V) identified 37 gene sequences as unique, Ras-dependent genomic targets of TGFβ1. TGFβ-regulation of connective tissue growth factor and vascular endothelial growth factor, two genes up-regulated in RIE-Ras cells and previously implicated in tumor promotion, was independently confirmed and further characterized by Northern analysis. Our data indicate that overexpression of oncogenic Ras in intestinal epithelial cells confers a significantly expanded repertoire of robust, early transcriptional responses to TGFβ via signaling pathways yet to be fully elucidated but including the canonical Raf-1/MAPK/Erk pathway. Loss of sensitivity to growth inhibition by TGFβ does not abrogate TGFβ signaling and actually expands the early transcriptional response to TGFβ1. Expression of some of these genes may confer to Ras-transformed cells characteristics favorable for tumor promotion.
Introduction
A large body of work during the past two decades has established transforming growth factor beta (TGFβ) as a potent suppressor of cellular proliferation in most experimental settings. Indeed, TGFβ is now distinguished as one of the most important tumor suppressors in human cancer biology [1–5]. Notwithstanding, for many years, it has been clear from in vitro studies that the contribution of TGFβ signaling to cell behavior is far more complex than negative regulation of cellular proliferation. In some contexts, TGFβ may contribute to the transformed phenotype. Several recent in vivo studies convincingly implicate TGFβ as a tumor promoter in transformed cells [6,7], leading to the hypothesis that the tumor-promoting effects of TGFβ increasingly predominate over growth-inhibitory effects during tumor progression. It seems that overexpression of genes associated with cellular proliferation cooperate with TGFβ to accelerate tumor formation and metastasis, presumably after cells have become resistant to TGFβ-mediated growth inhibition.
The most completely understood signal transduction pathway used by TGFβ is the canonical Smad pathway [8–10]. Transforming growth factor β receptor (TGFβR) activation results in serine phosphorylation of Smad2 and Smad3 and formation of heteromeric complexes with Smad4. The Smad complex accumulates in the nucleus by regulated nuclear import and binds to DNA in a sequence-specific manner in association with a large number of potential transcriptional coactivators and corepressors [11,12]. The specificity and complexity of the genomic response to TGFβ is attributable in part to the complex portfolio of coactivators and corepressors available under the specific conditions at the time of TGFβ exposure [3,10]. A large number of other signal transduction pathways, including Erk kinases, protein kinase C, Rho-like GTPases, p38, and c-Jun N-terminal kinases, have been identified as apparent Smad-independent signaling mechanisms, but activation of these depends to a significant extent on the cell system under study [13]. At present, it is not clear if tumor promotion by TGFβ occurs by these Smad-independent pathways or by a modification or attenuation of Smad signaling.
In intestinal neoplasia, loss of TGFβ tumor-suppressor activity occurs by a variety of well-described genetic and epigenetic defects. The small GTPase Ras is activated by mutation in many human cancers [14,15], including more than 50% of colorectal cancers [16]. Multiple cell culture studies have identified Ras activation as an epigenetic factor conferring resistance to growth inhibition by TGFβ [17]. We previously found that stable transformation of rat intestinal epithelial cells with oncogenic Ras(12V) did not attenuate Smad localization to the nucleus in response to TGFβ treatment but caused resistance to growth inhibition [18]. Furthermore, TGFβ activation of Smad binding element-driven reporter gene transcription in RIE-Ras(12V) cells was markedly reduced [19], indicating interference with Smad-dependent transcription. Notwithstanding this loss, or at least significant attenuation of Smad-mediated growth inhibitory signaling in the context of oncogenic Ras, it is clear that residual, albeit modified, TGFβ signaling continues to occur.
To examine more broadly the effects of Ras transformation on the early genomic response to TGFβ, we used DNA microarrays to compare induction of target gene transcription in nontransformed rat intestinal epithelial cells (RIE-1) and in cells stably transformed by Ras (12V) [RIE-Ras(12V)] 1 hour after treatment with TGFβ1. Cellular and specific Smad-responsive transcriptional responses to TGFβ have been extensively characterized in these cell lines, particularly in the context of Ras transformation [18–20]. Because growth inhibitory Smad signaling is markedly attenuated in RIE-Ras(12V) cells, we hypothesized that this apparent “resistance” to TGFβ would result in induction of fewer early transcriptional targets in Ras-transformed cells. However, to the contrary, we found that mutant oncogenic Ras expands the early genomic response to TGFβ, such that more than five times as many genes are activated in RIE-Ras(12V) cells than parental RIE cells at a cutoff threshold ≥2.6-fold induction. Microarray analysis of TGFβ-mediated gene transcription in RIE and RIE-Ras identifies a number of candidate genes, which may contribute to TGFβ actions as a tumor promoter, the setting of oncogenic Ras activation. Among these candidate genes, we examined in more detail the kinetics and signaling pathways involved in the induction of two differentially induced genes, vascular endothelial growth factor (VEGF) and connective tissue growth factor (CTGF).
Materials and Methods
Cell Lines and Reagents
RIE-1 rat intestinal epithelial cells (RIE) were obtained from Ken Brown (Cambridge, United Kingdom) and maintained in DMEM supplemented with 10% FCS. RIE cells are a diploid, nontransformed, TGFβ-growth-sensitive cell line derived from rat jejunum [18,20,21]. RIE-Ras cells were established by stable transfection of the parental cells with pSV2-H-Ras(12V) that contain human sequences encoding the constitutively active H-Ras(12V) protein [22]. The RIE-Ras transfectants were selected in G418 (Calbiochem, San Diego, CA). The cell line designated RIE-Ras(12V) in this study was established as a pooled population of stable clones and grown at all times in the presence of 50 µg/ml G418. For each experiment, RIE or RIE-Ras cells were plated in duplicate, grown to log phase, and treated with 2 ng/ml of TGFβ1 (BD Biosciences, Bedford, MA) or diluent for 1 hour, after which RNA was immediately collected.
RNA Isolation
For each array experiment, RNA was isolated from ∼ 107 cells growing in log phase on four separate 10-cm2 tissue culture dishes. RNA was isolated using Trizol (Invitrogen, Carlsbad, CA), followed by purification with the RNEasy system (Qiagen, Valencia, CA). Each collection yielded approximately 100 µg of RNA. RNA integrity was verified using the Agilent Bioanalyzer in the Microarray Core Facility at the Columbus Children's Research Institute.
Probe Preparation and Array Hybridization
Complementary DNA (cDNA) probe was prepared by the Microarray Core Facility (Columbus Children's Research Institute) with Superscript III using oligo-dT primers and dNTPs supplemented with amino-allyl-UTP to improve hybridization characteristics and stability. The DNA samples were labeled with Cy3 or Cy5 then were purified. Purified probe was hybridized to the Agilent Rat Olio Microarray (Agilent, Cincinnati, OH) for 14 hours at 48°C according to the manufacturer's protocol. This array (Agilent G4130A) is composed of 20,500 rat genes, ESTs, and EST clusters printed on a glass slide as 60-mers. The gene list can be downloaded from www.agilent.com. Slide images were acquired using an Affymetrix 428 scanner with gain settings set so that 95% of spots were below saturation to yield the maximum dynamic range within an experiment. Images were converted into “.gpr” files using GenePix software (Axon, Union City, CA). Image files (.tif ) were deposited onto the Gene Expression Omnibus Web site (http://www.ncbi.nlm.nih.gov/geo) with the Series Accession Number GSE2015.
Data Analysis
Four independent experiments from cell culture to chip hybridization were performed for both RIE and RIE-Ras. Data were analyzed using GeneTraffic 2.6 software (Iobion, La Jolla, CA). Lowess-global normalization was applied to all experiments. Flagging parameters were set as spot intensity lower than the intensity of local spot background, spot intensity lower than average background, and raw spot intensity less than 100. Flagged spots were not included in normalization or aggregate calculations. Only genes that were induced or repressed at a level of log2N > 1.4 (2.6-fold) in at least three of the four independent analyses and hybridizations were identified as TGFβ-regulated genes. The NIA Array Analysis Tool (http://lgsun.grc.nia.nih.gov) was used to determine the false discovery rate (FDR), which is equivalent to a P value in experiments with multiple hypothesis testing. The maximum FDR was set at .05. The NIA Array Analysis Tool was also used to determine the correlation coefficient matrix for each of the RIE and RIE-Ras experiment sets.
Real-time Polymerase Chain Reaction and Northern Blot Analysis
Real-time polymerase chain reaction (PCR) andNorthern blot analysis was used with a selected set of genes to verify results of the array experiments. RNA from two independent samples used to generate probes for the array experiments was used to generate cDNA for the real-time PCR reactions. First, 1 °g of total RNA was converted to cDNA using SuperScript II RT (Invitrogen). Serial 10-fold dilutions of the cDNA were used for each primer set in triplicate. Polymerase chain reaction reactions were carried out in 96-well plates with the with SybrGreen Taq polymerase (ABI, Foster City, CA) according to standard protocol with an ABI 7700 Sequence Detector machine. Primer sequences were determined using PrimerDesigner software (ABI) with published GenBank sequences. Cyclophilin and GAPDH were used as controls, and results are reported as ΔΔCt relative to GAPDH. For each experiment, the ΔCt was determined by the average of three identical samples. The values in Figure 1 represent the average ΔΔCt from two independent experiments. Primer sequences are listed below:
Figure 1.

Real-time PCR of TGFβ target genes identified by DNA array. The change in gene expression of genes in RIE-Ras(12V) cells treated for 1 hour with 2 ng/ml TGFβ1 is shown by the results from the array experiments as well as by real-time PCR experiments. The average change in gene expression determined by the four independent array experiments is shown in light gray. Real-time PCR was used with a selected set of genes to verify the results of the array experiments. The ΔΔCt for the genes relative to GAPDH represents fold-change after TGFβ treatment, shown in black. GAPDH and cyclophilin are housekeeping genes, the expression of which did not change significantly with TGFβ1 treatment. The genes identified by the array experiments as having increased transcription in RIE-Ras (12V) cells after TGFβ1 treatment were confirmed by real-time PCR experiments.
APC2-F: 5′-CGCTTCGGTACCTCAGACGA
APC2-R: 5′-TGTCAATCTGCTCCAGGCG
BHLHB2-F: 5′-CTGCCCAAAACGCCAGG
BHLHB2-R: 5′-CACTTGGTACATGTGGGCAAA
CYCLOPHILIN D-F: 5′-GGCCATGTATCCTTAGCAAGTGTC
CYCLOPHILIN D-R: 5′-GGTCAGCATTGCCGATGTC
ERBIN-F: 5′-CACTCTGTGGCACCCTAAACAA
ERBIN-R: 5′-CTGCACTCTCAGATCTTGGAGG
FMR2-F: 5′-TGGTTTTTCCACAGTTATGGCA
FMR2-R: 5′-CTGCAAAGACAGACCACCACAA
GADD45B-F: 5′-ACTCCCCTCTCCTCGTCTCAG
GADD45B-R: 5′-CTCAAAAGCTACCCTACCCGTG
GADD45G-F: 5′-CCAGTCCAGGCGGCC
GADD45G-R: 5′-GTGACTCAGCAAGCAGCCTTC
GAPDH-F: 5′-ACAAGATGGTGAAGGTCGGTGT
GAPDH-R: 5′-CAAGAGAAGGCAGCCCTGG
JUNB-F: 5′-GAGGAGCAGGAGGGCTT
JUNB-R: 5′-TCACGTGGTTCATCTTCTGCAG
LRF-F: 5′-TGTGCCACAGTGCGGC
LRF-R: 5′-GCACGGAAGTTCTTGCAGC
LY94-F: 5′-GGGATCACACAGCCCAGAAT
LY94-R: 5′-CAAAAGCCATACTAGAGCCATCAC
SMAD7-F: 5′-GCGAAAGTGGGTACCACCTTC
SMAD7-R: 5′-ATTCACGTACACCCCCCTCA
SNON-F: 5′-GATCGTGAAGTCGCCCAAGA
SNON-R: 5′-AGTCCTGCCAACCAAACACAG
VEGF-F: 5′-GCCCTGGAGTGCGTGC
VEGF-R: 5′-GTGAGGTTTGATCCGCATGA
Northern Blot Analysis
To generate nucleotide probe for FMR2 sequence, cDNA was first generated from RIE mRNA according to a standard protocol using Superscript II Reverse Transcriptase (Invitrogen). An FMR2 DNA probe was generated by amplification of FMR2 sequence using probes (FMR2F and FMR2R) designed to amplify from nucleotide 1250 to 1531 of the sequence published with the accession number XM_219832 using an annealing temperature of 60°C. RIE and RIE-Ras cells were treated with 2 ng/ml TGFβ (BD Biosciences) from 0 to 24 hours, and RNA was isolated using Trizol (Invitrogen) with subsequent purification using the Qiagen RNEasy kit (Qiagen). Total RNA samples (1 mg per lane) were run on agarose gels, then blotted onto Hybond-XL (Amersham, Piscataway, NJ) membrane, which was hybridized with the 281-bp 32P-labeled FMR2 probe. The membrane was then washed and exposed to film, scanned, and signal was quantitated with ImageQuant TL Image Analysis software (Amersham). The FMR2 PCR probes were as follows: FMR2F: 5′-TGCAAAGACAGACCACCACAA-3′; FMR2R: 5′-AAAGTGCGGGAGGGAACAG-3′.
For the expression of VEGF and CTGF, total cellular RNA was extracted using Trizol. RNA samples (20 µg per lane) were loaded into 1% agarose/formaldehyde gels, separated by electrophoresis, and blotted onto nitrocellose membranes. VEGF (a kind gift from Dr. Robert Coffey) and CTGF (a kind gift from Dr. David Brigstock) cDNA probes were labeled by random primer extension using Redivue with α-32 P-dCTP and the Rediprime DNA labeling system from Amersham Life Sciences (Arlington Heights, IL). After hybridization and washing, the membranes were subjected to autoradiography. Integrity and loading of the RNA samples were assessed by 18 rRNA signals or expression of cyclophilin, a constitutively expressed mRNA. For VEGF, mRNA results were confirmed by ELISA (VEGF Quantikine Kit; R&D Systems, Minneapolis, MN).
Results
Early Genomic Responses to Short-term TGFβ Exposure in RIE Cells
DNA microarray was used to quantitatively and qualitatively ascertain TGFβ1-regulated gene expression in RIE cells that are sensitive to growth inhibition by TGFβ and RIE cells stably transformed with oncogenic Ras(12V) that are resistant to growth inhibition by TGFβ [18]. We intentionally focused on early genomic responses because these most likely reflect the primary alterations in TGFβ signaling conferred by the expression of oncogenic Ras. Similarly, we imposed a stringent definition of a TGFβ1 target to identify as pure a profile of targets as possible.
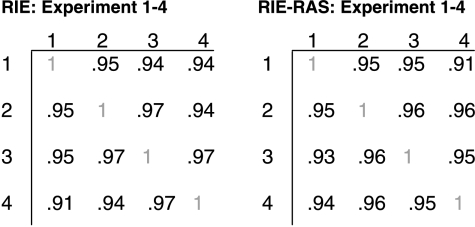
The Agilent Rat Oligo microarray glass slide system was used to compare expression of 20,500 genes in each cell line at baseline and 1 hour after exposure to 2 ng/ml TGFβ1. Significantly regulated genes were defined by a minimum change in expression of log2N = 1.4 (2.6-fold) in at least three of the four array slides used for each cell line. Data tables for all real genes on the Agilent Rat Oligo chip for each of the four independent experiments from both the RIE and RIE-Ras cell lines are included in Supplemental Data. Data including fold change and log ratios are also available on the Gene Expression Omnibus Web site (http://www.ncbi.nlm.nih.gov/geo) with the Series Accession Number GSE2015. Reproducibility of the data from the individual array slides was verified using the NIA Array Analysis Tool, and correlation coefficients are shown in Table 1.
Table 1.
Correlation Coefficient Matrices.
 |
In RIE cells, eight genes were up-regulated by TGFβ, and eight genes were down-regulated. In RIE-Ras(12V) cells, 42 genes were up-regulated and 3 genes were down-regulated (Table 2, A–D). Of the 42 genes up-regulated by TGFβ in RIE-Ras(12V) cells, 5 were also up-regulated in RIE cells, all of which have previously been described as transcriptional targets of TGFβ: GADD45β [23], VEGF [24], junB [25], BHLHB2 [26], and Smad7 [27]. In our experiments, 37 transcripts were uniquely up-regulated in RIE-Ras cells. Of these, seven have unknown functions. The remainder fall into diverse functional clusters including genes whose products function as proteases (n = 3), transcription factors (n = 4), structural proteins (n = 5), cell cycle regulators (n = 1), cell signaling factors (n = 5), regulator of cell-mediated immunity (n = 1), and proteins with other cellular functions (n = 11). These Ras-specific genes are ideal candidate pro-oncogenic genes that contribute to TGFβ-mediated tumor progression and metastasis [1].
Table 2.
Up-regulated and Down-regulated TGFβ-Responsive Genes in RIE and RIE-Ras Cells.
| Name | Accession Number | log2(N) | FDR | Function |
| (A) RIE-Ras: up-regulated | ||||
| Unknown | AI599739 | 2.86 | .000 | Unknown |
| GADD45β* | CA509894 | 2.64 | .000 | Cdc2/cyclin B1 kinase inhibitor |
| Unknown | BE117708 | 2.48 | .000 | Unknown |
| Bile acid CoA ligase | NM_024143 | 2.39 | .001 | Conjugates bile acid with amino acid |
| Endothelial differentiation, sphingolipid G-protein-coupled receptor, 3 | BF281109 | 2.26 | .000 | G-protein-coupled receptor with putative role in angiogenesis |
| Adenomatosis polyposis coli 2 | CB547568 | 2.24 | .000 | Homolog of tumor-suppressor APC, with putative roles in transcription and signal transduction |
| C1q-related factor | AI716054 | 2.07 | .000 | Unknown |
| Sciellin | AI717110 | 2.05 | .002 | Structural protein |
| Lymphocyte antigen 94 | NM_057199 | 2.03 | .017 | Lysis receptor of NK cells |
| Thyroglobulin* | NM_030988 | 1.95 | .000 | Catalyzes formation of thyroid hormone |
| Calcyon | NM_138915 | 1.9 | .000 | Membrane protein that interacts with dopamine receptor |
| Thrombospondin* | BE127095 | 1.87 | .000 | Extracellular matrix protein with putative role in angiogenesis |
| Fragile X mental retardation 2* | CB546478 | 1.85 | .000 | Putative transcription factor |
| Erbb2 interacting protein | CB546222 | 1.83 | .000 | Inhibit EGF signaling by preventing the activation of the Raf-1 kinase by Ras |
| Down syndrome cell adhesion molecule | NM_133587 | 1.72 | .000 | Ig superfamily cell-adhesion molecule thought to function in neuronal development |
| Unknown (serine/threonine kinase domain) | AW527300 | 1.7 | .000 | Unknown |
| Isopeptidase T-3 | AW521619 | 1.69 | .000 | Ubiquitin-specific protease |
| Jun B proto-oncogene* | NM_021836 | 1.69 | .000 | AP-1 family transcription factor |
| Basic helix-loop-helix domain containing, class B2* | NM_053328 | 1.68 | .000 | Transcription factor |
| MAD homolog 7* | NM_030858 | 1.68 | .000 | Inhibits Smad3/Smad4 signaling |
| FLJ00179 protein | BG673684 | 1.66 | .000 | Unknown |
| Myosin binding protein H | NM_031813 | 1.66 | .014 | Structural protein involved in myosin binding |
| Hypothetical C2H2 zinc finger protein | CB546651 | 1.65 | .000 | Putative transcription factor |
| Plasminogen activator inhibitor type 11 | NM_012620 | 1.62 | .000 | Major inhibitor of plasminogen activators |
| Gb3 synthase | NM_022240 | 1.62 | .004 | Catalyzes glycosphingolipid synthesis |
| Receptor type protein tyrosine phosphatase psi | U66566.1 | 1.6 | .000 | Transmembrane receptor-type protein tyrosine phosphatase with putative role in neuronal development |
| Neuraminidase 1 | NM_031522 | 1.59 | .000 | Removes sialic acids from glycoproteins and gangliosides |
| Preproprolactin-releasing peptide | NM_022222 | 1.56 | .000 | Stimulates stress hormone secretion by either direct pituitary or indirect hypothalamic actions |
| Leukemia/lymphoma-related factor | CB546785 | 1.54 | .000 | Zing finger transcription factor |
| Tenascin XB | CB544736 | 1.54 | .000 | Extracellular matrix glycoprotein |
| Connective tissue growth factor* | NM_022266 | 1.53 | .000 | Matricellular protein that plays an essential role in the formation of blood vessels, bone, and connective tissue |
| Unknown function | CB544526 | 1.52 | .000 | Unknown |
| Unknown function | AI409493 | 1.47 | .000 | Unknown |
| Ski-related novel protein N* | CB548407 | 1.46 | .000 | Smad 2/Smad 3 repressor |
| Hypothetical intermediate filament proteins containing protein | BM389145 | 1.45 | .002 | Putative structural protein in cytoskeleton or nuclear envelope |
| GADD45γ* | BF419904 | 1.45 | .000 | Cdc2/cyclin B1 kinase inhibitor |
| Olfactory protein | M64376.1 | 1.45 | .004 | Seven transmembrane domain protease |
| Eferin | CB545653 | 1.45 | .032 | ADP-ribosylation factor binding protein of unknown function |
| 45-kDa secretory protein | AJ132352.1 | 1.44 | .000 | Lipid binding protein of unknown function |
| Activator of G-protein signaling 3 | NM_144745 | 1.43 | .001 | Guanine nucleotide dissociation inhibitor that contains G protein regulatory motifs in its C-terminal domain |
| Vascular endothelial growth factor* | NM_031836 | 1.4 | .000 | Vascular growth factor |
| (B) RIE-Ras: down-regulated | ||||
| S100A6 | NM_053485 | -2.38 | .000 | Calcium binding protein of unknown function |
| Cadherin 15 | CB544279 | -2.15 | .020 | Cell-cell adhesion molecule |
| Stomatin-like 1 | CB544301 | -1.39 | .049 | Putative sterol-binding protein |
| (C) RIE: up-regulated | ||||
| MAD homolog 7* | NM_030858 | 2.16 | .000 | Inhibits Smad3/Smad4 signaling |
| GADD45β* | CA509894 | 2.03 | .000 | Cdc2/cyclin B1 kinase inhibitor |
| Basic helix-loop-helix domain containing, class B2* | NM_053328 | 1.87 | .000 | Transcription factor |
| Unknown | CB545096 | 1.77 | .002 | Unknown |
| Vascular endothelial growth factor* | NM_031836 | 1.64 | .000 | Vascular growth factor |
| Jun B proto-oncogene* | NM_021836 | 1.55 | .004 | AP-1 family transcription factor |
| Hairy and enhancer of split 1 | NM_024360 | 1.4 | .000 | Notch pathway homeobox transcription factor |
| Unknown | CB546780 | 1.4 | .000 | Zinc finger domain, unknown function |
| (D) RIE: Down-regulated | ||||
| Nuclear distribution gene C homolog | NM_017271 | -2.04 | .000 | Regulates movement of nuclei after mitosis |
| Ribosomal protein L27 | NM_022514 | -1.9 | .001 | Ribosomal protein |
| Ribosomal protein S29 | NM_012876 | -1.83 | .004 | Ribosomal protein |
| Glutathione S-transferase P subunit | NM_138974 | -1.7 | .002 | Component of glutathione S-transferase |
| Helicase DDX32 | CB548436 | -1.59 | .006 | DEAH family helicase |
| Hypothetical protein FLJ12800 | BE106894 | -1.56 | .009 | Unknown |
| Fragile X mental retardation 2* | CB546478 | -1.5 | .017 | Putative transcription factor |
| Unknown | BF390720 | -1.45 | .000 | Putative component of Golgi complex |
Previously identified as TGFβ-regulated.
Confirmation of TGFβ-Regulated Gene Expression by mRNA Analysis
Eleven genes up-regulated by TGFβ1 treatment of Ras-transformed cells were selected for confirmation by real-time PCR because they were prominently regulated or were putative tumor promoters. In each instance, genes identified as transcriptional targets of TGFβ1 by microarray were similarly up-regulated when tested by real-time PCR (Figure 1). Several of the genes induced in both the RIE (5/8) and RIE-Ras(12V) (10/42) cells have been identified as TGFβ-induced genes in previous studies (Table 2), further supporting the validity of our results. The differential expression of FMR2 was independently confirmed by a more extensive time course using Northern blot analysis. These results were also consistent with the results from the array experiments and were particularly interesting because expression was regulated in a binary fashion, being up-regulated in RIE-Ras cells treated with TGFβ1 and down-regulated in the parental cell line (Figure 2).
Figure 2.

RIE and RIE-Ras Northern blots show differential expression of FMR2 in response to TGFβ1. The RIE gene expression array showed inhibition of FMR2 expression after 1 hour of incubation with 2 ng/ml TGFβ1, whereas the RIE-Ras(12V) array showed activation of FMR2 gene expression. These findings were supported by Northern blot analysis of FMR2 gene expression as shown in the top panel. In the middle panel, equivalent RNA loading was confirmed verified by ethidium bromide staining of 18 and 28S ribosomal RNA. RNA induction by TGFβ1 was quantified by scanning the autoradiographs and analyzing band intensity with densitometry (bottom panel). Results from these Northern blots are consistent with FMR2 expression results obtained in the microarray experiments and show a marked differential effect on FMR2 expression, with 11-fold maximal induction in Ras-transformed cells and a fourfold repression in the parental cell line. In these graphs, band intensity is reported as the signal at any given time point relative to the signal of the control RNA or “0 hour” sample.
Regulation of VEGF and CTGF Expression by TGFβ in RIE-Ras Cells
Two genes, VEGF and CTGF, were selected for more intensive study because they were markedly induced in Ras-transformed RIE cells, they are abundantly expressed, and they are well known as TGFβ-induced genes. VEGF has been implicated as a potential contributor to the pro-oncogenic activities of TGFβ [5,28]. Northern blot analysis (Figure 3A) and signal quantification (Figure 3B, right panel) of an expanded time course of VEGF mRNA expression in intestinal epithelial cells lines show a fivefold induction in the TGFβ-growth-resistant, transformed RIE-Ras cell line as opposed to a twofold induction in the parental cell line. This more robust increase in Ras-transformed RIE-1 cells occurs despite a basal level of VEGF expression in RIE-Ras cells that significantly exceeds that in the RIE cells (Figure 3B, right panel). Similar observations were made for the VEGF protein measured by ELISA, again showing a more prominent induction of VEGF by TGFβ in the RIE-Ras cell line (Figure 3C). As prior work has found significantly reduced Smad-mediated transcription in Ras-transformed RIE cells [19,20], it is highly unlikely that TGFβ induction of VEGF in these cells occurs exclusively by Smad-dependent signaling. In support of this assertion, VEGF mRNA expression was also significantly induced in two human colon cancer cell lines, HT-29 and SW620, both of which have defective Smad-dependent signaling (not shown).
Figure 3.

Induction of VEGF by TGFβ in RIE and RIE-Ras(12V) cells. (A) Northern blots were prepared from RNA isolated from RIE and RIE-Ras(12V) cells treated with 2 ng/ml of TGFβ1 for the time intervals shown and probed for VEGF expression. Equivalent loading was confirmed by the signal intensity of IB15 (cyclophilin) a constitutively expressed gene. In (B), basal expression of VEGF is shown in RIE and RIE-Ras(12V) cells, confirming markedly increased basal expression in the Ras-transformed line. The bottom right shows densitometric values obtained from the Northern shown in the top panel with expression shown as a fold change overexpression at time 0. As shown in (C) VEGF protein was also differentially induced in RIE-Ras(12V) cells. Whole-cell protein lysates were obtained from RIE-1 and RIE-Ras(12V) cells treated with TGFβ for 24 hours. VEGF protein was quantified by ELISA, as described in the Materials and Methods section. (D) Effect of signaling pathway inhibitors on induction of VEGF by 2 ng/ml TGFβ1. Cells were treated with 10 µM SB203580 (p38 kinase inhibitor), 10 µM U0126 (a MEK inhibitor), 1 µM RafKI (BAY-439006, a Raf-1 kinase inhibitor), and 20 µM LY294002 (phosphoinositol 3-kinase inhibitor) for 24 hours and then treated with 2 ng/ml TGFβ1 for 1 hour before isolation of RNA according to procedures described in the Materials and Methods section. Equivalent loading was confirmed by hybridization with a cDNA probe complementary to cyclophilin. The results shown are representative of three separate experiments.
The potential contribution of alternate, Smad-independent signaling pathways in TGFβ-mediated VEGF induction was examined by use of specific small molecule inhibitors. Inhibition of the p38 MAPK pathway and the phosphoinositol 3-kinase pathway, two signaling cascades previously implicated in Smad-independent responses to TGFβ [13,29], did not attenuate induction of VEGF by TGFβ1 (Figure 3D). Inhibition of MAP kinase kinase (MEK) with U-0126 and Raf-1 kinase with BAY-439006 blocked induction of VEGF by TGFβ, implicating TGFβ activation of Raf-1/Erk signaling as a potential Smad-independent pathway for TGFβ signaling in Ras-transformed RIE-1 cells. Interestingly, in the setting of Raf-1 kinase inhibition, levels of VEGF were not further increased by TGFβ1, but basal levels of VEGF were elevated above control.
The role for CTGF in tumorigenesis is not clear, but in certain cellular contexts, CTGF is a pro-oncogenic, profibrogenic growth factor [30]. It is tumor-suppressive in other contexts [31]. CTGF is also frequently identified as a prominently regulated gene in genomic profiles of TGFβ action [32,33] a finding that we confirm herein. CTGF expression was also differentially and prominently up-regulated in RIE-Ras cells, with maximal expression 31-fold above basal levels occurring 2 hours after treatment with TGFβ in RIE-Ras cells compared with a 3.5-fold induction in the parental RIE-1 line (Figure 4A). CTGF expression was also robustly induced in HT-29 cells, a Smad4-deficient human colon carcinoma cell line that is resistant to growth inhibition by TGFβ (not shown), implying Smad independence. Figure 4B shows markedly reduced basal levels of CTGF in exponentially growing RIE-Ras(12V) cells compared with the parental line. The original of this differential expression is being explored separately. In contrast to the involvement of the canonical MAPK pathway in VEGF induction by TGFβ in Ras-transformed cells (Figure 3A), induction of CTGF was not blocked by inhibitors of the p38 MAPK, phosphoinositol 3-kinase, MEK, or Raf-1 kinase pathways (Figure 4C). Thus, the Smad4-independent signaling pathway responsible for TGFβ induction of CTGF in Ras-transformed intestinal remains unclear.
Figure 4.

Induction of CTGF by TGFβ in RIE and RIE-Ras(12V) cells. (A) Northern blots were prepared from RNA isolated from RIE and RIE-Ras(12V) cells treated with 2 ng/ml of TGFβ1 for the time intervals shown and probed for CTGF expression. Equivalent loading was confirmed by the signal intensity of IB15, a constitutively expressed gene. (B) Relative expression of CTGF in RIE and RIE-Ras (12V) cells shows markedly reduced basal expression in the Ras-transformed line. Equivalent loading is confirmed by expression of 1B15, a constitutively expressed RNA species. (C) Organized identically to Figure 3D, and the RNA samples were identical to those used in Figure 3D. Cells were treated with 10 µM SB203580 (p38 kinase inhibitor), 10 µM U0126 (a MEK inhibitor), 1 µM RafKI (BAY439006, a Raf-1 kinase inhibitor), and 20 µM LY294002 (phosphoinositol 3-kinase inhibitor) for 24 hours and then treated with 2 ng/ml TGFβ1 for 1 hour before isolation of RNA according to procedures described in the Materials and Methods section. Equivalent loading was confirmed by hybridization with a cDNA probe complementary to cyclophilin. The result shown is representative of three separate experiments.
Discussion
Until recent years, the pro-oncogenic effects of TGFβ could only be indirectly inferred [34–36] from studies that describe increased TGFβ levels in metastatic tumors [37,38] or report increased expression of TGFβ as a risk factor for recurrence and reduced survival [39]. For instance, colorectal cancers with mutant, inactivated TGFβRII have presumably no potential for pro-oncogenic signaling and a prognosis that is better than cancers with retention of one allele [40]. Conversely, restoration of TGFβRII expression in cells that carry a mutation in this gene increases cell invasiveness in vitro [35]. Thus, once tumors escape growth regulation by TGFβ, if the potential remains for residual TGFβ signaling, the result is an enhanced potential for tumorigenesis. Furthermore, the apparent duality of TGFβ signaling has been observed in vivo; in bitransgenic mice, mutant activated TGFβ is associated with delayed tumor onset in early stages, whereas in later stages, it leads to an increase in metastatic foci [7,41].
It is clear that oncogenic events such as transformation by mutant activated Ras may “adversely” interact with growth inhibitory TGFβ signaling. In Ras-transformed cells, loss of an autocrine growthinhibitory GFβ signaling occurs coincident with a synergistic stimulation of angiogenic molecules [42], matrix molecules [43], and epithelial to mesenchymal transition [35], each of which is believed to be important for tumor growth and metastasis in vivo. Inducible Ras in an in vitro model using the same rat intestinal epithelial cell line used in our current study conferred characteristics, such as invasiveness, consistent with tumor promotion and metastasis [44]. Ras overexpression also activates transcription of the TGFβ1 gene [45]. Increased levels of TGFβ in this context may further contribute to tumor-promoting signaling in growth-resistant cells.
In the present study, we found that oncogenic Ras confers to TGFβ a significant gain of robust early genomic responses in cultured rat intestinal epithelial cells that are resistant to growth inhibition. Although we found five immediate transcriptional targets of TGFβ activated in both RIE and RIE-Ras(12V) cells, a larger number of genes are upregulated by TGFβ only in the context of Ras activation. These findings imply a model in which Ras-dependent TGFβ early genomic targets may be responsible for pro-oncogenic activities of TGFβ and potentially contribute to resistance to growth inhibition as well.
The well-recognized reduction of Smad-dependent signaling that is characteristic of Ras-transformed epithelial cells [19,20,46,47] indicates that the expanded genomic response to TGFβ occurs by an as yet unidentified Smad-independent pathway or by an unrecognized modification or interaction with the canonical Smad signaling pathway. Several published reports compare transcriptional responses in epithelial cell lines that are sensitive and resistant to growth inhibition by TGFβ [32,48]. The conflicting findings in the aforementioned reports, both of which examined the consequences of silenced Smad4 on TGFβ-mediated gene expression, serve to further underscore the complexity of TGFβ signaling and reinforce the necessity for further investigation.
Although our own experimental approach was not designed to specifically determine functional profiles of genes induced by TGFβ1, it is unexpected that the GADD45β, a gene product generally associated with apoptosis and cell cycle arrest, was prominently induced in both TGFβ-growth-sensitive parental cells and growth-resistant Ras-transformed cells (Table 2), again emphasizing the complexity and intricacy of TGFβ signaling. Transforming growth factor β also regulates distinct genomic profiles in HaCaT cells with Smad2 and Smad3 individually attenuated by specific antisense molecules [49].
In a similar study examining transcriptional responses in a control cell line versus a Ras-transformed line, Chen et al. [33] found that four of the five genes up-regulated in our studies of RIE and RIE-Ras(12V) cells were also up-regulated by TGFβ exposure for 2 to 4 hours in a nontumorigenic mammary cell line (MCF-10A) that is sensitive to growth inhibition by TGFβ. Those same four genes were also up-regulated by TGFβ in MCF-10A cells dually transformed with activated c-Ha-ras and c-erbB2, as well as in a human breast cancer cell line with hyperactive Ras (MDA-MG-231) both of which are resistant to growth suppression by TGFβ [33]. The relative magnitude of the genomic response was not provided in the report by Chen et al, except to allude to the fact that many responses were generated in the Ras-transformed lines. These results, along with others [50,51], indicate that despite resistance to TGFβ growth inhibition, transformed epithelial cell lines from multiple epithelial lineages retain at least some common TGFβ transcriptional targets as well as an expanded repertoire of genomic responses. Thus, the results observed in the present study and others reported to date seem to be generalizable.
Individual Ras-specific, TGFβ up-regulated genes identified in our study have been implicated in tumor progression. For example, plasminogen activator inhibitor type 1 (PAI-1) is up-regulated in human cancer and correlates with metastatic behavior [52]. Thrombospondin and VEGF are associated with angiogenesis and metastasis (reviewed in Bergers and Benjamin [53]). The ErbB2-interacting protein (Erbin) is a PDZ domain-containing protein that binds the C-terminus of ErbB2 that is essential for epithelial integrity. Recent studies show that Erbin suppresses Ras activation of Erk signaling while preserving activation of other Ras effectors such as Akt. A recent expression profile of genes expressed in breast cancer includes Erbin as a “poor prognosis” marker [54].
A homolog of the tumor-suppressor gene adenomatosis polyposis coli (APC), APC2, was also specifically up-regulated in the RIE-Ras (12V) cell line. APC1 and APC2 have overlapping roles in Wingless pathway signaling in Drosophila and mice [55]. Loss of expression of APC has been associated with most sporadic colon adenocarcinomas leading to overactivation of the Wingless/Wnt signaling pathway (reviewed in Grady and Markowitz [56]). It is not clear how overexpression of APC2 might contribute to tumorigenesis; however, its expression has recently been found to be up-regulated in lung adenomas and lung adenocarcinomas compared to normal lung [57]. Neither APC nor APC2 has previously been identified as a transcriptional target of TGFβ.
An intriguing finding in our analysis is that fragile X mental retardation gene 2 (FMR2) was among the most repressed genes in RIE cells and among the most activated in RIE-Ras(12V) cells. Northern blot analysis of FMR2 expression at multiple time points verified the finding of the array that FMR2 is induced by TGFβ in RIE cells and is inhibited in RIE-Ras cells (Figure 2, A and B). FMR2 was originally cloned in an attempt to identify the basis for FRAXE-linked mental retardation. Subsequent analysis localized FMR2 to the nucleus and identified it as a transcriptional activator (reviewed in Gu and Nelson [58]). LAF4, an FMR2 family member, was identified through chromosomal translocations that resulted in Burkitt lymphoma [59]. Rearrangements of LAF4, as well as two other FMR2 family members, AF4 and AF5q21, with the mixed leukemia lineage (MLL) gene have been identified in infant acute lymphoblastic leukemia, indicating that this gene family may have a function in cell proliferation and oncogenesis [60–62]. Lilliputian, an FMR2 Drosophila homologue, is involved in both TGFβ [63] and Erk signaling [64], which is intriguing in light of our observation that FMR2 expression was repressed by TGFβ in RIE cells and induced by TGFβ in the setting of oncogenic Ras. The role of this gene in the cellular response to TGFβ merits further study.
VEGF and CTGF expression in response to TGFβ exposure was examined in more detail in our study. These genes are more robustly regulated in RIE-Ras cells than the parental line, raising the important question of the signaling mechanics involved. Because Smaddependent signaling is attenuated in Ras-transformed RIE cells [19], it is unlikely that this pathway accounts for the accentuated expression of these TGFβ-inducible genes. Our results indicate that activation of the canonical Raf-1/MAPK/Erk pathway may contribute to VEGF but not CTGF induction. Additional candidate pathways deserve further exploration.
In summary, ours is the first study designed to identify and characterize the TGFβ-induced transcriptome in intestinal epithelial cells transformed by oncogenic Ras. Despite the attenuation of Smaddependent, growth-inhibitory signaling in intestinal epithelial cells that overexpress oncogenic Ras, TGFβ induces a significantly expanded repertoire of highly regulated (≥2.6-fold induction) genomic targets in these cells. This finding is analogous to other existing work that shows an expanded genomic profile when specific components of the Smad pathway are abrogated. The biologic properties of many of these TGFβ-induced genes are consistent with a tumor-promoting role for TGFβ and deserve further scrutiny as pro-oncogenic factors. A critical consideration is the extent to which this unique transcriptome occurs because of Smad-independent signaling, Smad signaling that is modified in an unrecognized manner by hyperactive Ras activity, or both.
Supplemental Data
Raw Data
Raw image files (.tif ) for each experiment are available at the Gene Expression Omnibus Web site (http://www.ncbi.nlm.nih.gov/geo) with the Series Accession Number GSE2015. These images are the original chip scans used to generate the data in this study.
Complete Data Tables
Data tables are listed as RIE versus RIE-TGFβ x 1 hour and RAS versus RAS-TGFβ (for the RIE-Ras experiments), each with worksheets #1 to #4 corresponding to results from experiments #1 to 4. The tables summarize results including raw signal intensity from each channel (Lex.R corresponds to control, Lex.E corresponds to experimental), raw signal intensity with background subtracted, normalized signal intensity, fold change (experimental/control), flag status, Agilent gene description, Agilent probe identification, and GenBank Accession Number. Results are listed in ascending order of Agilent probe identification. Control spots on the Agilent Rat Oligo chip (Agilent G4130A) not corresponding to sequence from real genes were excluded from the table. Data including fold change and log ratios are also available at the NCBI GEO Web site (http://www.ncbi.nlm.nih.gov/geo) with the Series Accession Number GSE2015.
Acknowledgments
The authors thank the invaluable assistance of the Columbus Children's Research Institute Microarray Facility (Robert Munson, Robert Armbruster, Beth Baker, and Selnur Erdal). We also appreciate helpful discussions with David Cunningham.
Footnotes
This work was funded by National Institutes of Health Grant R01 DK57128 (J.A.B.).
References
- 1.Akhurst RJ, Derynck R. TGF-beta signaling in cancer—a double-edged sword. Trends Cell Biol. 2001;11:S44–S51. doi: 10.1016/s0962-8924(01)02130-4. [DOI] [PubMed] [Google Scholar]
- 2.Derynck R, Akhurst RJ, Balmain A. TGF-beta signaling in tumor suppression and cancer progression. Nat Genet. 2001;29:117–129. doi: 10.1038/ng1001-117. [DOI] [PubMed] [Google Scholar]
- 3.Massague J, Blain SW, Lo RS. TGFbeta signaling in growth control, cancer, and heritable disorders. Cell. 2000;103:295–309. doi: 10.1016/s0092-8674(00)00121-5. [DOI] [PubMed] [Google Scholar]
- 4.Roberts AB, Wakefield LM. The two faces of transforming growth factor beta in carcinogenesis. Proc Natl Acad Sci USA. 2003;100:8621–8623. doi: 10.1073/pnas.1633291100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bierie B, Moses HL. TGF-beta and cancer. Cytokine Growth Factor Rev. 2006;17:29–40. doi: 10.1016/j.cytogfr.2005.09.006. [DOI] [PubMed] [Google Scholar]
- 6.Muraoka RS, Koh Y, Roebuck LR, Sanders ME, Brantley-Sieders D, Gorska AE, Moses HL, Arteaga CL. Increased malignancy of Neu-induced mammary tumors overexpressing active transforming growth factor beta1. Mol Cell Biol. 2003;23:8691–8703. doi: 10.1128/MCB.23.23.8691-8703.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Deane NG, Lee H, Hamaamen J, Ruley A, Washington MK, LaFleur B, Thorgeirsson SS, Price R, Beauchamp RD. Enhanced tumor formation in cyclin D1 x transforming growth factor beta1 double transgenic mice with characterization by magnetic resonance imaging. Cancer Res. 2004;64:1315–1322. doi: 10.1158/0008-5472.can-03-1772. [DOI] [PubMed] [Google Scholar]
- 8.Derynck R, Zhang YE. Smad-dependent and Smad-independent pathways in TGF-beta family signalling. Nature. 2003;425:577–584. doi: 10.1038/nature02006. [DOI] [PubMed] [Google Scholar]
- 9.Massague J, Wotton D. Transcriptional control by the TGF-beta/Smad signaling system. EMBO J. 2000;19:1745–1754. doi: 10.1093/emboj/19.8.1745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Massague J, Gomis RR. The logic of TGF-beta signaling. FEBS Lett. 2006;580:2811–2820. doi: 10.1016/j.febslet.2006.04.033. [DOI] [PubMed] [Google Scholar]
- 11.Wu T, Rezai B, Rashid A, Michael CL, Cayouette MC, Kim C, Sani N. Genetic alterations and epithelial dysplasia in juvenile polyposis syndrome and sporadic juvenile polyps. Am J Pathol. 1997;150:939–947. [PMC free article] [PubMed] [Google Scholar]
- 12.Massague J, Seoane J, Wotton D. Smad transcription factors. Genes Dev. 2005;19:2783–2810. doi: 10.1101/gad.1350705. [DOI] [PubMed] [Google Scholar]
- 13.Yue J, Mulder KM. Transforming growth factor-beta signal transduction in epithelial cells. Pharmacol Ther. 2001;91:1–34. doi: 10.1016/s0163-7258(01)00143-7. [DOI] [PubMed] [Google Scholar]
- 14.Crespo P, Leon J. Ras proteins in the control of the cell cycle and cell differentiation. Cell Mol Life Sci. 2000;57:1613–1636. doi: 10.1007/PL00000645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Macaluso M, Russo G, Cinti C, Bazan V, Gebbia N, Russo A. Ras family genes: an interesting link between cell cycle and cancer. J Cell Physiol. 2002;192:125–130. doi: 10.1002/jcp.10109. [DOI] [PubMed] [Google Scholar]
- 16.Bos JL, Fearon ER, Hamilton SR, Verlaan-de Vries M, van Boom JH, van der Eb AJ, Vogelstein B. Prevalence of ras gene mutations in human colorectal cancers. Nature. 1987;327:293–297. doi: 10.1038/327293a0. [DOI] [PubMed] [Google Scholar]
- 17.Mulder KM. Role of Ras and MAPKs in TGFbeta signaling. Cytokine Growth Factor Rev. 2000;11:23–35. doi: 10.1016/s1359-6101(99)00026-x. [DOI] [PubMed] [Google Scholar]
- 18.Bulus NM, Sheng HM, Sizemore N, Oldham SM, Barnett JV, Coffey RJ, Beauchamp DR, Barnard JA. Ras-mediated suppression of TGFbetaRII expression in intestinal epithelial cells involves Raf-independent signaling. Neoplasia. 2000;2:357–364. doi: 10.1038/sj.neo.7900099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jiang B, Zhang JS, Du J, Urrutia R, Barnard J. Growth inhibitory signalling by TGFbeta is blocked in Ras-transformed intestinal epithelial cells at a post-receptor locus. Cell Signal. 2003;15:699–708. doi: 10.1016/s0898-6568(03)00010-x. [DOI] [PubMed] [Google Scholar]
- 20.Saha D, Datta PK, Beauchamp RD. Oncogenic ras represses transforming growth factor-beta/Smad signaling by degrading tumor suppressor Smad4. J Biol Chem. 2001;276:29531–29537. doi: 10.1074/jbc.M100069200. [DOI] [PubMed] [Google Scholar]
- 21.Winesett MP, Ramsey GW, Barnard JA. Type II TGF(beta) receptor expression in intestinal cell lines and in the intestinal tract. Carcinogenesis. 1996;17:989–995. doi: 10.1093/carcin/17.5.989. [DOI] [PubMed] [Google Scholar]
- 22.Du J, Jiang B, Coffey RJ, Barnard J. Raf and RhoA cooperate to transform intestinal epithelial cells and induce growth resistance to transforming growth factor beta. Mol Cancer Res. 2004;2:233–241. [PubMed] [Google Scholar]
- 23.Yoo J, Ghiassi M, Jirmanova L, Balliet AG, Hoffman B, Fornace AJ, Jr, Liebermann DA, Bottinger EP, Roberts AB. Transforming growth factor-beta-induced apoptosis is mediated by Smad-dependent expression of GADD45b through p38 activation. J Biol Chem. 2003;278:43001–43007. doi: 10.1074/jbc.M307869200. [DOI] [PubMed] [Google Scholar]
- 24.Benckert C, Jonas S, Cramer T, Von Marschall Z, Schafer G, Peters M, Wagner K, Radke C, Wiedenmann B, Neuhaus P, et al. Transforming growth factor beta 1 stimulates vascular endothelial growth factor gene transcription in human cholangiocellular carcinoma cells. Cancer Res. 2003;63:1083–1092. [PubMed] [Google Scholar]
- 25.Ohtsuki M, Massague J. Evidence for the involvement of protein kinase activity in transforming growth factor-beta signal transduction. Mol Cell Biol. 1992;12:261–265. doi: 10.1128/mcb.12.1.261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shen M, Yoshida E, Yan W, Kawamoto T, Suardita K, Koyano Y, Fujimoto K, Noshiro M, Kato Y. Basic helix-loop-helix protein DEC1 promotes chondrocyte differentiation at the early and terminal stages. J Biol Chem. 2002;277:50112–50120. doi: 10.1074/jbc.M206771200. [DOI] [PubMed] [Google Scholar]
- 27.Nagarajan RP, Zhang J, Li W, Chen Y. Regulation of Smad7 promoter by direct association with Smad3 and Smad4. J Biol Chem. 1999;274:33412–33418. doi: 10.1074/jbc.274.47.33412. [DOI] [PubMed] [Google Scholar]
- 28.Breier G, Blum S, Peli J, Groot M, Wild C, Risau W, Reichmann E. Transforming growth factor-beta and Ras regulate the VEGF/VEGF-receptor system during tumor angiogenesis. Int J Cancer. 2002;97:142–148. doi: 10.1002/ijc.1599. [DOI] [PubMed] [Google Scholar]
- 29.Bakin AV, Tomlinson AK, Bhowmick NA, Moses HL, Arteaga CL. Phosphatidylinositol 3-kinase function is required for transforming growth factor beta-mediated epithelial to mesenchymal transition and cell migration. J Biol Chem. 2000;275:36803–36810. doi: 10.1074/jbc.M005912200. [DOI] [PubMed] [Google Scholar]
- 30.Yang F, Tuxhorn JA, Ressler SJ, McAlhany SJ, Dang TD, Rowley DR. Stromal expression of connective tissue growth factor promotes angiogenesis and prostate cancer tumorigenesis. Cancer Res. 2005;65:8887–8895. doi: 10.1158/0008-5472.CAN-05-1702. [DOI] [PubMed] [Google Scholar]
- 31.Chien W, Yin D, Gui D, Mori A, Frank JM, Said J, Kusuanco D, Marchevsky A, McKenna R, Koeffler HP. Suppression of cell proliferation and signaling transduction by connective tissue growth factor in non-small cell lung cancer cells. Mol Cancer Res. 2006;4:591–598. doi: 10.1158/1541-7786.MCR-06-0029. [DOI] [PubMed] [Google Scholar]
- 32.Levy L, Hill CS. Smad4 dependency defines two classes of transforming growth factor {beta} (TGF-{beta}) target genes and distinguishes TGF-{beta}-induced epithelial-mesenchymal transition from its antiproliferative and migratory responses. Mol Cell Biol. 2005;25:8108–8125. doi: 10.1128/MCB.25.18.8108-8125.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chen CR, Kang Y, Massague J. Defective repression of c-myc in breast cancer cells: a loss at the core of the transforming growth factor beta growth arrest program. Proc Natl Acad Sci USA. 2001;98:992–999. doi: 10.1073/pnas.98.3.992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hocevar BA, Brown TL, Howe PH. TGF-beta induces fibronectin synthesis through a c-Jun N-terminal kinase-dependent, Smad4-independent pathway. EMBO J. 1999;18:1345–1356. doi: 10.1093/emboj/18.5.1345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Oft M, Heider KH, Beug H. TGFbeta signaling is necessary for carcinoma cell invasiveness and metastasis. Curr Biol. 1998;8:1243–1252. doi: 10.1016/s0960-9822(07)00533-7. [DOI] [PubMed] [Google Scholar]
- 36.Larsson J, Goumans MJ, Sjostrand LJ, van Rooijen MA, Ward D, Leveen P, Xu X, ten Dijke P, Mummery CL, Karlsson S. Abnormal angiogenesis but intact hematopoietic potential in TGF-beta type I receptor-deficient mice. EMBO J. 2001;20:1663–1673. doi: 10.1093/emboj/20.7.1663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tsushima H, Ito N, Tamura S, Matsuda Y, Inada M, Yabuuchi I, Imai Y, Nagashima R, Misawa H, Takeda H, et al. Circulating transforming growth factor beta 1 as a predictor of liver metastasis after resection in colorectal cancer. Clin Cancer Res. 2001;7:1258–1262. [PubMed] [Google Scholar]
- 38.Picon A, Gold LI, Wang J, Cohen A, Friedman E. A subset of human colon cancers expresses elevated levels of transforming growth factor beta 1. Cancer Epidemiol Biomarkers Prev. 1998;7:497–504. [PubMed] [Google Scholar]
- 39.Friedman E, Gold LI, Klimstra D, Zeng ZS, Winawer S, Cohen A. High levels of transforming growth factor beta 1 correlate with disease progression in human colon cancer. Cancer Epidemiol Biomarkers Prev. 1995;4:549–554. [PubMed] [Google Scholar]
- 40.Watanabe T, Wu TT, Catalano PJ, Ueki T, Satriano R, Haller DG, Benson AB, III, Hamilton SR. Molecular predictors of survival after adjuvant chemotherapy for colon cancer. N Engl J Med. 2001;344:1196–1206. doi: 10.1056/NEJM200104193441603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Siegel PM, Shu W, Cardiff RD, Muller WJ, Massague J. Transforming growth factor beta signaling impairs Neu-induced mammary tumorigenesis while promoting pulmonary metastasis. Proc Natl Acad Sci USA. 2003;100:8430–8435. doi: 10.1073/pnas.0932636100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rak J, Mitsuhashi Y, Sheehan C, Tamir A, Viloria-Petit A, Filmus J, Mansour SJ, Ahn NG, Kerbel RS. Oncogenes and tumor angiogenesis: differential modes of vascular endothelial growth factor up-regulation in ras-transformed epithelial cells and fibroblasts. Cancer Res. 2000;60:490–498. [PubMed] [Google Scholar]
- 43.Ellenrieder V, Hendler SF, Ruhland C, Boeck W, Adler G, Gress TM. TGF-beta-induced invasiveness of pancreatic cancer cells is mediated by matrix metalloproteinase-2 and the urokinase plasminogen activator system. Int J Cancer. 2001;93:204–211. doi: 10.1002/ijc.1330. [DOI] [PubMed] [Google Scholar]
- 44.Fujimoto K, Sheng H, Shao J, Beauchamp RD. Transforming growth factor-beta1 promotes invasiveness after cellular transformation with activated Ras in intestinal epithelial cells. Exp Cell Res. 2001;266:239–249. doi: 10.1006/excr.2000.5229. [DOI] [PubMed] [Google Scholar]
- 45.Geiser AG, Kim SJ, Roberts AB, Sporn MB. Characterization of the mouse transforming growth factor-beta 1 promoter and activation by the Ha-Ras oncogene. Mol Cell Biol. 1991;11:84–92. doi: 10.1128/mcb.11.1.84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Filmus J, Zhao J, Buick RN. Overexpression of H-ras oncogene induces resistance to the growth-inhibitory action of transforming growth factor beta-1 (TGF-beta 1) and alters the number and type of TGF-beta 1 receptors in rat intestinal epithelial cell clones. Oncogene. 1992;7:521–526. [PubMed] [Google Scholar]
- 47.Kretzschmar M, Doody J, Timokhina I, Massague J. A mechanism of repression of TGFbeta/Smad signaling by oncogenic Ras. Genes Dev. 1999;13:804–816. doi: 10.1101/gad.13.7.804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jazag A, Ijichi H, Kanai F, Imamura T, Guleng B, Ohta M, Imamura J, Tanaka Y, Tateishi K, Ikenoue T, et al. Smad4 silencing in pancreatic cancer cell lines using stable RNA interference and gene expression profiles induced by transforming growth factor-beta. Oncogene. 2005;24:662–671. doi: 10.1038/sj.onc.1208102. [DOI] [PubMed] [Google Scholar]
- 49.Kretschmer A, Moepert K, Dames S, Sternberger M, Kaufmann J, Klippel A. Differential regulation of TGF-beta signaling through Smad2, Smad3 and Smad4. Oncogene. 2003;22:6748–6763. doi: 10.1038/sj.onc.1206791. [DOI] [PubMed] [Google Scholar]
- 50.Grunert S, Jechlinger M, Beug H. Diverse cellular and molecular mechanisms contribute to epithelial plasticity and metastasis. Nat Rev Mol Cell Biol. 2003;4:657–665. doi: 10.1038/nrm1175. [DOI] [PubMed] [Google Scholar]
- 51.Qian J, Niu J, Li M, Chiao P, Tsao MS. In vitro modeling of human pancreatic duct epithelial cell transformation defines gene expression changes induced by K-ras oncogenic activation in pancreatic carcinogenesis. Cancer Res. 2005;65:5045–5053. doi: 10.1158/0008-5472.CAN-04-3208. [DOI] [PubMed] [Google Scholar]
- 52.Duffy MJ. The urokinase plasminogen activator system: role in malignancy. Curr Pharm Des. 2004;10:39–49. doi: 10.2174/1381612043453559. [DOI] [PubMed] [Google Scholar]
- 53.Bergers G, Benjamin LE. Tumorigenesis and the angiogenic switch. Nat Rev Cancer. 2003;3:401–410. doi: 10.1038/nrc1093. [DOI] [PubMed] [Google Scholar]
- 54.van 't Veer LJ, Dai H, van de Vijver MJ, He YD, Hart AAM, Mao M, Peterse HL, van der Kooy K, Marton MJ, Witteveen AT, et al. Gene expression profiling predicts clinical outcome of breast cancer. Nature. 2002;415:530–536. doi: 10.1038/415530a. [DOI] [PubMed] [Google Scholar]
- 55.Ahmed Y, Nouri A, Wieschaus E. Drosophila Apc1 and Apc2 regulate Wingless transduction throughout development. Development. 2002;129:1751–1762. doi: 10.1242/dev.129.7.1751. [DOI] [PubMed] [Google Scholar]
- 56.Grady WM, Markowitz SD. Genetic and epigenetic alterations in colon cancer. Annu Rev Genomics Hum Genet. 2002;3:101–128. doi: 10.1146/annurev.genom.3.022502.103043. [DOI] [PubMed] [Google Scholar]
- 57.Bonner AE, Lemon WJ, Devereux TR, Lubet RA, You M. Molecular profiling of mouse lung tumors: association with tumor progression, lung development, and human lung adenocarcinomas. Oncogene. 2004;23:1166–1176. doi: 10.1038/sj.onc.1207234. [DOI] [PubMed] [Google Scholar]
- 58.Gu Y, Nelson DL. FMR2 function: insight from a mouse knockout model. Cytogenet Genome Res. 2003;100:129–139. doi: 10.1159/000072847. [DOI] [PubMed] [Google Scholar]
- 59.Ma C, Staudt LM. LAF-4 encodes a lymphoid nuclear protein with transactivation potential that is homologous to AF-4, the gene fused to MLL in t(4;11) leukemias. Blood. 1996;87:734–745. [PubMed] [Google Scholar]
- 60.Hiwatari M, Taki T, Taketani T, Taniwaki M, Sugita K, Okuya M, Eguchi M, Ida K, Hayashi Y. Fusion of an AF4-related gene, LAF4, to MLL in childhood acute lymphoblastic leukemia with t(2;11)(q11;q23) Oncogene. 2003;22:2851–2855. doi: 10.1038/sj.onc.1206389. [DOI] [PubMed] [Google Scholar]
- 61.Imamura T, Morimoto A, Ikushima S, Kakazu N, Hada S, Tabata Y, Yagi T, Inaba T, Hibi S, Sugimoto T, et al. A novel infant acute lymphoblastic leukemia cell line with MLL-AF5q31 fusion transcript. Leukemia. 2002;16:2302–2308. doi: 10.1038/sj.leu.2402665. [DOI] [PubMed] [Google Scholar]
- 62.von Bergh AR, Beverloo HB, Rombout P, van Wering ER, van Weel MH, Beverstock GC, Kluin PM, Slater RM, Schuuring E. LAF4, an AF4-related gene, is fused to MLL in infant acute lymphoblastic leukemia. Genes Chromosomes Cancer. 2002;35:92–96. doi: 10.1002/gcc.10091. [DOI] [PubMed] [Google Scholar]
- 63.Su MA, Wisotzkey RG, Newfeld SJ. A screen for modifiers of decapentaplegic mutant phenotypes identifies lilliputian, the only member of the fragile-X/Burkitt's lymphoma family of transcription factors in Drosophila melanogaster. Genetics. 2001;157:717–725. doi: 10.1093/genetics/157.2.717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wittwer F, van der Straten A, Keleman K, Dickson B, Hafen E. Lilliputian: an AF4/FMR2-related protein that controls cell identity and cell growth. Development. 2001;128:791–800. doi: 10.1242/dev.128.5.791. [DOI] [PubMed] [Google Scholar]