

Abstract
Summary: Coenzyme M (2-mercaptoethanesulfonate; CoM) is one of several atypical cofactors discovered in methanogenic archaea which participate in the biological reduction of CO2 to methane. Elegantly simple, CoM, so named for its role as a methyl carrier in all methanogenic archaea, is the smallest known organic cofactor. It was thought that this cofactor was used exclusively in methanogenesis until it was recently discovered that CoM is a key cofactor in the pathway of propylene metabolism in the gram-negative soil microorganism Xanthobacter autotrophicus Py2. A four-step pathway requiring CoM converts propylene and CO2 to acetoacetate, which feeds into central metabolism. In this process, CoM is used to activate and convert highly electrophilic epoxypropane, formed from propylene epoxidation, into a nucleophilic species that undergoes carboxylation. The unique properties of CoM provide a chemical handle for orienting compounds for site-specific redox chemistry and stereospecific catalysis. The three-dimensional structures of several of the enzymes in the pathway of propylene metabolism in defined states have been determined, providing significant insights into both the enzyme mechanisms and the role of CoM in this pathway. These studies provide the structural basis for understanding the efficacy of CoM as a handle to direct organic substrate transformations at the active sites of enzymes.
INTRODUCTION
Over 40 years ago, the laboratory of Ralph Wolfe determined that coenzyme M (CoM) is a central cofactor in methanogenesis, serving as a key C1 carrier (43, 59, 60). Using a combination of 1H nuclear magnetic resonance and infrared spectroscopy, Taylor and Wolfe determined the structure of CoM to be 2,2′-dithiodiethanesulfonic acid. CoM is found in all methanogens and is involved in the final steps of methane formation, accepting methyl groups from methylcobalamin to form methylcoenzyme M, which is subsequently reductively demethylated, yielding methane (59, 60). While many methanogenic archaea can synthesize CoM, some methanogenic archaea, such as Methanobrevibacter ruminantium (former name, Methanobacterium ruminantium), have an obligate growth requirement for the coenzyme met only by supplementing growth medium with rumen fluid containing CoM synthesized by other methanogens (5, 58). Subsequent radiolabel experiments revealed that CoM was actively translocated across the membrane in M. ruminantium in a process which was inhibited by substrate analogs, including the potent inhibitor of methanogenesis bromoethanesulfonate (7).
Screens of eukaryotic tissues and cell extracts of a variety of prokaryotes in the late 1970s suggested that CoM was specific to methanogenesis (6). For the next 30 years, the paradigm of CoM being a unique cofactor in methanogens stood unchallenged until this small organic cofactor was found to play a key role in microbial alkene metabolism (1), a metabolism not sampled in previous CoM screens (6). Analysis of available enzyme structures containing various bound adducts of CoM exposed common themes linking the role of CoM in methanogenesis with its role in alkene metabolism. In addition, such structural examinations reveal a unique role for CoM as a handle in directing catalysis of small, chemically nondescript organic substrates.
CoM (2-mercaptoethanesulfonate) is one of several atypical cofactors discovered in methanogenic archaea that are essential for biological reduction of CO2 to methane (43, 60). CoM was first isolated and characterized by McBride and Wolfe in the early 1970s (43, 60) and was shown to function as a methyl group carrier in methanogenesis. CoM, the smallest organic cofactor presently known, is also the only known cofactor containing a sulfonic acid functional group. The sulfonic acid moiety in CoM is separated from the reactive thiol group by an ethyl group (Fig. 1).
FIG. 1.
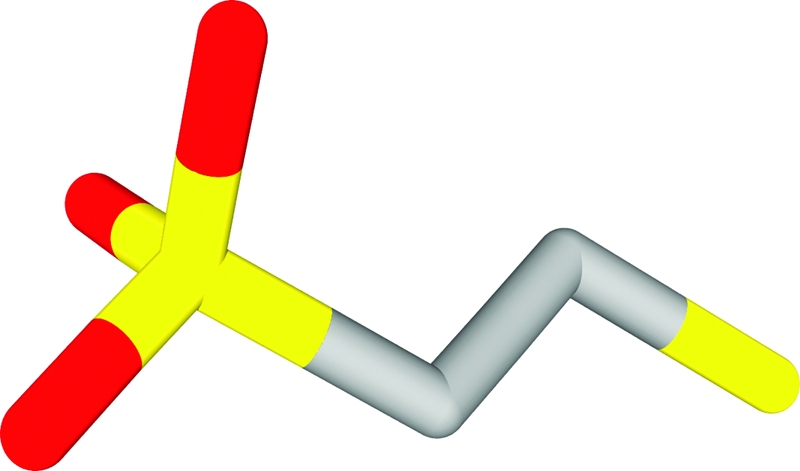
Structure of CoM (mercaptoethane sulfonic acid).
It was later found that CoM is a key cofactor in the pathway of propylene metabolism in Xanthobacter autotrophicus Py2 (1), Rhodococcus rhodochrous strain B276 (39), and Mycobacterium rhodesiae strain JS60 (13, 14). A four-step CoM-dependent pathway converts propylene and CO2 to acetoacetate, which feeds into central metabolism (2, 16). In this process, CoM is used to activate and convert highly electrophilic epoxypropane, formed from propylene epoxidation, into a nucleophilic species that undergoes carboxylation (1).
Crystal structures of CoM bound to three different enzymes have been solved, including methyl-CoM reductase (19), which liberates methane in methanogenesis, and two enzymes involved in propylene metabolism, R-hydroxypropylthioethanesulfonate dehydrogenase (R-HPCDH) (36) and 2-ketopropyl-CoM disulfide-oxidoreductase (2-KPCC) (48, 50). The latter structures provide templates for modeling CoM binding to two additional enzymes that utilize CoM as a cofactor, S-HPCDH and epoxyalkene CoM transferase (EaCoMT). Together, these structures provide significant insights into the enzyme mechanisms and the critical role(s) of CoM in catalysis.
CoM
CoM Biosynthesis
The enzymes necessary for CoM biosynthesis were originally identified based on isotope labeling patterns of CoM purified from three different methanogens grown on labeled acetate, sulfolactic acid, or sulfite and from examination of catalytic activities in cellular extracts (66, 69). In addition, radioactive labeling experiments identified cysteine as the source of the thiol group in enzymatic transformation of sulfoacetaldehyde to CoM, the final step of CoM biosynthesis (68). To date, four key enzymes from the methanogenic CoM biosynthetic pathway have been identified and characterized in vitro (25, 27, 70). For more information on methanogenic coenzyme biosynthesis, including CoM, the reader is referred to two recent and detailed reviews (26, 67).
While much is known concerning the biosynthesis and role of CoM in methanogens, less is known of CoM biosynthesis in bacteria. In Xanthobacter strain Py2, all of the CoM biosynthetic genes analogous to those found in methanogens reside on a linear megaplasmid along with genes for enzymes of aliphatic alkene and epoxide metabolism (38). Previous studies have shown that the biosynthesis of CoM is induced by propylene in both Xanthobacter strain Py2 and Rhodococcus rhodochrous B276 (39). Another recent study found the CoM pathway to be active in aerobic vinyl chloride and ethane assimilation pathways of Mycobacterium rhodesiae strain JS60 (14). CoM-dependent epoxyethane metabolism has been observed in all mycobacterial strains studied, and an in-depth analysis of 10 strains suggested that CoM-mediated pathways are universal in alkene-assimilating mycobacteria (13).
The involvement of CoM in these two different processes tempts speculation on the evolutionary origins of the genes involved in its biosynthesis. The fact that CoM is involved in alkene metabolism in bacteria and methanogenesis in archaea would lead one to consider that the biosynthetic genes evolved independently. Alternatively, given that homologs of genes involved in CoM biosynthesis are found on a megaplasmid in both Xanthobacter and Rhodococcus (38), one would be led to consider that the genes were acquired by lateral gene transfer. Further examination of CoM biosynthetic pathways in methanogenic archaea and in bacteria may provide insights into the evolutionary history of this process.
CoM in Methanogenesis
The metabolic pathways for methane production, regardless of growth substrate, converge via the activities of a variety of methyltransferases to a common metabolic reaction in which methylated CoM (methyl-CoM) is reductively cleaved by the activity of Ni-dependent methyl-CoM reductase (MCR) to form methane (61, 71). MCR, the primary enzyme responsible for microbial production of methane, catalyzes heterodisulfide formation between coenzyme B (CoB) and methyl-CoM, liberating methane from the latter in the process. Crystal structures of native and ligand-bound MCR from Methanothermobacter marburgensis (former name, Methanobacterium thermoautotrophicum), a 300-kDa heterohexamer, reveal an intersubunit nickel-porphinoid coenzyme F430 present at each active site, with a narrow methyl-CoM channel to the active site that is shielded from solvent upon binding of the “second substrate” CoB (19). Combining structural data with kinetic and electron paramagnetic resonance (EPR) data yields a mechanism for methane formation consisting of nucleophilic attack of Ni(I) on the methyl group of CoM, forming methyl-Ni(III). This mechanism represents an alternative to a previously proposed mechanism where Ni(I) attack on the thioether sulfur of CoM results in a Ni(II)-S-CoM intermediate (see reference 18 for a review of MCR mechanisms). Although the details of the intermediates are in dispute, methane and CoM-S-S-CoB are released as products.
Structural analysis of substrate-bound MCR reveals that CoM recognition is facilitated by direct interactions with the CoM sulfonate moiety, forming a salt bridge with Arg120 and hydrogen bonds to the peptide nitrogen of Tyr444 and a water molecule that bridges to the peptide oxygen of His364 (19). The ethylene portion of CoM lies in a hydrophobic region between the lactam ring of F430 and the phenyl ring of Phe443; the thiol sulfur is bonded to the porphinoid nickel atom. The recognition of the extended region connecting CoM and CoB in the heterodisulfide complex is facilitated by a ring of five Phe and Tyr side chains that form a hydrophobic “funnel” (19). It was also noted that the four known methylation sites occur at the active site, enhancing the hydrophobicity of this region. These relatively unreactive aromatic and aliphatic groups are postulated to help prevent damage from highly reactive radical intermediates upon reduction of methyl-CoM with CoB (19, 34). It has been proposed that the release of the heterodisulfide product may be promoted by disruption of MCR-CoM interactions, since in the CoM-S-S-CoB product complex, CoM and its sulfonate are translated out of the binding pocket (19).
In addition to MCR interaction, CoM is involved in a number of other enzymatic processes in methanogenic archaea. For example, most methanogens contain thio:fumarate reductase, which catalyzes the reduction of fumarate, with CoM and CoB as electron donors (8, 30). Similarly, many methanogens contain a heterodisulfide reductase that catalyzes the reduction of CoM-S-S-CoB, a product formed through the reduction of fumarate and the reduction of methyl-CoM during methanogenesis (29).
CoM in Propylene Metabolism
In the late 1990s, CoM was determined to be a cofactor in the pathway of propylene metabolism in Xanthobacter autotrophicus (1). In X. autotrophicus and other alkene-oxidizing bacteria, alkenes such as ethylene and propylene are oxidized to form aliphatic epoxides (16, 17). Aliphatic epoxides are toxic molecules, reacting indiscriminately with nucleophilic groups on proteins and DNA. Therefore, their synthesis and subsequent degradation must be tightly controlled. The detoxification of epoxides is accomplished by a series of reactions involving ring opening via nucleophilic attack of either glutathione or water to form covalent adducts by the activities of glutathione S-transferase and epoxide hydrolases (31, 63-65). While these detoxification strategies are nonproductive in some organisms, other organisms couple these processes to energy generation in reactions that require CoM (16, 17). In this process, alkene epoxidation is followed by conjugation with CoM and subsequent carboxylation to the metabolite acetoacetate in a pathway in which the chemical characteristics of CoM play a key role in binding and orienting conjugated substrates for catalysis (Fig. 2).
FIG. 2.

Comparison of CoM usage in epoxide carboxylation (A) and methanogenesis (B).
It is intriguing that CoM was found to be the cofactor for epoxide carboxylation, not only because CoM was thought to be an exclusively methanogenic coenzyme but also given the availability of other thiols, such as glutathione, lipoic acid, cysteine, or homocysteine, that could participate in this type of chemistry. CoM is not only the smallest organic cofactor known but also the only known cofactor containing a sulfonic acid functional group. Elegantly simple in structure, the sulfonic acid moiety is separated from the reactive thiol group by an ethyl group spacer (Fig. 1). The advantage of using CoM in the epoxide carboxylation pathway is twofold. First, the small size of CoM allows formation of 2-hydroxypropyl-CoM conjugates of manageable size (in contrast to bulky glutathione-alcohol conjugates formed by glutathione S-transferases) for the subsequent stereoselective dehydrogenation and reductive carboxylation steps in the pathway (Fig. 2). Second, the negatively charged sulfonate moiety of CoM provides a unique molecular handle that can be utilized to properly orient substrates for specific oxidation-reduction chemistry or stereospecific catalysis. This is a very important feature in the context of alkene metabolism because short-chain alkene and epoxide substrates lack distinctive chemical groups that could confer specificity.
Similarities in CoM Utilization during Methanogenesis and Propylene Metabolism
There are some interesting similarities between the pathways of epoxide carboxylation (Fig. 2A) and methanogenesis (Fig. 2B) with respect to CoM utilization. In both cases, the initial group transfer involves S-alkylation of CoM to form a thioether intermediate. In the case of methanogenesis, this reaction is carried out by vitamin B12-dependent methyltransferases. In methanogenic methyltransferases, zinc plays an integral role in activation of the thiol group of CoM for acceptance of a methyl group from various donors (22, 55). Analogously, the zinc atom in EaCoMT functions in the activation of CoM as well, in this instance for attack on the electrophilic epoxide substrate (9). In both methanogenesis and epoxide carboxylation, reductive dealkylation of a CoM thioether occurs, in the former system to generate methane and in the latter to form an enolate that undergoes carboxylation (11, 48, 50). In addition to Xanthobacter and Rhodococcus, other organisms where CoM-dependent alkene metabolism has been identified and studied include strains of Mycobacterium (14), Nocardioides (41), Pseudomonas, and Ochrobactrum (15). Several strains of these microorganisms are able to grow on and degrade ethene and vinyl chloride in a monoxygenase-catalyzed reaction, yielding chlorooxirane and ultimately epoxyethane (14). In Mycobacterium strain JS60, the epoxyethane ring is opened via the enzymatic activity of EaCoMT (14). While the downstream reactions are not well understood for these organisms, a hypothetical pathway has been proposed (16).
Our understanding of the role of CoM as a carrier molecule in alkene oxidation and methanogenesis has been facilitated by investigation of the three-dimensional structures of the key enzymes complexed with substrates, products, and/or inhibitors. Three-dimensional structures are a complement to detailed biochemical and kinetic studies and can be extremely informative for developing mechanistic models for enzymes that can be investigated further experimentally. The three-dimensional structures of two enzymes of the propylene oxidation pathway in Xanthobacter, namely, R-HPCDH and 2-KPCC, have been solved (36, 48). Ligand-bound crystal structures have revealed the molecular determinants for CoM recognition and its efficacy as a cofactor, defining a key role for CoM in specifically binding and orientating substrates for carboxylation and stereoselective catalysis. In both of these enzymes, strategically placed positively charged amino acids (arginine and lysine) interact with the negatively charged sulfonate group of the substrate, thereby orienting it properly for catalysis. A common pattern of interaction between these amino acids and substrate sulfonates suggests a structural signature for CoM binding. In the sections below, structures (where available) and newly created homology models of enzymes which bind CoM or its conjugates are analyzed in detail in order to better define the structural signature for CoM recognition.
EaCoMT: ZINC-MEDIATED ACTIVATION OF THE CoM THIOL
Reactions Catalyzed by EaCoMT and Related Alkyl Transferases
EaCoMT catalyzes the nucleophilic attack of CoM on epoxypropane (and other short-chain epoxides), thereby forming a hydroxyalkyl-CoM thioether conjugate (Fig. 2). EaCoMT belongs to a subset of the family of alkyl transferases for which zinc is crucial in activation of a thiol for nucleophilic attack (17, 42). Other examples of this family are the Ada protein, which is involved in DNA repair (44, 45), betaine:homocysteine methyltransferase (10), cobalamin-independent methionine synthases (MetE) (24, 42), and various methanogenic methyltransferases (MtaA, MtbA, and MtsA) (56).
EaCoMTs were first identified in X. autotrophicus (4) and Rhodococcus rhodochrous (3) as enzymes which catalyze the addition of CoM to R- or S-epoxypropane. As previously stated, EaCoMTs are also found in species such as Mycobacterium rhodesiae, Nocardioides sp. strain JS614, Pseudomonas putida, and Ochrobactrum sp. strain TD, all of which metabolize ethene and vinyl chloride (13, 14, 41). EaCoMTs from these organisms share high sequence identity and similarity (Fig. 3A), and a phylogram of these EaCoMT proteins indicates that the proteobacterial Pseudomonas putida and Ochrobactrum sp. strain TD genes cluster together in the same lineage as the genes from the high-GC, gram-positive bacterium Mycobacterium rhodesiae (Fig. 3B). EaCoMT amino acid sequences from the high-GC, gram-positive bacteria Nocardioides sp. strain JS614 and Rhodococcus rhodochrous also cluster together in a sister lineage to that in which Mycobacterium resides. It is also worth noting that the EaCoMT from the propylene-degrading bacterium X. autotrophicus clusters distinctly from those of the ethane- and/or vinyl chloride-degrading organisms. This may be a result of selective pressure for substrate specificity of EaCoMT for propylene as opposed to ethane or vinyl chloride in X. autotrophicus.
FIG. 3.

(A) Multiple-sequence alignment of EaCoMT protein sequences, performed using ClustalW (62). (B) Evolutionary relationships of six taxa. The evolutionary history was inferred using the neighbor-joining method (54). The bootstrap consensus tree, inferred from 500 replicates (20), is taken to represent the evolutionary history of the taxa analyzed (20). Branches corresponding to partitions reproduced in <50% of bootstrap replicates are collapsed. The percentages of replicate trees in which the associated taxa clustered together in the bootstrap test (500 replicates) are shown next to the branches (20). The tree is drawn to scale, with branch lengths in the same units as those of the evolutionary distances used to infer the phylogenetic tree. The evolutionary distances were computed using the Poisson correction method (73) and are given as numbers of amino acid substitutions per site. All positions containing alignment gaps and missing data were eliminated only in pairwise sequence comparisons. Phylogenetic analyses were conducted in MEGA4 (57).
Comparison with MetE (Zinc) Alkyl Transferase
Sequence alignment of EaCoMTs reveals the HXCXnC motif (Fig. 3A) shared by the “MetE” subclass of transferases, enzymes that use zinc to activate a thiol group (72). In contrast to Xanthobacter, Rhodococcus, Nocardioides, and Mycobacterium EaCoMTs, all of which have the full complement of the motif, the Pseudomonas and Ochrobactrum enzymes have only the histidine and the first cysteine (Fig. 3A). In addition to the zinc binding motif, there are other regions in the sequences that are highly conserved among these enzymes (data not shown).
The EaCoMT from Xanthobacter has been the subject of intensive characterization (4, 9, 37). Xanthobacter EaCoMT is a hexameric protein containing 1 Zn atom per subunit and is highly specific for CoM as the organic thiol substrate (37). When a range of other thiols were tested, only 3-mercaptopropionate, 2-mercaptoethanol, and cysteine served as substrates, but even these had very low affinities and specific activities relative to those observed for CoM (37).
EaCoMT, like MetE, is believed to coordinate the substrate thiol at a zinc center, thereby lowering the pKa of the thiol such that it can be deprotonated and serve as a nucleophile for attack on the second substrate, epoxypropane (9). For MetE, extended X-ray absorption fine structure analysis has shown that the zinc environment changes from a 2S + 2N/O environment to a 3S + 1N/O environment upon binding homocysteine (23, 52, 72). As noted above, two cysteine residues and a histidine serve as the protein ligands for the zinc center. Recent crystallographic studies of MetE enzymes from Thermotoga maritima (53) and Arabidopsis thaliana (21) have introduced some controversy as to the nature of the fourth ligand and the mechanism for thiol group activation. However, the structural studies of T. maritima MetE (53) support the extended X-ray absorption fine structure analysis of MetE from Escherichia coli (52, 72) and demonstrate that a Glu residue serves as the fourth exchangeable ligand for the Zn center of the enzyme. For T. maritima MetE, when the homocysteine thiol coordinates the zinc, the glutamate carboxylate moves away from the zinc, increasing the coordination distance (53).
Homology Modeling of EaCoMT Based on the Crystal Structure of T. maritima MetE
The crystal structure of EaCoMT has not yet been determined. However, the high similarity between Xanthobacter autotrophicus EaCoMT and T. maritima MetE sequences, depicted in the alignment shown in Fig. 4A together with the available structures of MetE (21, 53), provides the basis for constructing a homology model of EaCoMT (Fig. 4B) and rationalizing the zinc binding site in the X. autotrophicus enzyme (Fig. 4C).
FIG. 4.

(A) Sequence alignment of MetE (zinc-dependent methionine synthase) from T. maritima and XecA (EaCoMT) from X. autotrophicus Py2 with zinc binding motif, highlighted in green. (B) Homology model of the structure of XecA/EaCoMT from X. autotrophicus Py2, based on the structure of MetE generated using the CPH model (40) server. (C) Structural representation of the zinc binding site of EaCoMT, based on the homology model.
Similar to T. maritima MetE, the homology model suggests that X. autotrophicus EaCoMT has a specific glutamate (Glu275 in the case of EaCoMT) likely to serve as the exchangeable “N/O” ligand. Importantly, this Glu residue is conserved in EaCoMTs from all of the aforementioned bacteria, even though the second Cys residue believed to coordinate the zinc is missing in the EaCoMT enzymes from Pseudomonas and Ochrobactrum.
With the exception of the missing Cys in EaCoMTs in Pseudomonas and Ochrobactrum, homology models indicate that the substrate-binding site is conserved in all EaCoMTs. One end of the substrate-binding pocket contains zinc bound to the HXCXnC motif, while the other end consists of a hydrophobic pocket containing Trp, Phe, and Tyr. Also found in the substrate-binding site are two positively charged residues, arginine and lysine, both of which are conserved in EaCoMTs from Rhodococcus, Mycobacterium, Ochrobactrum, Pseudomonas, and Nocardioides. The details of the specific amino acid residues implicated in zinc and CoM binding in the various EaCoMTs are summarized in Fig. 5A. The CoM-binding arginine residue in the Rhodococcus, Mycobacterium, Ochrobactrum, Pseudomonas, and Nocardioides EaCoMTs is conserved with respect to both sequence and three-dimensional structure. Interestingly, the same arginine residue in Xanthobacter EaCoMT is structurally conserved but is distal in sequence from the conserved arginines present in other EaCoMTs. It is intriguing that the architecture of the substrate-binding site is conserved even though residues are contributed from different regions of sequence space.
FIG. 5.

(A) Amino acid residues involved in CoM and zinc binding in EaCoMTs from X. autotrophicus, R. rhodochrous, M. rhodesiae, Nocardioides sp. strain JS614, P. putida, and Ochrobactrum sp. strain TD. (B and C) Stereo views of the key residues of the zinc binding sites in X. autotrophicus, R. rhodochrous, M. rhodesiae, and Nocardioides sp. strain JS614 (B) and in P. putida and Ochrobactrum sp. strain TD (C). Docking of substrates was carried out using the program O (32), and in situ minimization of the models with the bound substrates was carried out using the DS modeling suite of programs, which uses a CHARMm minimization protocol.
In the crystal structure of T. maritima MetE, the homocysteine sulfhydryl group coordinates the zinc ion (53). The distance between the thiol and the zinc ion is ∼3.2 Å. In order to envision both substrate-enzyme interactions and the alkyl transfer reaction of EaCoMT, the homocysteine-bound MetE structure was superimposed on that of EaCoMT obtained from homology modeling. This superimposes the zinc exactly at the putative zinc binding site, consisting of His218, Cys220, Cys341, and Glu275. Also, the resulting position of homocysteine near zinc implies that it serves as a Zn ligand. The distances between the zinc ion and its ligands (namely, the two cysteines, histidine, and the thiol group) are consistent with those observed in the crystal structure (∼3.2 Å) of MetE. Using the position of the thiol group of homocysteine in the EaCoMT model as a starting point, a CoM molecule was modeled (with manual docking using the program O [32]) in the homocysteine binding site such that the CoM thiol is superimposed on the thiol of homocysteine. Subsequent rigid body rotation of CoM about the fixed thiol was performed to look for conserved basic residues that could provide ligands for the CoM sulfonate group. In situ ligand minimization was then initiated to remove any poor contacts in the models. The resulting generalized substrate-binding sites of Xanthobacter, Rhodococcus, Nocardioides, and Mycobacterium EaCoMTs with bound CoM are shown in Fig. 5B, and the zinc- and CoM-binding sites of the Pseudomonas and Ochrobactrum enzymes are shown in Fig. 5C. The minimized models reveal that arginine and lysine residues are positioned to form favorable electrostatic interactions with the sulfonate group of CoM. These residues approach CoM from opposing sides and place sulfonate oxygen atoms of CoM and side chain nitrogens of arginine and lysine residues in positions similar to those observed in crystal structures of R-HPCDH and 2-KPCC. These structures, which are discussed in depth in the following sections, reveal that these positively charged residues are positioned in such a way that their interaction with the sulfonate group of CoM facilitates proper binding and orientation of the alcohol and keto group substrates of R-HPCDH and 2-KPCC for dehydrogenation and thioether bond cleavage, respectively.
Model-Based CoM-Binding Determinants of EaCoMT
In EaCoMT, the distance between the CoM sulfonate group and the positively charged residues gives insight into the specificity of EaCoMT toward CoM and not other thiols. Studies have shown that when thiols such as 3-mercaptopropionate, 2-mercaptoethanol, and cysteine are compared with CoM for the ability to serve as substrates for alkyl transfer reactions catalyzed by EaCoMT, the enzyme shows very low levels of activity and high Km values, indicating low affinities for these thiols (37). This discrimination between thiols could be explained by the favorable interaction between the CoM sulfonate and the positively charged residues on EaCoMT. For effective zinc-mediated activation of the thiol group of CoM to take place, the electrostatic interaction between the thiol and zinc is critical. This implies that thiol activation is facilitated by interactions between EaCoMT and the rest of the substrate conjugate (i.e., the sulfonate moiety). Electrostatic interactions with the enzyme help to avoid translation of the substrate and subsequent disruption of the thiol-zinc interaction.
Although the EaCoMTs discussed above belong to a phylogenetically diverse group of microorganisms, their sequences are highly conserved. Analyses of the homology models not only have provided insights into the alkyl transfer reaction and substrate-binding modes but also have underscored the efficacy of the sulfonate group of CoM to act as an electrostatic handle for orienting conjugated substrates for catalysis. First, the homology models have revealed that Glu275 is likely to be the exchangeable ligand of the active-site zinc. Second, the above studies have yielded insight into the structural conservation of putative substrate-binding sites in EaCoMTs. The substrate-binding pockets of these enzymes feature conserved arginine and lysine residues that provide favorable electrostatic interactions with the sulfonate group of CoM. The arginine and lysine residues are placed strategically with respect to their distance and the way they approach the sulfonate to accommodate and orient the thiol for proper catalysis. As previously stated, the sulfonate group of CoM functions as a handle in key electrostatic interactions with conserved basic residues to ensure proper binding and orientation of the substrate for zinc-mediated thiol activation. The reaction carried out by the EaCoMT also ensures that the resultant alkyl groups have the sulfonate moiety of CoM conjugated to them. This is of great importance, since the alkyl groups themselves lack any special chemical groups which could aid in specific binding to enzymes that utilize them as substrates.
R- AND S-HPCDH: ROLE OF CoM IN STEREOSELECTIVE CATALYSIS
R- and S-HPCDH and the “Other” Stereoselective Enzyme Pair, Tropinone Reductases I and II
R- and S-HPCDH catalyze the NAD+-dependent oxidation of 2-R- and 2-S-hydroxypropyl CoM (R- and S-HPC), respectively, to the achiral product 2-ketopropyl CoM (2-KPC). These enzymes belong to the short-chain dehydrogenase reductase (SDR) family of enzymes and have the classic serine, tyrosine, and lysine catalytic triad (33, 35). The dehydrogenase pair of enzymes is one of only two pairs of stereoselective enzymes known to act on different substrate enantiomers in a common pathway. The other pair is the tropinone reductases I and II from the plant Datura stramonium, which catalyze the oxidation of the 3-keto group of tropine to tropinone and pseudotropine, respectively (46, 47). A model was proposed to explain how the two enzymes facilitate stereoselective catalysis. According to this model, differentially placed positively charged residues in the substrate-binding sites of these enzymes could provide the switch necessary to stereoselectively position and orient substrates in a correct fashion with respect to the active-site residues.
Roles of Specific Residues in R-HPCDH Binding and Catalysis
Biochemical and mechanistic studies of native and mutant R-HPCDH enzymes have identified two specific arginine residues (Arg152 and Arg196) that interact with the sulfonate moiety of 2-hydroxypropyl-CoM, thereby imparting specificity for the R-enantiomer (12). The elucidation of the three-dimensional structure of R-HPCDH confirmed these roles for Arg152 and Arg196 and provided additional mechanistic insights (36). Figure 6A shows the amino acid environment of the bound product 2-ketopropyl CoM at the substrate-binding site of R-HPCDH. The major interactions between the sulfonate group of CoM and the side chains of Arg196 and Arg152 are electrostatic, and Trp195 acts as a backstop to prevent translation of the substrate during catalysis. A very interesting feature of the substrate-binding pocket is the presence of two methionines flanking the substrate-binding pocket from two sides. These methionines appear to be shielding the substrate-binding site from the bulk solvent. The structure also suggests that upon substrate binding, these methionine residues could shield the electrostatic interactions between both the sulfonate group and the positively charged arginines and between the substrate hydroxyl group and the active site from competitive interactions with the solvent.
FIG. 6.

(A) Stereo views of key residues in the substrate-binding sites found in the structure of R-HPCDH (PDB accession no. 2CFC.pdb). (B) Superimposition of the structure of R-HPCDH (cobalt blue) and a homology model of S-HPCDH (orange) based on the R-HPCDH structure. The arrow indicates the difference in the substrate-binding site. (C) Key residues implicated in substrate binding in the S-HPCDH homology model.
Modeling S-HPCDH Based on the Structure of R-HPCDH: Deciphering CoM Specificity Determinants for HPCDHs
Effective hydride abstraction by short-chain dehydrogenases is dependent on the correct orientation of the hydride with respect to the nicotinamide and the correct orientation of the substrate hydroxyl group with respect to the Tyr of the catalytic triad (12). In order to provide the structural basis for stereoselectivity for both S- and R-HPCDH, a homology model of S-HPCDH was constructed. Comparison of the crystal structure of R-HPCDH and the homology model of S-HPCDH clearly shows that although the overall architectures of both enzymes are very similar, clear differences can be seen in the substrate-binding region (Fig. 6B). The structure of R-HPCDH, combined with the homology model of S-HPCDH, provides a structural basis for stereoselectivity and also reveals key enzyme-substrate interactions, especially with respect to the sulfonate-binding site. Although residues in the active site are superimposable, the Arg residues (Arg152 and Arg196) observed to be critical for binding the sulfonate moiety of CoM in R-HPCDH are replaced in S-HPCDH by Met147 and Gln191, respectively. These side chains would not support binding of substrate in the same manner as that observed for R-HPCDH. The absence of sulfonate-binding residues at the active site implies that the S-HPCDH substrate cannot be bound in the same mode as R-HPC bound to R-HPCDH.
Superimposition of S-HPC on R-HPC-bound R-HPCDH provided a starting point for modeling S-HPC bound to the active site of S-HPCDH. Rigid body reorientation of CoM places the sulfonate moiety within hydrogen bonding distance of two positively charged residues (Arg205 and Lys208) (Fig. 6C). As in the case of R-HPCDH, substrate binding is facilitated by two positively charged sulfonate-binding groups shielded by two flanking methionine residues (Met147 and Met188) (Fig. 6C). The sulfonate-binding sites in R- and S-HPCDH represent analogous sites involving different sets of amino acid residues at spatially disparate positions on the respective enzymes. Superimposition of R- and S-HPCDH predicts independent sulfonate-binding sites in these enzymes. As shown in Fig. 7, it is clear that although the orientations of active-site residues and NAD+ are the same, the sulfonate and methyl groups are switched to position the hydrogen and hydroxyl groups of the substrate for hydride and proton abstraction, respectively. These structures underscore the effectiveness of CoM conjugated to small substrate molecules that lack innate chemical groups for use as a chemical handle.
FIG. 7.

Stereo representation of superimposed R- and S-HPCDH to show the reorientation of the sulfonate-binding site. The R-HPCDH residues and R-HPC are shown in cobalt blue, while the S-HPCDH residues and S-HPC are shown in orange.
2-KPCC: ROLE OF CoM IN ALIGNING SUBSTRATE FOR ELECTRON TRANSFER AND THIOL-DEPENDENT REDUCTIVE CLEAVAGE
Reactions Catalyzed by 2-KPCC and Other DSORs
The final reaction in the pathway of epoxypropane ring opening and carboxylation is the CO2-dependent cleavage of 2-KPC to produce acetoacetate and CoM (11). 2-KPCC is a homodimeric protein composed of 57-kDa subunits containing one molecule of flavin adenine dinucleotide (FAD) per subunit (2, 4, 11). The enzyme belongs to the family of disulfide oxidoreductase (DSOR) enzymes, which catalyze diverse reactions involving the two-electron reduction of substrate. All members of this family of enzymes contain a redox-active cysteine pair which participates in the reduction of the substrate molecule (49).
The reaction catalyzed by 2-KPCC is initiated when NADPH reduces the enzyme-bound flavin cofactor, which then reduces the oxidized cysteine pair (Cys82 and Cys87) (11). 2-KPC then binds, followed by nucleophilic attack of the interchange cysteine thiol on the substrate thioether sulfur, resulting in heterolytic cleavage of the S-C bond. This step represents a fundamentally novel reaction with respect to the DSOR family, in that a thioether is attacked by cysteine rather than a disulfide. With the exception of mercuric reductase, all other known members of the DSOR family of enzymes attack a disulfide bond. The cleavage of thioether results in formation of a stabilized carbanion of acetone and a mixed disulfide of cysteine and CoM. Enolacetone then serves as a nucleophile for attack on CO2 in the carboxylation step. In the final step, CoM is regenerated by reduction of the mixed disulfide of CoM and the interchange thiol.
Crystal Structure of 2-KPCC and Conformational Changes upon Ligand Binding
The crystal structure of 2-KPCC has been solved (48), and it is structurally homologous with the DSOR family of enzymes. Relative to other DSOR enzymes, the most divergent parts of the 2-KPCC sequence provide two insertions that fill in the active-site region, providing a more compact, hydrophobic active site for recognition of a much smaller substrate than those used by other DSOR enzymes. Comparison of the substrate-bound and native structures reveals that substrate binding promotes a conformational change in which the substrate is encapsulated with the ketopropyl moiety in a hydrophobic environment devoid of bulk solvent (48, 50). This opens a hydrophobic channel where CO2 is postulated to enter and, upon binding, block access to bulk solvent. Encapsulation promotes formation of acetoacetate over the protonation and formation of the metabolically unproductive product acetone. The structure of substrate-bound 2-KPCC indicates that the substrate 2-KPC is bound to the active site primarily through electrostatic interactions between two Arg side chains (Arg56 and Arg365) of 2-KPCC and the sulfonate group of the substrate. The side chains of Arg56 and Arg365 approach from the sides and interact directly with the CoM sulfonate group through both side chain and main chain interactions. The ketopropyl moiety has a hydrogen bond to water, which in turn is hydrogen bonded to an adjacent histidine residue. This arrangement has implications about the mechanism of enolate stabilization (see below).
Structure-Based Catalytic Mechanism of 2-KPCC
The structure of CoM-bound 2-KPCC revealed that the formation and stabilization of an enolate intermediate may be facilitated by the interaction of a His-oriented water molecule with the oxo-group of the substrate. Recent structures, including mixed-disulfide- and CoM-disulfide-bound states of 2-KPCC, have led to a nearly complete structure-based catalytic mechanism (50). The detailed catalytic mechanism of 2-KPCC, including the roles of NAD and FAD, has been described previously (48, 50). It was found that the mixed disulfide state of the enzyme has the CoM bound to a sulfonate in a manner analogous to that of the substrate 2-KPC, an interaction driven primarily via electrostatic interactions with arginine residues. A well-ordered water molecule found within hydrogen bonding distance of the disulfide suggests a role in protonation of the mixed disulfide upon product reduction and release. The electron density features adjacent to the mixed disulfide are consistent with either acetone (the product) or acetate (a component of the crystallization buffer) and define an anion-binding pocket that could be important in stabilizing the developing charge during the formation of acetoacetate in the subsequent carboxylation reaction.
SIMILARITIES IN CoM-BINDING DETERMINANTS
Of particular relevance to the present study are detailed enzyme-substrate interactions that properly orient the substrate for thioether bond cleavage (Fig. 8). In all of the structures and homology models described above, the methanogen- and propylene-metabolizing CoM-utilizing enzymes recognize the CoM sulfonate by using one or more arginine residues, and the ethylene moiety of CoM is located in a hydrophobic region. Together, these features act to position the key reactive thiol at the respective enzyme active site. The substrate-binding pocket of 2-KPCC is strikingly similar to that of R- and S-HPCDH, which consists of two arginine residues approaching from opposing sides. In 2-KPCC, a phenylalanine side chain (Phe57) acts as a backstop, preventing further translation of the substrate. This is analogous to the role played by Trp195 in R-HPCDH. Also, as seen in R-HPCDH, 2-KPCC has two methionines (Met140 and Met361) flanking the substrate, thereby shielding the electrostatic interactions between the substrate and the enzyme from the surrounding environment. Homology modeling suggests that EaCoMT also recognizes CoM in a similar fashion, with two basic residues poised to form favorable electrostatic interactions with the CoM sulfonate.
FIG. 8.
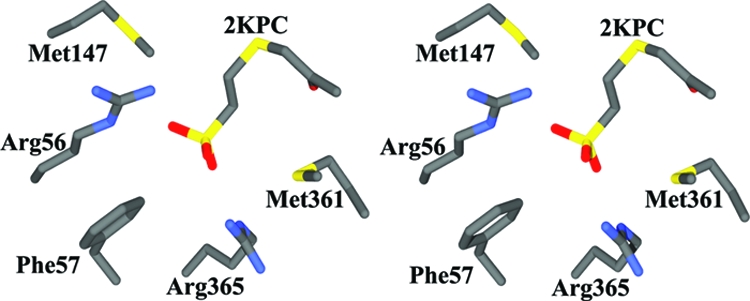
Stereo representation of the substrate-binding site of 2-KPCC, showing the bound substrate 2-KPC (PDB accession no. 1MO9.pdb). All figures were prepared using Swiss PDB viewer (28) and rendered using Povray (http://www.povray.org/povlegal.html).
In contrast with the propylene-metabolizing enzymes described above, the structure of CoM-complexed MCR reveals that the sulfonate interacts with only one basic residue, Arg120, but has additional hydrogen bonds to the peptide nitrogen of Tyr444 and a water molecule that bridges to the peptide oxygen of His364 (19). The CoM ethylene spacer is positioned in a hydrophobic region located between F430 and Phe443, positioning the thiol near the porphinoid nickel atom. This is reminiscent of the thiol ligation of Zn2+ observed in EaCoMT. It has been proposed that release of the heterodisulfide product of MCR may be promoted by disruption of CoM interactions, since in the CoM-S-S-CoB product complex, the CoM and its sulfonate are uprooted from the binding pocket (19). Product formation may also favor release in 2-KPCC, where the alternative anion-binding site postulated to bind acetoacetate shares an Arg residue with the CoM sulfonate-binding site. Product binding would result in two negative charges adjacent to a CoM sulfonate-binding Arg residue, triggering product release.
The nature of the substrate-binding pocket of 2-KPCC reflects the role of CoM in aligning the substrate for electron transfer and thiol-dependent reductive cleavage. The sulfonate moiety again, in this context, is used as a handle to properly orient the “business end” of the molecule toward the active-site cysteines for catalysis. It should be noted that proper alignment of the thioether bond of 2-KPC with respect to the interchangeable cysteine is the key to cleavage and reduction of the substrate. Similar to MCR in methanogenesis, this is brought about by positioning key basic residues such that they form favorable electrostatic interactions with the CoM sulfonate group, while the ethylene spacer is localized through van der Waals interactions with neighboring aliphatic or aromatic residues.
CONCLUSIONS
Specific binding and orientation of short-chain alkenes and their derivatives at the active sites of the enzymes that bind and transform them are a challenge due to the lack of innate chemical groups in these compounds. The above studies suggest that microorganisms have circumvented this problem by conjugating these reactants with a small but unique cofactor, CoM, whose use was previously thought to be limited to methanogenesis. CoM serves as a handle to bind and properly orient these substrates at the active sites of the enzymes, and its specific role is discussed in three different contexts. In EaCoMTs, the sulfonate group of CoM is utilized to align the thiol group of CoM for nucleophilic activation by zinc. The alignment of substrates is promoted by a CoM/sulfonate-binding motif containing arginine and lysine residues conserved across many genera of microorganisms, suggesting a common CoM-binding motif. In the stereospecific dehydrogenases, the substrate-binding site is structured such that the sulfonate groups of two very similar substrates are bound at different sites on structurally similar enzymes, allowing stereoselective catalysis. In 2-KPCC, the CoM-binding site has exploited interactions between the enzyme and sulfonate to align the substrate thioether linkage with the active-site cysteine. It should be noted that the CoM/sulfonate-binding sites in EaCoMT are less compact than those in R-HPCDH, S-HPCDH, and 2-KPCC. This could reflect the fact that the latter enzymes catalyze highly specific reactions, such as stereospecific catalysis and reductive cleavage/carboxylation, as opposed to the relatively simple group transfer reaction catalyzed by the former enzyme.
Acknowledgments
This work was supported by Department of Energy grant DE-FG02-04ER15563 (to J.W.P.) and National Institutes of Health grant GM51805 (to S.A.E.). Portions of this research were carried out at the Stanford Synchrotron Radiation Laboratory, a national user facility operated by Stanford University on behalf of the U.S. Department of Energy, Office of Basic Energy Sciences. The SSRL Structural Molecular Biology Program is supported by the Department of Energy, Office of Biological and Environmental Research, and by the National Institutes of Health, National Center for Research Resources, Biomedical Technology Program, and the National Institute of General Medical Sciences.
REFERENCES
- 1.Allen, J. R., D. D. Clark, J. G. Krum, and S. A. Ensign. 1999. A role for coenzyme M (2-mercaptoethanesulfonic acid) in a bacterial pathway of aliphatic epoxide carboxylation. Proc. Natl. Acad. Sci. USA 968432-8437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Allen, J. R., and S. A. Ensign. 1997. Characterization of three protein components required for functional reconstitution of the epoxide carboxylase multienzyme complex from Xanthobacter strain Py2. J. Bacteriol. 1793110-3115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Allen, J. R., and S. A. Ensign. 1998. Identification and characterization of epoxide carboxylase activity in cell extracts of Nocardia corallina B276. J. Bacteriol. 1802072-2078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Allen, J. R., and S. A. Ensign. 1997. Purification to homogeneity and reconstitution of the individual components of the epoxide carboxylase multiprotein enzyme complex from Xanthobacter strain Py2. J. Biol. Chem. 27232121-32128. [DOI] [PubMed] [Google Scholar]
- 5.Balch, W. E., and R. S. Wolfe. 1976. New approach to cultivation of methanogenic bacteria: 2-mercaptoethanesulfonic acid (HS-CoM)-dependent growth of Methanobacterium ruminantium in a pressurized atmosphere. Appl. Environ. Microbiol. 32781-791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Balch, W. E., and R. S. Wolfe. 1979. Specificity and biological distribution of coenzyme M (2-mercaptoethanesulfonic acid). J. Bacteriol. 137256-263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Balch, W. E., and R. S. Wolfe. 1979. Transport of coenzyme M (2-mercaptoethanesulfonic acid) in Methanobacterium ruminantium. J. Bacteriol. 137264-273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bobik, T. A., and R. S. Wolfe. 1989. An unusual thiol-driven fumarate reductase in Methanobacterium with the production of the heterodisulfide of coenzyme M and N-(7-mercaptoheptanoyl)threonine-O3-phosphate. J. Biol. Chem. 26418714-18718. [PubMed] [Google Scholar]
- 9.Boyd, J. M., and S. A. Ensign. 2005. Evidence for a metal-thiolate intermediate in alkyl group transfer from epoxypropane to coenzyme M and cooperative metal ion binding in epoxyalkane:CoM transferase. Biochemistry 4413151-13162. [DOI] [PubMed] [Google Scholar]
- 10.Breksa, A. P., III, and T. A. Garrow. 1999. Recombinant human liver betaine-homocysteine S-methyltransferase: identification of three cysteine residues critical for zinc binding. Biochemistry 3813991-13998. [DOI] [PubMed] [Google Scholar]
- 11.Clark, D. D., J. R. Allen, and S. A. Ensign. 2000. Characterization of five catalytic activities associated with the NADPH:2-ketopropyl-coenzyme M [2-(2-ketopropylthio)ethanesulfonate] oxidoreductase/carboxylase of the Xanthobacter strain Py2 epoxide carboxylase system. Biochemistry 391294-1304. [DOI] [PubMed] [Google Scholar]
- 12.Clark, D. D., J. M. Boyd, and S. A. Ensign. 2004. The stereoselectivity and catalytic properties of Xanthobacter autotrophicus 2-[(R)-2-hydroxypropylthio]ethanesulfonate dehydrogenase are controlled by interactions between C-terminal arginine residues and the sulfonate of coenzyme M. Biochemistry 436763-6771. [DOI] [PubMed] [Google Scholar]
- 13.Coleman, N. V., and J. C. Spain. 2003. Distribution of the coenzyme M pathway of epoxide metabolism among ethene- and vinyl chloride-degrading Mycobacterium strains. Appl. Environ. Microbiol. 696041-6046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Coleman, N. V., and J. C. Spain. 2003. Epoxyalkane: coenzyme M transferase in the ethene and vinyl chloride biodegradation pathways of Mycobacterium strain JS60. J. Bacteriol. 1855536-5545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Danko, A. S., C. A. Saski, J. P. Tomkins, and D. L. Freedman. 2006. Involvement of coenzyme M during aerobic biodegradation of vinyl chloride and ethene by Pseudomonas putida strain AJ and Ochrobactrum sp. strain TD. Appl. Environ. Microbiol. 723756-3758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ensign, S. A. 2001. Microbial metabolism of aliphatic alkenes. Biochemistry 405845-5853. [DOI] [PubMed] [Google Scholar]
- 17.Ensign, S. A., and J. R. Allen. 2003. Aliphatic epoxide carboxylation. Annu. Rev. Biochem. 7255-76. [DOI] [PubMed] [Google Scholar]
- 18.Ermler, U. 2005. On the mechanism of methyl-coenzyme M reductase. Dalton Trans. 20053451-3458. [DOI] [PubMed] [Google Scholar]
- 19.Ermler, U., W. Grabarse, S. Shima, M. Goubeaud, and R. K. Thauer. 1997. Crystal structure of methyl-coenzyme M reductase: the key enzyme of biological methane formation. Science 2781457-1462. [DOI] [PubMed] [Google Scholar]
- 20.Felsenstein, J. 1985. Confidence limits on phylogenies: an approach using the bootstrap. Evolution 39783-791. [DOI] [PubMed] [Google Scholar]
- 21.Ferrer, J. L., S. Ravanel, M. Robert, and R. Dumas. 2004. Crystal structures of cobalamin-independent methionine synthase complexed with zinc, homocysteine, and methyltetrahydrofolate. J. Biol. Chem. 27944235-44238. [DOI] [PubMed] [Google Scholar]
- 22.Gencic, S., G. M. LeClerc, N. Gorlatova, K. Peariso, J. E. Penner-Hahn, and D. A. Grahame. 2001. Zinc-thiolate intermediate in catalysis of methyl group transfer in Methanosarcina barkeri. Biochemistry 4013068-13078. [DOI] [PubMed] [Google Scholar]
- 23.Gonzalez, J. C., K. Peariso, J. E. Penner-Hahn, and R. G. Matthews. 1996. Cobalamin-independent methionine synthase from Escherichia coli: a zinc metalloenzyme. Biochemistry 3512228-12234. [DOI] [PubMed] [Google Scholar]
- 24.Goulding, C. W., and R. G. Matthews. 1997. Cobalamin-dependent methionine synthase from Escherichia coli: involvement of zinc in homocysteine activation. Biochemistry 3615749-15757. [DOI] [PubMed] [Google Scholar]
- 25.Graham, D. E., M. Graupner, H. Xu, and R. H. White. 2001. Identification of coenzyme M biosynthetic 2-phosphosulfolactate phosphatase. A member of a new class of Mg(2+)-dependent acid phosphatases. Eur. J. Biochem. 2685176-5188. [DOI] [PubMed] [Google Scholar]
- 26.Graham, D. E., and R. H. White. 2002. Elucidation of methanogenic coenzyme biosyntheses: from spectroscopy to genomics. Nat. Prod. Rep. 19133-147. [DOI] [PubMed] [Google Scholar]
- 27.Graupner, M., H. Xu, and R. H. White. 2000. Identification of an archaeal 2-hydroxy acid dehydrogenase catalyzing reactions involved in coenzyme biosynthesis in methanoarchaea. J. Bacteriol. 1823688-3692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Guex, N., and M. C. Peitsch. 1997. SWISS-MODEL and the Swiss-PdbViewer: an environment for comparative protein modeling. Electrophoresis 182714-2723. [DOI] [PubMed] [Google Scholar]
- 29.Hedderich, R., A. Berkessel, and R. K. Thauer. 1990. Purification and properties of heterodisulfide reductase from Methanobacterium thermoautotrophicum (strain Marburg). Eur. J. Biochem. 193255-261. [DOI] [PubMed] [Google Scholar]
- 30.Heim, S., A. Künkle, R. K. Thauer, and R. Hedderich. 1998. Thiol:fumarate reductase (Tfr) from Methanobacterium thermoautotrophicum identification of the catalytic sites for fumarate reduction and thiol oxidation. Eur. J. Biochem. 253292-299. [DOI] [PubMed] [Google Scholar]
- 31.Jacobs, M. H., A. J. Van den Wijngaard, M. Pentenga, and D. B. Janssen. 1991. Characterization of the epoxide hydrolase from an epichlorohydrin-degrading Pseudomonas sp. Eur. J. Biochem. 2021217-1222. [DOI] [PubMed] [Google Scholar]
- 32.Jones, T. A., J. Y. Zou, S. W. Cowan, and M. Kjeldgaard. 1991. Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Crystallogr. D 47110-119. [DOI] [PubMed] [Google Scholar]
- 33.Jornvall, H., B. Persson, M. Krook, S. Atrian, R. Gonzalez-Duarte, J. Jeffery, and D. Ghosh. 1995. Short-chain dehydrogenases/reductases (SDR). Biochemistry 346003-6013. [DOI] [PubMed] [Google Scholar]
- 34.Juan, B., and R. K. Thauer. 2007. Nickel and its surprising impact in nature. John Wiley and Sons, Chichester, United Kingdom.
- 35.Kallberg, Y., U. Oppermann, H. Jornvall, and B. Persson. 2002. Short-chain dehydrogenases/reductases (SDRs). Eur. J. Biochem. 2694409-4417. [DOI] [PubMed] [Google Scholar]
- 36.Krishnakumar, A. M., B. P. Nocek, D. D. Clark, S. A. Ensign, and J. W. Peters. 2006. Structural basis for stereoselectivity in the (R)- and (S)-hydroxypropylthioethanesulfonate dehydrogenases. Biochemistry 458831-8840. [DOI] [PubMed] [Google Scholar]
- 37.Krum, J. G., H. Ellsworth, R. R. Sargeant, G. Rich, and S. A. Ensign. 2002. Kinetic and microcalorimetric analysis of substrate and cofactor interactions in epoxyalkane:CoM transferase, a zinc-dependent epoxidase. Biochemistry 415005-5014. [DOI] [PubMed] [Google Scholar]
- 38.Krum, J. G., and S. A. Ensign. 2001. Evidence that a linear megaplasmid encodes enzymes of aliphatic alkene and epoxide metabolism and coenzyme M (2-mercaptoethanesulfonate) biosynthesis in Xanthobacter strain Py2. J. Bacteriol. 1832172-2177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Krum, J. G., and S. A. Ensign. 2000. Heterologous expression of bacterial epoxyalkane:coenzyme M transferase and inducible coenzyme M biosynthesis in Xanthobacter strain Py2 and Rhodococcus rhodochrous B276. J. Bacteriol. 1822629-2634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lund, O., M. Nielsen, C. Lundegaard, and P. Worning. 2002. X3M, a computer program to extract 3D models, abstr. A102. Abstr. CASP5 Conf.
- 41.Mattes, T. E., N. V. Coleman, J. C. Spain, and J. M. Gossett. 2005. Physiological and molecular genetic analyses of vinyl chloride and ethene biodegradation in Nocardioides sp. strain JS614. Arch. Microbiol. 18395-106. [DOI] [PubMed] [Google Scholar]
- 42.Matthews, R. G., and C. W. Goulding. 1997. Enzyme-catalyzed methyl transfers to thiols: the role of zinc. Curr. Opin. Chem. Biol. 1332-339. [DOI] [PubMed] [Google Scholar]
- 43.McBride, B. C., and R. S. Wolfe. 1971. A new coenzyme of methyl transfer, coenzyme M. Biochemistry 102317-2324. [DOI] [PubMed] [Google Scholar]
- 44.Myers, L. C., M. P. Terranova, A. E. Ferentz, G. Wagner, and G. L. Verdine. 1993. Repair of DNA methylphosphotriesters through a metalloactivated cysteine nucleophile. Science 2611164-1167. [DOI] [PubMed] [Google Scholar]
- 45.Myers, L. C., G. L. Verdine, and G. Wagner. 1993. Solution structure of the DNA methyl phosphotriester repair domain of Escherichia coli Ada. Biochemistry 3214089-14094. [DOI] [PubMed] [Google Scholar]
- 46.Nakajima, K., T. Hashimoto, and Y. Yamada. 1994. Opposite stereospecificity of two tropinone reductases is conferred by the substrate-binding sites. J. Biol. Chem. 26911695-11698. [PubMed] [Google Scholar]
- 47.Nakajima, K., A. Yamashita, H. Akama, T. Nakatsu, H. Kato, T. Hashimoto, J. Oda, and Y. Yamada. 1998. Crystal structures of two tropinone reductases: different reaction stereospecificities in the same protein fold. Proc. Natl. Acad. Sci. USA 954876-4881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Nocek, B., S. B. Jang, M. S. Jeong, D. D. Clark, S. A. Ensign, and J. W. Peters. 2002. Structural basis for CO2 fixation by a novel member of the disulfide oxidoreductase family of enzymes, 2-ketopropyl-coenzyme M oxidoreductase/carboxylase. Biochemistry 4112907-12913. [DOI] [PubMed] [Google Scholar]
- 49.Pai, E. F. 1991. Variations on a theme: the family of FAD-dependent NAD(P)H-(disulphide)-oxidoreductases. Curr. Opin. Struct. Biol. 1991796-803. [Google Scholar]
- 50.Pandey, A. S., B. Nocek, D. D. Clark, S. A. Ensign, and J. W. Peters. 2006. Mechanistic implications of the structure of the mixed-disulfide intermediate of the disulfide oxidoreductase, 2-ketopropyl-coenzyme M oxidoreductase/carboxylase. Biochemistry 45113-120. [DOI] [PubMed] [Google Scholar]
- 51.Reference deleted.
- 52.Peariso, K., Z. S. Zhou, A. E. Smith, R. G. Matthews, and J. E. Penner-Hahn. 2001. Characterization of the zinc sites in cobalamin-independent and cobalamin-dependent methionine synthase using zinc and selenium X-ray absorption spectroscopy. Biochemistry 40987-993. [DOI] [PubMed] [Google Scholar]
- 53.Pejchal, R., and M. L. Ludwig. 2005. Cobalamin-independent methionine synthase (MetE): a face-to-face double barrel that evolved by gene duplication. PLoS Biol. 3e31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Saitou, N., and M. Nei. 1987. The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol. Biol. Evol. 4406-425. [DOI] [PubMed] [Google Scholar]
- 55.Sauer, K., and R. K. Thauer. 2000. Methyl-coenzyme M formation in methanogenic archaea. Involvement of zinc in coenzyme M activation. Eur. J. Biochem. 2672498-2504. [DOI] [PubMed] [Google Scholar]
- 56.Tallant, T. C., L. Paul, and J. A. Krzycki. 2001. The MtsA subunit of the methylthiol:coenzyme M methyltransferase of Methanosarcina barkeri catalyses both half-reactions of corrinoid-dependent dimethylsulfide: coenzyme M methyl transfer. J. Biol. Chem. 2764485-4493. [DOI] [PubMed] [Google Scholar]
- 57.Tamura, K., J. Dudley, M. Nei, and S. Kumar. 2007. MEGA4: molecular evolutionary genetics analysis (MEGA) software version 4.0. Mol. Biol. Evol. 241596-1599. [DOI] [PubMed] [Google Scholar]
- 58.Taylor, C. D., B. C. McBride, R. S. Wolfe, and M. P. Bryant. 1974. Coenzyme M, essential for growth of a rumen strain of Methanobacterium ruminantium. J. Bacteriol. 120974-975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Taylor, C. D., and R. S. Wolfe. 1974. A simplified assay for coenzyme M (HSCH2CH2SO3). Resolution of methylcobalamin-coenzyme M methyltransferase and use of sodium borohydride. J. Biol. Chem. 2494886-4890. [PubMed] [Google Scholar]
- 60.Taylor, C. D., and R. S. Wolfe. 1974. Structure and methylation of coenzyme M (HSCH2CH2SO3). J. Biol. Chem. 2494879-4885. [PubMed] [Google Scholar]
- 61.Thauer, R. K. 1998. Biochemistry of methanogenesis: a tribute to Marjory Stephenson. Microbiology 1442377-2406. [DOI] [PubMed] [Google Scholar]
- 62.Thompson, J. D., D. G. Higgins, and T. J. Gibson. 1994. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 224673-4680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.van Hylckama Vlieg, J. E., J. Kingma, W. Kruizinga, and D. B. Janssen. 1999. Purification of a glutathione S-transferase and a glutathione conjugate-specific dehydrogenase involved in isoprene metabolism in Rhodococcus sp. strain AD45. J. Bacteriol. 1812094-2101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.van Hylckama Vlieg, J. E., J. Kingma, A. J. van den Wijngaard, and D. B. Janssen. 1998. A glutathione S-transferase with activity towards cis-1,2-dichloroepoxyethane is involved in isoprene utilization by Rhodococcus sp. strain AD45. Appl. Environ. Microbiol. 642800-2805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wade, D. R., S. C. Airy, and J. E. Sinsheimer. 1978. Mutagenicity of aliphatic epoxides. Mutat. Res. 58217-223. [DOI] [PubMed] [Google Scholar]
- 66.White, R. H. 1986. Biosynthesis of coenzyme M. Biochemistry 246487-6493. [Google Scholar]
- 67.White, R. H. 2001. Biosynthesis of the methanogenic cofactors. Vitam. Horm. 61299-337. [DOI] [PubMed] [Google Scholar]
- 68.White, R. H. 1988. Characterization of enzymatic conversion of sulfoacetaldehyde and l-cysteine into coenzyme M (2-mercaptoethanesulfonic acid). Biochemistry 277458-7462. [Google Scholar]
- 69.White, R. W. 1985. Intermediates in the biosynthesis of coenzyme M. Biochemistry 255304-5308. [Google Scholar]
- 70.Wise, E. L., D. E. Graham, R. H. White, and I. Rayment. 2003. The structural determination of phosphosulfolactate synthase from Methanococcus jannaschii at 1.7-A resolution: an enolase that is not an enolase. J. Biol. Chem. 27845858-45863. [DOI] [PubMed] [Google Scholar]
- 71.Wolfe, R. S. 1991. My kind of biology. Annu. Rev. Microbiol. 451-35. [DOI] [PubMed] [Google Scholar]
- 72.Zhou, Z. S., K. Peariso, J. E. Penner-Hahn, and R. G. Matthews. 1999. Identification of the zinc ligands in cobalamin-independent methionine synthase (MetE) from Escherichia coli. Biochemistry 3815915-15926. [DOI] [PubMed] [Google Scholar]
- 73.Zuckerkandl, E., and L. Pauling. 1965. Evolutionary divergence and convergence in proteins, p. 97-166. In V. Bryson and H. J. Vogel (ed.), Evolving genes and proteins. Academic Press, New York, NY.