

Abstract
Recent studies showed that enteric helminth infection improved symptoms in patients with inflammatory bowel disease as well as in experimental models of colitis. The aim of this study was to determine the mechanism of the protective effect of helminth infection on colitis-induced changes in immune and epithelial cell function. BALB/c mice received an oral infection of Heligmosomoides polygyrus third-stage larvae, were given intrarectal saline or trinitrobenzene sulfonic acid (TNBS) on day 10 postinfection, and were studied 4 days later. Separate groups of mice received intrarectal saline or TNBS on day 10 and were studied on day 14. Muscle-free colonic mucosae were mounted in Ussing chambers to measure mucosal permeability and secretion. Expression of cytokines was assessed by quantitative real-time PCR, and mast cells were visualized by immunohistochemistry. TNBS-induced colitis induced mucosal damage, upregulated Th1 cytokines, and depressed secretory responses. Heligmosomoides polygyrus elevated Th2 cytokine expression, increased mast cell infiltration and mucosal resistance, and also reduced some secretory responses. Prior H. polygyrus infection prevented TNBS-induced upregulation of Th1 cytokines and normalized secretory responses to specific agonists. TNBS-induced colitis did not alter H. polygyrus-induced mast cell infiltration or upregulation of Th2 cytokine expression. The results indicate that the protective mechanism of enteric nematode infection against TNBS-induced colitis involves prevention of Th1 cytokine expression and improved colonic function by a mechanism that may involve mast cell-mediated protection of neural control of secretory function. Similar response patterns could account for the clinical improvement seen in inflammatory bowel disease with helminthic therapy.
The vertebrate immune system has coevolved with enteric pathogens, leading to the elaboration of polarized Th1 or Th2 cytokine profiles. Disturbance of the balance between these immune pathways has deleterious consequences for the host. Inflammatory bowel disease (IBD), specifically Crohn's disease, is driven by a Th1-dominant immune response. The initiating factor is thought to be a dysregulated immune response to normal gut flora in genetically predisposed individuals, resulting in chronic gastrointestinal (GI) inflammation leading to significant host morbidity. The incidence and prevalence of IBD have increased substantially over the past 50 years in industrialized countries, but the incidence remains low in underdeveloped areas. This observation could not be explained completely by genetic factors, and therefore the “hygiene hypothesis” evolved as a result of the dramatic reduction in exposure of the gut to pathogens as a consequence of lifestyle and environmental changes designed to eliminate exposure to bacteria and parasites, such as nematodes (18). In this paradigm, the absence of helminth infections eliminates the normal upregulation of the Th2 and/or T regulatory immune response in childhood, culminating in a more Th1-prone immune response characteristic of autoimmune and inflammatory diseases (18).
The interaction between immune and structural cells, such as smooth muscle and epithelial cells, is critical to Th2 cytokine-mediated immunity. Infection-induced alterations in gut function play a major role in host resistance against enteric nematode infection. It is well established that induction of Th2 cytokines, including interleukin-4 (IL-4) and IL-13, is a prerequisite for expulsion. In contrast, the Th1 profile, featuring high levels of IL-12, tumor necrosis factor alpha (TNF-α), or gamma interferon (IFN-γ), promotes worm survival in the GI tract. Because of the ability of Th2 cytokines to modulate Th1 cytokines, there is considerable interest in their contribution, as well as the contributions of their individual receptors, to both Th1- and Th2-predominant pathologies.
The infectious third-stage larvae (L3) of Heligmosomoides polygyrus invade the submucosa of the duodenum for approximately 8 days before the adult stage emerges and persists in the proximal small intestine for weeks to months. Primary exposure results in a chronic infection with an upregulation of the Th2 cytokine response and minimal damage (45). Previous reports demonstrated that several different enteric pathogens ameliorated experimental Th1-driven colitis in mice (16, 26, 37), and H. polygyrus significantly reduced the inflammation seen in IL-10-deficient mice (17). More recently, in humans, administration of Trichuris suis eggs was shown to improve the clinical severity of Crohn's disease and ulcerative colitis (49, 50). The protective ability of nematode infection is thought to be immunomediated; however, there are few data showing changes in local colonic expression of cytokines.
It is known that Th1-driven inflammation is associated with alterations in mucosal function in both the small intestine and colon that contribute to the development of diarrhea, a major symptom of IBD. One of the major actions of the mucosal epithelium is to secrete a fluid that facilitates removal of noxious materials and agents and delivers immunoglobulin A into the gut lumen. Reflex circuits that control secretion are activated by a number of mechanical and chemical stimuli that originate in the lumen and involve serotonin (5-HT) and acetylcholine (13). Mast cells also alter secretion by increasing the sensitivity of nerves to stimulation that can lead to hypersecretory responses characteristic of allergy (3). It was of interest, therefore, that experimental models of IBD induce a stereotypic hyposecretion in response to a number of stimuli (41, 42, 61). This paradoxical effect on secretion suggests that the diarrhea observed in IBD is attributed to decreased sodium absorption due to loss or disruption of surface epithelial cells.
Mast cells are key mediators of allergic and other models of inflammation, including intestinal helminth infection (29). Mast cell mediators, including tryptase, 5-HT, and histamine, can modulate epithelial cell secretion (20). Tryptase binds to one of the protease-activated receptors (PARs) among the class of unique G protein-coupled receptors. Although at least four PARs have been identified, activation of PAR-1 or PAR-2 has important implications in the gut (62, 64). These receptors are autoactivated through their own “tethered ligand” upon enzymatic cleavage by thrombin (PAR-1), trypsin (PAR-2), or mast cell tryptase (PAR-2) (39, 56). Both PAR-1 and PAR-2 activation increases mucosal secretion and permeability and exerts proinflammatory effects in the GI tract (6-9, 11, 28, 55). Histamine is another major mast cell messenger that has numerous physiologic and pathophysiologic effects mediated through at least four distinct G protein-coupled receptors, represented by histamine receptors that include H1R, H2R, H3R, and H4R (25). In the gut, H1R is expressed on epithelial cells and is involved in chloride secretion (2). In contrast, the more recently discovered H4R (33, 38, 40, 65), preferentially expressed on immune cells of hematopoietic origin, is proposed to play a role in the regulation of immune and inflammatory processes (59).
The precise mechanism for the beneficial effects of nematode infection on Th1-driven inflammation remains unclear but may include alterations in the local immune response and/or epithelial cell function. The aims of this study were to determine (i) if the protective effects of intestinal nematode infection on colitis-induced alterations are due to local immune modulation, including colonic Th1 or Th2 cytokine expression, H4R expression, and mast cell activity; and (ii) if intestinal helminth infection affects colitis-induced alterations in epithelial cell function. Our results suggest that the protective effects of parasitic nematode infection are due to local immune modulation of trinitrobenzene sulfonic acid (TNBS)-induced inflammatory changes leading to attenuation or prevention of inflammation-induced changes in colonic mucosal function.
MATERIALS AND METHODS
Animals.
Female BALB/c mice (aged 8 to 10 weeks) were obtained from Charles River, Inc. (NCI, Frederick, MD). The mice were age matched with controls in all four experimental groups. These studies were conducted in accordance with principles set forth in the Guide for the Care and Use of Laboratory Animals (38a) and by the University of Maryland School of Medicine Institutional Care and Use Committee.
Treatment groups.
The timing of TNBS intrarectal injection and H. polygyrus inoculation was coordinated to ensure that mice from each treatment group were studied on the same day.
(i) TNBS-induced colitis.
Mice were fasted for 2 h prior to treatment but were allowed free access to water. Anesthetized mice received an intrarectal injection of TNBS (2 mg/mouse) in 40% ethanol or saline (total volume, 0.1 ml) and were held by the tail for 3 min to ensure continued exposure to the agent. Mice were studied 4 days later.
(ii) Heligmosomoides polygyrus infection.
Infective, ensheathed L3 of H. polygyrus (specimens on file at the U.S. National Helminthological Collection, USDA, Beltsville, MD) were propagated and maintained as described previously (53) and were stored at 4°C until use. BALB/c mice were inoculated orally, via an 18-gauge ball-tipped feeding tube, with 200 L3 on day 0 and were studied at 14 days postinfection. A primary H. polygyrus infection persists for a number of weeks.
(iii) Heligmosomoides polygyrus infection plus TNBS treatment.
At 10 days postinfection, one-half of the H. polygyrus-infected BALB/c mice received intrarectal TNBS and the other half received saline, as described above.
Histology.
Sections (1.5 cm) of distal colon were opened along the mesenteric border, rinsed promptly in saline, and placed in 4% paraformaldehyde. The tissue was embedded in paraffin, sectioned transversely (5 μm), and stained with Giemsa stain in order to visualize granulocytes. The degree of inflammation and tissue damage on microscopic cross sections of the colon was graded semiquantitatively from 0 to 10, using a standardized scoring system in which tissue was given 1 point for the presence of any of the following features: disruption of epithelial cells (loss of epithelial cells, mucosal lifting, mucosal ulceration, or distortion of epithelial crypts), loss of goblet cells or goblet cell mucus, mucosal edema, the presence of neutrophilic or lymphocytic infiltrate or granulomas, hemorrhage, or mucosal or smooth muscle hypertrophy. Two investigators who were unaware of the given treatment performed the grading; the scores were averaged for each slide to obtain a final slide score for each animal. These scores were then averaged for each treatment group.
Immunofluorescence staining.
Frozen blocks of colon were prepared by using the Swiss roll technique and were stored at −80°C as described previously (64). Tissue sections (5 μm) were cut from frozen blocks by use of an HM505E cryostat (Richard Allan Scientific). Slides were kept on dry ice and then stored at −80°C. For immunofluorescence staining, tissue slides were fixed in cold acetone for 30 min and subsequently blocked with 10% normal rabbit serum in phosphate-buffered saline for 1 h at room temperature. After the addition of 1:100 diluted primary goat antibodies for mast cell protease (MCP; Santa Cruz Biotechnology), slides were incubated overnight in a humidified chamber at 4°C. Slides were rinsed for 30 min in phosphate-buffered saline, followed by incubation with fluorescein isothiocyanate-conjugated rabbit anti-goat antibody (Jackson Immunoresearch Laboratories) for 1 h, using a 1:200 dilution (from the original stock solution). The slides were then coverslipped with Vectorshield (Vector Laboratories) and digitally photographed with a Nikon Eclipse 80i microscope using Image-Pro Plus 5.1 software. The intensity of staining was determined by establishing settings for the samples from the vehicle group and using the same conditions to evaluate the samples from the infected or treated groups. Comparisons were made only among the slides prepared on the same day.
Ussing chambers.
One-centimeter segments of mucosa were stripped of muscle and mounted in Ussing chambers that exposed 0.126 cm2 of tissue to 5 ml Krebs buffer. Agar-salt bridges and electrodes were used to measure potential difference. Every 50 s, the tissues were short circuited at 1 V (DVC 1000 voltage clamp; World Precision Instruments, Sarasota, FL), and the short circuit current (Isc) was continuously monitored. In addition, every 50 s, the clamp voltage was adjusted to 1 V for 10 s to allow for calculation of tissue resistance using Ohm's law.
After a 15-min period, concentration-dependent changes in Isc in separate tissues were determined following the cumulative addition of acetylcholine, histamine, or 5-HT (all at 1 nM to 1 μM) to the serosal side. Changes in Isc were also determined in response to PAR-1 (TFLLR; 100 μM) and PAR-2 (SLIGRL; 100 μM) agonists. After the peak response to the final concentration of each secretagogue was recorded, the Krebs buffer on each side of the chamber was replaced. To determine the contribution of enteric nerves to the response, tissues were incubated with the sodium channel blocker tetrodotoxin (TTX; 1 μM) for 15 min prior to the addition of secretagogues.
RNA extraction, cDNA synthesis, and real-time quantitative PCR.
Total RNAs were extracted from samples of mid-colon with TRIzol reagent (Invitrogen, Grand Island, NY) per the manufacturer's instructions. RNA integrity and quantity and genomic DNA contamination were assessed using an Agilent Bioanalyzer 2100 machine and an RNA 6000 Labchip kit (Agilent Technologies, Palo Alto, CA) as described previously (63). Only those RNA samples with 28S/18S ratios between 1.5 and 2 and no DNA contamination were studied. RNA samples (2 μg) were reverse transcribed to cDNAs by use of a first-strand cDNA synthesis kit (MBI Fermentas, Hanover, MD) with a random hexamer primer.
Real-time quantitative PCR was performed on an iCycler detection system. Primer sequences were designed by using Beacon Designer 4.0 (Premier Biosoft International, CA) and were synthesized by the Biopolymer Laboratory of the University of Maryland. The primer sequences for PAR-1, PAR-2, IFN-γ, TNF-α, IL-4, IL-13, H1R, H4R, and MCP-1 are listed in Table 1. PCR was performed in a 25-μl volume, using SYBR green Supermix (Bio-Rad, CA). Amplification conditions were as follows: 95°C for 3 min and 50 cycles of 95°C for 15 s, 60°C for 15 s, and 72°C for 20 s. The changes in mRNA expression for PAR-1, PAR-2, IFN-γ, TNF-α, MCP-1, IL-4, IL-13, H1R, and H4R were determined relative to the respective vehicle groups of mice after normalization to the 18S rRNA housekeeping gene.
TABLE 1.
Primer sequences for real-time quantitative PCR
| Gene | Primer orientation | Primer sequence (5′ to 3′) |
|---|---|---|
| PAR-1 | Forward | GCTGGAGGGTAGGGCAGTCT |
| Reverse | GTACACGGAGGGCATGAAGAG | |
| PAR-2 | Forward | CTGCATCTGTCCTCACTGGA |
| Reverse | ACAGAGAGGAGGTCAGCCAA | |
| IFNγ | Forward | GCATAGATGTGGAAGAAAAGAGTCTCT |
| Reverse | TGGCTCTGCAGGATTTTCATG | |
| TNF-α | Forward | CATCTTCTCAAAATTCGAGTGACAA |
| Reverse | CCAGCTGCTCCTCCACTTG | |
| IL-4 | Forward | CGGAGATGGATGTGCCAAAC |
| Reverse | GCACCTTGGAAGCCCTACAG | |
| IL-13 | Forward | GACCAGACTCCCCTGTGCAA |
| Reverse | TGGGTCCTGTAGATGGCATTG | |
| H1R | Forward | TCCGAAGACAAGATGTGTGAGC |
| Reverse | CACTGTGACCAGGAGATACTAC | |
| H4R | Forward | GGCTCCATACTGTCTGTTCAC |
| Reverse | CAGAAAGGGATTAACAAACGAATTG | |
| MCP-1 | Forward | TGGGAAGTTCCACAAAGTTAAAAAC |
| Reverse | GCCACACCAGCACACAGAAG |
Solutions and drugs.
Krebs buffer contained 4.74 mM KCl, 2.54 mM CaCl2, 118.5 mM NaCl, 1.19 mM NaH2PO4, 1.19 mM MgSO4, 25.0 mM NaHCO3, and 12 mM glucose. The tissues were allowed to equilibrate for 15 min in Krebs buffer. All drugs were obtained from Sigma (St. Louis, MO) unless stated otherwise. Stock solutions were prepared as follows. The PAR-1 peptide agonist TFLLR and the PAR-2 peptide agonist SLIGRL (10 mM; synthesized by the University Biomedical Instrumentation Center, Uniformed Services University of the Health Sciences) were dissolved in 20% dimethyl sulfoxide and stored at −70°C in aliquots. On the day of the experiment, PAR-1, PAR-2, 5-HT, and histamine were dissolved in water, and appropriate dilutions of 5-HT and histamine were made using distilled water.
Data analysis.
For each mouse, responses for each secretagogue were determined for a single tissue, and therefore the n for each group reflects the number of mice. Resistance was calculated for all tissue segments from each mouse and was averaged to yield one mean per animal. Statistical analysis was performed using one-way analysis of variance to compare inflammatory scores, resistances, and maximal Isc responses among groups. Cumulative dose responses were compared using multiple analysis of variance with post hoc analysis for multiple comparisons. P values of <0.05 were considered significant.
RESULTS
Prior H. polygyrus infection attenuated TNBS-induced colonic injury.
In control mice, the colonic mucosa exhibited intact epithelia, an even distribution of goblet cells, minimal infiltrate in the lamina propria and submucosa, and normal smooth muscle thickness (Fig. 1A). Heligmosomoides polygyrus infection did not cause mucosal damage (Fig. 1B) but induced goblet cell hyperplasia, lymphocytic infiltration, and smooth muscle thickening in the colon. Administration of TNBS to mice significantly increased tissue damage in the colon (Fig. 1C), including disruption of epithelial cells, dilation of crypts, widening of the submucosal space, and thickening of the smooth muscle. Separate sections of colon also showed increased lymphocytic infiltrate, loss of normal mucosal architecture, and granulomas (Fig. 1D). Interestingly, prior H. polygyrus infection markedly attenuated TNBS-induced colonic damage and inflammation, with an absence of granuloma formation and submucosal edema (Fig. 1E). There was a retention of smooth muscle hypertrophy and of the elevated number of goblet cells characteristic of infection alone (Fig. 1B), and thus the microscopic damage score remained elevated above that of controls. The mean damage score (Fig. 1F) demonstrates the protective effect of prior nematode infection on TNBS-induced colitis.
FIG. 1.

Samples of distal colon were fixed, paraffin embedded, sectioned (5 μm), and stained with Giemsa stain to visualize inflammatory infiltrate. Representative samples from controls (A) and the treatment groups are included. (B) H. polygyrus infection showed few changes, with the most notable being a goblet cell hyperplasia and thickening smooth muscle. (C) In contrast, TNBS administration induced numerous inflammatory changes, including crypt expansion (asterisk), neutrophil infiltration (arrowheads), and disruption of surface epithelial cells (arrow). (D) In addition, there were areas of graulomatous infiltration. (E) Infection with H. polygyrus prior to TNBS exposure markedly attenuated TNBS-induced inflammation, including protection against mucosal damage and neutrophil infiltration. (F) The mean scores for all groups demonstrate that prior H. polygyrus infection protects against the damage and inflammation induced by TNBS (*, P < 0.05 versus control; **, P < 0.01 versus control; φ, P < 0.05 versus TNBS group).
The protective effect of prior H. polygyrus infection on colitis was associated with a decrease in mRNA expression of Th1 cytokines.
It is well established that increased production of Th1 cytokines plays an important role in the pathogenesis of colitis. Consistent with previous studies, mice treated with TNBS exhibited an upregulation of the Th1 cytokines IFN-γ and TNF-α (Fig. 2A), but not of the Th2 cytokines IL-13 and IL-4 (Fig. 2B), in the colon. Heligmosomoides polygyrus preferentially colonizes the small intestine, and therefore the ability of the infection to upregulate expression of IL-4 and IL-13 (17.4- and 356.0-fold, respectively) was confirmed in this region. Heligmosomoides polygyrus infection also induced a significant increase in the expression of Th2 cytokines IL-4 and IL-13 in the colon (Fig. 2B) but had no effect on expression of the Th1 cytokines (Fig. 2A). Prior H. polygyrus infection prevented the TNBS-induced upregulation of Th1 cytokines (Fig. 2A). In contrast, the H. polygyrus infection-induced upregulation of Th2 cytokines in the colon was unaltered by administration of TNBS (Fig. 2B).
FIG. 2.
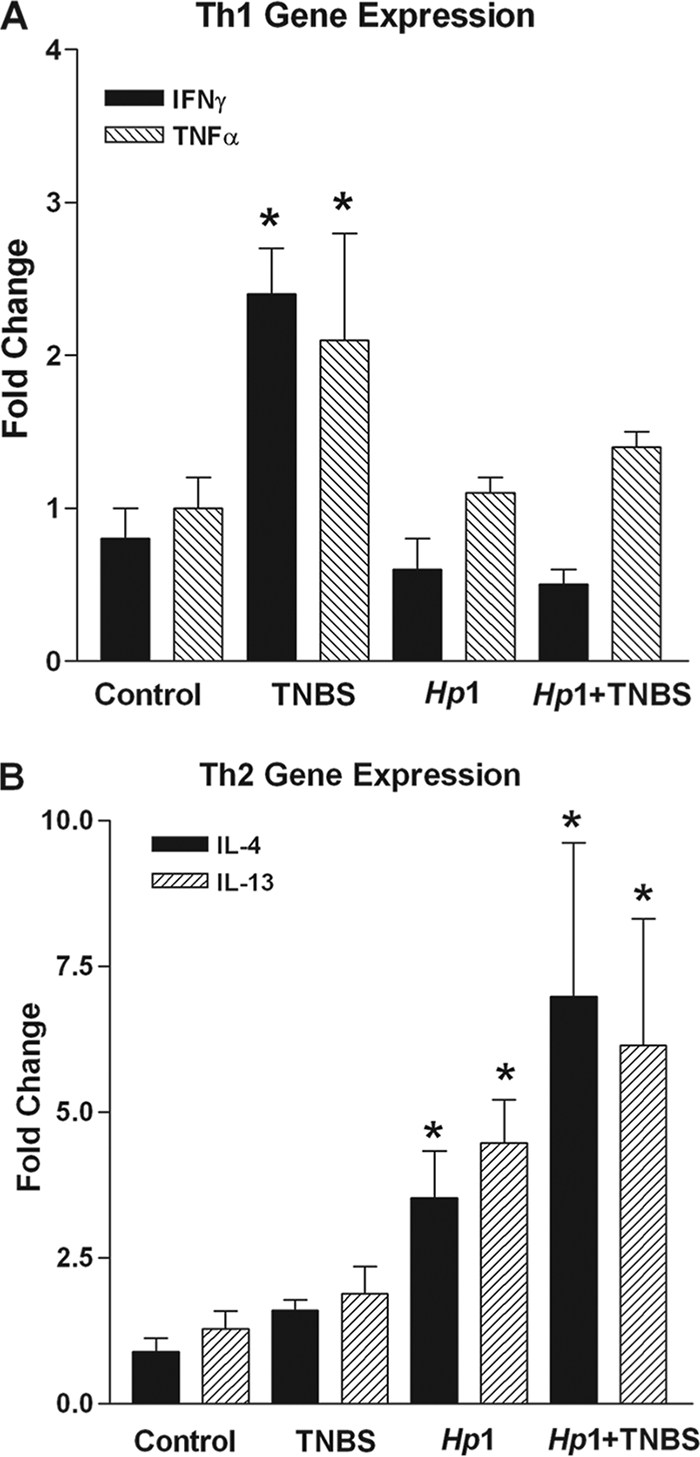
Samples of colon were taken from each group. Signature Th1 and Th2 cytokines were quantitated using real-time quantitative PCR on whole colonic tissue. (A) TNBS induced a significant increase in Th1 cytokine expression, while H. polygyrus significantly upregulated Th2 cytokine expression. Prior exposure to H. polygyrus prevented upregulation of Th1 cytokines. (B) TNBS had no effect on the upregulation of Th2 cytokines by H. polygyrus (*, P < 0.05 versus control).
Prior H. polygyrus infection improved colitis-induced changes in epithelial cell function.
Abnormal colonic epithelial cell function is implicated in the pathogenesis of colitis. To determine if the protective effect of prior H. polygyrus infection on TNBS-induced colitis is associated with the alteration of colonic epithelial cell function, muscle-free sections of mucosae were mounted in Ussing chambers. Colonic mucosal resistance, an index of permeability, was increased significantly by H. polygyrus infection (Fig. 3), indicating the beneficial effect of infection on barrier function. Although TNBS treatment alone did not alter mucosal resistance, the prior H. polygyrus infection-induced increase in mucosal resistance (decrease in permeability) remained even after the subsequent administration of TNBS (Fig. 3).
FIG. 3.
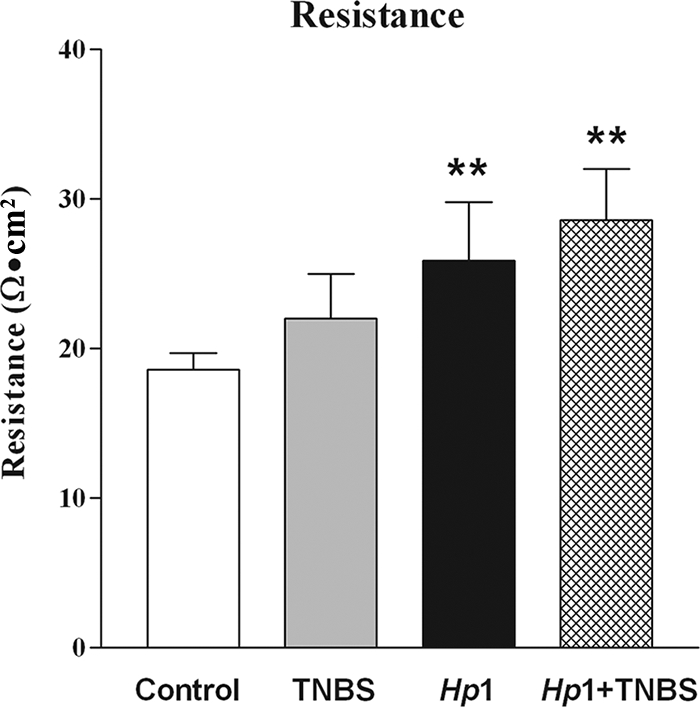
Resistance, an index of mucosal permeability, was determined with Ussing chambers, using muscle-free colonic mucosa. TNBS did not alter permeability, while H. polygyrus significantly increased resistance (Hp1). This increased resistance persisted after injection with TNBS (Hp1+TNBS) (**, P < 0.01 versus control).
We further assessed colonic epithelial cell secretary responses to various secretagogues. 5-HT, released from enterochromaffin-like cells, binds to receptors on sensory nerves forming the afferent arm of reflex-mediated secretion. Secretory responses to 5-HT were decreased significantly by TNBS but not by H. polygyrus (Fig. 4A). Prior enteric infection with H. polygyrus prevented the TNBS-induced inhibition of secretion in response to 5-HT. Acetylcholine, upon release from enteric nerves as part of the reflexive secretory arc, binds to muscarinic receptors on epithelial cells. Colonic secretory responses to acetylcholine were decreased by TNBS administration (Fig. 4B). Although H. polygyrus infection alone did not affect the response, it did prevent the hyposecretion in response to acetylcholine induced by TNBS (Fig. 4B). TTX, a neurotoxin, significantly reduced the response to acetylcholine, by 38%, in controls, indicating a significant contribution by enteric nerves (Fig. 4B). This neural contribution was enhanced in H. polygyrus-infected mice, as TTX decreased the response to acetylcholine by 73%. In contrast, TTX did not alter responses to acetylcholine in TNBS-treated mice, demonstrating a loss of the contribution of nerves to inflammation. Prior infection with H. polygyrus restored the TTX sensitivity to acetylcholine and rendered the responses nearly 100% dependent on enteric nerves (Fig. 4B).
FIG. 4.
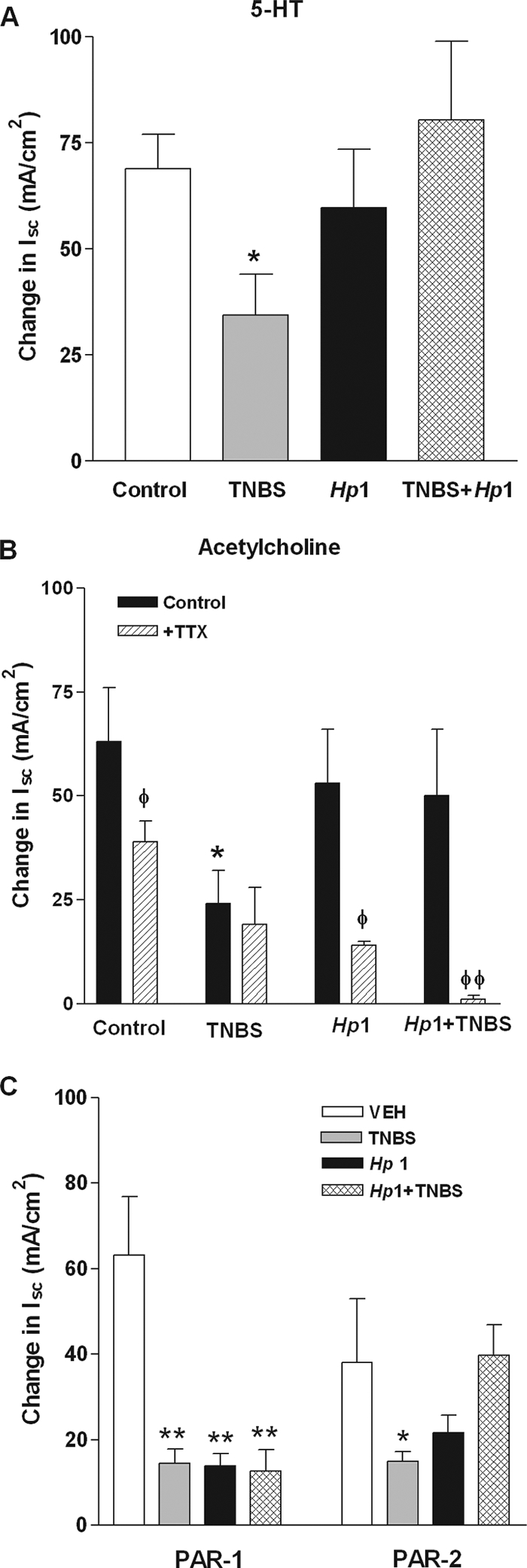
Secretion in response to 5-HT (A), acetylcholine (B), and the PAR-1 agonist TFLLR and PAR-2 agonist SLIGRL (C) was determined in muscle-free colonic mucosae by use of Ussing chambers. TNBS uniformly induced a hyposecretion in response to all secretagogues tested (A to C). Prior enteric infection with H. polygyrus (Hp1+TNBS) prevented the TNBS-induced hyposecretion in response to acetylcholine, 5-HT, and the PAR 2, but not PAR-1, agonist. Secretion in response to acetylcholine was reduced by TTX in control and H. polygyrus (Hp1)-infected mice (B). This sensitivity of acetylcholine-induced responses to TTX was lost in TNBS-treated mice but was restored in mice treated with TNBS after infection with H. polygyrus. *, P < 0.05 versus respective control; **, P < 0.01 versus respective control; φ, P < 0.05 versus respective vehicle; φφ, P < 0.01 versus respective vehicle.
We showed previously that a primary H. polygyrus infection induces a significant mastocytosis in BALB/c mice (45). Mast cells produce a number of proteases, and mast cell mediators can alter intestinal secretion, in part by increasing the sensitivity or responses to neural input (45). Endogenous ligands of PAR-1 (thrombin) and PAR-2 (tryptase and trypsin) have a number of effects on epithelial cell function. Colonic expression of PAR-1 and PAR-2 was unchanged by colonic inflammation or intestinal infection (Table 2). Colonic secretion in response to the PAR-1 agonist TFLLR was decreased in all groups (Fig. 4C). In contrast, there was a selective inhibition of the response to the PAR-2 agonist SLIGRL by TNBS that was completely prevented by prior infection with H. polygyrus (Fig. 4C).
TABLE 2.
Effects of H. polygyrus infection on TNBS-induced alterations in PAR expression and in secretory responses to acetylcholine
| Group | Fold change in expression
|
Response (Isc [μA/cm2]) to acetylcholinea | |
|---|---|---|---|
| PAR-1 | PAR-2 | ||
| Control | 1.0 ± 0.04 | 1.0 ± 0.1 | 63 ± 13 |
| TNBS | 1.2 ± 0.2 | 0.7 ± 0.1 | 24 ± 8* |
| H. polygyrus infection | 0.9 ± 0.2 | 1.0 ± 0.1 | 53 ± 13 |
| H. polygyrus infection + TNBS | 1.2 ± 0.2 | 1.0 ± 0.1 | 70 ± 24 |
*, P < 0.05 versus control group. The [acetylcholine] was 100 μM (n = 6 to 8).
Prior H. polygyrus infection altered TNBS-induced changes in histamine receptor expression.
Histamine is also secreted by mucosal mast cells and has direct prosecretory effects by binding to receptors on epithelial cells as well as indirect effects by increasing the sensitivity of enteric nerves to stimulation. In the present study, responses to histamine were inhibited by all treatments (Fig. 5A). Histamine increases secretion by binding to H1R located on epithelial cells. The decrease in H1R expression in the TNBS-treated, H. polygyrus-infected, and H. polygyrus-plus-TNBS-treated groups (Fig. 5B) is consistent with the hyposecretory response to histamine seen in these groups (Fig. 5A). H4R is an important proinflammatory mediator that is found on immune cells of hematopoietic lineage. TNBS significantly upregulated H4R expression (Fig. 5C); this effect was prevented completely by previous H. polygyrus infection.
FIG. 5.
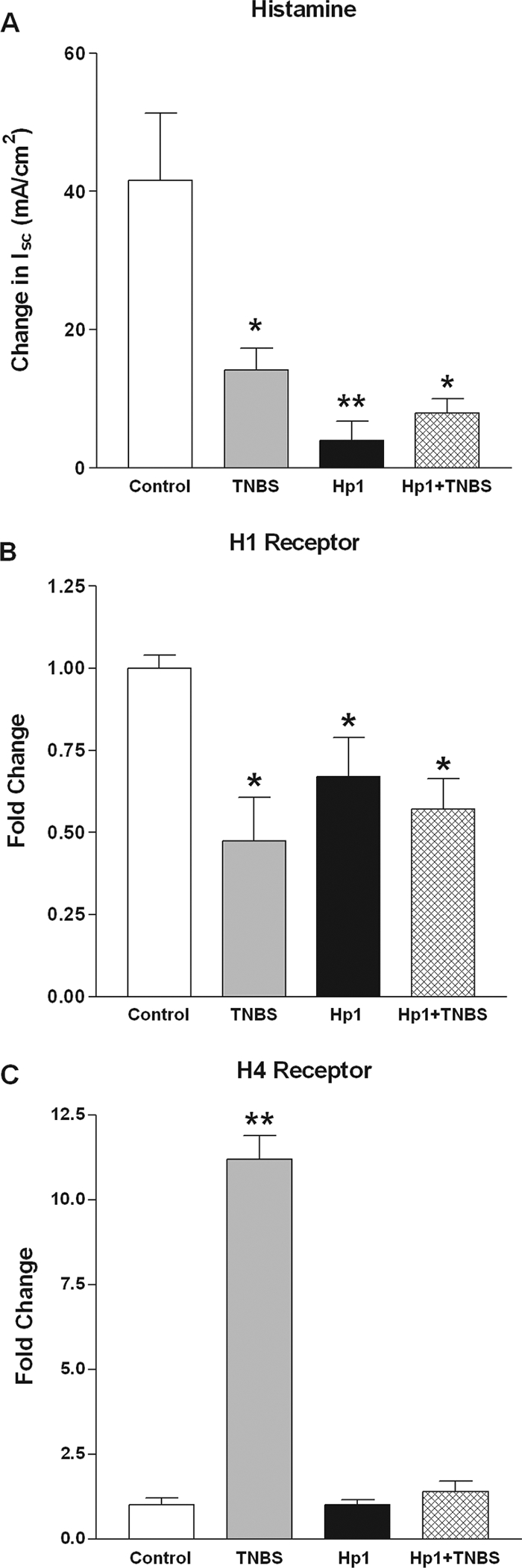
Secretory responses to histamine (changes in Isc) were determined in muscle-free colonic mucosae by use of Ussing chambers, and secretion was decreased significantly in all treatment groups. Expression of histamine receptors was determined by quantitative real-time PCR with colonic tissue. (A) TNBS inhibited secretion in response to histamine, and this was not prevented by prior enteric infection with H. polygyrus. (B) H1R expression was downregulated significantly in all groups. (C) H4R expression was increased significantly only in the TNBS group. Prior H. polygyrus infection completely prevented the TNBS-induced upregulation. *, P < 0.05 versus control or vehicle; **, P < 0.01 versus control or vehicle.
Mast cells have a role in the protective effect of H. polygyrus on TNBS-induced inflammation.
To explore the contribution of mast cells to the protective effect of H. polygyrus on Th1-driven inflammatory changes, we assessed expression of MCP-1 by real-time PCR and immunohistochemistry. MCP-1 gene expression was increased significantly by H. polygyrus but was unchanged by TNBS (Fig. 6A). In addition, the infection-induced increase in MCP-1 was not altered by TNBS administration. Controls showed modest immunostaining for MCP-1 (Fig. 6B) that was markedly increased in H. polygyrus infection (Fig. 6C). In contrast, immunostaining for MCP-1 was completely absent in the TNBS group (Fig. 6D), and TNBS decreased staining in mice with prior H. polygyrus infection (Fig. 6E). The effect of TNBS treatment in decreasing staining in uninfected mice (versus vehicle-treated controls) and infected mice (versus infected controls) is consistent with an inflammation-induced loss and/or degranulation of mast cells.
FIG. 6.

(A) MCP-1 mRNA expression was determined by real-time PCR. Expression was elevated significantly by H. polygyrus infection. **, P < 0.01 versus control. (B) Mast cell infiltration is indicated by immunofluorescent staining of MCP-1. Vehicle-treated control mice had some staining of mast cells (arrows). (C) Heligmosomoides polygyrus infection increased the marked mast cell infiltration. (D) In contrast, TNBS treatment caused a decrease in background mast cell staining. (E) Prior H. polygyrus infection partially stabilized the mast cell staining that was decreased by TNBS.
DISCUSSION
It is well known that the balance of immunoderived cytokines in vivo is critical to the control of immune function and that polarization toward the Th1 or Th2 cytokine profile can have consequences that are inappropriate to host defense. The increased Th1 response to TNBS is associated with moderate to severe granulomatous, and sometimes transmural, inflammation similar to that observed in Crohn's disease. The ability of infection-induced upregulation of Th2 cytokines to attenuate Th1-mediated inflammation in the GI tract is clear (16, 26, 37, 49, 50), but previous studies did not address immunomediated changes localized to the affected area or assess possible beneficial effects to gut function. In the present study, maintenance of the Th2-polarized response induced by H. polygyrus in the small intestine and colon effectively prevented TNBS-induced colonic upregulation of gene expression for the Th1 cytokines IFN-γ and TNF-α, pathological injury to the mucosa, and alterations in epithelial cell function. These data are consistent with the hypothesis that nematode infection prevents an exacerbated response to proinflammatory stimuli in the colon. Thus, the beneficial effects of nematode infection in the small intestine orchestrate immunomediated changes and protection from inflammation at a distant site in the colon.
The results of the present study are in contrast to those showing that helminth infection worsened colitis caused by infection with Citrobacter rodentium, an enteropathogen similar to Escherichia coli (10). It is well established that induction of the Th1 profile, featuring high levels of IL-12, TNF-α, or IFN-γ, is a prerequisite for expulsion of a number of enteric pathogens, including C. rodentium. In contrast, upregulation of Th2 cytokines, including IL-4 and IL-13, impairs its clearance. It is not surprising, therefore, that the ability of H. polygyrus to prevent upregulation of the requisite Th1 response would worsen C. rodentium-induced inflammation by prolonging exposure to unrestricted pathogen proliferation. It should be noted, however, that while the commensal bacteria play an important role in the pathogenesis of IBD (22) as well as other spontaneous animal models of inflammation (30), Crohn's disease and ulcerative colitis are considered to be immunomediated rather than infectious. The etiology of the TNBS-induced model of colitis may therefore be closer to clinical autoimmune disease in this regard.
The effects of Th1-dominant inflammatory processes are well documented and include reduced epithelial secretion in IBD patients as well as animal models of colitis (44, 60). This may be due to a direct effect of immune and/or inflammatory mediators on epithelial function or an indirect effect due to the loss of epithelial cells in inflammation. Secretion is an important part of mucosal defense because it facilitates removal of harmful substances and bacteria in the lumen away from the epithelial surface and reduces bacterial translocation (1). In the present study, TNBS uniformly inhibited secretion (Isc) in responses to all secretagogues tested, although it did not affect resistance, indicating a largely intact barrier function.
5-HT is critically involved in the afferent arm of the mucosal secretory reflex arc by transducing signals from the lumen to the afferent nerves (Fig. 7). Recent studies demonstrated alterations in 5-HT signaling in functional GI disorders, such as irritable bowel syndrome, chronic constipation, diarrhea, and functional dyspepsia (46). The gut is the largest source of 5-HT, whose effects are mediated through numerous 5-HT receptors. In response to these stimuli, enterochromaffin cells release 5-HT, which binds to specific receptors on primary afferent nerves or epithelial cells (13). In the current study, the decrease in 5-HT-stimulated secretion induced by TNBS is consistent with either loss or damage to epithelial cells and/or nerves.
FIG. 7.

Schematic showing the interaction between immune cells and epithelial cells during TNBS-induced colitis. (A) In this paradigm, secretion is elicited by luminal events that stimulate enterochromaffin cells (EC cells) to release 5-HT, which acts on receptors on sensory afferent nerves that may transmit information to the central nervous system (CNS) or act locally to release substance P (SP) at neurons within the enteric nervous system within the wall of the gut. This is part of a secretory reflex culminating in the release of neurotransmitters, such as acetylcholine (ACH), that bind to muscarinic receptors on epithelial cells to release chloride ions (Cl−) and fluid. TNBS upregulates Th1 cytokines, increases neutrophil infiltration, induces a loss of and/or degranulation of mast cells, and causes mucosal damage, including injury to enteric nerves (dashed lines). (B) In H. polygyrus infection, there is an increased production of Th2 cytokines that prevents TNBS-induced upregulation of Th1 cytokines. This is associated with an enhanced number of mast cells close to enteric nerves that increase the sensitivity of these nerves to stimulation and render the responses more dependent on enteric nerves. Prior infection with H. polygyrus prevents the development of the Th1 response, limits mucosal damage and inflammation and neutrophil infiltration, and enhances the contribution of nerves to secretory responses involving mast cells.
In the secretory reflex, 5-HT- or substance P-mediated activation of motor neurons results in release of neurotransmitters, such as acetylcholine (14, 15). Secretion in response to exogenous acetylcholine is a combination of effects mediated by binding to nicotinic receptors on enteric neurons or through muscarinic receptors on epithelial cells (Fig. 7). Secretion in response to acetylcholine was reduced by both H. polygyrus infection and TNBS. It is interesting that the contribution of enteric nerves to acetylcholine was lost after TNBS but was protected by prior infection with H. polygyrus, indicating that enteric infection protects against damage to the reflex secretory circuits. Mast cells approximate enteric nerves, and the mastocytosis of enteric infection enhances this interaction (45). These pathways play an important role in mucosal protection as part of the “weep and sweep” response to potential noxious stimuli (3). The increased mRNA expression and antibody staining for MCP-1 after H. polygyrus infection and H. polygyrus infection plus TNBS-induced colitis compared to those after TNBS induction alone indicate that an influx of mast cells may serve a protective role by increasing the sensitivity of sensory nerves to stimulation, effectively restoring secretion to control levels. This is consistent with the observation that responses to acetylcholine in the H. polygyrus infection and H. polygyrus infection-plus-TNBS groups were nearly abolished in the presence of TTX, indicating a high level of dependence on enteric nerves after infection (Fig. 7). Although MCP-1 mRNA expression was similar in H. polygyrus infection and H. polygyrus infection with TNBS induction, there was a reduction in antibody staining for MCP-1 for H. polygyrus infection with TNBS induction versus H. polygyrus infection. There was also a reduction in staining for baseline levels of mast cells in mice after treatment with TNBS alone compared to that for vehicle-treated control mice, suggesting that TNBS may degranulate mast cells even when they are activated by infection with H. polygyrus.
Colitis-induced abnormalities in enteric nerves may also alter responses to PAR agonists. PAR-2 is expressed along the entire length of the GI tract and has been identified in a number of cells, including enterocytes (48). The role of PAR-2 in inflammation is better defined than that of PAR-1 (23, 31). PAR-2 is also linked to sensitization of visceral afferents leading to hyperalgesia (12), and PAR-2 agonists increase epithelial secretion in both the small intestine and colon (4, 28, 36). In the present study, we showed that the TNBS-induced inhibition of responses to PAR-2 activation was normalized with prior exposure to H. polygyrus. MCPs (e.g., tryptase in humans) are important mediators of PAR-2 activation, suggesting that the restoration of secretion to PAR-2 may be mediated by an effect of H. polygyrus on mucosal mast cells and their effects on enteric nerves.
PAR-1 is expressed throughout the gut, having been identified in the smooth muscle, epithelial, nerve, and lamina propria cells (48). PAR-1 is overexpressed in the colons of IBD patients and 7 days after TNBS-induced colitis in mice due to the increased numbers of infiltrating inflammatory cells (55). Activation of PAR-1 leads to increased intestinal secretion, permeability, and leukocyte infiltration (6, 7, 11, 55). In the present study, however, PAR-1 expression was unchanged 4 days after TNBS administration. This may be due to the timing (4 versus 7 days) or method of analysis (real-time versus quantitative reverse transcription-PCR). Previous studies showed that PAR-1 agonists had no effect on colonic secretion in the unstripped murine colon (6) but had increased secretion in human epithelial cell lines (7). The uniform inhibition of responses to PAR-1 in all treatment groups indicates that the effect is not specific to inflammation but may be a generalized response to CD4+ T-cell-mediated immune activation.
Histamine is one of many mast cell products, and increased levels of histamine and its metabolites are reported in active IBD (27, 43, 57, 58). Much of the effect of histamine on epithelial cell secretion is mediated by H1R. In the current study, we observed an inhibition of histamine secretion in the colon for all treatment groups that was associated with a significant reduction in H1R expression. We showed previously that H. polygyrus had no effect on histamine responses in the small intestine (44), indicating a regional specificity governed by changes in H1R expression after infection.
H4R is preferentially expressed on hematopoietic cells, including mast cells, dendritic cells, T cells, eosinophils, and possibly neutrophils (33, 38, 40, 65). It exhibits numerous proinflammatory effects, including neutrophil, mast cell, and eosinophil chemotaxis (5, 21, 24, 32, 51, 52). Increased H4R expression was observed in the TNBS group, coincident with significant infiltration of immune/inflammatory cells and mucosal damage. The ability of H. polygyrus to block the increase in H4R expression suggests that its protective effect is related to limiting infiltration or activation of cells specific to Th1-dominant inflammation. Neutrophils are the most likely candidate, which is supported by a very recent study showing that an H4R antagonist significantly inhibited TNBS-induced injury and myeloperoxidase activity, an index of neutrophil infiltration, in the rat colon (54). These data indicate that H4R may be a novel therapeutic target in inflammatory pathologies.
Mucosal barrier function plays a critical role in the pathogenesis of a number of diseases, including IBD (19); however, mucosal permeability was unaltered 4 days after TNBS treatment. Previous studies using water flux and/or clearance of radioisotopes to measure changes in colonic permeability 4 h to 2 weeks after TNBS-induced colitis in rats showed increased permeability after TNBS treatment (47, 60). It should be noted, however, that these studies reflect permeability changes along the entire gut, and thus, increased permeability may be attributed to altered resistance in the small intestine rather than the colon. The ability of H. polygyrus infection to improve colonic resistance, even after administration of TNBS, is another benefit of the upregulation of Th2 cytokines. In contrast, we showed previously that IL-4 and IL-13 both decrease resistance in the small intestine (34, 35, 45). The noted increase in colonic resistance may serve to limit movement of antigens or toxins across the colon, an area of slow transit and high bacterial load relative to the small intestine.
In conclusion, these data demonstrate that H. polygyrus infection-induced upregulation of Th2 cytokines attenuated TNBS-induced changes in colonic epithelial cell function by attenuating the Th1 response, restoring normal secretory function to 5-HT, acetylcholine, and PAR2, and enhancing colonic resistance. Part of this effect may be mediated by a protective effect of infection on enteric nerves and/or facilitation of enteric nerve and mast cell interactions to maintain secretion. The upregulation of H4R in the local tissue may underscore an important histaminergic contribution to chronic colonic inflammation and may serve as a potential therapeutic target. Upregulation of Th2 cytokines through nematode infection prevents the development of the Th1 response, suggesting that elimination of helminth infection in industrialized countries exacerbates the response to proinflammatory stimuli and facilitates the development of immunogenic pathologies such as IBD.
Editor: W. A. Petri, Jr.
Footnotes
Published ahead of print on 21 July 2008.
REFERENCES
- 1.Asfaha, S., C. J. Bell, J. L. Wallace, and W. K. MacNaughton. 1999. Prolonged colonic epithelial hyporesponsiveness after colitis: role of inducible nitric oxide synthase. Am. J. Physiol. Gastrointest. Liver Physiol. 276G703-G710. [DOI] [PubMed] [Google Scholar]
- 2.Ash, A. S., and H. O. Schild. 1966. Receptors mediating some actions of histamine. Br. J. Pharmacol. Chemother. 27427-439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Berin, M. C., A. J. Kiliaan, P. C. Yang, J. A. Groot, Y. Kitamura, and M. H. Perdue. 1998. The influence of mast cells on pathways of transepithelial antigen transport in rat intestine. J. Immunol. 1612561-2566. [PubMed] [Google Scholar]
- 4.Bohm, S. K., W. Kong, D. Bromme, S. P. Smeekens, D. C. Anderson, A. Connolly, M. Kahn, N. A. Nelken, S. R. Coughlin, D. G. Payan, and N. W. Bunnett. 1996. Molecular cloning, expression and potential functions of the human proteinase-activated receptor-2. Biochem. J. 3141009-1016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Buckland, K. F., T. J. Williams, and D. M. Conroy. 2003. Histamine induces cytoskeletal changes in human eosinophils via the H(4) receptor. Br. J. Pharmacol. 1401117-1127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Buresi, M. C., A. G. Buret, M. D. Hollenberg, and W. K. MacNaughton. 2002. Activation of proteinase-activated receptor 1 stimulates epithelial chloride secretion through a unique MAP kinase- and cyclo-oxygenase-dependent pathway. FASEB J. 161515-1525. [DOI] [PubMed] [Google Scholar]
- 7.Buresi, M. C., E. Schleihauf, N. Vergnolle, A. Buret, J. L. Wallace, M. D. Hollenberg, and W. K. MacNaughton. 2001. Protease-activated receptor-1 stimulates Ca(2+)-dependent Cl(−) secretion in human intestinal epithelial cells. Am. J. Physiol. Gastrointest. Liver Physiol. 281G323-G332. [DOI] [PubMed] [Google Scholar]
- 8.Cenac, N., A. M. Coelho, C. Nguyen, S. Compton, P. Andrade-Gordon, W. K. MacNaughton, J. L. Wallace, M. D. Hollenberg, N. W. Bunnett, R. Garcia-Villar, L. Bueno, and N. Vergnolle. 2002. Induction of intestinal inflammation in mouse by activation of proteinase-activated receptor-2. Am. J. Pathol. 1611903-1915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cenac, N., R. Garcia-Villar, L. Ferrier, M. Larauche, N. Vergnolle, N. W. Bunnett, A. M. Coelho, J. Fioramonti, and L. Bueno. 2003. Proteinase-activated receptor-2-induced colonic inflammation in mice: possible involvement of afferent neurons, nitric oxide, and paracellular permeability. J. Immunol. 1704296-4300. [DOI] [PubMed] [Google Scholar]
- 10.Chen, C. C., S. Louie, B. McCormick, W. A. Walker, and H. N. Shi. 2005. Concurrent infection with an intestinal helminth parasite impairs host resistance to enteric Citrobacter rodentium and enhances Citrobacter-induced colitis in mice. Infect. Immun. 735468-5481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chin, A. C., N. Vergnolle, W. K. MacNaughton, J. L. Wallace, M. D. Hollenberg, and A. G. Buret. 2003. Proteinase-activated receptor 1 activation induces epithelial apoptosis and increases intestinal permeability. Proc. Natl. Acad. Sci. USA 10011104-11109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Coelho, A. M., N. Vergnolle, B. Guiard, J. Fioramonti, and L. Bueno. 2002. Proteinases and proteinase-activated receptor 2: a possible role to promote visceral hyperalgesia in rats. Gastroenterology 1221035-1047. [DOI] [PubMed] [Google Scholar]
- 13.Cooke, H. J. 1998. “Enteric tears”: chloride secretion and its neural regulation. News Physiol. Sci. 13269-274. [PubMed] [Google Scholar]
- 14.Cooke, H. J., M. Sidhu, P. Fox, Y. Z. Wang, and E. M. Zimmermann. 1997. Substance P as a mediator of colonic secretory reflexes. Am. J. Physiol. 272G238-G245. [DOI] [PubMed] [Google Scholar]
- 15.Cooke, H. J., M. Sidhu, and Y. Z. Wang. 1997. 5-HT activates neural reflexes regulating secretion in the guinea-pig colon. Neurogastroenterol. Motil. 9181-186. [DOI] [PubMed] [Google Scholar]
- 16.Elliott, D. E., J. Li, A. Blum, A. Metwali, K. Qadir, J. F. Urban, Jr., and J. V. Weinstock. 2003. Exposure to schistosome eggs protects mice from TNBS-induced colitis. Am. J. Physiol. Gastrointest. Liver Physiol. 284G385-G391. [DOI] [PubMed] [Google Scholar]
- 17.Elliott, D. E., T. Setiawan, A. Metwali, A. Blum, J. F. Urban, Jr., and J. V. Weinstock. 2004. Heligmosomoides polygyrus inhibits established colitis in IL-10-deficient mice. Eur. J. Immunol. 342690-2698. [DOI] [PubMed] [Google Scholar]
- 18.Elliott, D. E., J. F. Urban, Jr., C. K. Argo, and J. V. Weinstock. 2000. Does the failure to acquire helminthic parasites predispose to Crohn's disease? FASEB J. 141848-1855. [DOI] [PubMed] [Google Scholar]
- 19.Fasano, A., and T. Shea-Donohue. 2005. Mechanisms of disease: the role of intestinal barrier function in the pathogenesis of gastrointestinal autoimmune diseases. Nat. Clin. Pract. Gastroenterol. Hepatol. 2416-422. [DOI] [PubMed] [Google Scholar]
- 20.Frieling, T., J. M. Palmer, H. J. Cooke, and J. D. Wood. 1994. Neuroimmune communication in the submucous plexus of guinea pig colon after infection with Trichinella spiralis. Gastroenterology 1071602-1609. [DOI] [PubMed] [Google Scholar]
- 21.Gantner, F., K. Sakai, M. W. Tusche, W. W. Cruikshank, D. M. Center, and K. B. Bacon. 2002. Histamine h(4) and h(2) receptors control histamine-induced interleukin-16 release from human CD8(+) T cells. J. Pharmacol. Exp. Ther. 303300-307. [DOI] [PubMed] [Google Scholar]
- 22.Hanauer, S. B. 2006. Inflammatory bowel disease: epidemiology, pathogenesis, and therapeutic opportunities. Inflamm. Bowel. Dis. 12(Suppl. 1)S3-S9. [DOI] [PubMed] [Google Scholar]
- 23.Hansen, K. K., P. M. Sherman, L. Cellars, P. Andrade-Gordon, Z. Pan, A. Baruch, J. L. Wallace, M. D. Hollenberg, and N. Vergnolle. 2005. A major role for proteolytic activity and proteinase-activated receptor-2 in the pathogenesis of infectious colitis. Proc. Natl. Acad. Sci. USA 1028363-8368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hofstra, C. L., P. J. Desai, R. L. Thurmond, and W. P. Fung-Leung. 2003. Histamine H4 receptor mediates chemotaxis and calcium mobilization of mast cells. J. Pharmacol. Exp. Ther. 3051212-1221. [DOI] [PubMed] [Google Scholar]
- 25.Hough, L. B. 2001. Genomics meets histamine receptors: new subtypes, new receptors. Mol. Pharmacol. 59415-419. [PubMed] [Google Scholar]
- 26.Khan, W. I., P. A. Blennerhasset, A. K. Varghese, S. K. Chowdhury, P. Omsted, Y. Deng, and S. M. Collins. 2002. Intestinal nematode infection ameliorates experimental colitis in mice. Infect. Immun. 705931-5937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Knutson, L., O. Ahrenstedt, B. Odlind, and R. Hallgren. 1990. The jejunal secretion of histamine is increased in active Crohn's disease. Gastroenterology 98849-854. [DOI] [PubMed] [Google Scholar]
- 28.Kong, W., K. McConalogue, L. M. Khitin, M. D. Hollenberg, D. G. Payan, S. K. Bohm, and N. W. Bunnett. 1997. Luminal trypsin may regulate enterocytes through proteinase-activated receptor 2. Proc. Natl. Acad. Sci. USA 948884-8889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Krishnaswamy, G., J. Kelley, D. Johnson, G. Youngberg, W. Stone, S. K. Huang, J. Bieber, and D. S. Chi. 2001. The human mast cell: functions in physiology and disease. Front. Biosci. 6D1109-D1127. [DOI] [PubMed] [Google Scholar]
- 30.Kuhn, R., J. Lohler, D. Rennick, K. Rajewsky, and W. Muller. 1993. Interleukin-10-deficient mice develop chronic enterocolitis. Cell 75263-274. [DOI] [PubMed] [Google Scholar]
- 31.Lindner, J. R., M. L. Kahn, S. R. Coughlin, G. R. Sambrano, E. Schauble, D. Bernstein, D. Foy, A. Hafezi-Moghadam, and K. Ley. 2000. Delayed onset of inflammation in protease-activated receptor-2-deficient mice. J. Immunol. 1656504-6510. [DOI] [PubMed] [Google Scholar]
- 32.Ling, P., K. Ngo, S. Nguyen, R. L. Thurmond, J. P. Edwards, L. Karlsson, and W. P. Fung-Leung. 2004. Histamine H4 receptor mediates eosinophil chemotaxis with cell shape change and adhesion molecule upregulation. Br. J. Pharmacol. 142161-171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Liu, C., X. Ma, X. Jiang, S. J. Wilson, C. L. Hofstra, J. Blevitt, J. Pyati, X. Li, W. Chai, N. Carruthers, and T. W. Lovenberg. 2001. Cloning and pharmacological characterization of a fourth histamine receptor (H(4)) expressed in bone marrow. Mol. Pharmacol. 59420-426. [DOI] [PubMed] [Google Scholar]
- 34.Madden, K. B., L. Whitman, C. Sullivan, W. C. Gause, J. F. Urban, Jr., I. M. Katona, F. D. Finkelman, and T. Shea-Donohue. 2002. Role of STAT6 and mast cells in IL-4- and IL-13-induced alterations in murine intestinal epithelial cell function. J. Immunol. 1694417-4422. [DOI] [PubMed] [Google Scholar]
- 35.Madden, K. B., K. A. Yeung, A. Zhao, W. C. Gause, F. D. Finkelman, I. M. Katona, J. F. Urban, Jr., and T. Shea-Donohue. 2004. Enteric nematodes induce stereotypic STAT6-dependent alterations in intestinal epithelial cell function. J. Immunol. 1725616-5621. [DOI] [PubMed] [Google Scholar]
- 36.Mall, M., T. Gonska, J. Thomas, S. Hirtz, R. Schreiber, and K. Kunzelmann. 2002. Activation of ion secretion via proteinase-activated receptor-2 in human colon. Am. J. Physiol. Gastrointest. Liver Physiol. 282G200-G210. [DOI] [PubMed] [Google Scholar]
- 37.Moreels, T. G., R. J. Nieuwendijk, J. G. De Man, B. Y. De Winter, A. G. Herman, E. A. Van Marck, and P. A. Pelckmans. 2004. Concurrent infection with Schistosoma mansoni attenuates inflammation induced changes in colonic morphology, cytokine levels, and smooth muscle contractility of trinitrobenzene sulphonic acid induced colitis in rats. Gut 5399-107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Morse, K. L., J. Behan, T. M. Laz, R. E. West, Jr., S. A. Greenfeder, J. C. Anthes, S. Umland, Y. Wan, R. W. Hipkin, W. Gonsiorek, N. Shin, E. L. Gustafson, X. Qiao, S. Wang, J. A. Hedrick, J. Greene, M. Bayne, and F. J. Monsma, Jr. 2001. Cloning and characterization of a novel human histamine receptor. J. Pharmacol. Exp. Ther. 2961058-1066. [PubMed] [Google Scholar]
- 38a.National Research Council. 1996. Guide for the care and use of laboratory animals. National Academy Press, Washington, DC.
- 39.Nystedt, S., A. K. Larsson, H. Aberg, and J. Sundelin. 1995. The mouse proteinase-activated receptor-2 cDNA and gene. Molecular cloning and functional expression. J. Biol. Chem. 2705950-5955. [DOI] [PubMed] [Google Scholar]
- 40.Oda, T., N. Morikawa, Y. Saito, Y. Masuho, and S. Matsumoto. 2000. Molecular cloning and characterization of a novel type of histamine receptor preferentially expressed in leukocytes. J. Biol. Chem. 27536781-36786. [DOI] [PubMed] [Google Scholar]
- 41.Perez-Navarro, R., O. Martinez-Augustin, I. Ballester, A. Zarzuelo, and D. M. Sanchez. 2005. Experimental inflammation of the rat distal colon inhibits ion secretion in the proximal colon by affecting the enteric nervous system. Naunyn Schmiedebergs Arch. Pharmacol. 371114-121. [DOI] [PubMed] [Google Scholar]
- 42.Perez-Navarro, R., I. Ballester, A. Zarzuelo, and F. Sanchez de Medina. 2005. Disturbances in epithelial ionic secretion in different experimental models of colitis. Life Sci. 761489-1501. [DOI] [PubMed] [Google Scholar]
- 43.Raithel, M., M. Matek, H. W. Baenkler, W. Jorde, and E. G. Hahn. 1995. Mucosal histamine content and histamine secretion in Crohn's disease, ulcerative colitis and allergic enteropathy. Int. Arch. Allergy Immunol. 108127-133. [DOI] [PubMed] [Google Scholar]
- 44.Sandle, G. I. 2005. Pathogenesis of diarrhea in ulcerative colitis: new views on an old problem. J. Clin. Gastroenterol. 39S49-S52. [DOI] [PubMed] [Google Scholar]
- 45.Shea-Donohue, T., C. Sullivan, F. D. Finkelman, K. B. Madden, S. C. Morris, J. Goldhill, V. Pineiro-Carrero, and J. F. Urban, Jr. 2001. The role of IL-4 in Heligmosomoides polygyrus-induced alterations in murine intestinal epithelial cell function. J. Immunol. 1672234-2239. [DOI] [PubMed] [Google Scholar]
- 46.Spiller, R. C., D. Jenkins, J. P. Thornley, J. M. Hebden, T. Wright, M. Skinner, and K. R. Neal. 2000. Increased rectal mucosal enteroendocrine cells, T lymphocytes, and increased gut permeability following acute Campylobacter enteritis and in post-dysenteric irritable bowel syndrome. Gut 47804-811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Stein, J., J. Ries, and K. E. Barrett. 1998. Disruption of intestinal barrier function associated with experimental colitis: possible role of mast cells. Am. J. Physiol. 274G203-G209. [DOI] [PubMed] [Google Scholar]
- 48.Steinhoff, M., J. Buddenkotte, V. Shpacovitch, A. Rattenholl, C. Moormann, N. Vergnolle, T. A. Luger, and M. D. Hollenberg. 2005. Proteinase-activated receptors: transducers of proteinase-mediated signaling in inflammation and immune response. Endocr. Rev. 261-43. [DOI] [PubMed] [Google Scholar]
- 49.Summers, R. W., D. E. Elliott, J. F. Urban, Jr., R. Thompson, and J. V. Weinstock. 2005. Trichuris suis therapy in Crohn's disease. Gut 5487-90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Summers, R. W., D. E. Elliott, J. F. Urban, Jr., R. A. Thompson, and J. V. Weinstock. 2005. Trichuris suis therapy for active ulcerative colitis: a randomized controlled trial. Gastroenterology 128825-832. [DOI] [PubMed] [Google Scholar]
- 51.Takeshita, K., K. Sakai, K. B. Bacon, and F. Gantner. 2003. Critical role of histamine H4 receptor in leukotriene B4 production and mast cell-dependent neutrophil recruitment induced by zymosan in vivo. J. Pharmacol. Exp. Ther. 3071072-1078. [DOI] [PubMed] [Google Scholar]
- 52.Thurmond, R. L., P. J. Desai, P. J. Dunford, W. P. Fung-Leung, C. L. Hofstra, W. Jiang, S. Nguyen, J. P. Riley, S. Sun, K. N. Williams, J. P. Edwards, and L. Karlsson. 2004. A potent and selective histamine H4 receptor antagonist with anti-inflammatory properties. J. Pharmacol. Exp. Ther. 309404-413. [DOI] [PubMed] [Google Scholar]
- 53.Urban, J. F., Jr., I. M. Katona, and F. D. Finkelman. 1991. Heligmosomoides polygyrus: CD4+ but not CD8+ T cells regulate the IgE response and protective immunity in mice. Exp. Parasitol. 73500-511. [DOI] [PubMed] [Google Scholar]
- 54.Varga, C., K. Horvath, A. Berko, R. L. Thurmond, P. J. Dunford, and B. J. R. Whittle. 2005. Inhibitory effects of histamine H4 receptor antagonists on experimental colitis in the rat. Eur. J. Pharmacol. 522130-138. [DOI] [PubMed] [Google Scholar]
- 55.Vergnolle, N., L. Cellars, A. Mencarelli, G. Rizzo, S. Swaminathan, P. Beck, M. Steinhoff, P. Andrade-Gordon, N. W. Bunnett, M. D. Hollenberg, J. L. Wallace, G. Cirino, and S. Fiorucci. 2004. A role for proteinase-activated receptor-1 in inflammatory bowel diseases. J. Clin. Investig. 1141444-1456. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 56.Vu, T. K., D. T. Hung, V. I. Wheaton, and S. R. Coughlin. 1991. Molecular cloning of a functional thrombin receptor reveals a novel proteolytic mechanism of receptor activation. Cell 641057-1068. [DOI] [PubMed] [Google Scholar]
- 57.Weidenhiller, M., M. Raithel, S. Winterkamp, P. Otte, J. Stolper, and E. G. Hahn. 2000. Methylhistamine in Crohn's disease (CD): increased production and elevated urine excretion correlates with disease activity. Inflamm. Res. 49(Suppl. 1)S35-S36. [DOI] [PubMed] [Google Scholar]
- 58.Winterkamp, S., M. Weidenhiller, P. Otte, J. Stolper, D. Schwab, E. G. Hahn, and M. Raithel. 2002. Urinary excretion of N-methylhistamine as a marker of disease activity in inflammatory bowel disease. Am. J. Gastroenterol. 973071-3077. [DOI] [PubMed] [Google Scholar]
- 59.Xie, H., and S. H. He. 2005. Roles of histamine and its receptors in allergic and inflammatory bowel diseases. World J. Gastroenterol. 112851-2857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Yamada, Y., S. Marshall, R. D. Specian, and M. B. Grisham. 1992. A comparative analysis of two models of colitis in rats. Gastroenterology 1021524-1534. [DOI] [PubMed] [Google Scholar]
- 61.Zamuner, S. R., N. Warrier, A. G. Buret, W. K. MacNaughton, and J. L. Wallace. 2003. Cyclooxygenase 2 mediates post-inflammatory colonic secretory and barrier dysfunction. Gut 521714-1720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zhao, A., and T. Shea-Donohue. 2003. PAR-2 agonists induce contraction of murine small intestine through neurokinin receptors. Am. J. Physiol. Gastrointest. Liver Physiol. 285G696-G703. [DOI] [PubMed] [Google Scholar]
- 63.Zhao, A., J. F. Urban, Jr., M. Morimoto, J. E. Elfrey, K. B. Madden, F. D. Finkelman, and T. Shea-Donohue. 2006. Contribution of 5-HT2A receptor in nematode infection-induced murine intestinal smooth muscle hypercontractility. Gastroenterology 131568-578. [DOI] [PubMed] [Google Scholar]
- 64.Zhao, A., M. Morimoto, H. Dawson, J. E. Elfrey, K. B. Madden, W. C. Gause, B. Min, F. D. Finkelman, J. F. Urban, Jr., and T. Shea-Donohue. 2005. Immune regulation of protease-activated receptor-1 expression in murine small intestine during Nippostrongylus brasiliensis infection. J. Immunol. 1752563-2569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Zhu, Y., D. Michalovich, H. Wu, K. B. Tan, G. M. Dytko, I. J. Mannan, R. Boyce, J. Alston, L. A. Tierney, X. Li, N. C. Herrity, L. Vawter, H. M. Sarau, R. S. Ames, C. M. Davenport, J. P. Hieble, S. Wilson, D. J. Bergsma, and L. R. Fitzgerald. 2001. Cloning, expression, and pharmacological characterization of a novel human histamine receptor. Mol. Pharmacol. 59434-441. [DOI] [PubMed] [Google Scholar]