

Abstract
Bullous pemphigoid antigen 2 (BPAG2) is targeted by autoantibodies in patients with bullous pemphigoid (BP) and absent in patients with one type of epidermolysis bullosa (OMIM #226650). A keratin 14 promoter construct was used to produce transgenic (Tg) mice appropriately expressing human BPAG2 (hBPAG2) in murine epidermal basement membrane (BM). Grafts of Tg skin placed on gender-matched, syngeneic wild type (Wt) or MHC I -/- mice elicited IgG that bound human epidermal BM and BPAG2. Production of such IgG in grafted mice was prompt (detectable within 16±2 days), robust (titer ≥ 1280), durable (present ≥ 380 days), and correlated with the involution and loss of Tg skin grafts. MHC II -/- mice grafted with Tg skin did not develop anti-hBPAG2 IgG or graft loss indicating that MHC II:CD4+ T cell interactions were crucial for these responses. Tg skin grafts on Wt mice developed neutrophil-rich infiltrates, dermal edema, subepidermal blisters, and deposits of immunoreactants in epidermal BM. This model shows fidelity to alterations seen in patients with BP, has relevance to immune responses that may arise in patients with epidermolysis bullosa following BPAG2 gene replacement, and can be used to identify interventions that may block production of IgG against proteins in epidermal BM.
Keywords: animal model, bullous disease, epidermolysis bullosa, pemphigoid
Introduction
BPAG2 is a type II transmembrane collagen (specifically, type XVII collagen) associated with hemidesmosome-anchoring filament complexes in basal keratinocytes (Franzke et al., 2005; Franzke et al., 2003; Van den Bergh and Giudice, 2003). BPAG2 consists of an intracellular domain that associates with bullous pemphigoid antigen 1, integrin subunit β4, and plectin, a short, single pass transmembrane segment, and a long extracellular domain that consists of fifteen interrupted collagenous domains assembled into a homotrimeric triple helix. Rotary shadowing of purified BPAG2 images its intracytoplasmic region as a globular head and its extracellular region as a central rod with an adjoining flexible tail (Hirako et al., 1996). The latter extracellular regions correspond to the largest (i.e., domain 15) and the remaining (i.e., domains 1–14) collagenous segments of the protein, respectively. Immunoelectron microscopy studies indicate that the rod domain of BPAG2 spans the lamina lucida, while its carboxyl terminus loops through the lamina densa back into the lamina lucida (Masunaga et al., 1997; Nonaka et al., 2000). The carboxyl terminal portion of BPAG2 interacts with laminin 5 at the interface of the lamina lucida and the lamina densa (Masunaga et al., 1997; Tasanen et al., 2004).
Autoantibodies from patients with the autoimmune subepidermal blistering disease BP typically target the 16th noncollagenous (i.e., NC16A) domain of BPAG2 (Giudice et al., 1993). Passive transfer of experimental IgG (specifically, rabbit IgG) developed against the murine homolog of human NC16A elicits clinical, histologic, and immunopathologic alterations in neonatal BALB/c mice that mimic those seen in patients with BP (Liu et al., 1993). IgG-induced blister formation in this animal model is dependent upon complement activation, degranulation of dermal mast cells, and generation of neutrophil-rich infiltrates – the latter providing sufficient amounts of unrestrained neutrophil elastase to degrade BPAG2 and produce subepidermal blisters in vivo (Liu et al., 2000). This thoroughly characterized experimental murine model is widely regarded to provide proof of concept that IgG anti-NC16A autoantibodies in patients with BP are pathogenic (Yancey, 2005). While such acquired alterations in BPAG2 are held responsible for the pathogenesis of BP, patients with an inherited subepidermal blistering disease termed generalized atrophic benign epidermolysis bullosa (GABEB, OMIM 113811) typically possess null mutations in the gene encoding BPAG2 (i.e., COL17A1) (Darling et al., 1997b; McGrath et al., 1995; Pohla-Gubo et al., 1995). Patients with GABEB characteristically demonstrate a complete lack of BPAG2 in epidermal BM, skin fragility, subepidermal blisters at sites of friction or trauma, alopecia, dystrophic nails, and dental enamel hypoplasia – a series of clinical findings that further attest to the key role that BPAG2 plays in epidermal adhesion and homeostasis (Darling et al., 1997a; Van den Bergh and Giudice, 2003).
Given the pivotal role that BPAG2 plays in these inherited and acquired subepidermal blistering diseases, it has been hypothesized that successful gene replacement in patients with GAGEB may lead to unwanted immune responses to the transgene thus yielding a phenotype that mimics BP. To explore this hypothesis and develop an animal model in which experimental immune responses to hBPAG2 can be developed, modulated, and/or blocked in vivo, hBPAG2 Tg mice were created, characterized, and studied in a series of skin grafting experiments.
Results
Transient Transfection Studies
An immortalized GABEB keratinocyte line transfected with the hBPAG2 cDNA transgene construct showed bright, diffuse cytoplasmic staining for BPAG2 by immunofluorescence (IF) microscopy (Figure 1A). In contrast, GABEB keratinocytes transfected with the same construct lacking hBPAG2 cDNA (i.e., cells transfected with a “mock” construct and subjected to selection in the same manner) showed no staining for BPAG2 (Figure 1B). Immunoblot studies of extracts from transgene- and mock-transfected GABEB keratinocytes identified hBPAG2 as a 180 kD polypeptide in the former (Figure 1C, lane 4), but not the latter (Figure 1C, lane 2).
Figure 1.

The hBPAG2 cDNA transgene construct expressed protein of appropriate immunoreactivity and molecular weight in an immortalized GABEB keratinocyte line. a: An immortalized GABEB keratinocyte line transfected with the transgene construct showed bright, diffuse cytoplasmic staining for hBPAG2 in IF microscopy studies. b: In contrast, GABEB keratinocytes transfected with the same construct lacking hBPAG2 cDNA (i.e., cells transfected with a “mock” construct and subjected to selection in the same manner) showed no staining for hBPAG2. Nonimmune rabbit sera showed no reactivity to these cells (data not shown). c: Immunoblot studies of extracts from transgene- and mock-transfected GABEB keratinocytes identified hBPAG2 as a 180 kD polypeptide in the former (lane 4) but not the latter (lane 2). Immortalized GABEB keratinocytes transfected with the same hBPAG2 cDNA in an alternate vector (i.e., pcDNA3) served as an additional positive control (lane 3) as did extracts of normal HKs (lane 1). Nonimmune rabbit sera showed no reactivity to these extracts (data not shown). Ticks in the left margin correspond to 250 and 116 kD molecular weight markers; the arrow points to a 180 kD band corresponding to hBPAG2. d: Tg mice express hBPAG2 in epidermal BM. Cryosections of skin from man, a Tg mouse, and a Wt mouse (top, middle, and bottom panels, respectively) were studied by IF microscopy using rabbit antiserum developed against the carboxy terminus of hBPAG2 (Masunaga et al., 1997) (left panels) or normal rabbit serum (control, right panels). hBPAG2 was specifically expressed in the epidermal BMs of human and Tg skin (arrows, top and middle left panels); this antiserum showed no specific reactivity to the skin of a Wt mouse (bottom left panel). There was no cytosolic, suprabasal, or intradermal expression of hBPAG2 in any skin samples. Control IF microscopy studies using normal rabbit serum were negative (right panels).
Generation of hBPAG2 Tg Mice
Fifty-five mice representing candidates for chimeric status were screened by Southern blotting using a radiolabeled fragment of the hBPAG2 cDNA transgene construct. Five mice showed evidence of transgene integration; of these, four chimeras yielded founder lines for subsequent studies. DNA from the tails of founder mice was amplified by PCR using a forward primer specific to the human keratin 14 promoter and a reverse primer specific to hBPAG2 cDNA. Two founder lines (specifically lines D and E) expressed the transgene in murine epidermal BM at highest levels by IF microscopy. These two lines were expanded to yield numerous heterozygous (+/-) and homozygous (+/+) mice (i.e., generations ≥ F8). The genotype of initial +/+ Tg mice in lines D and E was confirmed by breeding these lines with Wt mice and showing that all progeny received a copy of the hBPAG2 transgene. These confirmatory experiments provided conclusive evidence that only +/+ mice were retained for expansion of our Tg colony. All Tg mice were phenotypically normal; all experiments reported herein used mice derived from founder line D. Fluorescent in situ hybridization studies showed that the hBAPG2 transgene was integrated into chromosome 15 (specifically, at 15F2-3) in mice of founder line D. This portion of the mouse genome is devoid of elements known to influence or govern immunologic or inflammatory reactions. Of note, the gene for murine COL17A1 is positioned on chromosome 19.
hBPAG2 Tg Mice Express the Transgene in Epidermal BM
Light microscopy studies of skin from +/- and +/+ Tg mice were identical to Wt controls. Biopsies of trunk, ear, and tail skin from these mice were examined in IF microscopy studies using: 1) rabbit antiserum specific for the carboxyl terminal portion of hBPAG2 (Masunaga et al., 1997); 2) normal rabbit serum (control); 3) HD18, a murine monoclonal antibody specific for the NC16A-2.5 region of hBPAG2 (Pohla-Gubo et al., 1995); and 4) an isotypematched, irrelevant murine monoclonal antibody (control). +/+ Tg mice showed expression of hBPAG2 in epidermal BM in the same manner this protein is expressed in normal human skin. Such reactivity in Tg skin was identified using both rabbit antiserum specific for hBPAG2 (Figure 1D) as well as a murine monoclonal antibody specific for its NC16A domain. IgG from patients with BP also bound the epidermal BM of Tg mice. hBPAG2 was not apparent in the cytosol of basal or suprabasal keratinocytes of Tg mice. Immunoblot studies of epidermal extracts from Tg and Wt mice found that hBPAG2 was expressed as a 180 kD protein in the former, but not the latter (data not shown).
Immunogold Studies
The same rabbit hBPAG2 antiserum shown in Figure 1D was used to perform immunogold electron microscopy studies of cryopreserved human (Figure 2A), Tg (Figure 2B), and Wt (Figure 2C) skin to assess the expression and localization of hBPAG2. The cryopreservation and cryofixation techniques applied in these studies retained the immunoreactivity of structural proteins with sufficient preservation of morphology to allow high resolution of epitopes in epidermal BM. In these studies, the carboxyl terminus of hBPAG2 in human and Tg skin was localized to the superior aspect of the lamina densa at its interface with the lamina lucida (Figures 2A and 2B). Expression of hBPAG2 in +/+ Tg skin was somewhat less than that in human skin. There was no ectopic localization or aggregation of hBPAG2 in Tg skin. Rabbit hBPAG2 antiserum showed no reactivity to Wt skin (Figure 2C).
Figure 2.

hBPAG2 displayed normal ultrastructural localization in the epidermal BM of Tg skin. Post-embedding immunogold electron microscopy studies localized 5 nm gold particles staining hBPAG2 within the superior aspect of the lamina densa in human (a) and Tg (b) skin (arrows). This staining was consistent with that reported for this antiserum (Masunaga et al., 1997). Expression of hBPAG2 in Tg skin was somewhat less than that in human skin. There was no ectopic localization or aggregation of hBPAG2 in Tg skin. Anti-hBPAG2 antiserum showed no specific reactivity to the skin of a Wt mouse (c). HD, hemidesmosome; LL, lamina lucida; LD, lamina densa.
Wt Mice Grafted with Tg Skin Develop Anti-hBPAG2 IgG
The studies described above indicated that hBPAG2 was expressed with appropriate localization and immunoreactivity in the epidermal BM of Tg mice and that such expression did not impair epidermal morphogenesis, adhesion, or differentiation. To determine if the skin of Tg mice would elicit immune reactions in Wt mice, full thickness 1 cm2 grafts from the tails of Tg mice were placed onto the flanks of gender-matched, syngeneic Wt recipients. As controls, tail skin from Wt donors was grafted onto the opposing flank of Tg graft recipients (or alternate gender-matched, syngeneic Wt mice). Tg and Wt skin (from tail or flank, the former used in the results presented herein) engrafted equally. To date over 100 individual skin grafts have been placed on various recipients; the experimental series shown herein are representative of results consistently documented in this body of work.
Longitudinal studies found that Wt mice grafted with Tg skin consistently developed IgG that bound epidermal BMs in human and Tg skin, but not skin from Wt mice (Figure 3A). Production of such IgG in Wt mice was prompt (detectable within 16±2 days), robust (titer ≥ 1280), and durable (present >380 days) (Figure 3B). This IgG was present in serum from each of 32 Wt mice grafted with Tg skin, exclusively reactive with the epidermal side of 1 M NaCl split human skin, and capable of fixing C3 to epidermal BM at dilutions as high as 160. Control experiments demonstrated that sera from Wt mice grafted only with gender-matched, syngeneic Wt skin had no evidence of IgG reactive with epidermal BMs in human, Tg, or Wt skin (Figure 3A, n=4, recipients followed for as long as 200 days after graft placement). These studies consistently demonstrated that Tg skin grafts elicited specific immune responses in Wt mice, that such reactions included humoral immune responses, and that such responses were characterized by immunoglobulin class switching.
Figure 3.
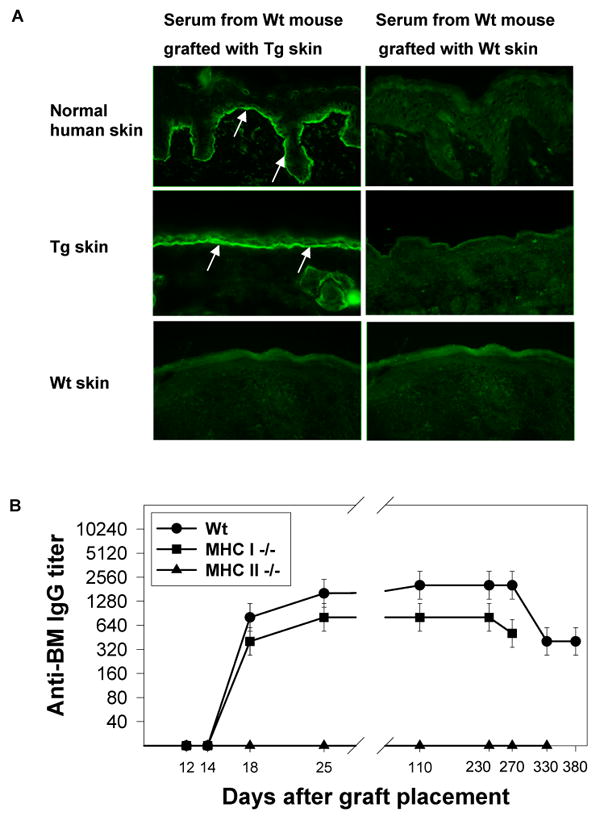
Tg skin grafts elicited anti-BM IgG in Wt and MHC I -/- mice. a. Tg skin grafts placed on Wt recipients elicited IgG that bound epidermal BMs in human and Tg skin (arrows, top and middle left panels), but not the skin of a Wt mouse (bottom left panel). Development of anti-BM IgG was unrelated to the grafting procedure (right panels). b: Longitudinal studies showed that Tg skin grafts placed on Wt (solid circles) and MHC I -/- (solid squares) recipients elicited high (640 to 2560) and durable (lasting ≥ 270 days) titers of anti-BM IgG that consistently appeared in the circulation of recipients 14 to 18 days after grafting. MHC II -/- mice (solid triangles) grafted with Tg skin did not develop anti-BM IgG. n=4/group in this longitudinal study that was representative of the aggregate work.
To determine if anti-BM IgG production was dependent upon MHC I:CD8+ T cell or MHC II:CD4+ T cell interactions, Tg skin was grafted onto gender-matched MHC I -/- and MHC II -/- mice, recipients otherwise syngeneic with donors. The kinetics and duration of anti-BM IgG production in MHC I -/- recipients was virtually identical to that seen in Wt recipients (Figure 3B). In contrast, MHC II -/- recipients did not develop anti-BM IgG (n=10, four recipients followed for as long as 330 days after graft placement) (Figure 3B). Anti-BM IgG also failed to develop in MHC II -/- mice that received two separate Tg skin grafts applied 30 days apart. These results indicated that anti-BM IgG production in grafted mice was dependent upon MHC II:CD4+ T cell interactions.
To define the precise reactivity of anti-BM IgG in Wt mice grafted with Tg skin, a series of immunoblot and immunoprecipitation experiments were performed. These experiments showed that the sera of these graft recipients contained IgG that bound hBPAG2 and that such reactivity included binding to NC16A (Figure 4A). Adsorption of immune mouse sera with recombinant NC16A removed all reactivity against this peptide, yet failed to impair the reactivity of immunoadsorbed sera with human epidermal BM by IF microscopy (data not shown) or full length hBPAG2 in HK extracts (Figure 4A). Sera from grafted mice showed no evidence of reactivity to other HK proteins including BPAG1 (Figure 4B).
Figure 4.

IgG in the sera of Wt mice grafted with Tg skin specifically bound hBPAG2. a. IgG in the sera of Wt mice grafted with Tg skin specifically bound hBPAG2 in extracts of cultured HKs as well as recombinant NC16A-GST (lanes 1 and 4, respectively). While preadsorption of immune sera with recombinant NC16A-GST completely removed all reactivity to this fusion protein (lane 5), immunoadsorbed immune sera still bound hBPAG2 (lane 2), indicating that epitopes other than NC16A are also targeted by such IgG. Normal mouse serum did not show any reactivity to hBPAG2 or NC16A-GST (lanes 3 and 6, respectively). Results were confirmed in three separate experiments. Markers in the left margins indicate Mr × 10-3. b. Immunoprecipitation studies of biosynthetically radiolabeled HK extracts showed that sera from Wt mice grafted with Tg skin (representative example, lane 1) contained IgG that bound BPAG2 but not BPAG1 – the former identified as a 180 kD protein that comigrated with BPAG2 immunoprecipitated by serum from a patient with BP (lane 3). Serum from a Wt mouse grafted with Wt skin (lane 2) as well as serum from a normal volunteer (lane 4) bound no specific HK proteins in these immunoprecipitation studies. Markers in the right margin indicate Mr × 10-3.
CD4+ T cells from Wt mice grafted with Tg skin demonstrated specific and dose-related proliferative responses to NC16A-GST in vitro
CD4+ T cells purified from axillary and inguinal lymph nodes of Wt mice 10 days previously grafted with Tg skin (both flanks) were assessed for secondary proliferative responses to recombinant NC16A-GST (Figure 5). CD4+ T cells from graft recipients (n=10) demonstrated dose-related proliferative responses to NC16A-GST, but not to equivalent amounts of GST alone or PBS. CD4+ T cells from Wt mice grafted with Wt skin (controls, n=10) did not show proliferative response to NC16A-GST, GST, or PBS (data not shown).
Figure 5.

CD4+ T cells from Wt mice grafted with Tg skin demonstrated specific and dose-related proliferative responses to NC16A-GST in vitro. Ten days after receiving grafts of Tg skin, axillary and inguinal lymph nodes of Wt mice were harvested and processed to yield CD4+ T cells that were placed in culture and assessed for secondary proliferative responses to various concentrations of recombinant NC16A-GST (solid circles), recombinant GST alone (solid squares), or PBS (solid triangles). γ-irradiated splenocytes from naïve, syngeneic Wt mice were used as stimulator cells. CD4+ T cells from graft recipients proliferated specifically to NC16A-GST but not to GST alone or PBS. CD4+ T cells from Wt mice grafted with Wt skin did not show proliferative response to NC16A-GST, GST, or PBS (data not shown). n=10/group.
Wt and MHC I -/- mice lost Tg skin grafts with identical kinetics, while MHC II -/- mice retained grafts of Tg skin
Grafts of Wt skin placed on gender-matched, syngeneic Wt recipients (controls) survived for their entire period of study (n=16, four recipients followed for as long as 200 days after graft placement, data not shown). In contrast, Tg skin grafts showed notable involution 15 to 23 days following placement on gender-matched, syngeneic Wt (n=12) and MHC I -/- (n=12) recipients (Figure 6). The kinetics of graft involution and loss were identical in Wt and MHC I -/- recipients (median graft survival time = 30±4 days). These studies showed that acceleration of graft involution consistently correlated with production of anti-hBPAG2 IgG and that graft loss in this model was not mediated by CD8+ T cells (Figure 6). Interestingly, Tg skin grafts placed on “immune” Wt mice (i.e., Wt mice previously grafted with Tg skin and displaying anti-hBPAG2 IgG) were typically lost within 10 to 15 days - a profile virtually identical to that observed once anti-hBPAG2 IgG first appeared in Wt mice grafted with Tg skin (Figure 6). Accelerated graft loss was not observed in Wt mice twice grafted with gender-matched, syngeneic Wt skin. Of note, Tg skin grafts placed on MHC II -/- mice retained their original viability and size for their entire period of study (n=10, four recipients followed for as long as 330 days) (Figure 6). These results indicated that Tg graft loss in this model was dependent upon MHC II:CD4+ T cell interactions just as was production of hBPAG2-specific IgG.
Figure 6.

Wt and MHC I -/- mice lost Tg skin grafts with identical kinetics, while MHC II -/- mice retained grafts of Tg skin. Tail skin was harvested from Tg and Wt mice and grafted onto the opposite flanks of age- and gender-matched Wt (solid circles), MHC I -/- (solid squares), MHC II -/- (solid triangles), and “immune” Wt (open circles) recipients. Bandages were removed on day 7; thereafter, mice were examined daily for 30 days then weekly to assess graft viability and size. Skin grafts were graded as “lost” if their area became ≤ 20% of their original size (McFarland et al., 2003). These longitudinal studies demonstrated that grafts of Tg skin on Wt and MHC I -/- recipients displayed accelerated involution at days 15 to 23 (i.e., the approximate time circulating anti-hBPAG2 IgG became detectable in recipients), and complete loss by days 28 to 30. Tg skin placed on “immune” Wt recipients (i.e., mice previously grafted with Tg skin and displaying anti-hBPAG2 in their circulation) were typically lost within 15 days – a profile like that observed once anti-hBPAG2 IgG first appeared in Wt mice grafted with Tg skin. Tg skin grafts placed on MHC II -/- recipients retained their viability and size for their entire period of study. n=4/group in this longitudinal study that was representative of the aggregate work.
Tg skin grafts on Wt mice developed in situ deposits of IgG and C3 in epidermal BM as well as neutrophil-rich leukocytic infiltrates and subepidermal blisters
Grafts of Tg and Wt skin were placed on opposite flanks of Wt, Tg, and MHC II -/- recipients (n=4 in each group of this representative experimental set) and sampled by 3 mm punch biopsies between 6 and 25 days after graft placement (i.e., prior to and during specific IgG production as well as during the time grafts displayed accelerated involution and loss). The corresponding skin samples were studied by light and IF microscopy as described previously. All specific alterations (i.e., those unrelated to wound reactions at the base and margins of the grafts) were confined to Tg grafts on Wt recipients. Such alterations were first noted approximately 17 days after graft placement and consisted of neutrophil-rich leukocytic infiltrates within the papillary dermis and along the epidermal BM (Figure 7A and Figure S1). Twenty to 25 days after Tg skin was placed on Wt mice, all grafts showed neutrophil-rich leukocytic infiltrates and edema within the papillary dermis; half of these grafts showed clefting within the epidermal BM and/or frank subepidermal blister formation (Figure 7B). Direct IF microscopy studies revealed in situ deposits of murine IgG and C3 in the epidermal BMs of Tg skin grafts on Wt recipients (Figure 7B). Immunoreactants were not identified in the epidermal BMs of recipient skin adjacent to, or at distance from, Tg grafts, or in control Wt grafts placed on the opposite flanks of Wt (Figure 7B), Tg, or MHC II -/- recipients. The accumulation of immunoreactants and neutrophil-rich leukocytic infiltrates in the epidermal BMs of Tg grafts on Wt mice displayed features characteristic of immune complex-mediated tissue injury, lesional skin from patients with BP, and neonatal murine skin exposed to experimental IgG directed against the murine homologue of the NC16A domain of hBPAG2.
Figure 7.

Tg skin grafts on Wt recipients developed neutrophil-rich leukocytic infiltrates and edema within the papillary dermis that eventuated in clefting and/or blister formation within the epidermal BM at sites where in situ deposits of IgG and C3 were localized. a: Tg and Wt skin was grafted onto the opposite flanks of age and gender-matched Wt, Tg, and MHC II -/- recipients; biopsies were obtained between 6 and 25 days after grafting for light and IF microscopy studies. All specific alterations (i.e., those unrelated to wounds at the base and margins of grafts) developed in Tg grafts on Wt recipients (shown here). Such alterations were evident as early as 17 days after grafting and consisted of neutrophil-rich infiltrates within the papillary dermis. b: Alterations in Tg skin grafts on Wt recipients progressed to cleft and/or subepidermal blister formation within 20 to 25 days (top panels). Direct IF microscopy revealed in situ deposits of murine IgG and C3 in epidermal BMs of such grafts (arrows, bottom panels). Immunoreactants were not identified in the epidermal BMs of recipient skin adjacent to, or at distance from, Tg grafts, or in control grafts of Wt skin placed on the opposite flanks of Wt (insets, bottom panels, dotted white lines outline epidermal BM), Tg, or MHC II -/- recipients. Scale bars, ∼25 μm.
Involution and loss of Tg skin grafts on CD4 -/- mice were delayed and correlated with production of anti-BM IgG rather than exposure to immune CD4+ T cells
Though light microscopy studies indicated that leukocytic infiltrates in involuting Tg skin grafts on Wt recipients were neutrophil-rich, it was of interest to explore the possibility that CD4+ T cells were contributing to the injury and loss of such grafts. To address this question, Tg skin was grafted onto CD4 -/- (and Wt, control) mice; recipients were followed for anti-BM IgG production and graft viability. Of note, prior studies showed that CD4 -/- mice possess a population of CD4-, CD8-, αβ+ T cells that provide “help” in development of specific antibody (Rahemtulla et al., 1994).
Longitudinal studies found that CD4 -/- mice grafted with Tg skin consistently developed anti-BM IgG directed against the epidermal side of 1 M NaCl split human skin (maximal titers, 160 to 1280) (Figure 8). In contrast to the kinetics of specific IgG production in controls, anti-BM IgG production in CD4 -/- mice was delayed, appearing between 42 and 52 days (mean, 47 days) after graft placement. Interestingly, 12 to 15 days following the detection of anti-BM IgG in CD4 -/- recipients, Tg skin grafts displayed prompt involution and loss (Figure 8). In sum, while Tg skin grafts placed on CD4 -/- mice survived longer than those placed on Wt controls, the profiles of graft loss in both CD4 -/- and Wt recipients displayed similar kinetics once IgG reactive with the epidermal side of 1 M NaCl split human skin was detected in their circulation.
Figure 8.

Involution and loss of Tg skin grafts on CD4 -/- mice correlated with production of anti-BM IgG rather than exposure to CD4+ T cells (naïve or “immune”). Longitudinal studies showed that Tg skin did elicit anti-BM IgG in CD4 -/- recipients (solid circles), but that its production was delayed (day 44±5) in comparison to development of anti-BM IgG in Wt mice grafted with Tg skin (day 16±2). Accelerated graft loss in CD4 -/- recipients correlated with production of anti-BM IgG and progressed to completion within 12 to 15 days, a kinetic profile like that seen once specific IgG appeared in the circulation of Wt mice grafted with Tg skin. Adoptive transfer studies showed that involution and loss of Tg skin grafts on CD4 -/- recipients infused with naïve (solid squares) or “immune” (solid triangles) CD4+ T cells correlated better with the development of anti-BM IgG than exposure to CD4+ T cells. Arrows and brackets indicate the time points anti-BM IgG appeared in the circulation of CD4 -/- mice engrafted with Tg skin.
To study further the potential role of CD4+ T cells in the loss of Tg skin grafts, a series of adoptive transfer studies were performed. In these studies, 5×106 naïve CD4+ T cells from Wt mice, or an equal number of “immune” CD4+ T cells purified from the draining lymph nodes of Wt mice grafted with Tg skin 10 days earlier were adoptively transferred to separate sets of CD4 -/- mice. One day later, these mice (n= 4 per group) were engrafted with Tg skin. Recipients of naïve or “immune” CD4+ T cells were then followed longitudinally for evidence of anti-BM IgG production and graft viability. CD4 -/- graft recipients infused with either naïve or “immune” CD4+ T cells developed anti-BM IgG with a kinetic profile like that seen in Wt mice grafted with Tg skin (maximal titers, 160 to 1280) (Figure 8). Graft loss in these experimental groups displayed similar kinetics and again followed the appearance of circulating anti-BM IgG by 12 to 15 days (Figure 8). The demonstration that anti-BM IgG production and graft loss were the same in CD4 -/- mice infused with either naïve or “immune” CD4+ T cells indicated that graft loss correlated better with the production of specific IgG rather than the direct exposure of grafts to CD4+ T cells (naïve or “immune”).
Discussion
hBPAG2 Tg mice displayed a normal phenotype, life span, and breeding capacity. Light, IF, and immunoelectron microscopy studies showed that these mice expressed hBPAG2 in murine epidermal BM with appropriate immunoreactivity and localization, and that this expression did not impair the homeostasis of murine epidermis or the ultrastructure of murine epidermal BM. Grafts of Tg skin placed on Wt and MHC class I -/- mice reliably elicited IgG that bound epidermal BMs in human and Tg skin, but not the skin of Wt mice. The production of anti-BM IgG in these mice consistently occurred 14 to 18 days after graft placement and was sustained for as long as 330 days. Grafts of Wt skin did not elicit production of anti-BM IgG in syngeneic, gender-matched recipients, showing that the grafting procedure itself did not account for production of anti-BM IgG in Wt and MHC class I -/- mice grafted with Tg skin. Immunoblot and immunoadsorption studies showed that the sera of Wt mice grafted with Tg skin contained IgG that bound the immunodominant NC16A domain as well as other epitopes within hBPAG2. Immunoblot and immunoprecipitation studies confirmed that IgG from Wt mice grafted with Tg skin bound hBPAG2 but no other keratinocyte proteins including BPAG1. Grafts of Tg skin did elicit production of low titers of anti-BM IgM (but not anti-BM IgA) in Wt mice (data not shown). Such IgM bound the epidermal side of 1 M NaCl split human skin (titers < 80), appeared in the circulation of graft recipients within 15 days, and deposited in the epidermal BMs of Tg skin grafts (data not shown). Tg skin grafts did not elicit production of anti-BM IgG (or IgM) in MHC class II -/- mice.
Tg skin grafts placed on Wt and MHC class I -/- recipients were consistently lost within 26 to 34 days. Accelerated graft involution in these mice directly correlated with production of anti-hBPAG2 IgG 14 to 18 days after graft placement. Longitudinal and internally controlled clinical as well as light and IF microscopy studies of Tg and Wt skin grafts placed on the opposite flanks of Wt, Tg, and MHC class II -/- recipients found that specific alterations were confined to Tg skin grafts on Wt recipients. Such alterations were noted 17 days after graft placement, localized within the papillary dermis, and consisted of neutrophil-rich leukocytic infiltrates and dermal edema. Twenty to 25 days following graft placement, light microscopy studies identified moderately dense neutrophil-rich leukocytic infiltrates within the papillary dermis, microscopic clefts within epidermal BM, and frank subepidermal blister formation in grafted skin. These alterations were not observed in: 1) Wt skin adjacent to, or at distance from, involuting Tg skin grafts; 2) Tg skin grafts placed on Tg or MHC class II -/- recipients; or 3) Wt skin grafts placed on gender-matched, syngeneic Wt, Tg, or MHC class II -/- recipients. All specific histologic alterations in involuting Tg skin grafts on Wt recipients were focused at sites where anti-BM IgG and C3 were deposited in graft epidermal BMs. Just as there were no specific alterations in recipient skin adjacent to, or distant from, involuting grafts, all in situ deposits of immunoreactants were localized within the epidermal BMs of Tg skin grafts. These histologic and immunopathologic findings were characteristic of immune complex-mediated tissue injury like those seen in lesional skin from patients with BP, neonatal mice challenged with experimental IgG directed against the murine homologue of human NC16A, and cryosections of normal human skin treated with IgG from patients with BP, a fresh source of complement, and viable human neutrophils (Gammon et al., 1982; Liu et al., 1993; Sitaru et al., 2002; Yancey and Egan, 2000).
MHC class I -/- mice grafted with Tg skin also developed hBPAG2-specific IgG and graft loss of the same character seen in Wt mice grafted with Tg skin. The lack of such alterations in MHC class II -/- mice grafted with Tg skin demonstrated that hBPAG2-specific IgG and graft loss were dependent upon MHC class II:CD4+ T cell interactions and occurred independent of CD8+ T cells. Grafting studies in CD4 -/- mice confirmed that such mice are capable of specific IgG production and that graft involution and loss in these recipients again correlated with development of anti-hBPAG2 IgG. This conclusion was substantiated by showing that the involution and loss of Tg skin grafts in CD4 -/- mice infused with naïve or immune CD4+ T cells correlated more closely with hBPAG2-specific IgG production than the administration of the T cells.
Prior studies demonstrated that skin allografts with a major histocompatibility antigen difference can be rejected within 14 days, while allografts differing in only a minor histocompatibility antigen tend to be rejected more slowly (Graff et al., 1966). The kinetics for loss of hBPAG2 Tg skin grafts in this model approximated the median survival time of skin grafts displaying experimental minor histocompatibility antigens such as β-galactosidase, HIV-1 genome, or ovalbumin (median skin graft survival times ranging from 25 to 33 days) (Dumois et al., 1995; Ehst et al., 2003; Valujskikh et al., 1999). Our experiments utilizing MHC I and II -/- mice demonstrated that CD8+ T cells were not responsible for graft loss and that presentation of hBPAG2 on host antigen presenting cells' MHC II molecules was required for initiation of specific IgG production and graft loss. In contrast to other experimental grafting paradigms exploring responses to other minor histocompatibility antigens, the loss of hBPAG2 Tg skin grafts was not dependent upon the action of both CD4+ and CD8+ T cells (Rees et al., 1990; Wettstein and Korngold, 1992). Moreover, since CD4 -/- recipients lost Tg grafts following development of hBPAG2-specigfic IgG, CD4+ T cells in this model appear to be primarily responsible for providing “help” for production of pathogenic antibody.
Studies in this and other experimental models suggest that ex vivo gene therapy approaches involving the introduction of BPAG2 into the skin of EB patients who are “null” for this protein may elicit humoral immune responses that are undesirable and/or pathogenic (Lu and Ghazizadeh, 2005). Findings in this model suggest that such responses would be specific to tissue bearing hBPAG2. There was no evidence that Tg skin grafts placed on Wt (or other) recipients led to a break in tolerance to any other murine (or human) skin protein. The consistent humoral immune response seen in Wt mice grafted with Tg skin suggests that this experimental model holds utility for the investigation of interventions that may block, attenuate, or silence production of antibodies directed against proteins in epidermal BM.
Methods
Generation of an hBPAG2 Transgene Vector
Total RNA from subconfluent monolayer cultures of human keratinocytes (HKs) was reverse transcribed using random hexamers. The resulting cDNA was amplified by PCR using 5′ and 3′ primers corresponding to regions of hBPAG2 cDNA (Genbank accession number M91669). Multiple cDNAs were produced and ligated to yield a 4.5 kb cDNA construct corresponding to the full length of hBPAG2. The latter was sequenced verified then cloned into a modified pCMVb vector 3′ to a 2.3 kb human keratin 14 promoter sequence (Shibaki et al., 2004). The resulting vector included: 1) a single SV40 intron positioned between the K14 promoter sequence and the hBPAG2 cDNA; and 2) a SV40 polyadenylation sequence following the 3′ end of the hBPAG2 cDNA. The function of this construct, as well as the integrity of its associated cDNA, was evaluated in transient transfection studies of a HK line known to harbor null mutations in COL17A1, the gene encoding BPAG2.
Transient Transfection Studies
Prior studies in our laboratory demonstrated that affected individuals in a large Austrian kindred with GABEB were homozygous for a two base pair deletion (4003delTC) in COL17A1 (Darling et al., 1997b; McGrath et al., 1996). This mutation results in a premature termination codon 86 base pairs downstream that in turn leads to nonsense-mediated mRNA decay. BPAG2 mRNA and protein were not detectable in cultured keratinocytes (or epidermal BMs) from such patients (Darling et al., 1997b). To develop a sustained (and genotype-defined) source of BPAG2-null keratinocytes, monolayer cultures of HKs derived from an affected member of this kindred were immortalized with an HPV16 E6/E7 construct. The resulting HK line was subsequently passaged greater than 50 times over a period of 12 months and stored in frozen aliquots. This immortalized BPAG2-null HK line served as a relevant cell source for characterization of our hBPAG2 transgene promoter and cDNA construct in transient transfection studies.
Generation of hBPAG2 Tg Mice
Following the verification experiments described above, the transgene construct was linearized then microinjected into fertilized oocytes of C57BL/6Ncr mice. The latter were subsequently transplanted into pseudopregnant C57BL/6Ncr females to yield chimeras. This approach was applied to yield Tg mice of a well-characterized strain that (other than the transgene itself) would be syngeneic to gender-matched wild type (Wt) C57BL/6Ncr mice. All experimental studies in animals were approved by the senior author's Institutional Review Board.
Immunogold Studies
Post-embedding immunoelectron microscopy of cryofixed and cryosubstituted human, Tg, and Wt murine skin was performed as described previously (Shimizu et al., 1992; Shimizu et al., 1989) with slight modification. Small pieces of fresh skin were cryoprotected with 20% glycerol/phosphate-buffered saline at 4°C for 30 min and cryofixed by plunging into liquid propane cooled to −190°C (KF80, Reichert-Jung, Vienna, Austria). Skin samples were cryosubstituted (CS-auto, Reichert-Jung) with methanol at −80°C for 120 h and embedded in Lowicryl K11M (Chemische Weke Lowi, Waldkraiburg, Germany) at −60°C. Specimens were polymerized by ultraviolet irradiation at −60°C for 72 hr and at room temperature for another 72 h. Ultra thin sections were incubated 1 h at 37°C with rabbit antiserum directed against the carboxyl terminus of hBPAG2 (or normal rabbit serum [control], both at 1:2 dilution) (Masunaga et al., 1997). After being washed, each section was placed on a drop of 5-nm-gold-labeled goat anti-rabbit IgG (1: 200 dilution) at 37°C for 1 h then washed with distilled water. The sections were counterstained with saturated uranyl acetate for 2 min.
Skin Grafting Studies
Tail skin was harvested from Tg and Wt (control) C57BL/6Ncr mice and grafted onto the backs of age- and gender-matched Wt and C57BL/6Ncr control recipients (Rosenberg et al., 1994). The latter included: B2m -/- mice created by inactivation of the beta-2 microglobulin gene, resultantly deficient in MHC I and lacking mature CD8+ T cells (hereafter referred to as MHC I -/-) (Taconic, Hudson, NY) (Zijlstra et al., 1990); Abb mice created by disruption of the H2-Ab1 gene, resultantly deficient in MHC II and lacking mature CD4+ T cells (hereafter referred to as MHC II -/-) (Taconic) (Grusby et al., 1991); and Cd4tm1Mak mice created by targeted mutation in the Cd4 gene, resultantly deficient in CD4+ T cells (hereafter referred to as CD4 -/- mice) (Jackson Laboratories, Bar Harbor, ME) (Rahemtulla et al., 1991). In some experiments, samples of Tg and Wt skin were grafted onto opposite flanks on the same recipient; in other experiments, recipients received only one graft. Bandages were removed at day 7 and grafts were inspected daily until day 30, then weekly, for viability and size. Skin grafts were graded as “lost” if their area became 20% or less of their original size (McFarland et al., 2003). Biopsies were obtained at various time points 6 to 25 days after grafting and placed in 4% buffered formalin. Such samples were embedded in paraffin, sectioned at 5 μm thickness, stained with hematoxylin and eosin, and studied by light microscopy. Matching biopsies were immediately placed in Tissue-Tek OCT Compound (Miles Inc., Elkhart, IN), frozen, and stored at -70°C; 6 μm cryosections of such samples were studied by direct IF microscopy.
IF Microscopy
Direct IF microscopy studies of murine skin cryosections were performed as described previously using fluorescein isothiocyanate- (FITC) conjugated goat anti-mouse Igs (reactive with IgG and IgM, cognate and crossreactive antigens, respectively; the latter displaying 50% of the reactivity of the former in comparative ELISAs [Biosource, Inc., Camarillo, CA]), FITC-conjugated rat anti-mouse IgG (Biosource, Inc.), and FITC-conjugated goat anti-mouse C3 (ICN Biomedicals, Aurora, OH) (Lazarova et al., 1996). Serial dilutions of murine sera were tested against cryosections of intact or 1 M NaCl split human skin (and in selected experiments, intact murine skin) by indirect IF microscopy as described previously (Lazarova et al., 1996). Second-step antibodies included FITC-conjugated goat anti-mouse Igs and FITC-conjugated rat anti-mouse IgG.
Complement fixation studies
Sera from Wt mice grafted with Tg skin (anti-BM IgG titers ranging from 640 to 2560) were tested for their ability to fix murine C3 to epidermal BM in 1M NaCl split skin as described previously (Jordon et al., 1976; Katz et al., 1976).
Immunoblotting
Cultured HKs and murine keratinocytes were extracted as described previously (Labib et al., 1986; Roh et al., 2000). Constructs encoding a NC16A-GST fusion protein (AA 490 to 562 of hBPAG2 [a gift of Dr. George Giudice, Medical College of Wisconsin]) or GST alone were expressed in Escherichia coli strain DH5α and purified by glutathione-agarose affinity chromatography using BugBuster GST Bind Purification Kit (Novagen, Madison, WI) (Giudice et al., 1993). Keratinocyte extracts were reduced and applied to 6% Novex Tris-Glycine SDS-polyacrylamide minigels (Invitrogen, Carlsbad, CA); NC16A-GST and GST were reduced and applied to 4-12% NuPage Bis-Tris SDS-polyacrylamide minigels (Invitrogen). Proteins were transferred to nitrocellulose paper (Invitrogen) by electrophoresis and immunoblotted as described previously (Lazarova et al., 1996; Roh et al., 2000). Alkaline phosphatase-conjugated goat anti-mouse, anti-rabbit, or anti-human IgG (all at 1:1000 dilution [Biosource, Inc.]) were used as second-step antibodies. Immunoblots were developed for 3 min (alkaline phosphatase-conjugate substrate kit; Bio-Rad Laboratories) and then washed extensively with Tris-buffer saline. Sera from patients with BP, normal volunteers, mice grafted with Tg or Wt skin, Wt mice, a rabbit immunized with recombinant hBPAG2, and a murine monoclonal antibody directed against human NC16A (HD-18) were studied by immunoblotting.
Immunoadsorption studies
Immunoadsorption was performed using a liquid phase protocol as described previously (Martins et al., 1990). Immune sera diluted in PBS were incubated with various concentrations of recombinant GST or NC16A-GST at room temperature for 2 h. Reaction mixtures were microcentrifuged at 10,000 × g for 15 min at 4 °C. Immunoadsorbed sera were tested in: 1) immunoblot studies of HK extracts and recombinant GST-NC16A; and 2) indirect IF microscopy studies of 1 M NaCl split human skin.
Immunoprecipitation studies
Immunoprecipitation studies of biosynthetically radiolabeled HK extracts were performed as described previously (Lazarova et al., 1996).
Quantitation of Antigen-Specific T Cell Proliferative Responses in Wt Mice Grafted with Tg skin
Tg skin was placed on both flanks of Wt recipients; 10 days later, axillary and inguinal lymph nodes were collected, pooled, put into suspension, and passed over a mouse CD4 subset column (R & D Systems, Minneapolis, MN) to yield CD4+ T cells. CD4+ T cells were plated at 2×105 cells/well in 96-well flat bottom microplates (Costar, Corning Inc., Corning, NJ) in cRPMI 1640 containing 10% heat-inactivated fetal calf serum supplemented with 50 μM 2-mercaptoethanol (Sigma), 10 mM HEPES, pH 7.4, 2 mM glutamine, 100 U/ml penicillin, 100 μg/ml streptomycin and 20 μg/ml gentamycin (all from Life Technologies, Chagrin Falls, OH). As stimulators, splenocytes from naïve C57BL/6Ncr mice were γ-irradiated with 3000r and added to the CD4+ T cells at the concentration of 1×106/well. Various concentrations of recombinant NC16A-GST, GST alone, or PBS were added to the wells. On day 3 of co-culture, [3H]thymidine (Amersham, Biosciences Corp., Piscataway, NJ) (1 μCi/well) was added and T cell proliferative responses were determined by the incorporation of [3H]thymidine during the last 16 h of culture using a gas ionization counter (Packard, Meriden, CT). Results are presented as the mean (± SEM) of assays performed in triplicates.
Adoptive transfer studies
CD4 -/- mice received by intravenous injection 5×106 CD4+ T cells (>95% purity, R & D Systems) from naïve or immune (i.e., 10 days previously grafted with Tg skin) Wt mice. The following day, Tg skin was placed on CD4 -/- recipients; mice were followed for anti-BM IgG production and graft loss.
Supplementary Material
Tg skin grafts on Wt recipients developed neutrophil-rich leukocytic infiltrates and edema within the papillary dermis. Tg and Wt skin was grafted onto the opposite flanks of age and gender-matched Wt, Tg, and MHC II -/- recipients (upper, middle, and lower panels, respectively). Biopsies were obtained 17 days after grafting for light microscopy studies. All specific alterations (i.e., those unrelated to wounds at the base and margins of grafts) developed in Tg grafts on Wt recipients and consisted of neutrophil-rich infiltrates within the papillary dermis (top left panel). Scale bars, ∼25 μm.
Acknowledgments
This work was supported in part by NIH grant RO1 AR048982 (KBY).
Abbreviations
- BPAG2
bullous pemphigoid antigen 2
- BP
bullous pemphigoid
- Tg
transgenic
- hBPAG2
human BPAG2
- BM
basement membrane
- Wt
wild type
- NC16A
the 16th noncollagenous domain of BPAG2
- GABEB
generalized atrophic benign epidermolysis bullosa
- IF
immunofluorescence
- HKs
human keratinocytes
- FITC
fluorescein isothiocyanate
- HD
hemidesmosome
- LL
lamina lucida
- LD
lamina densa
Footnotes
Conflict of Interest: The authors state no conflict of interest.
References
- Darling TN, Bauer JW, Hintner H, Yancey KB. Generalized atrophic benign epidermolysis bullosa. Adv Dermatol. 1997a;13:87–119. [PubMed] [Google Scholar]
- Darling TN, McGrath JA, Yee C, Gatalica B, Hametner R, Bauer JW, et al. Premature termination codons are present on both alleles of the bullous pemphigoid antigen 2/type XVII collagen gene in five Austrian families with generalized atrophic benign epidermolysis bullosa. J Invest Dermatol. 1997b;108:463–8. doi: 10.1111/1523-1747.ep12289718. [DOI] [PubMed] [Google Scholar]
- Dumois JA, VanderVegt FP, Kopp JB, Marinos NJ, Rooney JF, Notkins AL. Transplantation of skin from human immunodeficiency virus type 1-transgenic mice to normal congenic mice results in graft rejection. J Infect Dis. 1995;172:232–4. doi: 10.1093/infdis/172.1.232. [DOI] [PubMed] [Google Scholar]
- Ehst BD, Ingulli E, Jenkins MK. Development of a novel transgenic mouse for the study of interactions between CD4 and CD8 T cells during graft rejection. Am J Transplant. 2003;3:1355–62. doi: 10.1046/j.1600-6135.2003.00246.x. [DOI] [PubMed] [Google Scholar]
- Franzke CW, Bruckner P, Bruckner-Tuderman L. Collagenous transmembrane proteins: recent insights into biology and pathology. J Biol Chem. 2005;280:4005–8. doi: 10.1074/jbc.R400034200. [DOI] [PubMed] [Google Scholar]
- Franzke CW, Tasanen K, Schumann H, Bruckner-Tuderman L. Collagenous transmembrane proteins: collagen XVII as a prototype. Matrix Biol. 2003;22:299–309. doi: 10.1016/s0945-053x(03)00051-9. [DOI] [PubMed] [Google Scholar]
- Gammon WR, Merritt CC, Lewis DM, Sams WM, Jr, Carlo JR, Wheeler CE., Jr An in vitro model of immune complex-mediated basement membrane zone separation caused by pemphigoid antibodies, leukocytes, and complement. J Invest Dermatol. 1982;78:285–90. doi: 10.1111/1523-1747.ep12507222. [DOI] [PubMed] [Google Scholar]
- Giudice GJ, Emery DJ, Zelickson BD, Anhalt GJ, Liu Z, Diaz LA. Bullous pemphigoid and herpes gestationis autoantibodies recognize a common non-collagenous site on the BP180 ectodomain. J Immunol. 1993;151:5742–50. [PubMed] [Google Scholar]
- Graff RJ, Hildemann WH, Snell GD. Histocompatibility genes of mice. VI. Allografts in mice congenic at various non-H-2 histocompatibility loci. Transplantation. 1966;4:425–37. [PubMed] [Google Scholar]
- Grusby MJ, Johnson RS, Papaioannou VE, Glimcher LH. Depletion of CD4+ T cells in major histocompatibility complex class II-deficient mice. Science. 1991;253:1417–20. doi: 10.1126/science.1910207. [DOI] [PubMed] [Google Scholar]
- Hirako Y, Usukura J, Nishizawa Y, Owaribe K. Demonstration of the molecular shape of BP180, a 180-kDa bullous pemphigoid antigen and its potential for trimer formation. J Biol Chem. 1996;271:13739–45. doi: 10.1074/jbc.271.23.13739. [DOI] [PubMed] [Google Scholar]
- Jordon RE, Heine KG, Tappeiner G, Bushkell LL, Provost TT. The immunopathology of herpes gestationis. Immunofluorescence studies and characterization of “HG factor”. J Clin Invest. 1976;57:1426–31. doi: 10.1172/JCI108412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katz SI, Hertz KC, Yaoita H. Herpes gestationis. Immunopathology and characterization of the HG factor. J Clin Invest. 1976;57:1434–41. doi: 10.1172/JCI108413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Labib RS, Anhalt GJ, Patel HP, Mutasim DF, Diaz LA. Molecular heterogeneity of the bullous pemphigoid antigens as detected by immunoblotting. J Immunol. 1986;136:1231–5. [PubMed] [Google Scholar]
- Lazarova Z, Yee C, Darling T, Briggaman RA, Yancey KB. Passive transfer of anti-laminin 5 antibodies induces subepidermal blisters in neonatal mice. J Clin Invest. 1996;98:1509–18. doi: 10.1172/JCI118942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z, Diaz LA, Troy JL, Taylor AF, Emery DJ, Fairley JA, et al. A passive transfer model of the organ-specific autoimmune disease, bullous pemphigoid, using antibodies generated against the hemidesmosomal antigen, BP180. J Clin Invest. 1993;92:2480–8. doi: 10.1172/JCI116856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z, Zhou X, Shapiro SD, Shipley JM, Twining SS, Diaz LA, et al. The serpin alpha1-proteinase inhibitor is a critical substrate for gelatinase B/MMP-9 in vivo. Cell. 2000;102:647–55. doi: 10.1016/s0092-8674(00)00087-8. [DOI] [PubMed] [Google Scholar]
- Lu Z, Ghazizadeh S. Host immune responses in ex vivo approaches to cutaneous gene therapy targeted to keratinocytes. Exp Dermatol. 2005;14:727–35. doi: 10.1111/j.1600-0625.2005.00351.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martins CR, Labib RS, Rivitti EA, Diaz LA. A soluble and immunoreactive fragment of pemphigus foliaceus antigen released by trypsinization of viable human epidermis. J Invest Dermatol. 1990;95:208–12. doi: 10.1111/1523-1747.ep12478018. [DOI] [PubMed] [Google Scholar]
- Masunaga T, Shimizu H, Yee C, Borradori L, Lazarova Z, Nishikawa T, et al. The extracellular domain of BPAG2 localizes to anchoring filaments and its carboxyl terminus extends to the lamina densa of normal human epidermal basement membrane. J Invest Dermatol. 1997;109:200–6. doi: 10.1111/1523-1747.ep12319337. [DOI] [PubMed] [Google Scholar]
- McFarland HI, Hansal SA, Morris DI, McVicar DW, Love PE, Rosenberg AS. Signaling through MHC in transgenic mice generates a population of memory phenotype cytolytic cells that lack TCR. Blood. 2003;101:4520–8. doi: 10.1182/blood-2002-10-3265. [DOI] [PubMed] [Google Scholar]
- McGrath JA, Darling T, Gatalica B, Pohla-Gubo G, Hintner H, Christiano AM, et al. A homozygous deletion mutation in the gene encoding the 180-kDa bullous pemphigoid antigen (BPAG2) in a family with generalized atrophic benign epidermolysis bullosa. J Invest Dermatol. 1996;106:771–4. doi: 10.1111/1523-1747.ep12345821. [DOI] [PubMed] [Google Scholar]
- McGrath JA, Gatalica B, Christiano AM, Li K, Owaribe K, McMillan JR, et al. Mutations in the 180-kD bullous pemphigoid antigen (BPAG2), a hemidesmosomal transmembrane collagen (COL17A1), in generalized atrophic benign epidermolysis bullosa. Nat Genet. 1995;11:83–6. doi: 10.1038/ng0995-83. [DOI] [PubMed] [Google Scholar]
- Nonaka S, Ishiko A, Masunaga T, Akiyama M, Owaribe K, Shimizu H, et al. The extracellular domain of BPAG2 has a loop structure in the carboxy terminal flexible tail in vivo. J Invest Dermatol. 2000;115:889–92. doi: 10.1046/j.1523-1747.2000.00136.x. [DOI] [PubMed] [Google Scholar]
- Pohla-Gubo G, Lazarova Z, Giudice GJ, Liebert M, Grassegger A, Hintner H, et al. Diminished expression of the extracellular domain of bullous pemphigoid antigen 2 (BPAG2) in the epidermal basement membrane of patients with generalized atrophic benign epidermolysis bullosa. Exp Dermatol. 1995;4:199–206. doi: 10.1111/j.1600-0625.1995.tb00245.x. [DOI] [PubMed] [Google Scholar]
- Rahemtulla A, Fung-Leung WP, Schilham MW, Kundig TM, Sambhara SR, Narendran A, et al. Normal development and function of CD8+ cells but markedly decreased helper cell activity in mice lacking CD4. Nature. 1991;353:180–4. doi: 10.1038/353180a0. [DOI] [PubMed] [Google Scholar]
- Rahemtulla A, Kundig TM, Narendran A, Bachmann MF, Julius M, Paige CJ, et al. Class II major histocompatibility complex-restricted T cell function in CD4-deficient mice. Eur J Immunol. 1994;24:2213–8. doi: 10.1002/eji.1830240942. [DOI] [PubMed] [Google Scholar]
- Rees MA, Rosenberg AS, Munitz TI, Singer A. In vivo induction of antigen-specific transplantation tolerance to Qa1a by exposure to alloantigen in the absence of T-cell help. Proc Natl Acad Sci U S A. 1990;87:2765–9. doi: 10.1073/pnas.87.7.2765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roh JY, Yee C, Lazarova Z, Hall RP, Yancey KB. The 120-kDa soluble ectodomain of type XVII collagen is recognized by autoantibodies in patients with pemphigoid and linear IgA dermatosis. Br J Dermatol. 2000;143:104–11. doi: 10.1046/j.1365-2133.2000.03598.x. [DOI] [PubMed] [Google Scholar]
- Rosenberg AS, Sechler JM, Horvath JA, Maniero TG, Bloom ET. Assessment of alloreactive T cell subpopulations of aged mice in vivo. CD4+ but not CD8+ T cell-mediated rejection response declines with advanced age. Eur J Immunol. 1994;24:1312–6. doi: 10.1002/eji.1830240611. [DOI] [PubMed] [Google Scholar]
- Shibaki A, Sato A, Vogel JC, Miyagawa F, Katz SI. Induction of GVHD-like skin disease by passively transferred CD8(+) T-cell receptor transgenic T cells into keratin 14-ovalbumin transgenic mice. J Invest Dermatol. 2004;123:109–15. doi: 10.1111/j.0022-202X.2004.22701.x. [DOI] [PubMed] [Google Scholar]
- Shimizu H, Ishida-Yamamoto A, Eady RA. The use of silver-enhanced 1-nm gold probes for light and electron microscopic localization of intra- and extracellular antigens in skin. J Histochem Cytochem. 1992;40:883–8. doi: 10.1177/40.6.1375240. [DOI] [PubMed] [Google Scholar]
- Shimizu H, McDonald JN, Kennedy AR, Eady RA. Demonstration of intra- and extracellular localization of bullous pemphigoid antigen using cryofixation and freeze substitution for postembedding immunoelectron microscopy. Arch Dermatol Res. 1989;281:443–8. doi: 10.1007/BF00510078. [DOI] [PubMed] [Google Scholar]
- Sitaru C, Schmidt E, Petermann S, Munteanu LS, Brocker EB, Zillikens D. Autoantibodies to bullous pemphigoid antigen 180 induce dermal-epidermal separation in cryosections of human skin. J Invest Dermatol. 2002;118:664–71. doi: 10.1046/j.1523-1747.2002.01720.x. [DOI] [PubMed] [Google Scholar]
- Tasanen K, Tunggal L, Chometon G, Bruckner-Tuderman L, Aumailley M. Keratinocytes from patients lacking collagen XVII display a migratory phenotype. Am J Pathol. 2004;164:2027–38. doi: 10.1016/S0002-9440(10)63762-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valujskikh A, Matesic D, Heeger PS. Characterization and manipulation of T cell immunity to skin grafts expressing a transgenic minor antigen. Transplantation. 1999;68:1029–36. doi: 10.1097/00007890-199910150-00022. [DOI] [PubMed] [Google Scholar]
- Van den Bergh F, Giudice GJ. BP180 (type XVII collagen) and its role in cutaneous biology and disease. Adv Dermatol. 2003;19:37–71. [PubMed] [Google Scholar]
- Wettstein PJ, Korngold R. T cell subsets required for in vivo and in vitro responses to single and multiple minor histocompatibility antigens. Transplantation. 1992;54:296–307. doi: 10.1097/00007890-199208000-00020. [DOI] [PubMed] [Google Scholar]
- Yancey KB. The pathophysiology of autoimmune blistering diseases. J Clin Invest. 2005;115:825–8. doi: 10.1172/JCI24855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yancey KB, Egan CA. Pemphigoid: clinical, histologic, immunopathologic, and therapeutic considerations. Jama. 2000;284:350–6. doi: 10.1001/jama.284.3.350. [DOI] [PubMed] [Google Scholar]
- Zijlstra M, Bix M, Simister NE, Loring JM, Raulet DH, Jaenisch R. Beta 2-microglobulin deficient mice lack CD4-8+ cytolytic T cells. Nature. 1990;344:742–6. doi: 10.1038/344742a0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Tg skin grafts on Wt recipients developed neutrophil-rich leukocytic infiltrates and edema within the papillary dermis. Tg and Wt skin was grafted onto the opposite flanks of age and gender-matched Wt, Tg, and MHC II -/- recipients (upper, middle, and lower panels, respectively). Biopsies were obtained 17 days after grafting for light microscopy studies. All specific alterations (i.e., those unrelated to wounds at the base and margins of grafts) developed in Tg grafts on Wt recipients and consisted of neutrophil-rich infiltrates within the papillary dermis (top left panel). Scale bars, ∼25 μm.