

Abstract
Phthalate dioxygenase (PDO) and its reductase are parts of a two-component Rieske dioxygenase system that initiates the aerobic breakdown of phthalate by forming cis-4,5-dihydro-4,5-dihydroxyphthalate (DHD). Aspartate D178 in PDO, located near its ferrous mononuclear center, is highly conserved among Rieske dioxygenases. The analogous aspartate has been implicated in electron transfer between the mononuclear iron and Rieske center in naphthalene dioxygenase (Parales, R.E. et. al. (1999) J Bacteriol 181, 1831–1837) and in substrate binding and oxygen reactivity in anthranilate dioxygenase (Beharry, Z.M. et. al (2003), Biochemistry 42, 13625–13636). The effects of substituting D178 in PDO with alanine or asparagine on the reactivity of the Rieske centers, phthalate hydroxylation, and coupling of Rieske center oxidation to DHD formation were studied previously (Pinto, A., Tarasev, M., and Ballou, D. P. (2006) Biochemistry in press). This work describes effects that D178N and D178A substitutions have on the interactions between the Rieske and mononuclear centers in PDO. The mutations affected protonation of the Rieske center histidine and conformation of subunits within the PDO multimer to create a more open structure with more solvent-accessible Rieske centers. When the Rieske centers in PDO were oxidized, D178N and D178A substitutions disrupted communication between the Rieske and Fe-mononuclear centers. This was shown by the lack of perturbations of the UV-vis spectra on phthalate binding to the D178N and D178A variants, as opposed to that observed in WT PDO. However, when the Rieske center was in the reduced state, communication between the centers was not disrupted. Phthalate binding similarly affected the rates of oxidation of the reduced Rieske center in both WT and mutant PDO. Nitric Oxide binding at the Fe(II) mononuclear center, as detected by EPR spectrometry of the Fe(II) nitrosyl complex, was regulated by the redox state of the Rieske center. When the Rieske center was oxidized in either WT or D178N PDO, NO bound to the mononuclear iron in the presence or absence of phthalate. However, when the Rieske center was reduced, NO bound only when phthalate was present. These findings are discussed in terms of the “communication functions” performed by the bridging Asp-178.
Phthalate dioxygenase (PDO) and its reductase are parts of a two-component enzyme system (PDS) in Burkholderia cepacia DB01 that initiates the aerobic breakdown of phthalate by forming cis-4,5-dihydro-4,5-dihydroxyphthalate (DHD). PDS comprises the monomeric phthalate dioxygenase reductase (PDR), an enzyme that contains both FMN and a plant-type [2Fe–2S] ferredoxin, and a Rieske dioxygenase (PDO), an α6 multimer that contains Rieske and ferrous mononuclear centers. Mononuclear iron and Rieske centers on each subunit in Rieske oxygenases are shown to be separated by more than 40 Å (2), making direct electron transfer unfavorable (3). However, as shown by crystal structures of naphthalene (NDO) (2), biphenyl (BPDO) (4), nitrobenzene (NBDO) (5), carbazole 1,9a- (CarDO) (6), cumene (CumDO) (7) dioxygenases, and 2-oxoquinoline-8-monooxygenase (OMO) (8), the mononuclear centers are only about 12 Å from Rieske centers located on adjacent subunits of the enzyme. One histidine ligated to the Rieske center in these enzymes (His104 in NDO) is in van der Waals contact with a histidine ligated to the iron mononuclear center (His213 in NDO) on the adjacent subunit. These histidines are also in van der Waals contact with the so-called “bridging” aspartate (Asp-205 in NDO), as well as in van der Waals contact with the substrate bound at the mononuclear center (2) (Figure 1).
Figure 1.
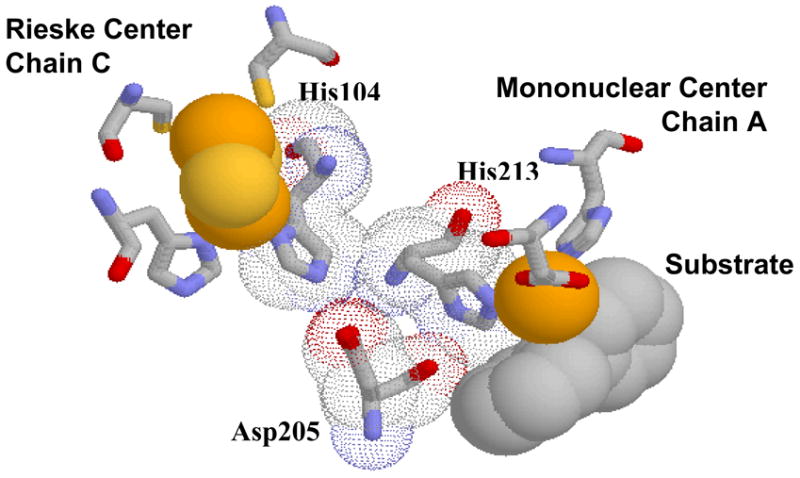
Proposed structure of the Rieske-mononuclear center interface of PDO based on substrate-bound NDO crystal structure. Picture built in Protein Explorer based on the 1NDO.pdb.
The reactivity of the mononuclear center is sensitive to the redox state of the Rieske center. Oxygen reacts poorly with the ferrous mononuclear center if the Rieske center is not reduced, and the presence of substrate greatly enhances oxygen reactivity (9). The binding of NO to ferrous sites has been previously used as a model for oxygen binding in heme as well as in Rieske proteins (9–11). Certain caveats should be noted concerning the exact geometry of binding in the non-heme enzymes; for example, while the binding of oxygen in NDO was shown to be side-on to the mononuclear iron (12), the binding to NO was shown to occur in an end-on fashion (13). Using NO as a limited model for oxygen binding at the Fe(II) of the mononuclear center , it was found that NO can bind to the mononuclear Fe(II) if the Rieske center is oxidized, but not when it is reduced, unless substrate is also bound at the mononuclear center (9, 14). Thus, there is important communication between the Rieske and mononuclear sites, and this communication has been proposed to be due to His-104, His-213, and Asp-205 in NDO (9). A similar type of regulation is thought to prevent reaction with oxygen unless substrate is present, thus inhibiting the production of reactive oxygen radicals by the enzyme.
Recent X-ray structural studies of the oxygenase component of 2-oxoquinoline-8-monooxygenase (OMO) have revealed how interactions between the analogous residues (His-108, His-221 and Asp-218 in OMO) can be modulated by the redox state of the Rieske center (8), thus providing a possible explanation for how NO (and by extension O2) binding at the mononuclear site is regulated by the redox state of the Rieske center and by substrate binding. The crystal structure of OMOox shows that while Asp-218 is within hydrogen bonding distance to the Nδ atom of His-221, it presumably is not involved in hydrogen bonds with the side chain of His-108. Binding of the substrate (2-oxoquinoline) to OMOox results in a closed conformation of the active site pocket with no pathway for dioxygen to diffuse to the mononuclear Fe(II). Reduction of the Rieske center, apparently, triggers the protonation of the mononuclear iron ligand His-l08, which then interacts with Asp-218, resulting in a displacement of both the mononuclear Fe(II) (possibly associated with the change in its coordination state detected previously (15–17)) and the Rieske center ligand, His-221. This conformational shift increases the accessible surface at the metal site and opens a pathway for oxygen to diffuse to the mononuclear center. It should be noted that these results correlate with the observed structures for other Rieske dioxygenases (2, 4, 5), in which the Rieske centers are likely to have been in the reduced state due to reduction that occurred in the electron beam, similar to that shown for NDO (18).
Consistent with its important role in the activation of the mononuclear site (8), the “bridging” aspartate was also found to be critically important for catalysis in PDO (1), NDO (19), AntDO (20), and TDO (21). Substitutions of this aspartate by other amino acids resulted in practically inactive enzymes (no turnover activity detected in AntDO and TDO, and the activity of both NDO and PDO was severely diminished), with the magnitude of the decrease in the turnover rates in some cases dependent on the nature of the substitution. This loss of activity can be partially attributed to poor iron binding in the mononuclear center for some AntDO and TDO variants (20, 21), whereas in other cases (NDO mutants D205A,E,N,Q as well as in D218A, N mutants in AntDO), the iron content of the mononuclear centers was essentially unchanged from that in the WT enzymes. In both NDO and AntDO, the Rieske centers could be reduced by their respective electron donor ferredoxins, but the rates of reduction were not measured. It was proposed for the NDO system (19) that the lack of activity of the “bridging” aspartate variants was due to impaired electron transfer between the Rieske center of one subunit and the mononuclear center located on another subunit. This, however, did not seem to be the case for the AntDO system, where Beharry et. al. (20) showed that the Rieske center oxidation in the dioxygenase remains similar to that in the WT enzyme. To account for decreased reactivity of the mutant AntDO they proposed that Asp-218 not only affects the protonation state of the Rieske center but also the oxygen reactivity at the mononuclear site and/or the binding of the substrate. The overall sequence of PDO is quite different from those of NDO, BDO, and OMO. However, the Rieske and the mononuclear domains of PDO are highly homologous in all these dioxygenases, which are structurally very similar in their domain interactions. Coupled with the fact that they all catalyze similar aromatic cis-dihydroxylations, it is likely that the structural basis for the cooperation between arrangements between the subunits within the Rieske center of PDO subunits also could be similar. Ligands for the Fe in both the Rieske center and in the mononuclear center, as well as the “bridging” aspartate (Asp-178 in PDO), are conserved, which appears to be a uniform feature for most Rieske oxygenases. A previously presented model for the subunit arrangement in the PDO multimer (11, 14) suggests that this aspartate can be a part of the electron transfer pathway from the Rieske center to the mononuclear site. Our previous work established that Asp-178 substitutions with alanine and glutamate result in weakened Fe(II) binding at the mononuclear center of PDO variants as well as in decreased rates of Rieske center reduction by PDR and Rieske center oxidation in the presence of oxygen. These changes accounted for the observed 200-fold decrease in the reactivity of the mutant PDO (1). D178 variants were found to be capable of hydroxylating phthalate; however, the efficiency of the process was severely hampered by the weaker binding of Fe(II) at the mononuclear centers and by the significantly decreased rates of catalytically relevant electron transfer to and from the Rieske center. The present study investigates the effects D178N and D178A substitutions have on the physical state of the Rieske center and on the interactions between the Rieske and Fe-mononuclear centers in PDO.
MATERIALS AND METHODS
Plasmids containing WT PDO and D178N and D178A variants genes were constructed and the proteins were expressed and purified as described previously (14). Concentrations of enzymes were determined spectrophotometrically using Δε575 = 2.38 mM−1cm−1 and Δε466= 17.54 mM−1cm−1 for the extinction coefficient differences between oxidized and reduced PDO and PDR, respectively. PDO activity was determined in steady-state assays as described previously (14). When necessary, Fe(NH4)2(SO4)2 (FAS), prepared as an anaerobic solution of known concentration, was added. PDO-APO, which lacks iron in the mononuclear center, and a less stringent preparation (PDO-APO20), which contained about 2.2 Fe/monomer, were prepared as described previously (14). Iron content in PDO was determined by Inductively Coupled Plasma High Resolution Mass Spectrometry (ICP) using a Finnegan Element Instrument from Thermo Finnegan Co. Samples were diluted with 1–5%(v/v) HNO3 containing an internal standard (In) to minimize matrix variations. Sample introduction was by low-flow (nominal 100–400 μL/min, pumped) concentric nebulizer with standard spray chamber.
PDO and PDR samples for stopped-flow studies (20–40 μM before mixing) were made anaerobic by vacuum/gas exchange (purified Ar) 10 times and overlaid with ~2 psi of purified argon. Enzyme reduction was achieved by anaerobic titration with a solution of sodium dithionite. The oxidation state of enzymes was monitored using a Shimadzu UV 2051PC spectrophotometer. Experiments were performed at 22 °C in 0.1 M HEPES, pH 7.8 containing 3 mM phthalate (if necessary). Kinetic data were acquired using a Kinetic Instruments, Inc. stopped-flow spectrophotometer in single-wavelength mode. Reactions were observed at 575 nm, a wavelength that corresponds to the maximum in the difference absorption spectrum between oxidized and reduced PDO. Data collection and analysis were performed using program A, which employs the Marquardt-Levenberg algorithm (22), and was developed in our laboratory by Chung-Jen Chiu, Rong Chang, Joel Dinverno, and David P. Ballou, University of Michigan. Rates and amplitudes obtained from curve fitting (mean values from at least 3 independent experiments were within 15% of each other unless specified otherwise). Experiments were performed at 22 °C, in 0.1 M HEPES pH 7.8 buffer containing 3 mM phthalate.
For the studies of nitric oxide complex formation, anaerobic PDO samples (100–300 μM) in 0.1 M HEPES, pH 7.8, supplemented with 5 mM phthalate when necessary, were reduced by the addition of a couple of grains of solid sodium dithionite and incubated with mixing for about 5 minutes under the slow flow of nitric oxide. Care was taken to avoid the degradation of the Rieske center that may occur during longer incubation with NO and/or if the sample contained any residual oxygen. EPR studies were performed using a Brücker EMX spectrometer equipped with Brücker 4102-ST general purpose cavity. Data were collected at 4 K and 15 K with a modulation amplitude of 10 Gauss, a microwave frequency of 9.426 GHz, and microwave power of 20 mW.
Protein film voltammetry (PFV) of PDO wild-type and mutant proteins was carried out using a standard three-electrode configuration, with a working cell compartment kept under a constant stream of nitrogen. A pyrolytic graphite edge electrode was used in all experiments, with a platinum wire as a counter electrode, and a resin-body saturated calomel electrode (SCE, Accumet) was used as a reference, located in a Luggin sidearm containing 100 mM KCl and maintained at room temperature. All potentials quoted herein are against the standard hydrogen electrode (SHE, E vs. SHE = E vs. SCE + 242 mV at 20 °C). Voltammetry was conducted with an Autolab electrochemical analyzer (PG-STAT 12, Eco. Chemie, Utrecht, The Netherlands), equipped with ECD and ADC modules, controlled by GPES software (Eco Chemie). All experiments were carried out using 10 mM HEPES, pH 7.5 containing 100 mM NaCl. Electroactive protein films were generated by directly painting ~0.5 μL of a concentrated protein solution upon the electrode surface, following polishing of the electrode with 1.0 μm alumina (Buehler). Excess protein was removed with washing with distilled water, and the electrode was then submerged in the pre-chilled cell solution. Voltammograms were collected at 100 mV/s, with an initial poise of +0.150 V. Data collected were then subjected to analysis by subtracting a polynomial baseline to remove the non-faradic current, using the SOAS software package (courtesy Dr. Christophe Léger).
Differential scanning calorimetry was performed using a N-DSC-2 from Calorimetry Sci. Corp. with a scanning rate of 1°C/min. All samples (1 μg/mL PDO in 0.1M HEPES, pH 7.8, 3 mM phthalate) were dialyzed overnight against the same buffer. Dialysis buffer was used as a baseline for DSC. For base titrations, PDO samples (20 μM) were dialyzed overnight against 1 mM HEPES pH 7.2 containing 3 mM phthalate. Titrations were performed by small additions (with mixing) of 0.3 M NaOH. Changes in pH were reversed by step-wise additions of 1.4 M HEPES pH 7.0. All chemicals were of analytical grade and used without further purification.
RESULTS
Metal content and steady-state activity
WT PDO as purified contained 2.85 ± 0.15 Fe/monomer and exhibited a steady-state turnover activity of about 4 s−1 under the standard assay conditions (1). WT PDO-APO, that lacks iron at the mononuclear center, had an iron content of 2.08 ± 0.10 Fe/monomer and an activity of 0.16 s−1. D178A and D178N variants as purified had about 2.2 Fe/monomer, determined by ICP, and a steady-state activity of about 0.04 s−1, demonstrating that aspartate replacement had major effects on both iron binding and activity. Preliminary gas phase electrophoretic mobility molecular analysis and electrospray ionization mass spectrometry experiments (Joseph A. Loo, personal communication) indicated that at neutral pH the molecular weight of both WT and the D178N and D178A mutants of PDO, is about 290 kDa, consistent with a hexameric structure with no evidence of other multimeric forms.
Spectroscopic properties
The absorption spectrum of PDO in the 350–650 nm region is due to the absorbance of the [2Fe-2S] Rieske center of the enzyme. Spectra of oxidized D178A and D178N mutants are very similar to each other, and exhibit the same general features in their spectra as the WT enzyme (Figure 2). However, the peaks at 464, 515, and 605 nm were slightly broader in the variants than in WT PDO. The insert of Figure 2 shows the results of subtracting the spectrum of WT PDO from those of the mutant enzymes. These spectral changes were not simply due to the lower Fe(II) content in the mononuclear center of the variants because the perturbations of the oxidized spectrum of PDO-APO due to metal binding at the mononuclear center are at least an order of magnitude smaller than those observed in the D178A and D178N variants. Nevertheless, these perturbations of the absorption spectra due to D178N and D178A substitutions are qualitatively similar to those observed in the “bridging” aspartate variants of AntDO (D218A and D218N) (20) as well as to those observed upon exposing the oxidized Rieske protein from Thermus thermophilus to basic pH (23). In particular, the difference spectra (PDO D178N and D178A minus PDO WT) presented in Figure 2, insert, show maxima at 420, 515 and 605 nm, similar to the pH-induced changes in the spectrum of Thermus Rieske protein (maxima of the difference spectrum pH 10.3 vs. pH 6.3 as reported by Kuila et. al. (23) are at 430, 515 and 620 nm), suggesting that these spectral effects with PDO could be due to changes in a pKa of the Rieske center caused by the mutations.
Figure 2.

Absorbance spectra of recombinant PDO (35 μM) in 0.1 M HEPES pH 7.5, 3 mM phthalate. WT PDO, PDO D178N and PDO D178A. Insert: Difference spectra of PDO D178N ( ▲ ) and PDO D178A spectra minus the WT PDO-Fe(II) spectrum.
Dependence of PDO spectrum on pH
The spectra of PDO D178N (Figure 3A) and of D178A (not shown, but similar to D178N) are markedly dependent on pH. The changes in spectra of D178N and D178A variants as pH is increased are shown in Figure 3B. In the pH range 7.2–9.5 the spectral changes of D178N and D178A (Figure 3C) were similar to those observed for the Thermus Rieske protein (23) and for AntDO (20) as a function of pH, and to the spectral difference between PDO WT and the D178N and D178A variants (Figure 2, insert). These changes were reversible by the addition of pH 7.0 HEPES buffer (as described in Materials and Methods). In contrast to the large effects of pH on D178N and D178A PDO variants and on WT AntDO (20), the spectral changes for WT PDO (both with and without iron in the mononuclear center) in the pH range 7.2–9.5 were barely detectable (Figure 3B and 3C). Further increases in pH, up to ~10.5, resulted in the development of a pronounced absorbance band at about 415–420 nm in both mutant and WT samples. Analysis by ICP showed that this increase did not affect the iron content of the PDO (zero net change) but brought about a 12% increase in sulfur content in the buffer, indicating that this spectral change correlates with the loss of sulfur from the Rieske center1. Increases in pH beyond 10.5 resulted in decreased UV-vis absorbance, and were very likely associated with degradation of the Rieske centers in PDO.
Figure 3.

A. UV-vis spectra of D178N (40 μM) at pH 7.2 (••••••), 9.5 (△), 9.7 (◆ ), 10.3 ( ▲ ) and 11.3 ( ● ). B. Effect of pH on the absorbance at 575 nm; PDO WT (◆ ), (PDO-APO20) ( △ ) and D178N (▲ ). C. Difference spectra (pH 9.5 vs. pH 7.2) for WT PDO (◆ ) and D178N ( ▲ ). Samples contained 0.7 mM HEPES, 0.1 M NaCl and 3 mM phthalate. Conditions as described in Materials and Methods.
Protein Electrochemistry
Protein film voltammetry (PFV) on pyrolytic graphite electrodes was used to determine the reduction potentials of the Rieske center of mutant and wild-type PDO. As shown in Figure 4, reversible electrochemical signals were observed by PFV, indicating fast electron transfer at the electrode. Comparison of peak height with scan rate revealed a direct linear relationship, indicating that PDO remains absorbed through the course of the PFV experiment. Peak separation in all experiments was between 10 and 30 mV, indicating good reversibility. Notably, the peak width at half-height (δ) was 70 mV in both the oxidative and reductive scans, while the expected value for an n =1 electrochemical process at 4 °C is 84 mV. The physical rational for the observed narrow peak widths is unclear presently, but it is consistent with the Rieske centers exhibiting cooperative redox chemistry at the electrode. In general, PDO wild-type protein gave less electroactive coverage (approximately 30%) than did either the D178N or D178A mutant; however, at pH 7.5 the redox potentials in both the mutants and the WT protein were found to be remarkably similar (−220 ± 5 mV for the WT PDO and −235 ± 5 mV for both D178A and D178N variants). The redox potential of the Rieske center in WT PDO was significantly lower than the redox potential observed in WT AntDO (−86 mV), indicating that the solvation environments and the proton-coupled properties of the two proteins must be quite different. In AntDO a substitution of the “bridging” aspartate with alanine resulted in a change in the redox potential of the Rieske center from −86 to −187 mV, in spite of the fact that a neutral residue had replaced an anionic one (20). The change seen with PDO due to an equivalent substitution is much smaller.
Figure 4.
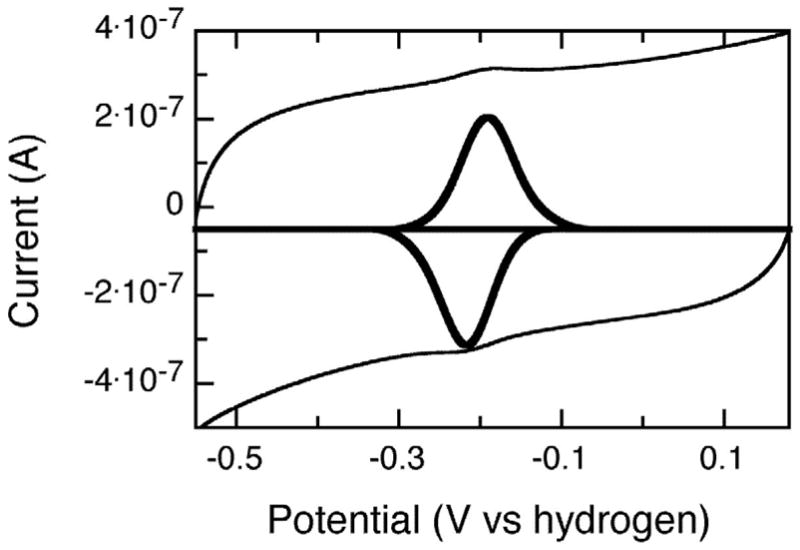
PFV response of D178N mutant of PDO adsorbed upon a graphite electrode, baseline subtracted data are inset into the voltammogram, and shown in bold. Conditions as described in Materials and Methods.
Reaction of dithionite with oxidized forms of PDO
The reduction of PDO (both WT and variants) by excess dithionite occurs in two kinetic phases (Figure 5 and Table 1). In general, reduction of the D178N and D178A variants was much faster than with WT PDO. About 85 percent of the reaction with the variants occurs at ~ 2.5 s−1 with the remaining 15 percent at < 0.1 s−1. In contrast, about 95% of the reaction with WT PDO occurs in a very slow phase (~ 3 ×10−3 s−1) (Fig. 5 and Table 1). It was also found that WT PDO-APO was reduced by dithionite more quickly than was WT PDO, but not as fast as the D178N and D178A variants (Table 1). This result suggests that the loss of iron from the mononuclear center makes the Rieske center of PDO more accessible to dithionite. However, the effect due to loss of iron is much smaller than that from the D178N and D178A substitutions. In can be noted that in contrast to the reactions with dithionite, in D178N and D178A the rates of reduction of the Rieske center by phthalate dioxygenase reductase were significantly (about 10-fold) decreased (1).
Figure 5.

Reaction of oxidized PDO (42 μM) with 1 mM sodium dithionite in 0.1 M HEPES pH 7.8, in the presence of 3 mM phthalate at 22 °C. WT PDO-Fe(II) ( ● ), WT PDO-APO ( ▲ ) and D178N ( ◆ ). Traces were recorded at 575 nm.
Table 1.
Reduction of the Rieske centers of the PDO and PDO variants by dithionite
|
k1dith, s−1 (A, %) |
k2dith, s−1 (A, %) |
|
|---|---|---|
| WT PDO-Fe(II) | 1.4
(6) |
0.003
(94) |
| WT PDO-APO | 0.06
(78) |
0.006
(23) |
| PDO D178A | 2.4
(82) |
0.013
(18) |
| PDO D178N | 2.5
(87) |
0.054
(13) |
k1dith and k2dith kinetic rates of PDO Riske center reduction by dithionite; A – Percentage contribution of the kinetic phase to the total absorbance change. Conditions as in Figure 7.
Additionally, as mentioned above, the redox potentials of the Rieske centers undergo a decrease of about 15 mV upon the substituting the “bridging” aspartate with either alanine or asparagine. This small decrease in the redox potential of the Rieske center is not likely to be responsible for such pronounced changes in the rates of reduction of the Rieske center. The fact that substitution of the “bridging” aspartate caused the rates of reduction of the Rieske center by dithionite and by PDR to change in opposite directions (increasing for dithionite and decreasing for PDR) indicates that these effects are not likely to be the result of changes in the redox state of the Rieske center. Thus, alanine and asparagine substitutions of Asp-178 appear to have resulted in conformational changes in the PDO multimer, such that the Rieske center becomes more accessible to dithionite, and presumably to solvent. The conformational changes due to the mutation appeared to be similar, but much greater than the changes in conformation due to the loss of iron from the mononuclear center.
Differential Scanning Calorimetry (DSC)
To further test if the increased solvent accessibility is related to a loosening of the PDO multimeric structure, we used DSC to determine molecular heat capacities (MHC) of WT and mutant PDO. The profile for WT protein (both with and without Fe(II) in the mononuclear center) shows a peak at about 60 °C that is probably related to the dissociation of the PDO multimer into individual monomers (Figure 6). By contrast, the DSC profile for both D178A and D178N variants shows two peaks, one at about 50 °C and the other at 55 °C, implying that the variants are less thermally stable than the WT PDO, possibly because in WT PDO, Asp-178 contributes to holding the two domains (Rieske and mononuclear iron on different subunits) together. The decreased thermal stability is also consistent with the postulated increased solvent accessibility of the Rieske centers in D178N and D178A variants that accounts for the more rapid reduction by dithionite. While the energies of the dissociation of all the individual subunits of the PDO WT multimer appear to be the same (single peak in the MHC profile), this does not appear to be the case for the mutants. Two peaks were present in the MHC profiles of the mutants, indicating the existence of two separate transitions. One possibility is hexamer dissociation into two trimers followed by dissociation into monomers.
Figure 6.

Relative molecular heat capacity as determined by differential scanning calorimetry. PDO WT ( ■ ), WT-APO20 ( ● ), D178A (▲ ) and D178N ( △ ). Conditions as described in Materials and Methods.
Perturbation of the Rieske center spectrum on substrate binding
Addition of phthalate to phthalate-free oxidized PDO WT or WT-APO results in small perturbations of the Rieske center absorbance (Figure 7). Similar additions caused no perturbations of the absorbance spectrum of the oxidized Rieske centers of the D178A and D178N variants (Figure 7). When the Rieske center was reduced, no changes in absorbance on substrate binding were detected for either WT or mutant PDO, probably due to drastically diminished extinction coefficients of the reduced enzymes, and thus, considerably changes in absorbance that would be induced by the interaction with the substrate.
Figure 7.

Effect of phthalate binding on the absorbance spectrum of PDO WT-APO (
 ) and of the D178N PDO variant ( – ). PDO (78 μM) in phthalate free buffer (0.1 M HEPES, pH 7.8) was mixed with an equal volume of buffer containing 10 mM phthalate. Presented are the difference spectra (with phthalate minus no phthalate).
) and of the D178N PDO variant ( – ). PDO (78 μM) in phthalate free buffer (0.1 M HEPES, pH 7.8) was mixed with an equal volume of buffer containing 10 mM phthalate. Presented are the difference spectra (with phthalate minus no phthalate).
Changes in Rieske center oxidation rates in the absence/presence of phthalate
In the absence of both phthalate and Fe(II) at the mononuclear center, reduced PDO variants (both D178A and D178N) reacted with oxygen extremely slowly with a rate of about 2 ×10−3 s−1 (1), similar to that of WT PDO-APO. D178A-APO and D178N-APO proteins, prepared as described in Materials and Methods, contained about 2.1 Fe/monomer while the “as purified” mutant proteins contained 2.2 Fe/monomer2. The rates of the Rieske center oxidation in the reaction of reduced D178A-APO and D178N-APO variants with oxygen were about the same as for the “as purified” enzymes (~ 1 ×10−3 s−1, Table 2 and Figure 8). In the presence of phthalate, the rate of oxidation of the Rieske center in both WT APO and D178A,N-APO enzymes increased slightly, as described previously for the WT PDO-APO (14). This was probably due to conformational changes brought about by the binding of phthalate and linked to the redox state of the Rieske center, possibly in a manner similar to that shown crystallographically for OMO (8) and by ENDOR for NDO (24). Addition of Fe(II) alone resulted in a significant increase in the rates of the Rieske center oxidation, and phthalate increased the rate further (Table 2 and Figure 8). The increased oxidation rates of the Rieske centers in PDO D178N and D178A variants, although pronounced (an increase of more than 1000-fold for the fastest phase up to about 7 s−1), were still less than that in WT PDO (up to 35 s−1 in the fast phase) (Table 2). Due to weaker binding of Fe(II) in the mononuclear center (1), a much higher iron concentration was necessary to induce this increase in the iron-depleted variants than in PDO WT-APO.
Table 2.
Reaction of reduced PDO with oxygen*.
| Phthalate | Fe per Monomer
σ = 0.1 |
Added Fe(II), μM |
k1, s−1 (A,%) |
k2, s−1 (A,%) |
k3, s−1 (A,%) |
k4, s−1 (A,%) |
|
|---|---|---|---|---|---|---|---|
| WT-APO | − | 2.08 | 0 | 1.4 ×10−3
(100) |
|||
| WT-APO | + | 2.08 | 0 | 1.8 ×10−3
(100) |
|||
| WT-APO | + | 2.08 | 20 | 35
(30) |
0.3
(70) |
||
| D178N-APO | − | 2.10 | 0 | 1.4 ×10−3
(100) |
|||
| D178N-APO | + | 2.10 | 0 | 2.3 ×10−3
(100) |
|||
| D178N-APO | − | 2.10 | 500 | 0.045 (22) | 1 ×10−2
(78) |
||
| D178N-APO | + | 2.10 | 500 | 7 ± 3
(20 ± 5) |
0.33 (26) | 0.045
(53) |
|
| D178A-APO | − | 2.15 | 0 | (1.0 ± 0.4)
×10−3 (100) |
|||
| D178A-APO | + | 2.15 | 0 | (1.5 ± 0.5)
×10−3 (100) |
|||
| D178A-APO | − | 2.15 | 500 | 0.015 ± 0.004
(30) |
0.4 ×10
−3 (70) |
||
| D178A-APO | + | 2.15 | 500 | 0.55 ± 0.1
(60) |
4 ×10
−3 (40) |
Blanks in the Table indicate that the relevant kinetic phase was not observed in the sample. Similar to the data presented in (1), k1 and k2 refer to the rates of oxidation of the Rieske centers when the reaction with oxygen occurs at the Fe(II) of the mononuclear center; k3 and k4 are characteristic of slow direct reactions of the reduced Rieske centers with oxygen. All data were fitted using a parallel reaction model, as described in Materials and Methods.
Figure 8.

Oxidation of reduced D178N-APO PDO by oxygen. D178N-APO (20 μM) was mixed with 125 μM O2 in 0.1M HEPES pH 7.8.0 at 22 °C ( ▲ ), in the presence of 3 mM phthalate in the oxygenated buffer (Δ), preincubated with 500 μM FAS (□) and in the presence of 3 mM phthalate, as well as preincubating with 500 μM FAS ( ■ ). Data for D178A are similar (not shown). All concentrations (except for the FAS) are those after mixing in the stopped-flow spectrophotometer. Traces were recorded at 575 nm.
EPR spectra of PDO-NO complexes
As mentioned in the introduction, NO mimics many of the binding properties of O2 to iron centers, and thus is often used as a model for oxygen interactions with the ferrous-containing centers (e.g., (10, 25)). Even though the mode of the binding in Rieske dioxygenases appears to be somewhat different for oxygen and NO (13), the general features of the regulation of oxygen binding deduced from the NO binding experiments are likely to be relevant for catalytic events. The complex of NO with ferrous iron bound at the mononuclear site is easily detectable by its distinct S=3/2 EPR spectrum (26, 27). Similar to NDO (9) and BZDO (28), when the Rieske center in PDO is reduced, NO binds to the mononuclear iron only when substrate is present (14). However, when the Rieske center is oxidized, NO can bind both in the presence and in the absence of the substrate (14). Such gating of NO (and by extension, possibly O2) binding by the redox state of the Rieske center and the availability of the substrate appears to be a common feature of this family of Rieske dioxygenases. At 15 K reduced samples of PDO exhibit a signal at ~350 mT characteristic for the reduced Rieske center. This signal was found to be identical for both WT and D178N variant PDO (not shown). An additional band at ~380 mT, similar to that observed previously in some incompletely Fe(II)reconstituted samples of WT PDO and for PDO WT-APO (14), was observed in all D178N samples, indicating that this enzyme has a significant portion of subunits with mononuclear centers that do not contain iron. This result correlates with the analysis of iron content in the variants that showed low levels of reconstitution of the mononuclear center. In the absence of phthalate no signal of the nitrosyl complex was detected in the EPR spectrum at 4 K for any of the PDO samples. Addition of phthalate resulted in the development of the characteristic signal around 170 mT (Figure 9). The magnitude of this signal in D178N was about 22 % of that in the WT PDO-APO20, indicating that only about 4% of the mononuclear centers in D178N actually contained Fe(II) and thus were able to form the nitrosyl complex (in WT PDO- APO20 about 20 percent of the mononuclear centers contained iron). This number seemingly contradicts the results obtained from the iron content analysis that indicated that the D178N variant contained about 2.2 Fe/monomer. However, it correlates with the amounts of iron in the mononuclear center estimated for D178N (about 4 %) based on the analysis of product in single turnover experiment (1). The presence of the nitrosyl complex in D178A was not verified because the estimated fraction of the Fe-mononuclear center that contained Fe(II) was less than 1 percent. Moreover, attempts to reconstitute the protein with iron were not successful (1), and it was not possible to significantly increase the concentration of D178A variant as the enzyme was precipitating at concentrations > 500–600 μM.
Figure 9.

EPR spectra of the reduced WT-APO20 (1) and reduced D178N (3), each incubated with NO in the presence of 5 mM phthalate. Spectra of reduced WT-APO20 and reduced D178N incubated with NO in the absence of phthalate were indistinguishable (2). Conditions as described in Materials and Methods. Data was collected at 4 K.
DISCUSSION
The recently published crystal structure of OMO (8) illustrates the importance of changes to the subunit interface in catalysis by Rieske dioxygenases. Conformational changes coupled to the redox state of the Rieske center in OMO suggest a possible mechanism for the redox-dependent regulation of catalysis at the Fe-mononuclear center of the enzyme. In particular, the structure shows that in oxidized OMO the main chain carbonyl oxygen of Asp-218, the analog of Asp-178 in PDO, is positioned within hydrogen bonding distance (2.5 Å) of the Nδ atom of His-221, one of the iron ligands in the mononuclear center. At the same time, the side chain carboxylate oxygens are about 3.6–3.8 Å away from the nearest Rieske center histidine ligand nitrogen on the neighboring subunit, so that any hydrogen bond between the two would be weak or non-existent. Reduction of the Rieske center is associated with a major conformational change in the mononuclear site helix. The main chain carbonyl oxygen of Asp-218 moves away from His-221, and the carboxylate group of Asp-218 swings 2 Å closer to the imidazole side chain (to 2.8 Å), allowing for formation of a new hydrogen bond across the dimer interface with His-108, the Rieske center ligand on the neighboring subunit. Thus, when the Rieske center is reduced, Asp-218 could provide a much stronger link for the interactions between the Rieske and the Fe-mononuclear centers.
It was surprising that substituting an anionic aspartate with a neutral residue decreased the redox potential of the Rieske center. Conformational change and solvent accessibility must have compensated the charge change brought about by the mutants. Whereas analogous substitutions of the “bridging” aspartate in AntDO result in a decrease of the redox potential of the Rieske center of about 100 mV (20), in PDO, at neutral pH, only a small decrease (about 15 mV) was observed. The decrease in redox potential for the mutant enzyme has been discussed for AntDO. It was proposed that the Rieske center of the D218A variant of AntDO, “unlike that of the wild-type protein, loses a proton from one of the histidine ligands upon oxidation at neutral pH” (20). If this concept is also true for PDO, it can be reasoned that the pKa of the histidines in the oxidized mutant protein could be shifted to a slightly lower value than that in the WT PDO.
In contrast to AntDO, the histidine ligands in WT PDO (independent of the presence of Fe(II) in the mononuclear center) are poorly accessible to solvent. That was evidenced by both the nearly invariant behavior of the Rieske center absorption spectra of WT PDO over the 7.0–9.5 pH range and by the low rates of Rieske center reduction by dithionite. Substitution of the “bridging” aspartate 178 in PDO with alanine or asparagine brought about a change in the Rieske absorption spectrum that suggested loss of a proton from the histidine Rieske center ligand. A similar conclusion was made for the “bridging” aspartate variants in AntDO (20). The absorption spectra of PDO variants, D178A and D178N, were markedly dependent on pH over the range of 7.0 to 9.5, suggesting further deprotonation of the Rieske center histidine ligands at higher pH. This effect was most likely made possible due to increased solvent access to the Rieske center relative to the WT.
Increased rates of reduction by dithionite of the D178N and D178A variants of PDO relative to that in the WT also is consistent with the hypothesis that in the mutant enzymes the Rieske centers are more accessible to solvent than in WT PDO. As discussed previously, this change in solvent accessibility is not due to the decrease in iron content of the mononuclear center, but is the result of the amino acid substitution itself. In alignment with this conclusion, WT PDO with and without iron in the mononuclear center had similar molecular heat capacity profiles, as determined by DSC. The molecular heat capacity profiles for the mutants showed overall lower temperature stability, indicating that the variant PDO multimers were less robust; in addition, the profiles indicated a possible development of an asymmetry in the arrangement of the subunits (see Figure 6). In hexameric WT PDO (e.g. with two α3 rings sitting on top of one another, as proposed in (1)) solvent access to the Rieske center is most likely severely restricted. In AntDO, NDO, and NBDO, where the three α subunits are arranged in a head-to tail, essentially planar ring, Rieske centers appear to be solvent accessible regardless of their redox state. In PDO, elimination of Fe(II) from the mononuclear center of WT PDO results in some relaxation of the multimer structure, but not enough to significantly affect the solvent accessibility of the Rieske centers. Substitution of Asp-178 with either alanine or asparagine appears to result in a major relaxation of the multimeric structure that exposes the histidine(s) that ligate iron in the Rieske center to solvent. Presumably, in analogy to OMO, this can be due, at least partially, to the elimination of the hydrogen bond between Asp-178 and the Rieske center ligand, His-92.
As mentioned above, the hydrogen bond between the “bridging” aspartate and the Rieske histidine observed in the reduced OMO is significantly weaker in OMOox than in OMOred (8). Assuming that this “bridging” aspartate is involved in the communication of conformational information between the Rieske and Fe-mononuclear centers, it might be expected that such an effect would be more pronounced when the Rieske center is reduced. In our experiments no substrate-induced perturbations of the Rieske center absorbance typical for WT PDO were observed in the D178N and D178A variants. Thus, when the Rieske center was oxidized, Asp-178 was essential for relaying of conformational information between the centers. On the other hand, when the Rieske center was reduced, substrate binding to WT and to mutant PDO initiated similar changes in the rates of oxidation of the Rieske centers. This seems to indicate that in reduced PDO the “bridging” aspartate is not critical for the communication between the subunits. This conclusion was further substantiated by the finding that the substrate regulated formation of the nitrosyl complex of NO with ferrous iron bound at the mononuclear site of reduced PDO was not lost in the Asp-178 variations.
Scheme 1.

Acknowledgments
We thank Dr. T. Huston, University of Michigan Geological Services for his help in obtaining the iron content data for PDO using ICP, James Windak, Supervisor of Instrument Services, Chemistry Department of the University of Michigan for his assistance in setting up the EPR experiments, and Dr. D. Arscott for many helpful discussions.
Supported by NIH grant (GM20877 to D.P.B.)
Abbreviations
- PDS
phthalate dioxygenase system
- PDO
phthalate dioxygenase
- WT PDO
enzyme with no substitutions in the sequence, “as purified” unless otherwise stated
- PDO-APO
phthalate dioxygenase that lacks iron in the mononuclear center
- WT PDO-APO20
WT PDO with 20% of the mononuclear centers containing Fe(II)
- PDR
phthalate dioxygenase reductase
- DHD
cis-4,5-dihydrodiol of phthalate
- FAS
ferrous ammonium sulfate
- NDO
naphthalene dioxygenase
- NDF
naphthalene dioxygenase ferredoxin
- NDS
naphthalene dioxygenase system
- TDO
toluene dioxygenase
- AntDO
anthranilate dioxygenase
- BPDO
biphenyl dioxygenase
- BZDO
benzoate dioxygenase
- NBDO
nitrobenzene dioxygenase
- CarDO
carbazole 1,9a-dioxygenase
- CumDO
cumene dioxygenase
- OMO
2-Oxoquinoline-8-monooxygenase
- OMOred and OMOox
reduced and oxidized forms of OMO
- DSC
differential scanning calorimetry
- MHC
molecular heat capacity
Footnotes
Release of sulfur into the solution was attributed to a partial degradation of the [2S-2Fe] Rieske centers in PDO. The only other potential source of sulfur would be Cys/Met residues, but it is unlikely that iron would remain bound at the Rieske center under the conditions necessary to release sulfur from cysteinyl and methionyl residues.
As discussed previously ((1), due to the weak binding of iron at the mononuclear site, most of the iron in excess of 2 Fe/monomer of the Rieske center was adventitious Fe(III), similar to that detected previously (16). Thus, changes in measured Fe content in D178N and D178A -APO enzymes, are likely to reflect not the actual changes in the Fe-mononuclear center iron content, but the decrease in the adventitious iron concentration.
References
- 1.Pinto A, Tarasev M, Ballou DP. Substitutions of the “Bridging” Aspartate 178 Result in the Profound Changes in the Reactivity of the Rieske Center of Phthalate Dioxygenase. Biochemistry. 2006 doi: 10.1021/bi060216z. in press. [DOI] [PubMed] [Google Scholar]
- 2.Kauppi B, Lee K, Carredano E, Parales RE, Gibson DT, Eklund H, Ramaswamy S. Structure of an aromatic-ring-hydroxylating dioxygenase-naphthalene 1,2-dioxygenase. Structure. 1998;6:571–586. doi: 10.1016/s0969-2126(98)00059-8. [DOI] [PubMed] [Google Scholar]
- 3.Moser CC, Keske JM, Warncke K, Farid RS, Dutton PL. Nature of biological electron transfer. Nature. 1992;355:796–802. doi: 10.1038/355796a0. [DOI] [PubMed] [Google Scholar]
- 4.Furusawa Y, Nagarajan V, Tanokura M, Masai E, Fukuda M, Senda T. Crystal structure of the terminal oxygenase component of biphenyl dioxygenase derived from Rhodococcus sp. strain RHA1. Journal of Molecular Biology. 2004;342:1041–1052. doi: 10.1016/j.jmb.2004.07.062. [DOI] [PubMed] [Google Scholar]
- 5.Friemann R, Ivkovic-Jensen MM, Lessner DJ, Yu CL, Gibson DT, Parales RE, Eklund H, Ramaswamy S. Structural insight into the dioxygenation of nitroarene compounds: the crystal structure of nitrobenzene dioxygenase. Journal of Molecular Biology. 2005;348:1139–1151. doi: 10.1016/j.jmb.2005.03.052. [DOI] [PubMed] [Google Scholar]
- 6.Nojiri H, Ashikawa Y, Noguchi H, Nam JW, Urata M, Fujimoto Z, Uchimura H, Terada T, Nakamura S, Shimizu K, Yoshida T, Habe H, Omori T. Structure of the terminal oxygenase component of angular dioxygenase, carbazole 1,9a-dioxygenase. Journal of Molecular Biology. 2005;351:355–370. doi: 10.1016/j.jmb.2005.05.059. [DOI] [PubMed] [Google Scholar]
- 7.Dong X, Fushinobu S, Fukuda E, Terada T, Nakamura S, Shimizu K, Nojiri H, Omori T, Shoun H, Wakagi T. Crystal structure of the terminal oxygenase component of cumene dioxygenase from Pseudomonas fluorescens IP01. Journal of Bacteriology. 2005;187:2483–2490. doi: 10.1128/JB.187.7.2483-2490.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Martins BM, Svetlitchnaia T, Dobbek H. 2-Oxoquinoline 8-monooxygenase oxygenase component: active site modulation by Rieske-[2Fe-2S] center oxidation/reduction. Structure (Camb) 2005;13:817–824. doi: 10.1016/j.str.2005.03.008. [DOI] [PubMed] [Google Scholar]
- 9.Wolfe MD, Parales JV, Gibson DT, Lipscomb JD. Single turnover chemistry and regulation of O2 activation by the oxygenase component of naphthalene 1,2-dioxygenase. J Biol Chem. 2001;276:1945–1953. doi: 10.1074/jbc.M007795200. [DOI] [PubMed] [Google Scholar]
- 10.Yang TC, Wolfe MD, Neibergall MB, Mekmouche Y, Lipscomb JD, Hoffman BM. Substrate binding to NO-ferro-naphthalene 1,2-dioxygenase studied by high-resolution Q-band pulsed 2H-ENDOR spectroscopy. Journal of the American Chemical Society. 2003;125:7056–7066. doi: 10.1021/ja0214126. [DOI] [PubMed] [Google Scholar]
- 11.Tarasev M, Ballou DP. Chemistry of the catalytic conversion of phthalate into its cis-dihydrodiol during the reaction of oxygen with the reduced form of phthalate dioxygenase. Biochemistry. 2005;44:6197–6207. doi: 10.1021/bi047724y. [DOI] [PubMed] [Google Scholar]
- 12.Karlsson A, Parales JV, Parales RE, Gibson DT, Eklund H, Ramaswamy S. Crystal structure of naphthalene dioxygenase: side-on binding of dioxygen to iron. Science. 2003;299:1039–1042. doi: 10.1126/science.1078020. [DOI] [PubMed] [Google Scholar]
- 13.Karlsson A, Parales JV, Parales RE, Gibson DT, Eklund H, Ramaswamy S. NO binding to naphthalene dioxygenase. J Biol Inorg Chem. 2005;10:483–489. doi: 10.1007/s00775-005-0657-1. [DOI] [PubMed] [Google Scholar]
- 14.Tarasev M, Rhames F, Ballou DP. Rates of the Phthalate Dioxygenase Reaction with Oxygen Are Dramatically Increased by Interactions with Phthalate and Phthalate Oxygenase Reductase. Biochemistry. 2004;43:12799–12808. doi: 10.1021/bi0490587. [DOI] [PubMed] [Google Scholar]
- 15.Gassner GT, Ballou DP, Landrum GA, Whittaker JW. Magnetic circular dichroism studies on the mononuclear ferrous active site of phthalate dioxygenase from Pseudomonas cepacia show a change of ligation state on substrate binding. Biochemistry. 1993;32:4820–4825. doi: 10.1021/bi00069a017. [DOI] [PubMed] [Google Scholar]
- 16.Pavel EG, Martins LJ, Ellis WRJ, Solomon EI. Magnetic circular dichroism studies of exogenous ligand and substrate binding to the non-heme ferrous active site in phthalate dioxygenase. Chemistry & Biology. 1994;1:173–183. doi: 10.1016/1074-5521(94)90007-8. [DOI] [PubMed] [Google Scholar]
- 17.Bertini I, Luchinat C, Mincione G, Parigi G, Gassner GT, Ballou DP. NMRD studies on phthalate dioxygenase: evidence for displacement of water on binding substrate. J Biol Inorg Chem. 1996:468–475. [Google Scholar]
- 18.Karlsson A, Parales JV, Parales RE, Gibson DT, Eklund H, Ramaswamy S. The reduction of the Rieske iron-sulfur cluster in naphthalene dioxygenase by X-rays. J Inorg Biochem. 2000;78:83–87. doi: 10.1016/s0162-0134(99)00213-5. [DOI] [PubMed] [Google Scholar]
- 19.Parales RE, Parales JV, Gibson DT. Aspartate 205 in the catalytic domain of naphthalene dioxygenase is essential for activity. J Bacteriol. 1999;181:1831–1837. doi: 10.1128/jb.181.6.1831-1837.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Beharry ZM, Eby DM, Coulter ED, Viswanathan R, Neidle EL, Phillips RS, Kurtz DM., Jr Histidine ligand protonation and redox potential in the rieske dioxygenases: role of a conserved aspartate in anthranilate 1,2-dioxygenase. Biochemistry. 2003;42:13625–13636. doi: 10.1021/bi035385n. [DOI] [PubMed] [Google Scholar]
- 21.Jiang H, Parales RE, Lynch NA, Gibson DT. Site-directed mutagenesis of conserved amino acids in the alpha subunit of toluene dioxygenase: potential mononuclear non-heme iron coordination sites. J Bacteriol. 1996;178:3133–3139. doi: 10.1128/jb.178.11.3133-3139.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bevington PR. Data Reduction and Error Analysis for the Physical Sciences. McGraw-Hill Inc; New York: 1996. pp. 235–242. [Google Scholar]
- 23.Kuila D, Fee JA. Evidence for a redox-linked ionizable group associated with the [2Fe- 2S] cluster of Thermus Rieske protein. J Biol Chem. 1986;261:2768–2771. [PubMed] [Google Scholar]
- 24.Yang TC, Wolfe MD, Neibergall MB, Mekmouche Y, Lipscomb JD, Hoffman BM. Modulation of substrate binding to naphthalene 1,2-dioxygenase by rieske cluster reduction/oxidation. Journal of the American Chemical Society. 2003;125:2034–2035. doi: 10.1021/ja028781m. [DOI] [PubMed] [Google Scholar]
- 25.Orville AM, Lipscomb JD. Cyanide and nitric oxide binding to reduced protocatechuate 3,4-dioxygenase: insight into the basis for order-dependent ligand binding by intradiol catecholic dioxygenases. Biochemistry. 1997;36:14044–14055. doi: 10.1021/bi970609l. [DOI] [PubMed] [Google Scholar]
- 26.Arciero DM, Lipscomb JD, Huynh BH, Kent TA, Munck E. EPR and Mossbauer studies of protocatechuate 4,5-dioxygenase. Characterization of a new Fe2+ environment. J Biol Chem. 1983;258:14981–14991. [PubMed] [Google Scholar]
- 27.Chen VJ, Orville AM, Harpel MR, Frolik CA, Surerus KK, Munck E, Lipscomb JD. Spectroscopic studies of isopenicillin N synthase. A mononuclear nonheme Fe2+ oxidase with metal coordination sites for small molecules and substrate. J Biol Chem. 1989;264:21677–21681. [PubMed] [Google Scholar]
- 28.Wolfe MD, Altier DJ, Stubna A, Popescu CV, Munck E, Lipscomb JD. Benzoate 1,2-dioxygenase from Pseudomonas putida: single turnover kinetics and regulation of a two-component Rieske dioxygenase. Biochemistry. 2002;41:9611–9626. doi: 10.1021/bi025912n. [DOI] [PubMed] [Google Scholar]