

Abstract
Bacterial pathogenicity is often manifested by the expression of various cell-associated and secreted virulence factors, such as exoenzymes, protease, and toxins. In Pseudomonas aeruginosa, the expression of virulence genes is coordinately controlled by the global regulatory quorum-sensing systems, which includes the las and rhl systems as well as the Pseudomonas quinolone signal (PQS) system. Phenazine compounds are among the virulence factors under the control of both the rhl and PQS systems. In this study, regulation of the phzA1B1C1D1E1 (phzA1) operon, which is involved in phenazine synthesis, was investigated. In an initial study of inducing conditions, we observed that phzA1 was induced by subinhibitory concentrations of tetracycline. Screening of 13,000 mutants revealed 32 genes that altered phzA1 expression in the presence of subinhibitory tetracycline concentrations. Among them, the gene PA0964, designated pmpR (pqsR-mediated PQS regulator), has been identified as a novel regulator of the PQS system. It belongs to a large group of widespread conserved hypothetical proteins with unknown function, the YebC protein family (Pfam family DUF28). It negatively regulates the quorum-sensing response regulator pqsR of the PQS system by binding at its promoter region. Alongside phzA1 expression and phenazine and pyocyanin production, a set of virulence factors genes controlled by both rhl and the PQS were shown to be modulated by PmpR. Swarming motility and biofilm formation were also significantly affected. The results added another layer of regulation in the rather complex quorum-sensing systems in P. aeruginosa and demonstrated a clear functional clue for the YebC family proteins.
Pseudomonas aeruginosa is a major opportunistic pathogen capable of causing a variety of soft tissue infections in susceptible hosts. P. aeruginosa chronic infection and associated pulmonary inflammation are ultimately responsible for the majority of morbidity and mortality of patients with cystic fibrosis. In P. aeruginosa, global regulation through the quorum-sensing systems controls population behaviors and plays fundamental roles in bacterial pathogenicity (47, 51). A Pseudomonas quinolone signal (PQS) system mediated by 2-heptyl-3-hydroxy-4-quinolone (41) and two N-acyl-homoserine lactone (HSL)-mediated quorum-sensing systems, the las and rhl systems (27, 36, 39, 40), are present in this bacterium. The synthesis of the PQS is carried out by enzymes encoded by the pqsABCDE and phnAB operons as well as pqsH (19). Transcription of the pqsABCDE and phnAB operons is regulated by the transcriptional regulator PqsR (11, 19), also known as MvfR (3). In the HSL-mediated systems, the transcriptional regulators LasR and RhlR and the cognate autoinducers, N-(3-oxododecanoyl)-l-HSL (3-oxo-C12-HSL) and N-butyryl-l-HSL (C4-HSL), constitute the las and rhl system, respectively. LasI and RhlI are responsible for the synthesis of 3-oxo-C12-HSL and C4-HSL, respectively. Expression of numerous cellular and secreted virulence factors, such as protease and pyocyanin, is regulated by quorum sensing in P. aeruginosa (47, 51).
Pyocyanin is the major phenazine compound produced by P. aeruginosa and functions both as an important virulence factor (28, 29) and as a signal molecule (12). Pyocyanin produced by P. aeruginosa has been shown to inhibit the ciliary function of respiratory epithelial cells in vitro (50) and can alter the host immune and inflammatory response (1, 10). Because they are bioactive secondary metabolites, the role of phenazine compounds as signal molecules has recently been explored (12, 42). It has been shown that pyocyanin acts as the final signal in the quorum-sensing system cascade and regulates at least 22 genes in P. aeruginosa PA14 (12).
Two homologous operons are involved in the synthesis of phenazine compounds in P. aeruginosa, phzA1B1C1D1G1 (phzA1) and phzA2B2C2D2G2 (phzA2) (34). The expression of phzA1 accounts for the majority of phenazine production (6, 49). In addition, the gene products of phzM, phzH, and phzS are required to convert the intermediate phenazine-1-carboxylic acid to other final products, including phenazine-1-carboxamide, 1-hydroxphenazine, and pyocyanin (33, 34, 38). Production of phenazine compounds is tightly regulated by quorum-sensing systems in P. aeruginosa. Both the PQS and rhl systems positively regulate phzA1 expression (13, 27), and the orphan LuxR-type quorum-sensing regulator QscR negatively regulates the expression of phzA1 and phzA2 as well as the elastase gene lasB (31).
Recently, another group of molecules, antibiotics, have been shown to play important roles in regulating bacterial gene expression when present at subinhibitory concentrations (9, 21, 32). Antibiotics are bioactive compounds that are generally believed to be weapons in intermicrobial interactions due to their killing or inhibitory activity toward other microorganisms. They have been used to treat bacterial infections for decades, exploiting the inhibitory and killing capability of these molecules toward pathogens. However, the ecological roles of antibiotics in natural communities have remained ambiguous (7). Only recently has their role as signaling agents emerged (7-9, 32, 53). Despite these observations, however, knowledge of the mechanisms in signaling and gene regulation is currently very limited, and the relationship of subinhibitory antibiotic signaling with other bacterial signal pathways remains unclear.
In this study we took a new approach to identify genes involved in phzA1 expression by examining the expression of P. aeruginosa virulence factors in the presence of subinhibitory concentrations of antibiotics. We identified 32 genes that influence the expression of phzA1. Among them, PA0964, encoding a protein that belongs to a conserved hypothetical protein family, was shown to be a regulator of pqs gene expression and phenazine production.
MATERIALS AND METHODS
Bacterial strains and culture conditions.
The bacterial strains and plasmids used in this study are listed in Table 1. P. aeruginosa PAO1 and derivatives were routinely grown at 37°C on LB agar plates or in broth (LB broth or M9 minimal medium supplemented with 0.1% Casamino Acids and 0.5% glucose) with shaking at 200 rpm. Antibiotics were used at the following concentrations where appropriate: for Escherichia coli, kanamycin (Kn) at 50 μg/ml, tetracycline (Tet) at 25 μg/ml, and ampicillin (Ap) at 100 μg/ml; for P. aeruginosa, gentamicin (Gm) at 75 μg/ml in LB and 150 μg/ml in Pseudomonas isolation agar (PIA), Tet at 75 μg/ml in LB and 200 μg/ml in PIA, carbenicillin (Cb) at 500 μg/ml in PIA, and trimethoprim (Tmp) at 300 μg/ml.
TABLE 1.
Bacterial strains and plasmids used
| Bacterial strain or plasmid | Genotype or phenotype | Reference |
|---|---|---|
| E. coli strains | ||
| DH5α | F−mcrA Δ(mrr-hsdRMS-mcrBC) 80dlacZΔM15 ΔlacX74 deoR recA1 endA1 araD139 Δ(ara leu)7697 galU galK λ−rpsL nupG | Invitrogen |
| SM10 | Mobilizing strain, RP4 integrated in the chromosome; Knr | 46 |
| P. aeruginosa strains | ||
| PAO1 | Wild type, lab strain | |
| NH003 | A0964 transposon mutant of PAO1; Gmr | This study |
| PAO1(Δ0964) | PA0964 replacement mutant of PAO1; PA0964::Gmr | This study |
| NH004 | PAO1(Δ0964) complemented strain; PAO1(Δ0964) attB::PA0964 | This study |
| Plasmids | ||
| pMS402 | Expression reporter plasmid carrying the promoterless luxCDABE; Knr Tmpr | 15 |
| pBT20 | Mini-TnM delivery vector; Apr Gmr | 25 |
| pEX18Ap | oriT+sacB+ gene replacement vector with multiple-cloning site from pUC18; Apr | 22 |
| pZ1918-lacZGm | Source plasmid of Gmr cassette | 44 |
| pRK2013 | Broad-host-range helper vector; Tra+ Knr | 14 |
| Mini-CTX1 | Integration plasmid; Tetr | 23 |
| pKD-0964K | PA0964 knockout plasmid pEX18Ap with 596-bp upstream region, Gmr-lacZ fragment from pZ1918Gm-lacZ, and 617 bp downstream of PA0964 cloned between PstI and EcoRI sites; Apr (Cbr) Gmr | This study |
| pKD-0964C | Complementation plasmid, mini-CTX1 with a 962 PCR fragment covering the entire PA0964 gene and 167-bp upstream and 52-bp downstream regions between PstI and BamHI sites; Tetr | This study |
| pKD-phzA1 | pMS402 containing 844-bp promoter region of the phzA1 operon cloned between XhoI and BamHI sites; Knr Tmpr | 15 |
| pKD-pqsH | pMS402 containing a 826-bp promoter region of the pqsH gene between XhoI and BamHI sites; Knr Tmpr | This study |
| pKD-pqsR | pMS402 containing 940-bp promoter region of pqsR cloned between XhoI and BamHI sites; Knr Tmpr | This study |
| pKD-pqsA | pMS402 containing 421-bp promoter region of pqsA cloned between XhoI and BamHI sites; Knr Tmpr | This study |
| pKD-rhlI | pMS402 containing rhlI promoter region | 16 |
| pKD-rhlR | pMS402 containing rhlR promoter region | 16 |
| pKD-lasI | pMS402 containing lasI promoter region | 16 |
| pKD-lasR | pMS402 containing lasR promoter region | 16 |
| pKD-lasB | pMS402 containing lasB promoter region | 15 |
| pKD-rhlA | pMS402 containing rhlA promoter region | 15 |
| pProEX-HTb | Protein expression vector | Invitrogen |
| pProEX-0964 | Protein expression construct, PA0964/pmpR cloned in pProEX-HTb vector | This study |
Monitoring gene expression.
The plasmid pMS402 carrying a promoterless luxCDABE reporter gene cluster was used to construct promoter-luxCDABE reporter fusions of the phzA1 operon and other genes as reported previously (15). The promoter regions of pqsR, pqsA, and pqsH were PCR amplified using high-fidelity Pfx DNA polymerase (Invitrogen), and primers were designed according to the PAO1 genome data (48). The primer pairs used were as follows: for pqsR, 5′-GGAAGATCTGCGGTGTGCGACTTGCGC-3′ and 5′-ATACTCGAGCGATGCTCGGCGCTCTCC-3′; for pqsH, 5′-CGACGGATCCTGGCCCGCTCGACCAGCAG-3′ and 5′-GCAGCTCGAGCTCGGCAGCCGAACCCT-3′; and for pqsA, 5′-CGACCTCGAGCATGCTGCCAAAGAATCGC-3′ and 5′-TGTAGGTCCGGCATTGCAG-3′. Restriction sites (underlined) were incorporated into the PCR primers. The promoter regions were cloned into the BamHI site or BamHI-XhoI site upstream of the lux genes on pMS402. The constructs were transformed into PAO1 by electroporation. DNA manipulation, PCR, and transformation were performed by following general procedures. Cloned promoter sequences were confirmed by DNA sequencing.
Using these lux-based reporters, gene expression in liquid cultures was measured as light production (in counts per second) in a Victor3 multilabel plate reader (Perkin-Elmer). Overnight cultures of the reporter strains were diluted to an optical density at 620 nm (OD620) of 0.2 and cultivated for two additional hours before being used as inoculants. The cultures were inoculated into parallel wells on a 96-well black plate with a transparent bottom. Fresh culture (5 μl) was inoculated into the wells containing a total of 95 μl medium plus other components, and the OD620 in the wells was ∼0.07. Filter-sterilized mineral oil (60 μl) was added to prevent evaporation during the assay. Promoter activities were measured every 30 min for 24 h. Bacterial growth was monitored at the same time by measuring the OD595 in the Victor3 multilabel plate reader. Expression on solid medium was measured by plating the reporter strains in soft top LB agar and imaging in a LAS300 imaging system (Fuji Corp.).
Quantification of phenazine production.
Phenazine antibiotics were extracted from P. aeruginosa and quantified by a UV-visible-light spectrophotometer as described previously (5). Briefly, triplicate 5-ml cultures were grown overnight at 37°C with orbital shaking at 200 rpm/min in LB broth or LB broth with 0.625 μg/ml Tet (1/16 of the MIC) (1/16-MIC Tet). The cultures were centrifuged, and the supernatants were collected and acidified to pH 2.0 with concentrated HCl. Phenazines were extracted twice with an equal volume of benzene for 1 h. Following evaporation of the benzene under air, the phenazine was resuspended in 0.5 ml of 0.1 N NaOH, and serial dilutions were quantified by absorbance at 367 nm. The absorbance for each sample was normalized to total absorbance per initial 5-ml culture. The measurements were repeated three times, and the averaged values were reported.
Measurement of pyocyanin production.
Pyocyanin was extracted from culture supernatants and measured by a previously reported method (17, 26). Briefly, 3 ml of chloroform was added to 5 ml of culture supernatant. After extraction, the chloroform layer was transferred to a fresh tube and mixed with 1 ml of 0.2 N HCl. After centrifugation, the top layer (0.2 M HCl) was removed and its absorption measured at 520 nm. Concentrations, expressed as micrograms of pyocyanin produced per ml of culture supernatant, were calculated using an extinction coefficient at 520 nm of 17.072 (26).
Transposon mutagenesis.
The donor strain (E. coli SM10), containing pBT20 (25), and the recipient, PAO1(pKD-phzA1), containing the phzA1-lux reporter fusions, were scraped from overnight-incubated plates and resuspended separately in 0.2 ml of LB broth. The bacterial suspensions were adjusted to an OD600 of 40 for the donors and an OD600 of 20 for the recipient. Twenty-five-microliter portions of each donor and recipient were mixed together, spotted on an LB agar plate, and incubated for 8 h at 37°C. The mating mixtures were scraped and resuspended in 14 ml of M9 medium. A 200-μl portion of this suspension was plated on PIA plates containing Gm at 150 μg/ml and Tmp at 300 μg/ml. A transposon mutant library was constructed by picking 13,056 colonies grown on these selective plates and placing them in individual wells of 34 384-well plates.
To screen for genes involved in phzA1 expression in response to Tet, the random mutant library was cultured overnight in LB medium supplemented with Tmp at 300 μg/ml. The library was then inoculated into the appropriate medium (with or without Tet) for the screen in multiwell plates with a 384-pin plate replicator. Inoculated plates were incubated at 37°C, and luminescence was measured. In the initial screens, readings were taken at 16 h and 24 h. A subset of clones with expression differing by a factor of two or more in the presence and absence of 1/16-MIC Tet (0.625 μg/ml) were identified and collected. To eliminate false-positive clones, three additional rescreens were performed. During rescreening, the overnight cultures were diluted 1:300 in the appropriate media in 96-well clear-bottom black plates (Costar 9520; Corning Inc.) and were assayed for both luminescence and absorbance over the experimental time course. Confirmed mutant clones with altered phzA1 expression profiles in the presence of 1/16-MIC Tet were selected for further characterization.
Determination of transposon insertion site by arbitrary PCR and sequencing.
The transposon insertion sites of the selected mutants were determined by arbitrary primed PCR and subsequent sequencing of the PCR product (18). Arbitrary primed PCR was performed in two steps with minor modifications as outlined below. The first-round reaction was run under the following conditions: 95°C for 5 min followed by six cycles of 95°C for 30 s, 30°C for 30 s, and 72°C for 1.5 min; 30 cycles of 95°C for 30 s, 45°C for 30 s, and 72°C for 2 min; and 72°C for an additional 5 min. Chromosomal DNA from the mutants was used as the PCR template, and the two primers used were P7-1 (5′-CTAACAATTCGTTCAAGCCG-3′), reading out from one sides of the transposon, and the arbitrary primer arb1 (5′-GGCCACGCGTCGACTAGTACNNNNNNNNNNGATAT-3′). In the second-round reaction, 2 μl of the first-round PCR product was used as a template and the PCR was carried out as follows: 95°C for 1 min followed by 30 cycles of 95°C for 30 s, 52°C for 30 s, and 72°C for 2 min; 72°C for 5 min. The primers used were P7-2 (5′-GGATGCGTCTAAAAGCCTGC-3′), a second outward transposon primer downstream to the first primer, and arb2 (5′-GGCCACGCGTCGACTAGTAC-3′), corresponding to the constant region of the original arbitrary primers. This secondary reaction specifically amplifies the products of the first PCR, which include transposon junctions. PCR products were purified using QIAquick PCR purification kits (Tiangen, Beijing, China), and DNA sequencing was performed using primer P7-2.
DNA sequencing was performed by using an automated DNA sequencer (model 373; Applied Biosystems), and DNA sequences obtained were compared with the P. aeruginosa chromosome sequences (48) to localize the transposon insertion sites.
Gene replacement mutagenesis.
For gene replacement, the previously described sacB-based strategy was employed (22). The suicide plasmid pKD0964K for gene replacement was constructed by inserting the BamHI-digested Gmr-lacZ cassette derived from pZ1918-lacZ (44) between two PCR fragments of PA0964 cloned in pEX18Ap. The two PCR fragments are upstream and downstream fragments of the PA0964 gene. The 596-bp upstream fragment of PA0964 was amplified using primer NH1 (5′-CGGCTGCAGTTCGGCTTCCTGCTCGAC-3′), containing a PstI restriction site, and NH2 (5′-GCCGGATCCGTCAGCGCCTTGTCCACG-3′), containing a BamHI site (restriction sites are underlined); the 617-bp downstream region of PA0964 was generated using the primers NH3 (5′-CGCGGATCC TATGAAGGTTATGCGC-3′), containing a BamHI site, and NH4 (5′-CGCGAATTCGAACGCATCAAGCGT-3′), containing an EcoRI site.
pKD0964K was transferred to PAO1 using a triparental mating procedure employing the helper vector pRK2013 (14) Briefly, overnight cultures (25 ml each) of E. coli strains containing plasmids pKD0964K and pRK2013 and P. aeruginosa PAO1 were centrifuged, and the pellets were resuspended in phosphate-buffered saline, mixed in equal proportions, and spotted onto an LB agar plate. Following incubation overnight at 37°C, bacteria were removed from the agar surface and resuspended in 1 ml of phosphate-buffered saline. Appropriate dilutions were spread on PIA plates containing Gm at 150 μg/ml. Subsequently, strains that had undergone a second crossover event were selected for by plating on LB supplemented with 5% sucrose plus Gm. A PA0964 gene replacement mutant, designated PAO1(Δ0964), was verified by assessing Gm resistance and Cb sensitivity and by PCR analysis.
Complementation of PA0964 knockout mutants.
To circumvent the use of antibiotic selection during the complementation experiments, the gene PA0964 was integrated into the attB site on the chromosome using the mini-CTX1 system (23). The entire gene was PCR amplified together with a 167-bp upstream promoter region and a 52-bp downstream region using the primers NH09 (5′-GCCGGATCCGTCTATCCGCGAAGTGATCG-3′) and NH10 (5′-TAACTGCAGCTTGAGCA CCTTCTGCG-3′), containing a BamHI and a PstI site (underlined), respectively. The product was ligated into mini-CTX1 to generate plasmid pKD0964C, which was transferred into E. coli SM10-λpir. Transfer of pKD0964C into the PAO1(Δ0964) mutant was carried out by a biparental mating. Integrants were selected on PIA with Tet at 200 μg/ml plus Gm at 50 μg/ml. The CTX integrase of pKD0964C promoted integration of the vector into the attB site of the P. aeruginosa genome. The plasmid portion of pKD0964C was resolved from the chromosome using the Flp recombinase encoded by pFLP2 (23). The final resultant strain was designated as NH004.
Reverse transcription-PCR.
P. aeruginosa cells at the mid-logarithmic to early stationary growth phase were collected by centrifugation at 4°C for 5 min at 10,000 × g. The supernatant was removed, and total RNA was isolated by using a Trizol Max bacterial RNA isolation kit (Invitrogen) according to the manufacturer's instructions. Residual DNA was eliminated by DNase treatment using 20 U of RQ1 RNase-free DNase, followed by phenol-chloroform extraction. The RNA was precipitated with ethanol and resuspended in RNase-free water. cDNA was synthesized from 1 μg of RNA using reverse transcriptase (Fermentas) and the primers RH (5′-ATGACCGTTCTTATCCAGG-3′) and RR (5′-AACATGTTCCTCCAGGTCAT-3′) for pqsH and pqsR, respectively. The 16S rRNA was used as a control with the primer 519R (5′-ATTACCGCGGCTGG-3′). Reverse transcriptase was inactivated by incubation at 70°C for 15 min, and the cDNAs were quantitatively measured using a PCR kit (Tiangen Biotech, Beijing, China). The primer pairs used for the subsequent PCR were 5′-ATGACCGTTCTTATCCAGG-3′ and 5′-ACTGGAAGGCATCGACAT-3′ for pqsH and 5′-AACATGTTCCTCCAGGTCAT-3′ and 5′-GTTGAGATTGAAGGCGATGT-3′ for pqsR. D88F (5′-AGAGTTTGATYMTGGCTCAG-3′) and 519R (5′-ATTACCGCGGCTGG-3′) were used for the 16S RNA.
The PCR product was analyzed on a 10% nondenaturing polyacrylamide-bis-acrylamide gel, and the bands were quantified with a LAS-3000 imaging system (Fujifilm Life Science) and its accompanying software, Multi-Gauge version 3.0.
In vitro expression and purification of PA0964 protein.
PA0964 protein was expressed as a His tag fusion protein using the pProEX-HT protein expression system (Invitrogen). PA0964 was amplified by PCR with the primers 5′-GCCGGATCCATGGCTGGTCATTCTAAATG-3′ and 5′-CTGAAGCTTCGAACGCATCAAGC-3′. Underlining indicates the BamHI and HindIII restriction sites, added to facilitate cloning. The 801-bp PCR product obtained was cloned between the BamHI and HindIII sites of the pProEX-HTb vector to form pProEX-0964, which was then transferred to E. coli 10B. The PA0964 sequence was verified by sequencing.
For PA0964 protein expression, E. coli carrying pProEX-0964 was grown overnight in LB broth supplemented with 100 μg/ml Ap at 37°C. Fresh LB broth (100 ml) supplemented with 100 μg/ml Ap was then inoculated with 1 ml of the overnight culture, and the culture was grown at 37°C with agitation. When the culture reached an OD600 of 0.6, IPTG (isopropyl-β-d-thiogalactopyranoside) was added at 0.6 mM to induce protein expression. The cells were harvested 2 h later by centrifugation and used immediately or stored at −70°C for later use.
The pellets from 100 ml of culture were resuspended in 10 ml LE buffer (50 mM NaH2PO4, 300 mM NaCl [pH 8.0]) with 1 mM phenylmethylsulfonyl fluoride and 1 mg/ml lysozyme. Cells were ruptured by sonication using a Hielscher Sonifier. The insoluble material was removed by centrifugation (10,000 × g for 30 min). The supernatant was added to a high-affinity nickel-nitrilotriacetic acid resin column (Qiagen) equilibrated with LE buffer. The column was washed with eight bed volumes of wash buffer (50 mM NaH2PO4, 300 mM NaCl, 10 mM imidazole [pH 8.0]). Finally, the polyhistidine-tagged protein was eluted with five bed volumes of elution buffer (50 mM NaH2PO4, 300 mM NaCl, 250 mM imidazole). Collected elute was dialyzed against 10 mM Tris-HCl, 1 mM EDTA, 10% glycerol. The purified protein was stored at −70°C. The process was monitored by sodium dodecyl sulfate-polyacrylamide gel electrophoresis. The PA0964 protein preparation was also subjected to sodium dodecyl sulfate-polyacrylamide gel electrophoresis to check purity. Protein concentrations were estimated by comparison to protein standards and by measurement at A280 using a U-3310 spectrophotometer (Hitachi).
Electrophoretic mobility shift assay.
The promoter regions of pqsR and lasR were PCR amplified with the following primers: for pqsR, 5′-GCGGTGTGCGACTTGCGC-3′ and 5′-CGATGCTCGGCGCTCTCC-3′, and for lasR, 5′-CCGGGCTCGGCCTGTTCT-3′ and 5′-GGATGGCGCTCCACTCCA-3′. The template used was P. aeruginosa genomic DNA. The sizes of the PCR products were 940 bp and 591 bp, respectively. The promoter regions of the other quorum-sensing genes were amplified from the respective reporter plasmids using pZE05 and pZE.06 primers (15). The 1,204-bp PCR product of PA1243 was used as a negative control. It was amplified using the primers 5′-GGAATTCGTCGCCCTGTA-3′ and 5′-TTCGAACTGGTGCGGATC-3′. The PCR product (50 ng) and the PA0964 protein were incubated for 20 min at room temperature in binding buffer (40 mM Tris-HCl [pH 7.5], 0.1 mM EDTA, 70 mM KCI, 0.1 mM dithiothreitol, 4 mM MgCl2) and subjected to electrophoresis on a 6% native polyacrylamide gel. The gel was stained with SYBR green (Invitrogen) and photographed using a LAS-3000 imaging system.
Swarming motility assay.
Swarm medium was based on M8 minimal medium (24), supplemented with MgSO4 (1 mM), glucose (0.2%), and Casamino Acids (0.5%), and solidified with agar (0.5%). Bacteria were spot inoculated onto swarm agar plates as 2.5-μl aliquots taken directly from overnight LB cultures. Swarm agar plates were incubated for 24 h at 37°C and then incubated an additional 24 h at room temperature for a total of 48 h. Photographs were taken with the LAS-3000 imaging system.
Biofilm formation assay.
Biofilm formation was measured in a static system as described by O'Toole and Kolter (37) with minor modifications. Cells from overnight cultures were inoculated at 1:100 dilutions into LB medium in 96-well polystyrene microtiter plates (Costar) and grown at 30°C for 10 h. A 25-μl volume of 1% crystal violet was added to each well and stained for 15 min prior to removal by aspiration. Wells were rinsed three times by submerging the plates in distilled water, and the remaining crystal violet was dissolved in 200 μl of 95% ethanol. A 125-μl portion of this solution was transferred to a new polystyrene microtiter plate, and the absorbance was measured at 595 nm.
N-Acyl-HSL measurement.
In order to measure HSL in bacterial cultures, we used methods based on an Agrobacterium tumefaciens system (4) and the rhlA-luxCDABE reporter (16) as described previously. Briefly, an overnight culture of the reporter strains was diluted 1:300 in LB medium, and 90 μl was added to the wells of a 96-well microtiter plate. A 10-μl portion of the samples or medium control was added to the wells; the luminescence and OD values were measured every half hour for a total of 24 h in a Victor3 1420 multilabel plate reader, and relative levels of HSL were calculated from the maximal values (counts per second) minus the medium control.
RESULTS
Regulation of the phzA1 operon by subinhibitory concentrations of Tet.
It was recently suggested that in addition to their roles as growth inhibitors, antibiotics can serve as signal molecules to regulate gene expression in bacteria (9, 21, 32). To investigate the effect of subinhibitory concentrations of antibiotics on P. aeruginosa gene expression, a set of 31 virulence-related and quorum-sensing genes (15, 16) were examined in the presence of polymyxin, Tet, and rifampin at concentrations below their MICs.
A disk diffusion method was initially used to visualize response to subinhibitory antibiotic concentrations. The reporter strain carrying a specific promoter inserted upstream of the promoterless luxCDABE on a plasmid (15) was spread in top agar on a petri dish, and the test antibiotics were spotted on spaced filter discs laid on top of the agar. A positive response was observed as an increase in light emission from the reporter strain beyond the zone of inhibition. Likewise, a negative response was observed as a decrease in light production beyond the zone of inhibition. As shown in Fig. 1, phzA1 was activated by Tet, repressed by rifampin, and unaffected by polymyxin or Kn. Beyond the zone of growth inhibition, enhanced expression of the phzA1 reporter was observed with Tet. In contrast, reduced expression was observed beyond the zone of growth inhibition of rifampin. Significantly, this pattern of response was not observed with all promoters tested, indicating that the observed expression pattern was not an indirect effect of the antibiotics on the luciferase reporter system but specific to transcriptional activity of the cloned promoter.
FIG. 1.

Regulation of PhzA1 expression by different antibiotics. (A) Image of reporter strain on plate under white light; (B) light production of the reporter strain imaged in dark. Pol, polymyxin; Rif, rifampin. The reporter plasmid carries a Kn resistance gene, and therefore the effect of Kn on phzA1 is negated.
Further analysis of gene regulation by subinhibitory antibiotics was carried out in multiwell plates, and expression was measured as light production (counts per second) in a multilabel plate reader. Subinhibitory concentrations of Tet (1/16-MIC Tet) activated the expression of phzA2, rhlA, lasB, exoS, exoY, and rhlR in addition to the phzA1 operon more than fourfold, measured as the ratio of maximal levels of expression in the absence and presence of 0.625 μg/ml Tet. No effect was observed with polymyxin on any of these genes tested, while 1/4-MIC rifampin (0.15 μg/ml) reduced the expression of rhlA, lasB, and phzA at least threefold.
The phzA1 operon encodes products responsible for the synthesis of phenazine compounds in P. aeruginosa (6, 34, 49). It is one of the reporters that showed the greatest activation in our assay and was selected for further investigation. As shown in Fig. 2A, phzA1 expression was activated in a dose-dependent manner and increased more than 20-fold in the presence of 1/16-MIC Tet where bacterial growth was not affected.
FIG. 2.

Activation of phzA1 expression by subinhibitory Tet. (A) phzA1expression profile and bacterial growth in the absence of Tet (triangles) and in the presence of 1/16-MIC (squares) and 1/32-MIC (diamonds) Tet. The assays were independently repeated at least three times, and the data shown are representative of comparable results. (B) Phenazine production in the absence and presence of 1/16-MIC Tet. cps, counts per second.
To verify that the effect of subinhibitory concentrations of antibiotics on phzA1 at the transcriptional level actually resulted in an altered phenotype, phenazine production was measured in the presence of 0.625 μg/ml Tet. As shown in Fig. 2B, phenazine production increased significantly in the presence of subinhibitory Tet concentrations (P < 0.0001, unpaired t test).
Identification of genes involved in the Tet regulation of phzA1 by transposon mutagenesis.
To identify genes involved in regulating phzA1 expression and investigate the mechanisms of the observed gene regulation by subinhibitory concentration of Tet, phzA1 was used as the reporter and transposon mutagenesis was conducted. The P. aeruginosa strain PAO1 (pKD-phzA1) was subjected to random transposon mutagenesis, and 13,056 mutants were screened. The screening was carried out in the presence of 0.625 μg/ml Tet (1/16-MIC Tet) to identify mutants exhibiting different profiles of phzA1 expression activation by Tet (more than a twofold difference) compared to the wild type. After three rounds of rescreening in 96-well plates, 32 positive clones were selected. In several of the mutants, the changes in the expression of the phzA1 operon resulted in colony color changes because phenazine compounds are the major contributors to the color of P. aeruginosa cultures. Examples of these mutants producing distinct pigments are shown in Fig. 3. P. aeruginosa produces several pigmented compounds in addition to phenazine. The final greenish color is the combination of these pigments. The loss of phenazine production changes the balance and therefore changes the colony appearance but does not necessarily lead to a nonpigmented phenotype.
FIG. 3.
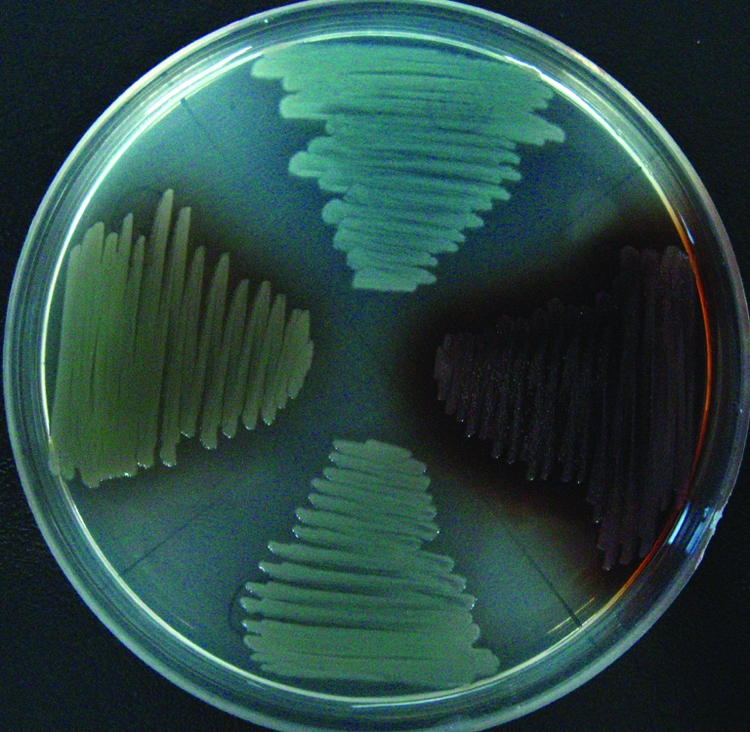
Changes in pigmentation patterns of transposon mutants with altered phzA1 expression. (Top) Transposon inserted in PA0648. (Right) Transposon inserted in PA0487. (Bottom) PAO1. (Left) Transposon inserted in PA1196. The purple color in the mutants arises from the production of other pigmented compounds.
In order to rule out the possibility that the observed changes in phzA1 expression resulted from mutations on the reporter plasmid independent of transposon mutagenesis, all the mutants were cured of plasmids by growing at 40°C overnight without antibiotic and selecting for Tmp-sensitive clones. pKD-phzA1 was reintroduced into these mutants, and expression profiles with and without the presence of subinhibitory concentrations of Tet were reexamined. Two clones that had altered plasmids were discarded.
To identify the genes mutated by transposon insertion, the insertion sites were determined by arbitrary primed PCR and DNA sequencing. The mutated genes identified are listed in Table 2. Transposon insertion in 17 genes caused increased expression of phzA1, and 13 resulted in decreased expression. Of the 32 genes identified, 13 fall into the class of hypothetical or conserved hypothetical proteins. Two of them, PA1921 and PA2372, have been identified previously as belonging to the las and rhl quorum-sensing regulon (43). Five genes belong to the membrane protein class; four are components of ABC transporters, and one is a siderophore receptor protein. Three genes encoding transcriptional regulators were identified. qscR encodes a LuxR-type quorum-sensing regulator that represses lasI (6) and directly controls phzA1 (30). PA0487 and PA1196 encode a probable molybdenum transport regulator and a putative transcriptional regulator, respectively. Interruption of PA0487 and PA1196 abolished phzA1 expression, indicating that these genes are essential for phzA1 expression. The distinctive color of these mutants (Fig. 3) is indicative of altered phenazine compound production. The remaining genes encode known or probable enzymes and cytoplasmic proteins. In this group is pqsA, which is involved in PQS synthesis, indicating that the PQS influences phzA1 regulation, in agreement with previous reports (19).
TABLE 2.
Mutated genes identified that caused alterations in phzA1 expression
| Category and PA no. | Gene name | Insertion site | Operon context | Fold changea | Protein description |
|---|---|---|---|---|---|
| Increased expression | |||||
| PA4456 | 4988244 | PA4456-PA4450 | 10.0 | Probable ATP-binding component of ABC transporter | |
| PA1921 | 2096823 | 9.9 | Hypothetical protein | ||
| PA0011 | 14518 | 8.5 | Probable 2-OH-lauroyltransferase | ||
| PA5570-Pb | rpmH | 6264404 | PA5570-PA5569 | 7.7 | 50S ribosomal protein L34 |
| PA0964 | 1050694 | 6.7 | Conserved hypothetical protein | ||
| PA0649-Pb | 703088 | PA0649-PA0651 | 6.4 | Anthranilate synthase component II | |
| PA0931 | pirA | 1019566 | 5.2 | Ferric enterobactin receptor | |
| PA5217 | 5874931 | PA5217-PA5216 | 4.8 | Probable binding protein component of ABC iron transporter | |
| PA3658 | glnD | 4096731 | PA3658-PA3659 | 4.8 | Protein-PII uridylyltransferase |
| PA5045 | ponA | 5681281 | 2.9 | Penicillin-binding protein 1A | |
| PA0292 | aguA | 329322 | PA0293-PA0292 | 2.7 | Agmatine deiminase |
| PA5043 | pilN | 5679309 | PA5044-PA5036 | 2.7 | Type 4 fimbrial biogenesis protein PilN |
| PA1801 | clpP | 1954518 | PA1800-PA1804 | 2.6 | ATP-dependent CIP protease proteolytic subunit |
| PA0053 | 69466 | 2.5 | Hypothetical protein | ||
| PA2372 | 2623766 | 2.3 | Hypothetical protein | ||
| PA4462-Pb | rpoN | 4992777 | 2.5 | RNA polymerase sigma-54 factor | |
| PA5410 | 6086130 | 2.4 | Probable ring hydroxylating dioxygenase alpha-subunit | ||
| PA1898 | qscR | 2069713 | 2.0 | Quorum-sensing control repressor | |
| Decreased expression | |||||
| PA1829 | 1987935 | −6.5 | Hypothetical protein | ||
| PA1938/PA1939 | 2120223 | −5.0 | Intergenic region | ||
| PA5263 | argH | 5925147 | −5.7 | Argininosuccinate lyase | |
| PA2610 | 2951949 | −3.7 | Conserved hypothetical protein | ||
| PA3979 | 4459677 | −3.2 | Hypothetical protein | ||
| PA4312 | 4840986 | PA4311-PA4313 | −3.1 | Conserved hypothetical protein | |
| PA4732 | pgi | 5314851 | −2.7 | Glucose-6-phosphate isomerase | |
| PA2649 | nuoN | 2997261 | PA2637-PA2650 | −2.4 | NADH dehydrogenase I chain N |
| PA0996 | pqsA | 1079234 | PA0996-PA1000 | −2.2 | Probable coenzyme A ligase |
| PA4047-Pb | ribA | 4530692 | PA4047-PA4045 | −2.2 | GTP cyclohydrolase II |
| PA3992 | 4472652 | −1.7 | Hypothetical protein | ||
| PA3258 | 3645894 | −1.6 | Hypothetical protein | ||
| No expression | |||||
| PA0487-Pb | 549239 | NA | Probable molybdenum transport regulator | ||
| PA1196 | 1297441 | PA1196-PA1197 | NA | Probable transcriptional regulator |
Maximal ratio of expression (measured in counts per second) between the mutant and the wild type in the presence of 1/16-MIC Tet. The ratio is reverted and marked with a minus sign when a decreased in expression was recorded in the mutant. NA, no expression of phzA1 was observed in the mutant.
Transposon inserted at the promoter region of the gene or operon.
Mutation of PA0964 by gene replacement and complementation analysis.
Among the genes identified, the PA0964 mutation resulted in one of the largest increases in the expression of phzA1 operon. PA0964 encodes an unknown protein belonging to one of the most widespread groups of conserved hypothetical proteins in bacteria and yeasts (20). Even though this family has been characterized by crystal structure analysis, remarkably, there is no function assigned to any of the members (20). To verify that the observed phenotype is due to the transposon insertion in this gene, gene replacement was carried out by inserting a Gmr cassette into PA0964. When pKD-phzA1 was introduced into this mutant, an expression profile similar to that in the transposon mutant was observed (Fig. 4). A complementation study was also carried out to confirm the functionality of PA0964. The intact PA0964 gene together with its promoter region was PCR amplified and integrated as a single copy at the attB site on the P. aeruginosa chromosome via CTX integrase. The expression of phzA1 was restored to the wild-type level (Fig. 4). These results provide clear evidence that PA0964 functions to repress the expression of the phzA1 operon.
FIG. 4.

Complementation of phzA1 expression in P. aeruginosa PA0964 mutant. The expression of phzA1 in PAO1(Δ0964) (squares), wild type PAO1 (triangles), and PAO1(Δ0964) complemented by an intact copy of PA0964 together with its promoter region inserted at the attB site on the chromosome (diamonds) is shown. No Tet was added to the media. cps, counts per second.
Modulation of P. aeruginosa quorum sensing by PA0964.
It is known that phzA1 is controlled by both the rhl acyl-HSL quorum-sensing system and the PQS system (13, 27). To investigate the possible connection between PA0964 and the quorum-sensing systems, the expression of the quorum-sensing genes was tested in the PA0964 mutant. Three genes involved in PQS synthesis, pqsR, pqsA, and pqsH, were all upregulated in the mutant (Fig. 5). The upregulation of pqsH and pqsR expression was confirmed by reverse transcription-PCR analysis (data not shown). rhlR (Fig. 5) was also elevated in the PA0964 mutant. In contrast, lasI and rhlI were not affected and lasR was affected slightly in the late stage of growth (data not shown). In agreement with the unchanged synthase gene expression, the level of acyl-HSLs in the mutant was unchanged compared to that in the wild type (data not shown). It is likely that the observed activation of phzA1 in the PA0964 mutant was due to the activation of either the PQS system or rhlR in this mutant. Because the PQS positively regulates the rhl system (27) and the rhl system negatively regulates the PQS system (13), the observed activation of rhlR in PA0964 mutant (Fig. 5D) could result from the increased expression of PQS genes, but the activation of the PQS could not be due to activation of rhlR. These results suggest that PA0964 probably negatively regulates the PQS or both the PQS and rhlR.
FIG. 5.

Activation of PQS quorum-sensing genes and rhlR in the PA0964 mutant. Expression profiles and corresponding growth curves are shown for pqsR, pqsA, pqsH, and rhlR. The data from the wild type are shown by triangles and those from the mutant by diamonds. The assays were independently repeated at least three times, and the data shown are representative of comparable results. No Tet was added to the media. cps, counts per second.
Repression of pqsR by PA0964 via binding at the promoter region.
To examine whether PA0964 regulated the PQS and/or the rhl systems and whether the regulation was direct or indirect, the PA0964 protein was expressed and DNA binding assays were carried out. The PA0964 gene was PCR amplified, cloned in pProEX-HTb vector, and expressed in vitro. The purified PA0964 was used to examine interactions with DNA fragments containing the promoter regions of the PQS and rhl genes, including pqsR, pqsA, pqsH, rhlI, rhlR, lasR, and las by electrophoretic mobility shift assays. As shown in Fig. 6, PA0964 bound to the promoter region of pqsR, resulting in a gel mobility shift. No binding was observed with the DNA fragments containing promoters of other quorum-sensing-related genes (data not shown). The result indicates that the PA0964 directly represses pqsR by binding at its promoter region. PA0964 was therefore designated pmpR (pqsR-mediated PQS regulator).
FIG. 6.

Binding of PA0964 at the pqsR promoter region, shown by electrophoretic mobility shift assay. PCR products containing the PpqsR and PlasR promoter regions and PPA1243 were added to the reaction mixtures at 50 ng each. PA0964 protein was added to the reaction mixtures in lanes B to D at 320 ng, 640 ng, and 960 ng, respectively. No protein was added in lane A. DNA of the PpqsR region was specifically bound by PmpR (PpqsR-PmpR).
Pyocyanin production, biofilm formation and swarming ability affected in the PA0964 mutant.
Since expression of the PQS quorum-sensing system was elevated in the PA0964 pmpR mutant, phenazine production, biofilm formation, and swarming motility were tested, as they are known to be controlled by quorum-sensing systems in P. aeruginosa. The expression of rhlA, which is regulated by both the PQS and rhl, and the expression of lasB, regulated by rhl and las systems, were also tested. Figure 7 shows the effect of PmpR on these genes and phenotypes. Increased expression of both rhlA and lasB was observed in the PA0964 deletion mutant compared to the PAO1 parent strain (Fig. 7A and B). Similarly, increased pyocyanin (Fig. 7C), biofilm formation (Fig. 7D), and swarming motility (Fig. 7E) were observed in the mutant. The results confirm that PA0964 negatively regulates the PQS quorum-sensing system in P. aeruginosa, and its mutation alters quorum-sensing-regulated phenotypes. Because of the relationship between the PQS and rhl quorum-sensing systems, the changes are probably caused by the PQS directly and by rhl indirectly.
FIG. 7.

Changed gene expression and quorum-sensing-regulated phenotypes in the PAO1(Δ0964) mutant compared to wild-type PAO1. (A) Expression profiles and corresponding growth curves for rhlA (A) and lasB (B). The data from PAO1 are shown by triangles and those from PAO1(Δ0964) by diamonds. (C) Pyocyanin production. (D) Biofilm formation. (E) Swarming mobility. The average diameter of the swarm trajectory was about 2.6 cm for PAO1 and 5 cm for PAO1(Δ0964). Pyocyanin production in the mutant is very significantly different from that in the wild type (P < 0.001, unpaired t test); biofilm formation is also significantly different (P < 0.005, unpaired t test).
DISCUSSION
Phenazine compounds have been shown to be important virulence factors in P. aeruginosa (28, 29). In addition, they act as cell-cell signaling molecules and have inhibitory activity against other bacteria (12, 35). This report presents the first evidence that the phzA1 operon is activated by subinhibitory concentrations of Tet. The identification of several new genes involved in phenazine regulation was somewhat surprising, considering that a number of previous studies focused on this pathway (19, 52). Since the mutants were initially selected when phzA1 expression was activated by subinhibitory Tet concentrations, these mutants might be missed by studies under other conditions. Additionally, the mutant library was screened in liquid with a sensitive transcriptional reporter. As expected, some of the genes affecting phzA1 expression in the presence of subinhibitory Tet are known regulators of the operon. The LuxR-type transcriptional regulator QscR is known to negatively control phzA1 (6) and lasB (30) expression and interacts with LasR and RhlR directly (30). The identification of pqsA confirms the requirement for the PQS system in phzA1 expression (19). It is worth noting that two genes (PA1921 and PA2372) encoding hypothetical proteins have been previously identified as quorum-sensing-regulated genes (43). Since their mutations also affected phzA1 expression, it suggests that the control of phzA1 by quorum-sensing systems is likely to be more complex than single-layer, one-way transcriptional regulation. PA0487 and PA1196 appear to be novel regulators of the phzA1 operon that are required for phenazine production. These are being investigated further.
PA0964 belongs to a large group of widespread conserved hypothetical proteins, the YebC protein family (Pfam family DUF28). Neither predicted biological functions nor biochemical clues exist for this widespread family of the “unknown unknowns” (20). However, this group of proteins has been characterized from a structural perspective. The X-ray structures of YebC proteins from Aquifex aeolicus (45), E. coli, and Helicobacter pylori have been solved (Protein Data Bank entries 1lfp, 1kon, and 1mw7, respectively). Structural analysis revealed a large cavity with a predominance of negatively charged residues on the surface of this protein (45). Intriguingly, all three crystal structure solved proteins have a putative DNA binding function and a domain sharing topological similarity with the Tet repressor class D variant (Protein Data Bank [http://www.rcsb.org/pdb]). The results presented here indicate that the YebC class protein PA0964 in P. aeruginosa negatively regulates phzA1 through binding to the promoter region of the pqsR gene. This result should provide useful information on the functions of proteins of this conserved family as a whole.
The search for mutants in which the activation of phzA1 by subinhibitory concentrations of Tet was eliminated was, however, not conclusive. The abolition of subinhibitory Tet regulation in PA0487 and PA1196 mutants is complicated by their role as an activator in phzA1 expression under the experimental conditions. It was not possible to determine whether the regulation of phzA1 by subinhibitory antibiotic concentrations was mediated through these regulators. The test of phzA1 expression in PAO1(Δ0964) indicate that Tet regulation was unaffected (data not shown), suggesting that pmpR was not part of the regulatory pathway. In agreement with this observation, the binding of PmpR to the pqsR promoter region was not affected by the presence of Tet (data not shown).
The results also add another layer of regulation in the complex quorum-sensing networks in P. aeruginosa. There is a complex regulatory network that controls the quorum-sensing systems at both the transcriptional and posttranscriptional levels. Multiple signals, such as temperature, pH, stress, and nutrient depletion, are integrated into the important global regulatory systems through these regulators. Bacteria integrate multiple stimuli to orchestrate optimal responses and survival strategies. We suspect that all the major regulatory pathways in bacteria are connected in some way (16). These regulators are known to regulate the las or the rhl system either positively or negatively. A hierarchy in which the las system controls the rhl system is believed to exist within the acyl-HSL-based quorum-sensing system; however, this is dependent on specific growth conditions (16). The PQS system is intricately connected to the AHL systems. The rhl and las systems, respectively, place negative and positive regulation on the PQS while the PQS positively influences the rhl system. PqsR (MvfR) is the response regulator of the PQS system. The binding of PA0964 to the pqsR promoter region indicates that the effect of PA0964 on phenazine production and swarming motility was through the PQS, thus adding to a new repressor to the list of regulators of quorum-sensing systems in P. aeruginosa.
Antibiotics are bioactive compounds that can serve as weapons in microbial communities at high concentrations due to their inhibitory activity toward other microorganisms. In ecological environments, antibiotics may be at lower concentrations and likely play additional roles as signaling molecules (7, 53). Phenazine compounds, such as pyocyanin produced by P. aeruginosa, are antibiotics in their own right that can function as competitive agents in microbial communities. The regulation of phenazine compound synthesis and exoenzyme and protease expression by subinhibitory concentrations of Tet suggests that in a microbial community, the antibiotics secreted by other microorganisms could serve as a signal to alert P. aeruginosa to the existence or aggression of other bacteria, and the subsequent increased pyocyanin production would help P. aeruginosa to compete with the other microbes. It has been estimated that 0.1% of soil actinomycetes produce Tet; it is therefore a prevalent antibiotic in this community (2). As a soil microbe, P. aeruginosa would have been exposed to these compounds throughout its evolution and not only recently because of the clinical use of Tet. Genes such as efflux pumps were also upregulated by subinhibitory concentrations of Tet (unpublished data), probably to expel toxic compounds in this competitive polymicrobial environment. All of these intrinsic regulatory systems contribute to the effectiveness of P. aeruginosa as an opportunistic pathogen.
Acknowledgments
This study was supported by grants from the National Natural Science Foundation of China (no. 30470098; 30611120520), the Major State Basic Research Program Preliminary Program (no. 2004CCA01700), and the Canadian Cystic Fibrosis Foundation. M.G.S. is a Canadian Research Chair in Microbial Gene Expression and Alberta Heritage Foundation for Medical Research Scientist.
Footnotes
Published ahead of print on 18 July 2008.
REFERENCES
- 1.Allen, L., D. H. Dockrell, T. Pattery, D. G. Lee, P. Cornelis, P. G. Hellewell, and M. K. Whyte. 2005. Pyocyanin production by Pseudomonas aeruginosa induces neutrophil apoptosis and impairs neutrophil-mediated host defenses in vivo. J. Immunol. 1743643-3649. [DOI] [PubMed] [Google Scholar]
- 2.Baltz, R. H. 2007. Antimicrobials from Actinomycetes: back to the future. Microbes 2125-131. [Google Scholar]
- 3.Cao, H., G. Krishnan, B. Goumnerov, J. Tsongalis, R. Tompkins, and L. G. Rahme. 2001. A quorum sensing-associated virulence gene of Pseudomonas aeruginosa encodes a LysR-like transcription regulator with a unique self- regulatory mechanism. Proc. Natl. Acad. Sci. USA 9814613-14618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chambers, C. E., M. B. Visser, U. Schwab, and P. A. Sokol. 2005. Identification of N-acylhomoserine lactones in mucopurulent respiratory secretions from cystic fibrosis patients. FEMS Microbiol. Lett. 244297-304. [DOI] [PubMed] [Google Scholar]
- 5.Chancey, S. T., D. W. Wood, and L. S. Pierson, 3rd. 1999. Two-component transcriptional regulation of N-acyl-homoserine lactone production in Pseudomonas aureofaciens. Appl. Environ. Microbiol. 652294-2299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chugani, S. A., M. Whiteley, K. M. Lee, D. D'Argenio, C. Manoil, and E. P. Greenberg. 2001. QscR, a modulator of quorum-sensing signal synthesis and virulence in Pseudomonas aeruginosa. Proc. Natl. Acad. Sci. USA 982752-2757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Davies, J. 2006. Are antibiotics naturally antibiotics? J. Ind. Microbiol. Biotechnol. 33496-499. [DOI] [PubMed] [Google Scholar]
- 8.Davies, J. 2007. Small molecules: the lexicon of biodiversity. J. Biotechnol. 1293-5. [DOI] [PubMed] [Google Scholar]
- 9.Davies, J., G. B. Spiegelman, and G. Yim. 2006. The world of subinhibitory antibiotic concentrations. Curr. Opin. Microbiol. 9445-453. [DOI] [PubMed] [Google Scholar]
- 10.Denning, G. M., L. A. Wollenweber, M. A. Railsback, C. D. Cox, L. L. Stoll, and B. E. Britigan. 1998. Pseudomonas pyocyanin increases interleukin-8 expression by human airway epithelial cells. Infect. Immun. 665777-5784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Deziel, E., F. Lepine, S. Milot, J. He, M. N. Mindrinos, R. G. Tompkins, and L. G. Rahme. 2004. Analysis of Pseudomonas aeruginosa 4-hydroxy-2-alkylquinolines (HAQs) reveals a role for 4-hydroxy-2-heptylquinoline in cell-to-cell communication. Proc. Natl. Acad. Sci. USA 1011339-1344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dietrich, L. E., A. Price-Whelan, A. Petersen, M. Whiteley, and D. K. Newman. 2006. The phenazine pyocyanin is a terminal signalling factor in the quorum sensing network of Pseudomonas aeruginosa. Mol. Microbiol. 611308-1321. [DOI] [PubMed] [Google Scholar]
- 13.Diggle, S. P., K. Winzer, S. R. Chhabra, K. E. Worrall, M. Camara, and P. Williams. 2003. The Pseudomonas aeruginosa quinolone signal molecule overcomes the cell density-dependency of the quorum sensing hierarchy, regulates rhl-dependent genes at the onset of stationary phase and can be produced in the absence of LasR. Mol. Microbiol. 5029-43. [DOI] [PubMed] [Google Scholar]
- 14.Ditta, G., S. Stanfield, D. Corbin, and D. R. Helinski. 1980. Broad host range DNA cloning system for gram-negative bacteria: construction of a gene bank of Rhizobium meliloti. Proc. Natl. Acad. Sci. USA 777347-7351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Duan, K., C. Dammel, J. Stein, H. Rabin, and M. G. Surette. 2003. Modulation of Pseudomonas aeruginosa gene expression by host microflora through interspecies communication. Mol. Microbiol. 501477-1491. [DOI] [PubMed] [Google Scholar]
- 16.Duan, K., and M. G. Surette. 2007. Environmental regulation of Pseudomonas aeruginosa PAO1 Las and Rhl quorum-sensing systems. J. Bacteriol. 1894827-4836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Essar, D. W., L. Eberly, A. Hadero, and I. P. Crawford. 1990. Identification and characterization of genes for a second anthranilate synthase in Pseudomonas aeruginosa: interchangeability of the two anthranilate synthases and evolutionary implications. J. Bacteriol. 172884-900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Friedman, L., and R. Kolter. 2004. Genes involved in matrix formation in Pseudomonas aeruginosa PA14 biofilms. Mol. Microbiol. 51675-690. [DOI] [PubMed] [Google Scholar]
- 19.Gallagher, L. A., S. L. McKnight, M. S. Kuznetsova, E. C. Pesci, and C. Manoil. 2002. Functions required for extracellular quinolone signaling by Pseudomonas aeruginosa. J. Bacteriol. 1846472-6480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Galperin, M. Y., and E. V. Koonin. 2004. ‘Conserved hypothetical’ proteins: prioritization of targets for experimental study. Nucleic Acids Res. 325452-5463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Goh, E. B., G. Yim, W. Tsui, J. McClure, M. G. Surette, and J. Davies. 2002. Transcriptional modulation of bacterial gene expression by subinhibitory concentrations of antibiotics. Proc. Natl. Acad. Sci. USA 9917025-17030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hoang, T. T., R. R. Karkhoff-Schweizer, A. J. Kutchma, and H. P. Schweizer. 1998. A broad-host-range Flp-FRT recombination system for site-specific excision of chromosomally-located DNA sequences: application for isolation of unmarked Pseudomonas aeruginosa mutants. Gene 21277-86. [DOI] [PubMed] [Google Scholar]
- 23.Hoang, T. T., A. J. Kutchma, A. Becher, and H. P. Schweizer. 2000. Integration-proficient plasmids for Pseudomonas aeruginosa: site-specific integration and use for engineering of reporter and expression strains. Plasmid 4359-72. [DOI] [PubMed] [Google Scholar]
- 24.Kohler, T., L. K. Curty, F. Barja, C. van Delden, and J. C. Pechere. 2000. Swarming of Pseudomonas aeruginosa is dependent on cell-to-cell signaling and requires flagella and pili. J. Bacteriol. 1825990-5996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kulasekara, H. D., I. Ventre, B. R. Kulasekara, A. Lazdunski, A. Filloux, and S. Lory. 2005. A novel two-component system controls the expression of Pseudomonas aeruginosa fimbrial cup genes. Mol. Microbiol. 55368-380. [DOI] [PubMed] [Google Scholar]
- 26.Kurachi, M. 1958. Studies on the biosynthesis of pyocyanine. Isolation and determination of pyocyanine. Bull. Inst. Chem. Res. Kyoto Univ. 36163-173. [Google Scholar]
- 27.Latifi, A., M. K. Winson, M. Foglino, B. W. Bycroft, G. S. Stewart, A. Lazdunski, and P. Williams. 1995. Multiple homologues of LuxR and LuxI control expression of virulence determinants and secondary metabolites through quorum sensing in Pseudomonas aeruginosa PAO1. Mol. Microbiol. 17333-343. [DOI] [PubMed] [Google Scholar]
- 28.Lau, G. W., D. J. Hassett, H. Ran, and F. Kong. 2004. The role of pyocyanin in Pseudomonas aeruginosa infection. Trends Mol. Med. 10599-606. [DOI] [PubMed] [Google Scholar]
- 29.Laursen, J. B., and J. Nielsen. 2004. Phenazine natural products: biosynthesis, synthetic analogues, and biological activity. Chem. Rev. 1041663-1686. [DOI] [PubMed] [Google Scholar]
- 30.Ledgham, F., I. Ventre, C. Soscia, M. Foglino, J. N. Sturgis, and A. Lazdunski. 2003. Interactions of the quorum sensing regulator QscR: interaction with itself and the other regulators of Pseudomonas aeruginosa LasR and RhlR. Mol. Microbiol. 48199-210. [DOI] [PubMed] [Google Scholar]
- 31.Lequette, Y., J. H. Lee, F. Ledgham, A. Lazdunski, and E. P. Greenberg. 2006. A distinct QscR regulon in the Pseudomonas aeruginosa quorum-sensing circuit. J. Bacteriol. 1883365-3370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Linares, J. F., I. Gustafsson, F. Baquero, and J. L. Martinez. 2006. Antibiotics as intermicrobial signaling agents instead of weapons. Proc. Natl. Acad. Sci. USA 10319484-19489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mavrodi, D. V., N. Bleimling, L. S. Thomashow, and W. Blankenfeldt. 2004. The purification, crystallization and preliminary structural characterization of PhzF, a key enzyme in the phenazine-biosynthesis pathway from Pseudomonas fluorescens 2-79. Acta Crystallogr. D 60184-186. [DOI] [PubMed] [Google Scholar]
- 34.Mavrodi, D. V., R. F. Bonsall, S. M. Delaney, M. J. Soule, G. Phillips, and L. S. Thomashow. 2001. Functional analysis of genes for biosynthesis of pyocyanin and phenazine-1-carboxamide from Pseudomonas aeruginosa PAO1. J. Bacteriol. 1836454-6465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mazzola, M., R. J. Cook, L. S. Thomashow, D. M. Weller, and L. S. Pierson III. 1992. Contribution of phenazine antibiotic biosynthesis to the ecological competence of fluorescent pseudomonads in soil habitats. Appl. Environ. Microbiol. 582616-2624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ochsner, U. A., A. Fiechter, and J. Reiser. 1994. Isolation, characterization, and expression in Escherichia coli of the Pseudomonas aeruginosa rhlAB genes encoding a rhamnosyltransferase involved in rhamnolipid biosurfactant synthesis. J. Biol. Chem. 26919787-19795. [PubMed] [Google Scholar]
- 37.O'Toole, G. A., and R. Kolter. 1998. Flagellar and twitching motility are necessary for Pseudomonas aeruginosa biofilm development. Mol. Microbiol. 30295-304. [DOI] [PubMed] [Google Scholar]
- 38.Parsons, J. F., K. Calabrese, E. Eisenstein, and J. E. Ladner. 2003. Structure and mechanism of Pseudomonas aeruginosa PhzD, an isochorismatase from the phenazine biosynthetic pathway. Biochemistry 425684-5693. [DOI] [PubMed] [Google Scholar]
- 39.Passador, L., J. M. Cook, M. J. Gambello, L. Rust, and B. H. Iglewski. 1993. Expression of Pseudomonas aeruginosa virulence genes requires cell-to- cell communication. Science 2601127-1130. [DOI] [PubMed] [Google Scholar]
- 40.Pearson, J. P., L. Passador, B. H. Iglewski, and E. P. Greenberg. 1995. A second N-acylhomoserine lactone signal produced by Pseudomonas aeruginosa. Proc. Natl. Acad. Sci. USA 921490-1494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pesci, E. C., J. B. Milbank, J. P. Pearson, S. McKnight, A. S. Kende, E. P. Greenberg, and B. H. Iglewski. 1999. Quinolone signaling in the cell-to-cell communication system of Pseudomonas aeruginosa. Proc. Natl. Acad. Sci. USA 9611229-11234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Price-Whelan, A., L. E. Dietrich, and D. K. Newman. 2006. Rethinking ‘secondary’ metabolism: physiological roles for phenazine antibiotics. Nat. Chem. Biol. 271-78. [DOI] [PubMed] [Google Scholar]
- 43.Schuster, M., C. P. Lostroh, T. Ogi, and E. P. Greenberg. 2003. Identification, timing, and signal specificity of Pseudomonas aeruginosa quorum-controlled genes: a transcriptome analysis. J. Bacteriol. 1852066-2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Schweizer, H. P. 1993. Two plasmids, X1918 and Z1918, for easy recovery of the xylE and lacZ reporter genes. Gene 13489-91. [DOI] [PubMed] [Google Scholar]
- 45.Shin, D. H., H. Yokota, R. Kim, and S. H. Kim. 2002. Crystal structure of conserved hypothetical protein Aq1575 from Aquifex aeolicus. Proc. Natl. Acad. Sci. USA 997980-7985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Simon, R., U. Priefer, and A. Pühler. 1983. A broad host range mobilization system for in vivo genetic engineering: transposon mutagenesis in gram negative bacteria. Bio/Technology 1784-791. [Google Scholar]
- 47.Smith, R. S., and B. H. Iglewski. 2003. P. aeruginosa quorum-sensing systems and virulence. Curr. Opin. Microbiol. 656-60. [DOI] [PubMed] [Google Scholar]
- 48.Stover, C. K., X. Q. Pham, A. L. Erwin, S. D. Mizoguchi, P. Warrener, M. J. Hickey, F. S. Brinkman, W. O. Hufnagle, D. J. Kowalik, M. Lagrou, R. L. Garber, L. Goltry, E. Tolentino, S. Westbrock-Wadman, Y. Yuan, L. L. Brody, S. N. Coulter, K. R. Folger, A. Kas, K. Larbig, R. Lim, K. Smith, D. Spencer, G. K. Wong, Z. Wu, I. T. Paulsen, J. Reizer, M. H. Saier, R. E. Hancock, S. Lory, and M. V. Olson. 2000. Complete genome sequence of Pseudomonas aeruginosa PAO1, an opportunistic pathogen. Nature 406959-964. [DOI] [PubMed] [Google Scholar]
- 49.Whiteley, M., K. M. Lee, and E. P. Greenberg. 1999. Identification of genes controlled by quorum sensing in Pseudomonas aeruginosa. Proc. Natl. Acad. Sci. USA 9613904-13909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wilson, R., T. Pitt, G. Taylor, D. Watson, J. MacDermot, D. Sykes, D. Roberts, and P. Cole. 1987. Pyocyanin and 1-hydroxyphenazine produced by Pseudomonas aeruginosa inhibit the beating of human respiratory cilia in vitro. J. Clin. Investig. 79221-229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Winzer, K., and P. Williams. 2001. Quorum sensing and the regulation of virulence gene expression in pathogenic bacteria. Int. J. Med. Microbiol. 291131-143. [DOI] [PubMed] [Google Scholar]
- 52.Xu, H., W. Lin, H. Xia, S. Xu, Y. Li, H. Yao, F. Bai, X. Zhang, Y. Bai, P. Saris, and M. Qiao. 2005. Influence of ptsP gene on pyocyanin production in Pseudomonas aeruginosa. FEMS Microbiol. Lett. 253103-109. [DOI] [PubMed] [Google Scholar]
- 53.Yim, G., H. H. Wang, and J. Davies. 2007. Antibiotics as signalling molecules. Philos. Trans. R. Soc. Lond. B 3621195-1211. [DOI] [PMC free article] [PubMed] [Google Scholar]