

Abstract
The hairpin ribozyme requires functional group contributions from G8 to assist in phosphodiester bond cleavage. Previously, replacement of G8 by a series of nucleobase variants showed little effect on interdomain docking, but a 3- to 250-fold effect on catalysis. To identify G8 features that contribute to catalysis within the hairpin ribozyme active site, structures for five base variants were solved by X-ray crystallography in a resolution range between 2.3 to 2.7 Å. For comparison, a native all-RNA “G8” hairpin ribozyme structure was refined to 2.05 Å resolution. The native structure revealed a scissile bond angle (τ) of 158°, which is close to the requisite 180° ‘in-line’ geometry. Mutations G8(inosine), G8(diaminopurine), G8(aminopurine), G8(adenosine) and G8(uridine) folded properly, but exhibited non-ideal scissile bond geometries (τ ranging from 118° to 93°) that paralleled their diminished solution activities. A superposition ensemble of all structures, including a previously described hairpin ribozyme-vanadate complex, indicated the scissile bond can adopt a variety of conformations resulting from perturbation of the chemical environment, and provided a rationale for how the exocyclic amine of nucleobase 8 promotes productive, in-line geometry. Changes at position 8 also caused variations in the A−1 sugar pucker. In this regard, variants A8 and U8 appeared to represent non-productive ground-states in which their 2'-OH groups mimicked the pro-R, non-bridging oxygen of the vanadate transition-state complex. Finally, the results indicated that ordered water molecules bind near the 2'-hydroxyl of A−1, lending support to the hypothesis that solvent may play an important role in the reaction.
Keywords: hairpin ribozyme, RNA enzyme, inosine, aminopurine, diaminopurine, crystal structure, phosphoryl transfer, catalysis
The hairpin ribozyme is a small ribozyme whose family members catalyze a reversible, site-specific phosphodiester bond cleavage reaction within a complementary RNA substrate strand (1, 2). Cleavage proceeds through deprotonation of the 2'-OH group at position A−1 leading to nucleophilic attack on the G+1 phosphorus (Scheme 1). The reaction proceeds via a trigonal bipyramidal intermediate followed by formation of a cyclic-2',3' phosphodiester and a free 5'-OH group as products (3, 4). This pathway plays a central role in the replication of several plant viroids (5) and serves as a model system to understand more complex biological catalysts of RNA origin (6-8).
Scheme 1.
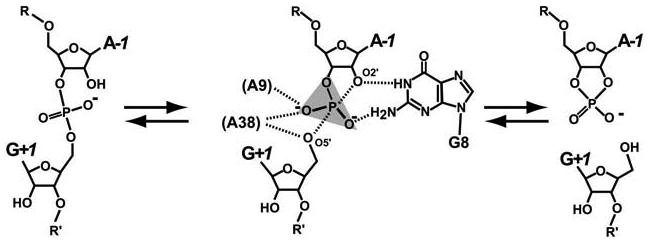
Transesterification Reaction of the Hairpin Ribozyme
At present, complete three-dimensional structures of the hairpin ribozyme motif have been reported by two independent laboratories. The first structures described were those of the naturally occurring 4WJ1 form, which revealed the global RNA architecture, as well as stereochemical details at the active site, including the location of functional groups from G8, A9 and A38 at the scissile bond (9, 10). More recently, the structure of a junctionless, all-RNA variant was reported (11). This minimal construct was derived from studies showing that separate loop A and B domains of the hairpin ribozyme could be reconstituted in solution to form a ‘docked’, catalytically competent enzyme (12, 13). The minute size of this 61-mer (Figure 1A) enabled the facile incorporation of single-atom point alterations into the sequence by use of solid support chemical synthesis. This approach led to the identification of conformational heterogeneity at position U37 that could be eliminated by incorporation of a U39C point mutant (11). Overall, the structure of the minimal all-RNA variant (Figure 1B) exhibited a modest 1.3 Å r.m.s.d. from the equivalent core atoms of the 4WJ-U1A complex, suggesting that two different ribozyme constructs can adopt the same global fold despite the contextual differences of their conserved domains.
FIGURE 1.
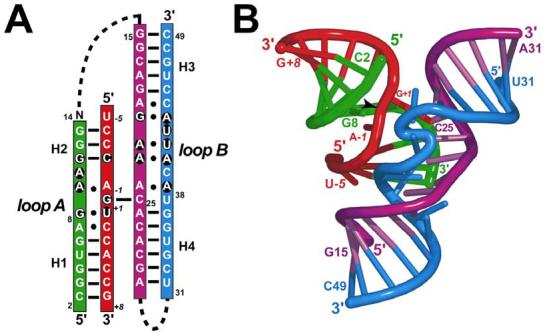
Schematic diagrams of the minimal all-RNA hairpin ribozyme used in this and the previous study (ref. 11). (A) Secondary structure depiction based on the known structure. The substrate strand is red; other ribozyme strands are colored green, purple or blue. This color scheme is preserved throughout the figures. Lines indicate W-C H-bond pairs; filled circles represent non-canonical interactions. Dashed lines indicate covalent connections that were excluded from this construct. The loop A and B domains are labeled with conserved residues on black circles. Helices 1-4 are labeled (H1 to H4). Position 14 was omitted in this study. (B) Ribbon diagram of the refined 2.05 Å resolution G8 native hairpin ribozyme of this study, which is representative of the core fold. The scissile bond is indicated by an arrowhead. Positions A−1, G+1 and G8 are labeled and represent the active site.
Biochemical, structural and kinetic studies of the hairpin ribozyme suggested an important role for G8 in the chemical reaction (Scheme 1). In one study, a series of base variants introduced at position 8 indicated a correlation between N1 imino protonation and cleavage proficiency with G > DAP > I > AP > A > U (Scheme 2) (14); in that study, G8 cleaved at 0.236 min−1 whereas U8 functioned at 0.001 min−1. In addition, each variant was shown to affect a step after interdomain docking, suggesting an effect on catalysis not folding. These results also suggested an important role for the N2 exocylic amine at position 8, which was corroborated by crystallographic observations (10). Further evidence suggesting the importance of G8 was derived from studies of a 4WJ hairpin ribozyme in which G8 was made abasic. This modification resulted in a 350-fold loss in catalytic activity, whereas abasic substitutions at neighboring A9 and A10 positions resulted in <10-fold losses, respectively (15). Subsequent studies suggested that nucleobase rescue of abasic G8 ribozymes was consistent with electrostatic stabilization of the transition state (16). Most recently, the observation that abasic substitution of A38 reduced cleavage and ligation rates by 104 and 105, respectively, (17) demonstrated that the contribution of the G8 base is modest at best. Collectively, these data support the proposal that G8 may be engaged in a more structural role in the reaction, thereby revitalizing the concept that water may serve as a proton acceptor at the 2'-OH of A−1 during the cleavage reaction (10).
Scheme 2.
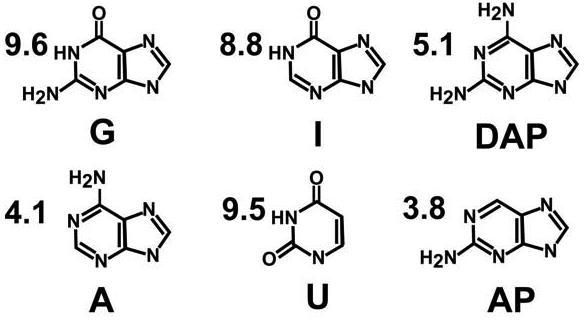
Structures and pKa values of free bases in solution
For efficient phosphoryl transfer to occur during the bond breaking step of the hairpin ribozyme reaction, the apical 2'-OH must be ‘in-line’ with the electrophilic phosphorus and O5'-leaving group (18, 19). Specifically, the O2'•P•O5' angle, τ, must be close to 180° (Scheme 1), which in the case of the hairpin ribozyme is dependent upon the influence of the local RNA environment, since metal ions play only a passive structural role in the chemical reaction (20-22). To address the functional importance of G8, we solved the crystal structure of five hairpin ribozyme variants (Scheme 2) as a means to correlate solution studies with structural information. The variants are analyzed and discussed with regard to their deviation from the necessary catalytic geometry, as established by the hairpin-ribozyme-vanadate complex, which serves as a transition state mimic (23). Additionally, the structure of the native G8 hairpin ribozyme in a ‘pre-catalytic’ state was refined to 2.05 Å resolution, making it possible to identify ordered water molecules in the active site and to assess the possibility that solvent participates directly in the catalytic reaction.
Experimental Procedures
The Minimal All-RNA Hairpin Ribozyme Construct and Nucleotide Modifications
Methods for the design and structural optimization of a 61-nucleotide minimal, junctionless, all-RNA hairpin ribozyme construct were described previously (11). The sequence of the native (G8) hairpin ribozyme of this study is described in Figure 1A. For all structures, position U39 was changed to a cytidine to avoid conformational heterogeneity in the S-turn observed for the native, U39 structure (11). Position 14 was removed to uncouple the loop A and loop B domains, which can be reconstituted (docked) into an active form as described (12). The RNA stem-loop at helix 4 was also removed to promote blunt-ended crystal packing. These modifications made it possible to produce all four hairpin ribozyme strands (Figure 1A) by chemical synthesis and to incorporate desired mutations at any location. This construct will be referred to as the ‘native’ G8 hairpin ribozyme throughout this study. All RNA strands, including mutants, were obtained from Dharmacon, Inc. and subjected to in-house deprotection, HPLC purification and domain-docking as described (11, 24, 25). The mutations in the loop A ribozyme strand (Figure 1A, green) were introduced at position 8 and included: G8I, G8DAP, G8AP, G8A and G8U. Mutations to the substrate strand (Figure 1A, red) were: dA−1 and 2'-OMeA−1, which were shown to inactivate the ribozyme from cleavage without disrupting the tertiary fold (26-28). Under high pH conditions, the G8DAP and G8AP substitutions cleaved slowly (14) and were used in this study at pH > 8.5 in combination with an A−1 ribonucleotide. Due to its light sensitive properties, AP oligonucleotides were purified and handled under light restricted conditions.
Crystallization and X-ray Diffraction Experiments
Crystals of the native G8, all-RNA hairpin ribozyme were identified by screening about conditions established previously for the all-RNA 61-mer (11); crystallization was by the hanging-drop vapor-diffusion method. Superior crystals of the native G8 hairpin ribozyme were obtained from solutions of 21.5% (w/v) PEG 2K MME, 0.25 M Li2SO4, 0.10 M Na-cacodylate pH 6.0, 2 mM spermidine-HCl, and 2.5 mM Co(NH3)6Cl3. Hexagonal prisms grew at 20° C and reached a size of 0.40 mm × 0.40 mm × 0.35 mm over 2 months. Optimization of crystallization conditions was necessary to produce well-diffracting crystals of the G8 variants of this study. Empirically formulated changes to mother liquors included: growth of G8I crystals from 22.5% (w/v) PEG 2K MME; growth of G8I/2'-deoxy A−1 crystals from 24.5% (w/v) PEG 550 MME, 0.10 M Na-HEPES buffer pH 7.2 and 1 mM Co(NH3)6Cl3; growth of G8DAP crystals from 26% (w/v) PEG 2K MME, 0.30 M Li2SO4, 0.10 M TRIS-Cl buffer pH 8.6 and 1 mM Co(NH3)6Cl3; growth of G8AP crystals from 25.5% (w/v) PEG 2K MME, 0.275 M Li2SO4, 0.10 M TRIS-Cl buffer pH 8.8 and 1 mM Co(NH3)6Cl3; growth of G8A crystals from 24% (w/v) PEG 2K MME and 0.275 M Li2SO4; and growth of G8U crystals from 24.3% (w/v) PEG 2K MME, 0.320 M Li2SO4 and 0.5 mM Co(NH3)6Cl3. All crystals grew to > 0.25 mm × 0.25 mm × 0.2 mm in 3 weeks and were harvested shortly afterwards for X-ray diffraction analysis. Crystals were cryoprotected by stepwise transfers of 3 min duration into synthetic mother liquor solutions containing 5%, 10%, 15% or 17.5% (v/v) glycerol. Crystals were then captured in thin rayon loops (Hampton Research) and flash cooled in a stream of cold nitrogen gas at −180° C generated by an X-Stream (Rigaku/MSC). Samples were tested for X-ray diffraction in-house as described (11) and then stored in liquid nitrogen dewars until retrieval for data collection using synchrotron radiation. All diffraction data were recorded on a Quantum210 CCD (ADSC) at the Cornell High Energy Synchrotron Source (CHESS) station A1; see ref. (11) for details. Data sets were reduced with Crystal Clear software (Rigaku-MSC) (29). Intensity and data reduction statistics are provided in Table 1.
Table 1.
X-ray data collection and refinement statistics
| Sample Name | G8, 2'-OMe A−1 | G8I, 2'-OMe A−1 | G8I, 2'-deoxy A−1 | G8DAP, 2'-OH A−1 | G8AP, 2'-OH A−1 | G8A, 2'-OH A−1 | G8U, 2'-OH A−1 |
|---|---|---|---|---|---|---|---|
| Unit Cell (Å) | a = 93.3, c = 131.3 | a = 93.4, c = 131.5 | a = 94.5, c = 127.6 | a = 93.8, c = 127.6 | a = 93.9, c = 129.2 | a = 93.5, c = 124.6 | a = 93.4, c = 123.6 |
| Resolution (Å) | 2.05 to 30 | 2.33 to 47 | 2.40 to 44 | 2.40 to 29 | 2.70 to 38 | 2.40 to 44 | 2.38 to 47 |
| Total reflections | 178,418 | 122,779 | 110,424 | 128,453 | 87,945 | 155,415 | 116,261 |
| Unique reflections | 21,669 | 15,007 | 13,689 | 12,161 | 9,763 | 13,112 | 13,252 |
| Redundancya | 8.2 (4.7) | 8.2 (7.0) | 6.3 (5.9) | 10.6 (9.3) | 6.9 (8.4) | 11.9 (11.0) | 8.7 (8.5) |
| Completeness (%)a | 99.3 (99.1) | 99.6 (99.9) | 99.2 (98.7) | 89.8 (92.8) | 97.9 (99.9) | 99.8 (100) | 99.2 (99.9) |
| <I / σ(I) >a | 24.0 (3.3) | 26.2 (4.2) | 19.1 (4.3) | 34.8 (5.1) | 14.7 (4.0) | 27.4 (4.9) | 29.9 (5.2) |
| Rsym (%)a,b | 3.0 (43.2) | 4.0 (44.5) | 4.3 (43.0) | 3.4 (45.8) | 5.8 (45.7) | 4.5 (42.8) | 3.6 (42.9) |
| Num. RNA Atoms | 1312 | 1311 | 1309 | 1311 | 1310 | 1310 | 1308 |
| Num. Waters | 84 | 33 | 5 | 7 | 11 | 34 | 21 |
| Num. Ions | 2 Co(NH3)6(III), 1 SO42− |
2 Co(NH3)6(III), 1 SO42− |
2 Co(NH3)6(III), 1 SO42− |
1 Co(NH3)6(III) | 1 Co(NH3)6(III) | 1 Co(NH3)6(III), 1 SO42− |
2 Co(NH3)6(III), 1 SO42− |
| avg. B RNA/Wat (Å2) | 69.5/74.0 | 66.5/65.5 | 76.7/73.9 | 85.0/77.5 | 81.3/73.7 | 75.0/67.6 | 69.6/73.9 |
| Rcryst/Rfree (%)c,d | 25.4/26.9 | 22.7/24.2 | 24.0/27.0 | 23.9/26.0 | 24.5/26.8 | 24.3/25.3 | 23.7/25.7 |
| r.m.s.d Bonds (Å) | 0.006 | 0.006 | 0.009 | 0.009 | 0.009 | 0.006 | 0.006 |
| r.m.s.d Angles (°) | 1.40 | 1.40 | 1.50 | 1.72 | 1.69 | 1.50 | 1.30 |
| coord. error (Å)e | 0.40 | 0.43 | 0.47 | 0.56 | 0.53 | 0.49 | 0.47 |
Highest resolution shells are: 2.05-2.12 Å, 2.33-2.41 Å, 2.40-2.48 Å, 2.40-2.49 Å, 2.70-2.80 Å, 2.40-2.49 Å and 2.38-2.47 Å for statistics noted parenthetically (left to right).
Rsym = Σ|Ij - <Ij>|/ Σ |Ij| × 100
Rcryst = Σhkl|Fo − kFc|/Σhkl|Fo| × 100, where k is a scale factor.
Rfree is defined as the Rcryst calculated using 6 to 10% of the data selected randomly and excluded from refinement.
Based upon cross-validated Luzzati plots calculated in CNS for the full-resolution range.
Structure Determination and Refinement
All mutants of the hairpin ribozyme crystallized in space group P6122 with a single 61-mer molecule per asymmetric unit. The initial native G8 structure was solved by rigid body refinement at 3.0 Å resolution as implemented in CNS (30) using initial coordinates from the all-RNA hairpin ribozyme 61-mer refined previously to 2.19 Å resolution (PDB entry 1X9C). Rigid body refinement of the entire structure was conducted first, followed by independent treatment of the respective loop A and B domains. The ensuing refinement was conducted to 2.05 Å resolution using cycles of positional minimization and individual atomic B-factor refinement. Manual adjustments were performed using the interactive graphics program O (31). Upon completion of refinement, these coordinates were used to solve all ensuing structures whose base compositions are depicted in Scheme 2. This method was successful, even though the unit cell dimensions of some mutants differed by as much as 7.7 Å along the c-axis (Table 1). The locations of position 8 variants were verified by use of simulated annealing omit maps that were employed to reduce model bias (Figure 2). All structures exhibited an alternate conformation for position U−5 (Figure 1A) that engages in a (designed) dyad related crystal packing interaction that forms a U•U pair. One or two Co(NH3)63+ ions were observed consistently in all structures (Table 1). These ions at G12/G13 (site I) and G21/A40 (site II) were documented previously (11). In cases where only a single ion was observed, it was located at site II.
FIGURE 2.
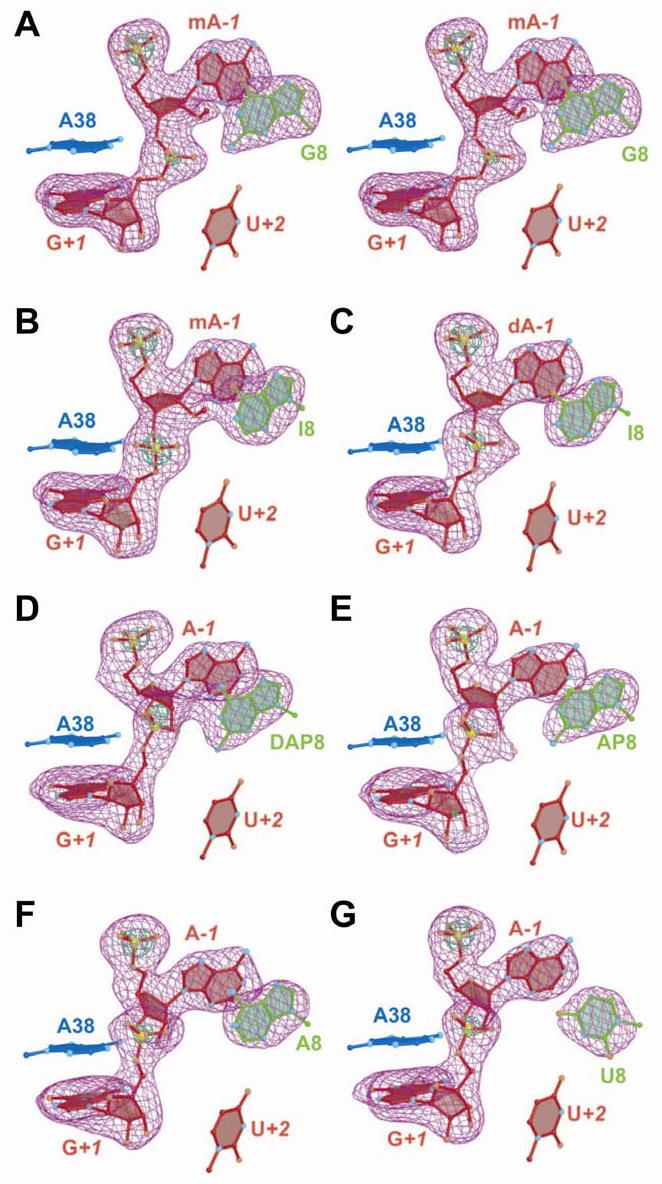
Representative simulated annealing omit electron density maps for the scissile bond and position 8 of the hairpin ribozyme. Maps have σA coefficients of type (m|Fo| - D|Fc|). Nucleotides A−1 and G+1, as well as the base moiety at position 8, were removed from the phase calculation. The orientation is rotated 180o about the z-axis compared to Figure 1B. Bases of A38 and U+2 are shown for perspective. Electron density is contoured at the 3σ (purple) and 9σ levels (green) unless specified otherwise. (A) Stereo diagram of the native structure at 2.05 Å resolution. The label mA−1 indicates a 2'-OMe group at this position. The green contour level is 10σ. (B) The G8I structure at 2.33 Å resolution. The green contour level is 10.5 σ. (C) The G8I/2'-deoxy A−1 structure at 2.40 Å resolution with a 2'-deoxy substitution at position A−1. (D). The G8DAP structure at 2.40 Å resolution. The A−1 position is a ribonucleotide. The green contour level is 6.0 σ. (E) The G8AP structure at 2.70 Å resolution. The green contour level is 7.0 σ. (F) The G8A structure at 2.40 Å resolution. (G) The G8U structure at 2.38 Å resolution The green contour level is 10.0 σ.
Assignment of Waters and Analysis Methods
Atoms representing waters were added manually to each structure. A solvent molecule was assigned if it exhibited electron density in σA weighted (reduced bias) electron density maps (32) with coefficients (2Fo-Fc) and (Fo-Fc), at levels ≥1σ and ≥3σ, respectively. Refined solvent molecules met the criteria that they were between 2.6 to 3.5 Å from a suitable H-bond donor or acceptor group with atomic temperature factors representative of those of their neighboring RNA or solvent partners. In the native structure, the average RNA B-factor was 69 Å2 (on a scale of 1 to 400 Å2), whereas that of water was 74 Å2 (Table 1). The possibility that ions were present in the active site in lieu of water was possible, but unlikely because the crystallization medium comprised 0.5 M Li+ (which is electron deficient) with a SO42− counter ion. Sulfate was assigned in the structures (Table 1), but it is chemically and sterically unsuited to occupy the solvent peaks observed in the active site (Figure 3A). Although 1.0 to 2.5 mM Co(NH3)6Cl3 was present, this ion complex was excluded as a possible source for the solvent electron density at the active site because it is too electron dense, exhibits a large van der Waals radius and its ligands are relatively inert to exchange (33). The locations of 1-2 Co(NH3)63+ sites were assigned for each hairpin ribozyme structure of this study (Table 1 and ref. (11)), and these sites were inconsistent with the size, shape and coordination geometry observed in the active site; no anomalous diffraction signal for cobalt was detected in the active site. Efforts to replace the observed active site solvent peaks by co-crystallization or crystal soaking in salts including K+ or NH4+ were unsuccessful (data not shown). Sodium is present in the mother liquors at 0.10 M levels. Although Na+ would be isoelectronic with water in electron density maps (10 e-), it prefers a trigonal bipyramidal coordination geometry with ligand distances of 2.4 Å (34). Na+ also prefers negatively charged functional groups, such as oxygen, but not the exocyclic amine interactions observed here (Figure 3A). Furthermore, the assigned waters exhibited nearly tetrahedral coordination geometry with no more than 2 H-bond donors and/or 2 acceptors, which is a characteristic of water. Whereas waters frequently engage in H-Bond networks (Figure 3A), ions of similar polarity do not form close contacts with each other as observed here. Notably, the same active site waters were observed in the previous allRNA hairpin ribozyme structure refined to 2.19 Å resolution (11). In both studies, the temperature factors of active site waters matched those of neighboring RNA ligand atoms, which were 59.7 Å2 versus 56.7 Å2, respectively, for the current study. These values were somewhat lower compared to the same RNA atoms of the 4WJ-U1A complex, which were 73.9 Å2 on average (for both molecules in the asymmetric unit) in the pre-catalytic complex (10). Although waters were assigned in the crystal structure of the G8I and G8U mutants, which were the highest resolution structures after G8 (Table 1), no comparable solvent molecules were detectable in their active sites. This may have been caused by lower resolution, as well as greater apparent flexibility of the scissile bond in these structures, which had fewer H-bonds to the G+1 phosphate. Waters in the active site of the G8 native structure were corroborated by simulated annealing omit maps as implemented in CNS (Figure 3A). Superpositions for structures were performed for all matching atoms using LSQKAB as implemented in CCP4 (35). Superpositions with the 4WJ hairpin ribozyme-vanadate complex were performed using chains D and E of PDB entry 1M5O. Statistics for the refined structures are reported in Table 1. Figures 2 through 6B were made by use of Bobscript (36). Figure 1B was produced with Pymol (37) and NUCCYL (38).
FIGURE 3.
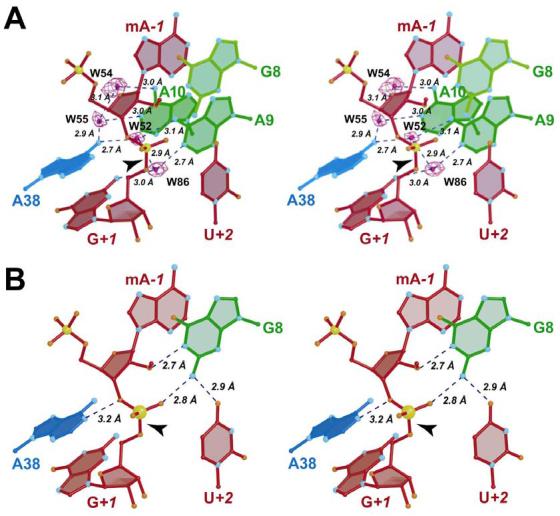
Stereo view of ball-and-stick diagrams of the hairpin ribozyme active site for the G8 native structure. Putative H-bond interaction distances are depicted with broken blue lines. (A) Schematic diagram of the native active site depicting solvent atoms (W) in contact with bases G+1, A9, A10 and A38. A simulated annealing omit electron density map with coefficients (m|Fo| - D|Fc|) is shown in which the four waters were eliminated from the phase calculation. The map is contoured at the 6σ level. (B) Schematic view of the native G8 structure with contacts to the scissile bond from neighboring residues. These coordinates provide the reference orientation for all other superpositions of this study except where noted.
FIGURE 6.
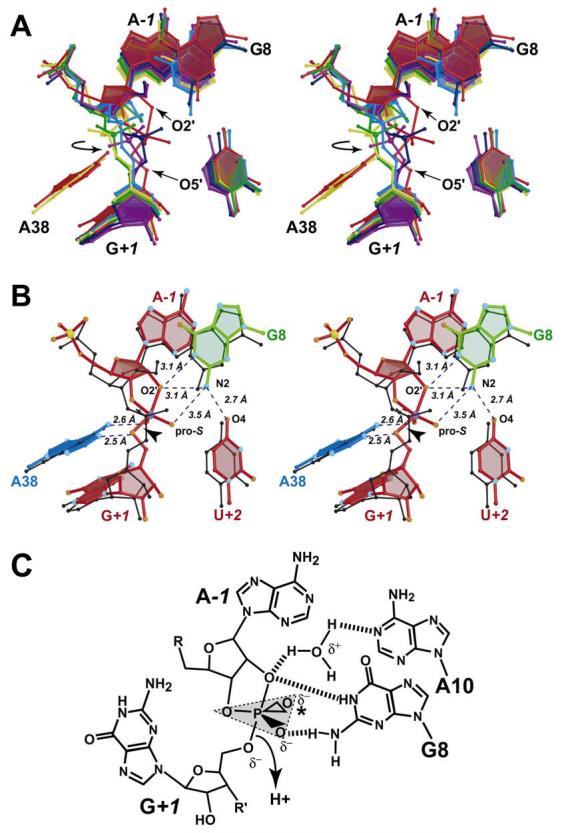
Superposition ensemble of structures representing the native G8 structure, the four purine variants at position 8, and the 4WJ hairpin ribozyme-vanadate complex (10), as well as a proposed role for water. (A) Stereo diagram for the least squares superposition ensemble The structures are: G8A (yellow), G8AP (green), G8DAP (blue), G8I (purple), G8 native (dark blue) and the hairpin ribozyme-vanadate complex (red). A circular arrow indicates the dihedral angle rotation required to produce a more in-line angle. (B) Stereo diagram showing a superposition between the hairpin ribozyme-vanadate complex (ball-and-stick model) with the native G8 structure (black). Atoms O4 of U+2, N2, of G8, O2' of A−1 and the pro-S oxygen of G+1 are labeled for clarity. (C) Schematic diagram indicating a proposed role for water in the cleavage reaction Dashed lines indicate possible H-bonds. The asterisk indicates the location of the pro-R oxygen, which coordinates to the exocyclic amines of A9 and A38 in the transition-state.
Results
Quality of Native Hairpin Ribozyme and Structures of Position 8 Variants
Previous studies described the overall fold of the hairpin ribozyme in the form of a 4WJ-U1A complex (9, 10, 23), as well as a minimal, junctionless all-RNA construct (11). Structures of the hairpin ribozyme in this study represent the minimal all-RNA form, and each is reminiscent of previous crystallographic results in the sense that no major conformational differences arose in the core fold (Figure 1B). As a basis for comparison in the current study, the resolution of the native G8 hairpin ribozyme was extended to 2.05 Å (Table 1). This structure, as well as the accompanying G8-variants, exhibited reasonable refinement statistics including Rcryst at or below 25%, with Rfree not more than 3% greater than Rcryst (Table 1). To further corroborate that the structural changes at G8 and the scissile bond were unbiased by model coordinates, σA weighted simulated annealing omit electron density maps (32) were calculated (Figure 2). At 2.05 Å resolution (Figure 2A), the sugar pucker and orientation of the 2'-OMe moiety at A−1 in the native G8 structure are somewhat better defined than previous studies (9-11). As such, these coordinates provided a model of high stereochemical quality that served as a guide during the model building and refinement of lower resolution molecules (Table 1). Indeed, the resolution of diffraction for the G8-variants ranged from 2.33 to 2.40 Å with the exception of G8AP, which diffracted to 2.7 Å resolution (Table 1). Overall, all RNA atoms for each structure were well-ordered in the electron density, although the quality of maps was noticeably poorer for nucleotides of H1 (Figure 1A). The positions of solvent atoms were not always consistent among structures and water molecules were added independently to each model.
Fit of Models to Electron Density Maps
At 2.4 Å resolution or better, four aspects of the electron density maps were utilized to facilitate interpretation of how each active site modification influenced the scissile bond geometry. These features included: (i) definition of the base and exocyclic groups at position 8; (ii) identification of exo versus endo sugar puckers for ribose; (iii) differentiation among 2'-modifications at position A−1, (i.e. 2'-deoxy, 2'-hydroxyl or 2'-OMe groups); and (iv) localization of the phosphorus atom of G+1 based on its electron density. In the first category, the electron density for each cleavage site and base at position 8 was well-defined (Figure 2). In the second category, the atoms at the 2'-position of A−1 were readily discernable (refer to the Supporting Information Figure 1). For example, compare the sugar at A−1 representing 2'-OMe modifications (Figures 2A & 2B) to that of the 2'-deoxy (Figure 2C) or 2'-hydroxyl modifications (Figure 2F & 2G). Similarly, there is a tangible effect resulting from removal of a base functional group, such as the N2 exocyclic amine of guanosine (i.e. inosine, Figure 2B) or the O6 group of guanosine (i.e. AP, Figure 2E). Lastly, the phosphorus atom of G+1 could be localized by contouring maps to higher signal-to-noise levels (Figure 2, green peaks). In general, the electron density was well defined for the active sites of all structures, and the orientation of the pro-R and pro-S oxygens at G+1 could be assigned based on the complementary fit of these atoms to the asymmetric electron density of this region (Figure 2 and Supporting Information Figure 1).
The Active Site of the Hairpin Ribozyme is Wet
A noticeable difference between the native G8 hairpin ribozyme of this study and that of previous studies of the 4WJ-U1A complex (9, 10) was the presence of ordered water molecules in the active site in the current study (Figure 3A). Four water molecules form a distinctive H-bond network to the W-C faces of A9 and A10. Each of these tandem adenine bases contacts a water at both its N6 amino and N1 imino positions (Figure 3A). Specifically, W86 mediates an interstrand contact between the imino group of A9 and the N2 exocyclic amine of G+1. Similarly, W52 mediates an interdomain contact between the exocyclic amines of A9 and A38. A comparable interdomain contact is mediated by W55, which is positioned between the imino moiety of A10 and the anti side of the A38 exocyclic amine (Figure 3A). W54 mediates a 3.0 Å contact between the O4' atom of A−1 and W55 (not shown), and directly contacts both W55 and the exocyclic amine of A10 (Figure 3A). Distances for the water-RNA interactions ranged from 2.7 to 3.1 Å, consistent with previous studies of small ribozymes (39). The average trigonal angle comprising solvent atoms and RNA functional groups was 113o, which was close to ideal tetrahedral values. No large conformational differences were present in the active site compared to previous hairpin ribozyme structures, indicating that a molecular “breathing” phenomenon was not required to accommodate the observed waters (9). It is also conceivable that the waters were sequestered during interdomain docking. However, the G8 native structure reveals a pathway for solvent entry into, or egress from, the active site at a corridor located between the ribose of A40 and the base of A38. This suggests the ability of active site water molecules to exchange with bulk solvent. A plausible explanation for the observation of the waters in the current study is that the resolution of the native structure is higher, and the overall B-factors are lower than reported previously for the best 4WJ-U1A complex at 2.2 Å resolution (10). In addition, the asymmetric unit is substantially smaller in the current study, comprising ∼1400 atoms (Table 1) versus ∼6300 atoms (10). This is significant since a smaller asymmetric unit leads to greater diffraction sampling for crystals of the same size (40).
Details of the G8 Native Active Site Structure with a 2'-OMe Modification at A−1
Crystal structures of the G8 native hairpin ribozyme have been reported with a 2'-OMe modification at A−1 that inhibits catalysis (9-11). In each case, the value of τ was observed to be nearly in-line, ranging from 160 to 164o, and these structure have been dubbed a ‘pre-catalytic’ state (23). In this study, τ = 158o (Table 2) and most of the interactions observed previously between the neighboring RNA and the scissile bond have been preserved. For example, the N1 atom of G8 is within 2.7 Å of the 2'-oxygen of A−1 (Figure 3B). This distance implies an H-bond between these atoms (23), which is reasonable given that the structure of the present study was determined from 100 mM Na-cacodylate buffered at pH 6.0 and that the solution pKa of guanine is 9.6 (Scheme 2). Protonation at the N1 position of G8 is inconsistent with the role of G8 as a general base catalyst since the guanosine would be negatively charged leading to repulsion between the N1 and 2'-OMe positions. A comparable analysis of base A38 reveals that its N1 protonation state cannot be surmised readily from pre-catalytic structures. The distance observed here between N1 of A38 and the 3'-oxygen of A−1 was 3.2 Å (Figure 3B), but the average distance for this interaction in comparable 4WJ-U1A complexes was 3.7 Å (10), making the assignment of an H-bond somewhat ambiguous. One explanation for the disparity in distances at this position was that mother liquors used to grow crystals of 4WJ-U1A complexes did not contain buffer (9, 10, 41), making it difficult to correlate the pH values of respective crystal forms to changes associated with specific functional groups. The previous structural study of the 4WJ-U1A complex does suggest that the imino group of A38 may be protonated in the transition state due to its 2.7 Å distance from the O5' leaving group in the hairpin ribozyme vanadate complex (10, 23). This observation provided support for the idea that the N1 group of A38 exhibited an exceptionally elevated pKa relative to the free base in solution (Scheme 2), possibly consistent with a role for A38 as a general acid catalyst in cleavage (8, 17).
Table 2.
Relative differences in the A−1:G+1 linkage due to active site modifications
| 2'-group at A−1 |
G8 Base |
τ (°) | 2'-O-to-P (Å)a |
rmsd vanadate (Å)b |
Position 8 contacts to the scissile bond (Å)c |
|---|---|---|---|---|---|
| 2'-OMe | G | 158 | 2.9 | 1.5 (−) | N1/2'-O (2.7); N2/pro-R (2.8) |
| 2'-O-vanadate | G | 144 | 2.0 | − (1.5) | N1/2'-O (3.1); N2/pro-S (3.5); N2/2'-O (3.1) |
| 2'-OMe | I | 118 | 3.3 | 2.3 (1.0) | N1/2'-O (2.8) |
| 2'-deoxy | I | - | - | 2.6 (1.3) | none |
| 2'-OH | DAP | 98 | 3.1 | 2.9 (1.9) | N2/2'-O (3.3) |
| 2'-OH | AP | 112 | 3.2 | 3.0 (2.0) | N2/2'-O (3.4) |
| 2'-OH | A | 112 | 3.9 | 3.3 (2.3) | none |
| 2'-OH | U | 93 | 3.8 | 3.1 (2.1) | none |
This value represents the distance between the 2'-O-nucleophile and the phosphorus of G+1.
This value indicates the deviation of the G+1 phosphorus of a given structure from the position of vanadium in the 4WJ-U1A-vanadate complex based on minimization of all core atoms. The value in parentheses is the rmsd relative to the G+1 phosphorus of the native G8 structure of this study.
Other conserved active site interactions include an apparent 2.9 Å H-bond between the exocyclic amine of G8 and the O4 keto group of U+2, a conserved core residue (Figure 1A) (42). The O4 atom helps align the G8 exocyclic amine such that it contacts the pro-R oxygen of G+1 (Figure 3B). The fact that a trigonal angle of 100o can be measured for the O4•N2•pro-R oxygen interaction suggests that any lateral movement of the G8 base about the N-glycosidic linkage would break the planarity of this interaction, thereby disrupting this H-bond network. Interaction of the pro-R oxygen and the G8 exocyclic amine appears to be critical for promoting the in-line geometry in the docked state (9). The structural consequences of eliminating this functional group were explored with the various base modifications, such as G8I.
The G8I Modification with a 2'-OMe Group at A−1
The base substitution of G with I at position 8 was designed to reveal structural effects caused by the elimination of the 2 amino group (Scheme 2). Like G8 native crystals, those of the G8I variant were grown at pH 6, which should produce a protonated imino group. This expectation is consistent with the 2.8 Å distance between the 2'-oxygen of A−1 and the N1 atom of I8 (Figure 4A). In contrast, absence of the I8 exocyclic amine resulted in loss of an H-bond to both the pro-R oxygen of G+1 and the O4 keto group of U+2. Consequently, the purine base of I8 shifts toward the vacant N2 pocket causing a slight base unstacking from A−1 (Figure 4A). Most noticeably, the scissile bond (τ = 118o) rotates away from I8, displacing the G+1 phosphorus atom by a distance of 1 Å relative to the native structure (Figure 4A).
FIGURE 4.
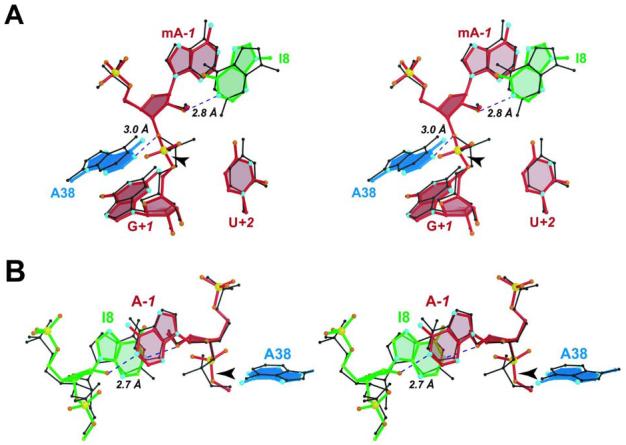
Stereographic views of the G8I structure. (A) Schematic view of the G8I structure (ball-and-stick) superimposed upon the native G8 structure (black ball-and-stick model). (B) Reverse orientation of the G8I structure superimposed upon the G8 native structure showing a new interaction between the 2'-OH of I8 and the imino group of A−1. This view is rotated 180o about the y-axis relative to Figure 4A. The ribose and base of G+1 have been omitted for clarity.
Remarkably, very little of the scissile bond geometry of the I8 structure appeared to be the result of H-bonds donated from inosine. In fact, removal of the sole H-bond between A−1 and the I8 base was accomplished by substituting the 2'-OMe modification at A−1 with dA (Supporting Information Figure 2). This change appeared to have almost no effect on the conformation of the scissile bond, the sugar pucker at A−1, or the position of the I8 base. Notably, all three structures (G8, G8I and G8I/2'-deoxy A−1) maintained a very similar ∼3 Å distance between the N1 atom of A38 and O3' atom of A−1 (Figures 3B & 4A and Supporting Information Figure 2), which appeared to support protonation of the A38 imino group as observed for the vanadium transition-state mimic (10, 23).
G8I Promotes a Cross-Strand Loop A Domain Interaction
Substitution of the 2'-OMe moiety at A−1 with a dA caused little change between structures of the I8 variant (Supporting Information Figure 2). However, a notable feature associated only with the G8I substitutions was formation of a new ∼2.7 Å H-bond between the N1 atom of A−1 and the 2'-OH of I8 (Figure 4B). It is feasible that this interaction occurs at least transiently in the wild-type ribozyme, although it was not observed in other hairpin ribozyme native structures or within other G8 base variants of this study. This interaction involves a distinct change in the sugar pucker at position 8 from a twisted C3'-endo to a C2'-endo conformation (43), as compared to the G8 native structure (Figure 4B, green bonds versus black stick model). This change in furanose pucker occurred regardless of 2'-OMe versus 2'-deoxy substitutions at A−1 (data not shown). Interestingly, removal of the 2'-OH from G8 of the native structure was reported to produce a ΔΔG of +0.7 kcal/mol, corresponding to a weak NAIM effect that primarily effected the KM for the reaction (44). These observations suggest the loss of a single H-bond that could be explained if the cross-strand interaction between I8 and A−1 represents the actual native hairpin ribozyme conformation (Figure 4B). In the native structures, the 2'-OH group of G8 does not H-bond strongly to any functional groups. The closest potential H-bond partner is the O4' oxygen of residue A9, but this group is ∼3.6 Å away in the native and variant structures (on average). At present, it is unclear why the native G8 structure does not corroborate the solution data, although a previous structural study of the hairpin ribozyme supported the possibility that the S-turn motif in the crystal structure represented a folding intermediate (11). The G8I substitution itself was reported previously to cause a +2.2 kcal/mol increase in ΔΔG of the hairpin ribozyme during catalysis, as well as a ‘strong’ effect by NAIM (45, 46). Such a free energy difference is equivalent to the loss of ∼4 H-bonds (47) with a 50-fold effect on kcat (45). Although loss of 2 H-bonds can be accounted for by this study, the additional destabilization is likely attributable to more complex (enthalpic) interactions that are not readily discernable in this study.
pH Dependent Cleavage Effects of AP8 and DAP8 Variants
Well-diffracting crystals of the G8AP and G8DAP variants with a 2'-OMe A−1 could not be prepared for structural studies. Therefore, a strategy was adopted to prepare these variants with unprotected 2'-OH groups at position A−1 under conditions of reduced activity. Reduction of the catalytic turnover rates during crystallization was accomplished by growing the AP8 and DAP8 crystals at pH 8.8 and 8.6, respectively, since previous studies indicated these variants displayed bell-shaped pH rate profiles centered at 6.5 and 6.9. In contrast, the native G8 hairpin ribozyme exhibited an ascending rate profile between pH 4 and neutral, followed by a flat slope through pH 9 (14). At the high pH values used for crystallization, the AP8 and DAP8 variants are approximately 15-fold slower than their optimal rates, which are still 20- and 10-fold diminished, respectively, compared to the activity of the native G8 enzyme. To verify that the electron density of crystals was not an equilibrium mixture of substrate and product, the composition of crystals was assessed by analytical HPLC methods. The results revealed that AP8 and DAP8 crystals prepared at high pH did not exhibit appreciable levels of cleaved product (Supporting Information Figure 3); AP8 showed ∼6% product formation, whereas DAP8 showed ∼15% product formation. Products were not detected in electron density maps. Analysis of DAP8 crystals that were several months old indicated both product strands were present with little substrate, consistent with cleavage in the crystal lattice (Supporting Information Figure 3G). Overall, the results provided confidence that modeled substrate structures were not biased significantly by products and that the hairpin ribozymes of this study are active in the crystal lattice at elevated pH.
The G8DAP Variant with a 2'-OH at A−1
The DAP8 variant was reported to be nearly 10-fold more reactive in cleavage than G8I in solution (14). However, an inspection of the DAP8 scissile bond geometry (Figure 5A) revealed τ = 98o (Table 2). This deviation from in-line geometry was a likely result of imino deprotonation at DAP8. At present, a systematic structural study of the hairpin ribozyme has not been conducted as a function of pH. However, both G8 and A38 abasic variants shifted pH rate profiles, suggesting that each possesses titratable functional groups that affect a chemical step or a rate limiting conformational change (14, 17). In this study, it is probable that the structures of both DAP8 and AP8 deviate from the in-line geometry due to a pH dependent conformational change, rather than replacement of the G8 base itself. Nonetheless, a comparison to the native structure still provides insight into how their respective functional groups contribute to the active site geometry, and how pH changes alter the hairpin ribozyme structure.
FIGURE 5.
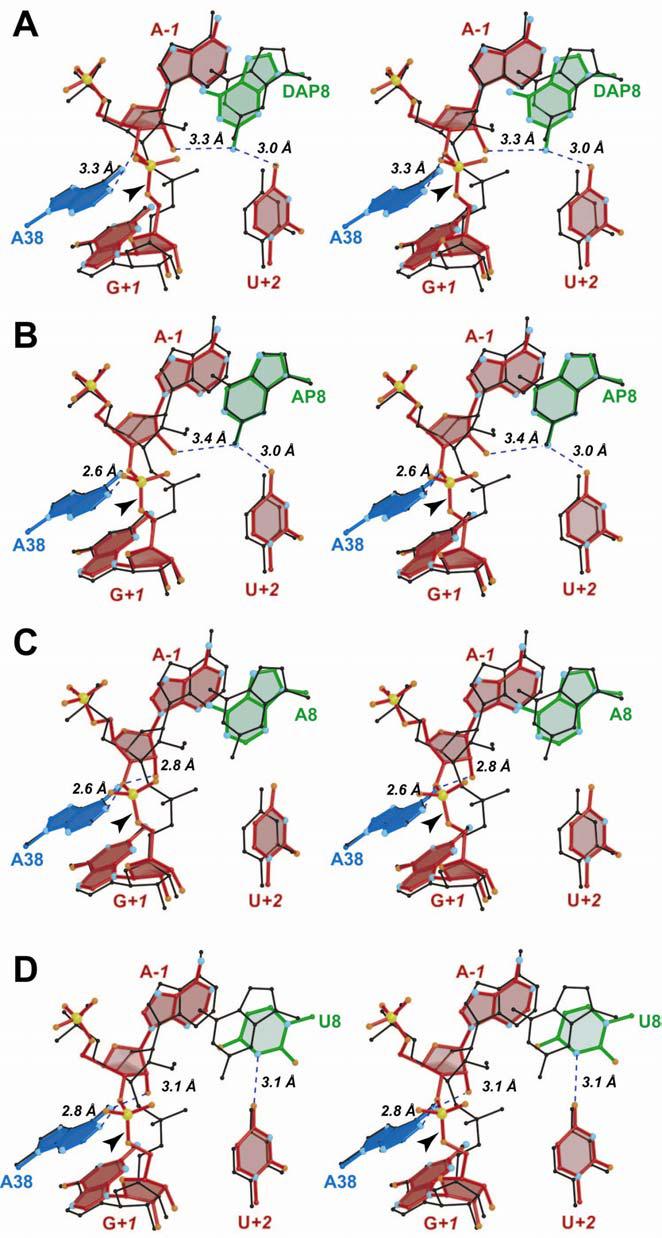
Stereographic pairwise superpositions of the respective DAP, AP, A8 and U8 ball-and-stick models with the native G8 hairpin ribozyme (black). (A) DAP8 and the native G8 structure. (B) AP8 and the native G8 structure. (C) A8 and the native G8 structure. (D) U8 and the native G8 structure.
Although an H-bond is maintained between the N2 exocyclic amine of DAP8 and the O4 keto group of U+2, the long 4.1 Å distance between the N2 amine of DAP8 and the pro-R oxygen of G+1 precludes a direct H-bond interaction (48), as compared to the native structure (Figure 5A versus 3B). However, the N2 exocyclic amine of DAP8 is within 3.3 Å of the 2'-OH group of A−1. This interaction is absent in the native, pre-catalytic state since the 2'-position of A−1 is protected by a methyl group. Removal of the 2'-OMe group at A−1 in the DAP8 structure yielded an altered sugar pucker with a twisted C3'-endo/C2'-exo conformation (43). The H-bond between the A−1 2'-oxygen and the N2 exocyclic amine of DAP8 appears to restrict the ability of the 2'-OH at A−1 to serve as a proton donor to the unprotonated N1 atom of DAP8. The latter interaction would be sterically and electrostatically unfavorable because it would require close contact (2 Å) between the hydrogen of the A−1 2'-hydroxyl group and that of the syn hydrogen on the N2 amine at position 8 (based on modeling of the hydrogens). To avoid this clash, it is reasonable that the hydrogen of the A−1 2'-hydroxyl group would pivot about the C2'-O2'-bond while the N2 amine remains stationary; this configuration seems plausible since the exocyclic amine cannot rotate, due to its partial resonance character associated with the purine base. In this manner, the hydrogen of the A−1 2'-OH group would project into the solvent exposed pocket adjacent to A10 (Figure 3A). This orientation has implications for the geometry needed for proton transfer during cleavage and suggests an important role for the N2 group of position 8, beyond stabilization of the transition state. Comparable observations were made for the G8AP variant.
The G8AP Variant
The active site structure of the G8AP modification prepared at pH 8.8 is most similar to that of G8DAP. The AP8 variant exhibited a τ value of 112°, which is somewhat more in-line at the scissile bond than DAP8, but less than ideal compared to the G8 native structure (Table 2 & Figure 5B). The AP8 variant revealed similar H-bond patterns compared to DAP8, including a 3.0 Å distance between the N2 exocyclic amine and the O4 keto group of U+2 (Figure 5B). Likewise, the N2 group of AP8 was 3.4 Å from the 2'-OH of A−1, but the distance between N1 of AP8 and the 2'-OH of A−1 was 3.7 Å. The latter distance suggests the absence of an H-bond donated by the 2'-OH of A−1 to the imino group of position 8, as deduced for DAP8. Hence, the N1 group of AP8 appears to be deprotonated in the crystal structure, in accord with its pH rate profile measured in solution (14).
Previously, the AP8 base variant proved to be a useful spectroscopic probe of the hairpin ribozyme active site. A fluorescence analysis indicated a pH dependent transition from 5.0 to 5.7 under conditions that promoted the undocked versus docked complex, respectively. This folding transition suggested that apposition of the docked loop A and B domains influences the pKa of the base at position 8 (14). These studies were conducted in the presence of a 2'-deoxy A−1 modification and reported a pKa for AP8 nearly one log unit lower than that determined from the pH rate dependence of cleavage. These data may be interpreted in the context of the current structure as being consistent with a pH-dependent conformational change at the A−1 ribose. Specifically, at lower pH values where the imino group of position 8 is protonated, a direct H-bond forms to the 2'-oxygen of A−1 (e.g. as in Figures 3B & 4A). This interaction, which occurs upon docking, influences the imino pKa value at position 8, elevating it towards neutral as observed for DAP8 and AP8 (14). At higher, non-reactive pH levels, the imino groups of these bases are deprotonated, which appears to be associated with a flatter sugar pucker at A−1 compared to the native G8 conformation (Figures 5A & 5B). Overall, changes in the active site resulting from N1 deprotonation at position 8 in this study help account for the structural transitions measured by fluorescence, as well as the sharp decline in activity in the pH rate profile following loss of AP8 (or DAP8) imino protons.
A8 and U8 variants with a 2'-OH at A−1
Structures of G8A and G8U prepared at pH 6.0 represent the least active conformations of the hairpin ribozyme (Table 2). Their respective activities in solution were approximately 100- and 250-fold lower than then the native G8 ribozyme (14). Inspection of their scissile bonds revealed τ values of 112° and 93°, respectively. A comparison of ribose puckers among position 8 variants with unprotected 2'-OH groups at A−1 revealed that the U8 and DAP8 structures exhibited 2'-OH groups in a twisted exo conformation (Figures 5D & 5A), whereas those of A8 and AP8 where somewhat flatter (Figures 5C & 5B). This comparison shows that at least two classes of sugar puckers can be accommodated when the 2'-OH at A−1 is unprotected. Interestingly, neither the A8 nor the U8 structure exhibited contacts to the scissile bond from the base at position 8 (Figures 5C & 5D). These structures lack a suitable N2 exocylic amine, which contacts the 2'-OH of A−1 in the DAP8 and AP8 structures (Figures 5A & 5B). The absence of H-bond contacts from position 8 to the cleavage site was a unique feature of the A8 and U8 structures compared to other position 8 variants (Figures 3B, 4A, 5A & 5B). In addition, the A8 and U8 structures were the only ones to exhibit close contacts between their 2'-OH groups at A−1 and the N6 exocyclic amine of A38 (Figures 5C & 5D). This putative H-bond, along with the presumed interaction between the N1 position of A38 and the O3' atom of A−1 (Figures 5C & 5D), appears to stabilize substrate binding in a novel way. Significantly, the displacement of the 2'-oxygens of A−1 at A8/U8, relative to those of the G8/I8 and DAP8/AP8 structures, was 1.6 Å and 3.8 Å, respectively. Once again, these differences emphasize the important role of the exocyclic amine at position 8 as a means to influence the A−1 sugar pucker and possibly prevent formation of an overly stable ground state complex (Figures 5C & 5D).
The A8 and U8 variants were the least reactive in solution assays and their structures suggest they represent non-catalytic states. Although protonation of the N1 imino group at position 8 appears important as a means to orient the 2'-nucleophile in the native, pre-catalytic structure (Figure 3B), the low pKa of A8 (Scheme 2) and pyrimidine base orientation of U8 (Figure 5D) appear to preclude this function. Nonetheless, both variants cleaved substrate in solution assays, corroborating the observation that an abasic G8 hairpin ribozyme could still function, albeit at a 350-fold reduced rate (15, 16). Differences between the A8 and U8 structures include the observation that the O4 keto of U8 occupies the same spatial location as N1 of G8 (Figure 5D), and its N3 imino group forms an H-bond to the O4 keto moiety of U+2 (Figure 5D); the A8 structure exhibits no comparable interactions to the ribozyme strand (Figure 5C). It has been demonstrated previously by FRET that G8U was still capable of docking despite nearly complete inhibition of catalysis (49). These observations suggest that docking of the loop A and B domains is related to the number of interactions that the position 8 base makes with its surroundings, but catalytic proficiency correlates with the number of H-bonds formed between the base at position 8 and the cleavage site (Table 2). G8A is the only structure of this study with a purine base at position 8 that does not make an H-bond contact to the scissile bond and its catalytic geometry is poorest among purine variants of this study. However, the nucleotide base at A8 stacks with A−1, conferring some cross-strand stability not observed for G8U (Figures 5C & 5D), which exhibits the least solution activity of the variants in this study. Of the position 8 variants analyzed, G8C was the only one that did not crystallize, presumably because it did not fold properly, as indicated by native gel electrophoresis (14). Given the plasticity of the scissile bond observed here, it is not possible to explain why G8C destabilizes the native fold, although its inability to base stack with A−1 and lack of an imino proton at N3 may be underlying factors.
Discussion
Previously, a kinetic analysis of various base substitutions at position 8 of the hairpin ribozyme produced the following reactivity profile: G > DAP > I > AP > A > U, where differences between G8 and U8 spanned ∼250-fold (14). Since in-line geometry provides at most a 10 to 20-fold rate acceleration over the uncatalyzed cleavage reaction, chemical attributes beyond the local secondary structure must play a critical role in RNA catalyzed phosphoryl transfer (50, 51). At present, there are two proposals for how the hairpin ribozyme achieves this rate enhancement. The first entails acid/base catalysis involving G8 and/or A38 (14, 23, 52). The second calls for electrostatic stabilization involving localized nucleobase charge dissipation (15-17, 23, 53). Although X-ray crystallography cannot differentiate between these proposals, it can provide additional stereochemical information relevant to understanding the principles by which the enzyme promotes catalytically productive or non-productive states. The goal of this study is to relate structure to function by exploring changes in the scissile bond that result from changes in the chemical environment at G8.
Chemical Changes at Position 8 Alter the Scissile Bond Geometry
In this study, a minimal all-RNA variant of the hairpin ribozyme was solved and refined to 2.05 Å resolution. This native molecule possesses G8 at the active site and a 2'-OMe at A−1 to prevent catalysis in the crystal lattice. For comparison, the structures of five hairpin ribozymes containing modifications (Scheme 2) at position 8 of the ribozyme strand (Figure 1A) were solved to resolutions between 2.3-2.7 Å (Table 1). These structures were then used to correlate changes at the A−1 to G+1 internucleotide linkage with activity levels observed in solution (14). The base modifications were chosen because they were demonstrated to be competent in interdomain docking, but catalytically impaired over a broad range. The results revealed that scissile bond geometry is highly dependent on functional group interactions with the base at position 8. The trend towards in-line geometry followed the series: G8 > I8 ≥ DAP8 > AP > A8 > U8 (Figure 6A & Table 2). Deviation of DAP8 from the activity series in solution may be the result of a conformational change resulting from the use of high pH in structural analysis. AP8 was solved under similar conditions, therefore caution must be exercised when considering both DAP8 and AP8 structures relative to other variants of the series that were solved at pH 6.0. In the final analysis, assignment of the structural trend towards reactivity was not based solely on τ, since other factors appeared important as well (Table 2). For example, DAP8 displayed a relatively close 3 Å distance between the 2'-nucleophile and the scissile phosphorus, as well as contacts to the pro-R oxygen of G+1 (Figure 5A & Table 2). Indeed, both DAP8 and AP8 variants exhibited modest levels of cleavage in crystals (Supporting Information Figure 3). A complete list of interactions for position 8 variants is provided in Table 2.
The superposition of G8 variants revealed an ensemble of structures representing a continuum of conformations (Figure 6A). This analysis indicated the scissile bond could rotate by as much as 180° from a non-productive conformation, in which the non-bridging oxygens reside above the plane of the scissile bond, to a productive conformation in which they lie below (Figure 6A, yellow versus red structures). Changes to the scissile bond appear to originate mostly from base substitutions at position 8, although the effects of modifications at the 2'-position of A−1 cannot be ruled out entirely (e.g. recall G8I/2'-OMe A−1 and G8I/2'-dA−1 revealed little change). Overall, subtle changes were observed in the conformation of the A38 base, which mirrored the results of a prior study in which U39 was replaced by a propyl linker (11). A comparison of the N1 positions of A38 from the most active conformation (i.e. the 4WJ-vanadate complex) and the least active structure, U8 of this study, revealed an ∼0.8 Å shift of the latter away from the scissile bond. This result indicates that the W-C face of A38 remains largely unchanged regardless of the scissile bond geometry. In contrast, the O4 position of conserved base U+2 varied widely (Figure 6A). This change was not a direct indicator of in-line geometry, but rather that the O4 oxygen at U+2 preferred H-bonding to the exocyclic amine at position 8. In the most extreme comparison, the O4 keto atom of U+2 changed by as much as 1.7 Å between the G8I and DAP structures (Figure 6A, purple versus blue structures) indicating the range of motion associated with N2 removal.
Comparison of Position 8 Variants to the Hairpin Ribozyme Vanadate Complex
Vanadium oxide has been cited as a structural mimic of the oxyphosphorane intermediate of phosphoryl transfer reactions (Scheme 1). As such, it has been introduced into the active sites of numerous protein enzymes (54). The 4WJ form of the hairpin ribozyme has been solved in complex with vanadate, thereby providing a spatial analogue for the trigonal bipyramidal transition state of this and other ribozyme reactions (10, 23, 39). A comparison of the vanadate complex to the native G8 , pre-catalytic structure of this and prior studies (9-11), revealed that τ = 144° for the vanadate mimic (Figure 6B & Table 2). Although this angle is not ideal for phosphoryl transfer, several features suggest it is the closest available structure to the actual geometry of the reaction intermediate. First, the 2'-nucleophile and phosphorus are within a distance of 2.0 Å (Table 2). Second, the vanadium ion exhibits a nearly trigonal bipyramidal coordination sphere (Figures 6A & 6B). Third, the 5'-oxo leaving group is within 2.7 Å of the vanadium ion. Therefore, for the purposes of this study, the locations and interactions of neighboring functional groups with vanadate, such as G8 and A38, are of great interest since they reveal potentially important features regarding how localized modifications predispose the scissile bond to cleavage or ligation.
The 4WJ hairpin ribozyme-vanadate complex revealed that the N1 and N2 positions of the G8 base are within H-bonding distance of the 2'-oxo nucleophile of A−1 (Scheme 1 & Figure 6B). These interactions originate from the ability of the imino group to donate a proton in H-bonding, as observed for G8 and I8 of this study (Figures 3B & 4A). Conversely, AP8 and A8 exhibited distances inconsistent with H-bonding at this position (Figures 5B & 5C), and DAP8, AP8 and A8 are expected to be deprotonated at the pH values chosen for crystallization (Scheme 2). However, unlike the imino group of a purine base, the N2 exocyclic amine is not subject to the same pH dependence. Consequently, this functional group is able to fulfill two H-bonding roles from both its syn and anti hydrogens. All bases of this study that possess an N2 or equivalent H-bond donor at position 8, including G, DAP, AP and U, formed an H-bond contact to the O4 keto oxygen of U+2 (Figures 3B, 5A, 5B & 5D). Similarly, a syn H-bond was noted to the pro-S oxygen of the vanadate complex (Figure 6B). The latter interaction was ascribed an important role in transition state stabilization (10, 23). However, another facet of this interaction seems plausible. The trigonal angle for the (pro-S)O-N2-O4 interaction in the vanadate complex (Figure 6B) is ∼80°, which is suitable for H-bonding. However, the O2'-N2-O4 angle is nearly 120°, which is optimal. Hence, the 3.1 Å distance between the N1 exocyclic amine of G8 to the 2'-oxygen of A−1, as well as a 3.5 Å contact to the pro-S oxygen of G+1, suggested the possibility of a bifurcated H-bond interaction in the transition state. In the pre-catalytic structure, this dual interaction orients the A−1 sugar upon docking, thereby preventing it from adopting a non-productive C2'-exo conformation, as observed for the A8 and U8 modifications (Figures 5C & 5D), which dock, but are impaired catalytically (14, 49). In contrast, the DAP8 and AP8 modifications, despite being deprotonated at N1, maintain close distances between the 2'-OH nucleophile and the reactive G+1 phosphorus (Figure 6A & 6B and Table 2). The close proximity between the 2'-OH of A−1, the imino proton of position 8, as well its N2 exocylic amine, should help lower the pKa of the 2'-OH nucleophile. Rescue of a G8 abasic hairpin ribozyme by a positively charged nucleobase (15, 16) also appears consistent with this role.
A8 & U8 Variants as Non-Productive Ground States
Further comparison of the G8 variant structures in this study to the 4WJ-vanadate complex revealed two key H-bond interactions at the W-C face of A38 (Figure 6B). The first is an N6 H-bond from A38 to the pro-R oxygen of G+1. This contact has been cited as a mode of transition state stabilization in a manner similar to the interaction between the exocyclic amine of G8 and the substrate pro-S oxygen (10, 23). To the contrary, crystal structures of U8 and A8 both oriented their 2'-OH groups of A−1 in a manner consistent with an H-bond to the N6 amine of A38 (Figures 5C & 5D). This observation suggested that the A−1 hydroxyl of these variants could represent a spatial mimic for the pro-R oxygen of G+1 in the transition state (Figure 6A, yellow versus red structures). Consequently, the A8 and U8 variants appear to represent non-productive ground states that may account for their more precipitous 100- to 250-fold losses in activity, respectively, compared to other variants (14). Another key H-bond at A38 of the vanadate structure occurs between its imino group and the O5'-leaving group of G+1 (Figure 6B). The close proximity of these two atoms led to the suggestion that N1 of A38 was protonated at neutral pH implying that its pKa was shifted towards neutrality (10). The proposal for an elevated pKa at A38 is consistent with the structures of this study given the distances between respective O3' atoms of A−1 and the exocyclic amines of A38 (Figures 3B & 5). However, a note of caution should be exercised given the observation that the DAP and AP modifications where both analyzed at pH >8.5, but their respective distances are 3.3 and 2.6 Å. Given the ∼0.4 to 0.5 Å coordinate error of these structures (Table 1), it is risky to infer nucleobase protonation states at this position without further experimental information.
Implications for Water in the Active Site
An intriguing observation of this study was the presence of four ordered water molecules in the active site of the 2.05 Å resolution G8 native structure (Figure 3A). Water was not observed in the active site of previous 4WJ hairpin ribozyme structures, although solvent has been invoked to account for both structural and biochemical observations regarding the mechanism of action (10, 15-17). In earlier structural studies, it was noted that the N1 imino position of G8 appeared to function as an obligate proton donor to the 2'-oxygen of the nucleophile, and not as a proton acceptor as corroborated here for AP8 and DAP8. This insightful observation was supported by structures of the native G8 precursor with a 2'-OMe modification at A−1 (e.g. Figure 3B), as well as a product complex containing a cyclic-2',3' phosphodiester (10). Of equal importance was the observation that the rate of cleavage was greatest when the N1 group at position 8 was protonated (14). However, given the modest effects of G8 on catalysis, based on abasic substitution at this position (15-17), it is more likely that G8 plays a structural role rather than serving as a general base (10). Consequently, the protonation and deprotonation of the 2'-oxygen at A−1 was speculated to occur via donation of the hydroxyl proton to water. Although the conformation of the native G8 structure of this study does not show water in direct contact with the 2'-OH group of A−1, the structure of the 4WJ hairpin ribozyme-vanadate complex provides some clues as to how this might occur. A superposition of the vanadate and native G8 structures (Figure 6B) suggests that W52 (Figure 3A) occupies the same position as the pro-R oxygen of the scissile bond during the transition state. This molecular mimicry phenomenon by water has been documented for other phosphoryl transfer enzymes solved in the presence and absence of oxyphosphorane analogues (55). Likewise, the pro-R oxygen atom of G+1 in the vanadate complex appears to be spatially equivalent to the 2'-OH group of A−1 in the A8 and U8 structures of this study (Figure 6A). As such, W52 would be expected to shift its location in the transition state, pushing it towards W54 (Figure 3A). One possible outcome of this interaction is depicted in Figure 6C. In this scenario, the proton from the 2'-OH of A−1 is accepted by a water to form a hydronium ion. This proposal is consistent with the principle of microscopic reversibility in cleavage and ligation, and is supported by NAIM observations by Ryder & Strobel, who showed that A10 favored protonation during ligation (53). The native G8 structure of this study indicates that A10 has the capacity to interact with at least two solvent molecules on its W-C face (Figure 3A). Furthermore, although it is distant from the scissile bond, conversion of A10 to an abasic residue has a modest <10-fold effect on activity (15) consistent with the minor structural role ascribed here.
Summary and Conclusion
The structure of an all-RNA, minimal hairpin ribozyme has been solved in its native form and refined to 2.05 Å resolution. This structure served as a basis for comparison to five other hairpin ribozyme variants in which G8 was changed to I, DAP, AP, A or U. Further structural comparisons to the previously determined 4WJ hairpin ribozyme in complex with vanadate revealed a wide variation in the scissile bond geometry, with significant deviations from the inline attack configuration resulting from subtle changes in the chemical environment. This work also revealed a new role for the 2-amino group at position 8. Namely, it appears to be required to prevent non-productive ground-state conformations, as observed for the G8A and G8U substitutions. Finally, this study provided strong evidence that at least four ordered water molecules occupy the hairpin ribozyme active site in the pre-catalytic state. Water has been invoked recently in the ribosome mechanism of action of peptidyl transfer, suggesting an important means to diversify the functionality of RNA enzymes through substrate-assisted proton transfer networks (56, 57). The presence of solvent molecules in the hairpin ribozyme active site supports existing proposals that water serves as a proton acceptor or donor for the 2'-oxygen of A−1 (10). This idea is consistent with the 3- to 4-fold rate reduction in D2O over a broad pH range and that 2 protons are in flight in the transition state (58). Given the recent observation that G8 plays a relatively modest role in the reaction chemistry (15-17), the results suggest a new path for exploration centered on specific acid/base catalysis.
Supplementary Material
For Table of Contents Use Only
Acknowledgment
The authors thank S. Strobel, S. Ryder, J. Burke, K. Hampel, D. Lambert, D. Mathews, M. Fedor and A. Mondragón for their insightful comments. We thank N. Walters, C. MacElrevey and A. Torelli for critical remarks about the manuscript. We appreciate the assistance from staff members at CHESS, which is supported by the NSF under award DMR-0225180 and NIH through NCRR award RR-01646.
Footnotes
This work was supported by NIH Grant GM63162 to J.E.W.
Abbreviations: 4WJ, four-way helical junction; H-bond, hydrogen bond; W-C, Watson-Crick; I, inosine; DAP, 2,6-diaminopurine; AP, 2-aminopurine; dA, 2'-deoxyadenosine; U, uridine; 2'-OMeA, 2'-deoxy 2'-O-methyl adenosine; PEG 2K MME, poly(ethylene) glycol 2000 mono methyl ether; TEAA, triethylammonium acetate; TEA, triethylamine; NAIM, nucleotide analog interference mapping; rmsd, average root mean square XYZ displacement.
Supporting Information Available
A stereo diagram of representative electron density maps is provided for the native and position 8 variants of this study (Figure 1). A diagram comparing the G8I/2'-OMe A−1 and G8I/2'-deoxy A−1 variants is also available (Figure 2). The method for HPLC composition analysis of AP8 and DAP8 crystals is reported, as well as elution profiles for the separated hairpin ribozyme strands (Figure 3). This information is provided free of charge at http://pubs.acs.org
References
- 1.McKay DB, Wedekind JE. In: The RNA World. Cech T, editor. Cold Spring Harbor Laboratory Press; Cold Spring Harbor: 1999. pp. 265–286. [Google Scholar]
- 2.Doherty EA, Doudna JA. Ribozyme structures and mechanisms. Annu. Rev. Biophys. Biomol. Struct. 2001;30:457–75. doi: 10.1146/annurev.biophys.30.1.457. [DOI] [PubMed] [Google Scholar]
- 3.Buzayan JM, Hampel A, Bruening G. Nucleotide sequence and newly formed phosphodiester bond of spontaneously ligated satellite tobacco ringspot virus RNA. Nucleic Acids Res. 1986;14:9729–43. doi: 10.1093/nar/14.24.9729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Long DM, Uhlenbeck OC. Self-cleaving catalytic RNA. FASEB J. 1993;7:25–30. doi: 10.1096/fasebj.7.1.8422971. [DOI] [PubMed] [Google Scholar]
- 5.Symons RH. Small catalytic RNAs. Annu. Rev. Biochem. 1992;61:641–71. doi: 10.1146/annurev.bi.61.070192.003233. [DOI] [PubMed] [Google Scholar]
- 6.Moore PB, Steitz TA. After the ribosome structures: how does peptidyl transferase work? RNA. 2003;9:155–9. doi: 10.1261/rna.2127103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Doudna JA, Lorsch JR. Ribozyme catalysis: not different, just worse. Nat. Struct. Mol. Biol. 2005;12:395–402. doi: 10.1038/nsmb932. [DOI] [PubMed] [Google Scholar]
- 8.Fedor MJ, Williamson JR. The catalytic diversity of RNAs. Nat. Rev. Mol. Cell. Biol. 2005;6:399–412. doi: 10.1038/nrm1647. [DOI] [PubMed] [Google Scholar]
- 9.Rupert PB, Ferre-D'Amare AR. Crystal structure of a hairpin ribozyme-inhibitor complex with implications for catalysis. Nature. 2001;410:780–6. doi: 10.1038/35071009. [DOI] [PubMed] [Google Scholar]
- 10.Rupert PB, Massey AP, Sigurdsson ST, Ferre-D'Amare AR. Transition state stabilization by a catalytic RNA. Science. 2002;298:1421–4. doi: 10.1126/science.1076093. [DOI] [PubMed] [Google Scholar]
- 11.Alam S, Grum-Tokars V, Krucinska J, Kundracik ML, Wedekind JE. Conformational heterogeneity at position U37 of an all-RNA hairpin ribozyme with implications for metal binding and the catalytic structure of the S-turn. Biochemistry. 2005;44:14396–14408. doi: 10.1021/bi051550i. [DOI] [PubMed] [Google Scholar]
- 12.Butcher SE, Heckman JE, Burke JM. Reconstitution of hairpin ribozyme activity following separation of functional domains. J. Biol. Chem. 1995;270:29648–51. doi: 10.1074/jbc.270.50.29648. [DOI] [PubMed] [Google Scholar]
- 13.Hampel KJ, Walter NG, Burke JM. The solvent-protected core of the hairpin ribozyme-substrate complex. Biochemistry. 1998;37:14672–82. doi: 10.1021/bi981083n. [DOI] [PubMed] [Google Scholar]
- 14.Pinard R, Hampel KJ, Heckman JE, Lambert D, Chan PA, Major F, Burke JM. Functional involvement of G8 in the hairpin ribozyme cleavage mechanism. EMBO J. 2001;20:6434–42. doi: 10.1093/emboj/20.22.6434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lebruska LL, Kuzmine II, Fedor MJ. Rescue of an abasic hairpin ribozyme by cationic nucleobases: evidence for a novel mechanism of RNA catalysis. Chem. Biol. 2002;9:465–73. doi: 10.1016/s1074-5521(02)00130-8. [DOI] [PubMed] [Google Scholar]
- 16.Kuzmin YI, Da Costa CP, Fedor MJ. Role of an active site guanine in hairpin ribozyme catalysis probed by exogenous nucleobase rescue. J. Mol. Biol. 2004;340:233–51. doi: 10.1016/j.jmb.2004.04.067. [DOI] [PubMed] [Google Scholar]
- 17.Kuzmin YI, Da Costa CP, Cottrell JW, Fedor MJ. Role of an active site adenine in hairpin ribozyme catalysis. J. Mol. Biol. 2005;349:989–1010. doi: 10.1016/j.jmb.2005.04.005. [DOI] [PubMed] [Google Scholar]
- 18.Westheimer FH. Pseudo-rotation in the hydrolysis of phosphate esters. Acc. Chem. Res. 1968;1:70–78. [Google Scholar]
- 19.Usher DA, McHale AH. Hydrolytic stability of helical RNA: a selective advantage for the natural 3',5'-bond. Proc. Natl. Acad. Sci. U.S.A. 1976;73:1149–53. doi: 10.1073/pnas.73.4.1149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Young KJ, Gill F, Grasby JA. Metal ions play a passive role in the hairpin ribozyme catalysed reaction. Nucleic Acids Res. 1997;25:3760–6. doi: 10.1093/nar/25.19.3760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Murray JB, Seyhan AA, Walter NG, Burke JM, Scott WG. The hammerhead, hairpin and VS ribozymes are catalytically proficient in monovalent cations alone. Chem. Biol. 1998;5:587–95. doi: 10.1016/s1074-5521(98)90116-8. [DOI] [PubMed] [Google Scholar]
- 22.Nesbitt S, Hegg LA, Fedor MJ. An unusual pH-independent and metal-ion-independent mechanism for hairpin ribozyme catalysis. Chem. Biol. 1997;4:619–30. doi: 10.1016/s1074-5521(97)90247-7. [DOI] [PubMed] [Google Scholar]
- 23.Ferre-D'Amare AR. The hairpin ribozyme. Biopolymers. 2004;73:71–8. doi: 10.1002/bip.10516. [DOI] [PubMed] [Google Scholar]
- 24.Grum-Tokars V, Milovanovic M, Wedekind JE. Crystallization and X-ray diffraction analysis of an all-RNA U39C mutant of the minimal hairpin ribozyme. Acta Crystallogr. 2003;D59:142–5. doi: 10.1107/s0907444902019066. [DOI] [PubMed] [Google Scholar]
- 25.Wedekind JE, McKay DB. Purification, crystallization, and X-ray diffraction analysis of small ribozymes. Methods Enzymol. 2000;317:149–68. doi: 10.1016/s0076-6879(00)17013-2. [DOI] [PubMed] [Google Scholar]
- 26.Chowrira BM, Burke JM. Binding and cleavage of nucleic acids by the “hairpin” ribozyme. Biochemistry. 1991;30:8518–22. doi: 10.1021/bi00099a003. [DOI] [PubMed] [Google Scholar]
- 27.Walter NG, Burke JM. Real-time monitoring of hairpin ribozyme kinetics through base-specific quenching of fluorescein-labeled substrates. RNA. 1997;3:392–404. [PMC free article] [PubMed] [Google Scholar]
- 28.Walter NG, Hampel KJ, Brown KM, Burke JM. Tertiary structure formation in the hairpin ribozyme monitored by fluorescence resonance energy transfer. EMBO J. 1998;17:2378–91. doi: 10.1093/emboj/17.8.2378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pflugrath JW. The finer things in X-ray diffraction data collection. Acta Crystallogr. 1999;D55:1718–25. doi: 10.1107/s090744499900935x. [DOI] [PubMed] [Google Scholar]
- 30.Brünger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, Read RJ, Rice LM, Simonson T, Warren GL. Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr. 1998;D54:905–21. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 31.Jones TA, Zou JY, Cowan SW, Kjeldgaard M. Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Crystallogr. 1991;A47:110–9. doi: 10.1107/s0108767390010224. [DOI] [PubMed] [Google Scholar]
- 32.Pannu NS, Read RJ. Improved Structure Refinement through maximum likelihood. Acta Crystallogr. 1996;A52:659–668. [Google Scholar]
- 33.Fedor MJ. The role of metal ions in RNA catalysis. Curr. Opin. Struct. Biol. 2002;12:289–95. doi: 10.1016/s0959-440x(02)00324-x. [DOI] [PubMed] [Google Scholar]
- 34.Glusker JP. In: Advances in Protein Chemistry. Anfinsen CB, Richards FM, Eisenberg DS, editors. Academic Press; Orlando: 1991. pp. 1–76. [Google Scholar]
- 35.Collaborative Computational Project, N. The CCP4 Suite: Programs for Protein Crystallography. Acta Crystallogr. 1994;D50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 36.Esnouf RM. Further additions to MolScript version 1.4, including reading and contouring of electron-density maps. Acta Crystallogr. 1999;D55:938–40. doi: 10.1107/s0907444998017363. [DOI] [PubMed] [Google Scholar]
- 37.DeLano WL. The PyMOL Molecular Graphics System. DeLano Scientific; San Carlos, California: 2002. [Google Scholar]
- 38.Jovine L. Nuccyl. 2003 http://www.mssm.edu/students/jovinl02/research/nuccyl.html.
- 39.Wedekind JE, McKay DB. Crystal structure of the leadzyme at 1.8 Å resolution: metal ion binding and the implications for catalytic mechanism and allo site ion regulation. Biochemistry. 2003;42:9554–63. doi: 10.1021/bi0300783. [DOI] [PubMed] [Google Scholar]
- 40.Eisenberg DS, Crothers DM. Physical Chemistry with Applications to the Life Sciences. Benjamin Cummings Inc.; Menlo Park: 1979. [Google Scholar]
- 41.Rupert PB, Ferre-D'Amare AR. Crystallization of the hairpin ribozyme: illustrative protocols. Methods Mol. Biol. 2004;252:303–11. doi: 10.1385/1-59259-746-7:303. [DOI] [PubMed] [Google Scholar]
- 42.DeYoung M, Siwkowski AM, Lian Y, Hampel A. Catalytic properties of hairpin ribozymes derived from Chicory yellow mottle virus and arabis mosaic virus satellite RNAs. Biochemistry. 1995;34:15785–91. doi: 10.1021/bi00048a024. [DOI] [PubMed] [Google Scholar]
- 43.Saenger W. Principles of Nucleic Acid Structure. Springer-Verlag; New York: 1984. [Google Scholar]
- 44.Chowrira BM, Berzal-Herranz A, Keller CF, Burke JM. Four ribose 2'-hydroxyl groups essential for catalytic function of the hairpin ribozyme. J. Biol. Chem. 1993;268:19458–62. [PubMed] [Google Scholar]
- 45.Grasby JA, Mersmann K, Singh M, Gait MJ. Purine functional groups in essential residues of the hairpin ribozyme required for catalytic cleavage of RNA. Biochemistry. 1995;34:4068–76. doi: 10.1021/bi00012a025. [DOI] [PubMed] [Google Scholar]
- 46.Ryder SP, Strobel SA. Nucleotide Analog Interference Mapping of the Hairpin Ribozyme: Implications for Secondary and Tertiary Structure Formation. J. Mol. Biol. 1999;291:295–311. doi: 10.1006/jmbi.1999.2959. [DOI] [PubMed] [Google Scholar]
- 47.Klostermeier D, Millar DP. Energetics of hydrogen bond networks in RNA: hydrogen bonds surrounding G+1 and U42 are the major determinants for the tertiary structure stability of the hairpin ribozyme. Biochemistry. 2002;41:14095–102. doi: 10.1021/bi025551b. [DOI] [PubMed] [Google Scholar]
- 48.McDonald IK, Thornton JM. Satisfying hydrogen bonding potential in proteins. J. Mol. Biol. 1994;238:777–93. doi: 10.1006/jmbi.1994.1334. [DOI] [PubMed] [Google Scholar]
- 49.Wilson TJ, Zhao ZY, Maxwell K, Kontogiannis L, Lilley DM. Importance of specific nucleotides in the folding of the natural form of the hairpin ribozyme. Biochemistry. 2001;40:2291–302. doi: 10.1021/bi002644p. [DOI] [PubMed] [Google Scholar]
- 50.Soukup GA, Breaker RR. Relationship between internucleotide linkage geometry and the stability of RNA. RNA. 1999;5:1308–25. doi: 10.1017/s1355838299990891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mikkola S, Kaukinen U, Lonnberg H. The effect of secondary structure on cleavage of the phosphodiester bonds of RNA. Cell. Biochem. Biophys. 2001;34:95–119. doi: 10.1385/CBB:34:1:95. [DOI] [PubMed] [Google Scholar]
- 52.Bevilacqua PC. Mechanistic considerations for general acid-base catalysis by RNA: revisiting the mechanism of the hairpin ribozyme. Biochemistry. 2003;42:2259–65. doi: 10.1021/bi027273m. [DOI] [PubMed] [Google Scholar]
- 53.Ryder SP, Oyelere AK, Padilla JL, Klostermeier D, Millar DP, Strobel SA. Investigation of adenosine base ionization in the hairpin ribozyme by nucleotide analog interference mapping. RNA. 2001;7:1454–63. [PMC free article] [PubMed] [Google Scholar]
- 54.Davies DR, Hol WG. The power of vanadate in crystallographic investigations of phosphoryl transfer enzymes. FEBS Lett. 2004;577:315–21. doi: 10.1016/j.febslet.2004.10.022. [DOI] [PubMed] [Google Scholar]
- 55.Gil-Ortiz F, Ramon-Maiques S, Fita I, Rubio V. The course of phosphorus in the reaction of N-acetyl-L-glutamate kinase, determined from the structures of crystalline complexes, including a complex with an AlF(4)(−) transition state mimic. J. Mol. Biol. 2003;331:231–44. doi: 10.1016/s0022-2836(03)00716-2. [DOI] [PubMed] [Google Scholar]
- 56.Weinger JS, Parnell KM, Dorner S, Green R, Strobel SA. Substrate-assisted catalysis of peptide bond formation by the ribosome. Nat. Struct. Mol. Biol. 2004;11:1101–6. doi: 10.1038/nsmb841. [DOI] [PubMed] [Google Scholar]
- 57.Schmeing TM, Huang KS, Kitchen DE, Strobel SA, Steitz TA. Structural Insights into the Roles of Water and the 2′ Hydroxyl of the P Site tRNA in the Peptidyl Transferase Reaction. Mol. Cell. 2005;20:437–48. doi: 10.1016/j.molcel.2005.09.006. [DOI] [PubMed] [Google Scholar]
- 58.Pinard R, Lambert D, Walter NG, Heckman JE, Major F, Burke JM. Structural basis for the guanosine requirement of the hairpin ribozyme. Biochemistry. 1999;38:16035–9. doi: 10.1021/bi992024s. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.