

Abstract
The class II fusion proteins of the alphaviruses and flaviviruses mediate virus infection by driving the fusion of the virus membrane with that of the cell. These fusion proteins are triggered by low pH, and their structures are strikingly similar in both the prefusion dimer and the postfusion homotrimer conformations. Here we have compared cholesterol interactions during membrane fusion by these two groups of viruses. Using cholesterol-depleted insect cells, we showed that fusion and infection by the alphaviruses Semliki Forest virus (SFV) and Sindbis virus were strongly promoted by cholesterol, with similar sterol dependence in laboratory and field isolates and in viruses passaged in tissue culture. The E1 fusion protein from SFV bound cholesterol, as detected by labeling with photocholesterol and by cholesterol extraction studies. In contrast, fusion and infection by numerous strains of the flavivirus dengue virus (DV) and by yellow fever virus 17D were cholesterol independent, and the DV fusion protein did not show significant cholesterol binding. SFV E1 is the first virus fusion protein demonstrated to directly bind cholesterol. Taken together, our results reveal important functional differences conferred by the cholesterol-binding properties of class II fusion proteins.
Alphaviruses and flaviviruses are transmitted in nature by mosquito and tick vectors and can cause severe human and animal illnesses, such as encephalitis and hemorrhagic fever. These viruses include a number of important human pathogens, such as the alphaviruses chikungunya virus (CHIKV) and Venezuelan equine encephalitis virus and the flaviviruses dengue virus (DV), yellow fever virus (YFV), tick-borne encephalitis virus (TBE), and West Nile virus (WNV). CHIKV caused a recent outbreak in Réunion, Mauritius, and southern India (48), with an estimated 1.4 million infections in India (12). DV is a major human health problem and currently causes ∼50 million to 100 million infections and ∼500,000 cases of dengue hemorrhagic fever per year (8, 15). Although an effective vaccine against YFV has been in use since the 1930s, there are an estimated 200,000 cases of yellow fever and 30,000 deaths annually (3). Vaccine strategies and antiviral therapies for flaviviruses and alphaviruses are urgently needed. An important target of these approaches is the virus entry pathway, and thus, a molecular understanding of virus entry is critical.
Alphaviruses and flaviviruses are small, enveloped RNA viruses. These viruses enter cells by endocytosis and deliver their RNA genomes into the cytoplasm via a membrane fusion reaction triggered by endosomal low pH (16, 20). Alphavirus and flavivirus membrane fusion is mediated by transmembrane proteins, termed class II fusion proteins. The membrane fusion proteins E1 of the alphaviruses and E of the flaviviruses have strikingly similar elongated three-domain structures (31, 40, 45, 47, 61). The domains are composed primarily of β-strands, with a hydrophobic fusion loop at one end of the molecule and the stem and transmembrane region connecting to domain III at the other end. During fusion, the E1 and E proteins insert into the target membrane via the fusion loop, trimerize, and refold to form hairpinlike structures, with the domain III and stem regions folded back to pack against the trimer core (see Fig. 4A) (4, 13, 40). This rearrangement produces a rodlike homotrimer, with the transmembrane domains and fusion loops at the same end. The energetically unfavorable membrane fusion reaction is driven by the refolding of the metastable fusion protein to the final highly stable hairpin.
FIG. 4.

Photoaffinity labeling reveals the specific cholesterol interaction of the SFV but not DV2 fusion protein. (A) Fusion protein structure and schematics. The structure of the SFV E1*HT is shown with one monomer in color and the others in gray. The color monomer shows the positions of the three domains (red, DI; yellow, DII; and blue, DIII), the fusion peptide (green), and the ij loop (cyan). Linear representations of the E1 and E sequences are shown below the structure. (B) Liposome coflotation of SFV E1′ and DV2 E′. E1′ and E′ proteins were mixed with liposomes containing [3H]photocholesterol and treated at the indicated pH for 30 min at 25°C. Aliquots of the samples were tested for liposome binding by analysis on sucrose flotation gradients. The protein-liposome mixture was loaded on the bottom of a sucrose step gradient and centrifuged to float the liposomes to the top of the gradient. The proteins in the top and bottom fractions were quantitated by SDS-PAGE and Western blotting, and the total liposome-bound protein in each sample was calculated. (C) Formation of SFV E1′ and DV2 E′ homotrimers. Aliquots of the samples described in the legend for panel B were tested for trimerization of the fusion protein by solubilization with 1% β-OG, analysis on sucrose sedimentation gradients, and quantitation as for Fig. 4B. Sedimentation is to the right, and the positions of the SFV E1′ monomer (M), the DV2 E′ dimer (D), and the SFV and DV homotrimers (T) are indicated. (D) Photoaffinity labeling of SFV E1′ and DV2 E′. Aliquots of the samples were exposed to UV light for 20 min as indicated, acid precipitated, washed with acetone, and analyzed by SDS-PAGE and fluorography. In a separate reaction mixture, purified PFO was incubated at 37°C for 10 min with liposomes containing [3H]photocholesterol, exposed to UV light, and analyzed as above. Panels B to D are representative examples of two independent experiments.
The formation of the homotrimer can be blocked by mutations in the fusion protein (22). The folding back of domain III to form the hairpin can be specifically inhibited by the addition of exogenous domain III as a competitor for binding to the trimer core (33). In each of these cases, virus fusion and infection are blocked, suggesting that inhibitors of trimerization would be potent antiviral compounds. Another critical step in the fusion pathway is the initial interaction of the fusion loop with the target membrane prior to trimer formation. Antibody binding to the fusion loop or the insertion of charged amino acids in this region blocks its interaction with the target membrane and inhibits fusion (2, 51). Conversely, membrane insertion of the fusion loops of the alphaviruses Semliki Forest virus (SFV) and Sindbis virus (SIN) is strongly promoted by the presence of cholesterol in the target membrane (1, 23, 49).
An important role for cholesterol in the initial interaction of the fusion loop with the target membrane is in keeping with studies of the fusion properties of both SFV and SIN. In vitro, the low-pH-dependent fusion of both viruses is strongly promoted by the presence of cholesterol in liposome target membranes (23, 49, 60). In vivo, SFV and SIN fusion and infection are 3 to 5 logs higher in control cholesterol-containing cells than in cholesterol-depleted cells (35, 43, 57). This cholesterol dependence is reduced in SFV srf mutants selected for growth on cholesterol-depleted cells, and the decreased sterol dependence is conferred by single-point mutations on the srf E1 protein (6, 57). Interestingly, once the SFV E1 ectodomain (termed E1*) is inserted into the target membrane at a low pH, it is strongly associated with cholesterol-enriched membrane domains (1), suggesting a possible interaction of the protein with cholesterol.
In contrast, the role of cholesterol in flavivirus fusion is unclear. Cholesterol promotes the low-pH-triggered interaction of the TBE fusion protein ectodomain with liposomes (52). However, the virus itself is not strongly cholesterol dependent when tested in lipid mixing assays for the initial steps of fusion (10, 52). WNV fusion with liposomes is somewhat more efficient when the membranes contain cholesterol (14). An earlier study showed that the pretreatment of mammalian cells with the cholesterol-extracting compound methyl-β-cyclodextrin (MβCD) inhibits DV production (46), but this study did not localize inhibition to a specific step(s) in the virus life cycle, such as entry or fusion. Given these inconclusive results, it is not clear if the structurally similar alphavirus and flavivirus fusion proteins differ in their cholesterol dependence and, if so, what the mechanism of such a difference might be. The fact that the alphaviruses and flaviviruses readily adapt to growth in tissue culture (26, 36) also raises important questions about the role of adaptation in cholesterol dependence, since the studies to date have used primarily laboratory strains.
Here we have addressed the role of cellular cholesterol in fusion and infection by alphaviruses and flaviviruses. We correlated these results with studies of fusion protein-cholesterol interaction using photoactivatable cholesterol and cholesterol extraction. Our results demonstrate, for the first time, direct cholesterol binding by a virus membrane fusion protein. We identify significant differences between the cholesterol-binding activities of the SFV and DV fusion proteins, which are reflected in the fusion mechanisms of these class II proteins. More broadly, we demonstrate that photocholesterol can be used in liposome systems to define cholesterol-membrane protein interactions.
MATERIALS AND METHODS
Cells.
BHK-21 cells were cultured at 37°C in Dulbecco's modified Eagle's medium (DMEM) containing 100 U of penicillin/ml and 100 μg of streptomycin/ml (P/S), 5% fetal calf serum (FCS), and 10% tryptose phosphate broth (43). Control cholesterol-containing C6/36 mosquito cells were maintained at 28°C in DMEM with P/S and 10% heat-inactivated FCS (43). Where indicated, C6/36 cells were cholesterol depleted by 5 to 15 passages at 28°C in DMEM containing P/S and 10% delipidated, heat-inactivated FCS (37, 43). Previous studies showed that these conditions reduced the levels of both free and esterified cholesterol to less than 2% of those of control cells (43).
Viruses.
The wild-type (wt) SFV used in this study was a previously characterized plaque-purified isolate (57). The srf-3 mutant was isolated from this parental virus by selection for growth on cholesterol-depleted cells and has a single amino acid change of E1 P226S (57). This mutant shows increased infection and fusion on cholesterol-depleted cells, although its fusion with cells and liposomes is still strongly promoted by the presence of cholesterol (7, 57). Robert Tesh of the World Reference Center for Emerging Viruses and Arboviruses provided field isolates of SFV and SIN that were passaged four to seven times by the intracerebral inoculation of mice and stored as lyophilized stocks. SFVA was a prototype strain isolated in Uganda in 1942 from Aedes abnormalis mosquitoes, SFVB was the Kumba strain isolated from Eretmapodites mosquitoes in Cameroon in 1952, and SIN was strain UG MP 6440, isolated from Mansonia fuscopennata mosquitoes in Uganda in 1958. DV strains were the New Guinea C strain of DV2 (provided by John Roehrig, Centers for Disease Control, Fort Collins, CO), the DV2 16681 strain originally isolated from a dengue hemorrhagic fever patient (provided as an infectious clone by Richard Kinney, Centers for Disease Control, Fort Collins, CO) (24), the DV1 Western Pacific strain (provided by Richard Stockert, Albert Einstein College of Medicine), the DV1 and DV2 high-passage reference strains (D1rs and D2rs, both originally from Robert Shope of the WHO arbovirus reference collection at UTMB-Galveston), and the low-passage clinical isolates (D1ci, strain no. 206077; D2ci, strain no. 271220; both originally from Duane Gubler of the Centers for Disease Control, Fort Collins, CO) (all provided by Aravinda de Silva, University of North Carolina School of Medicine). YFV was the 17D vaccine strain (provided by Richard Kuhn, Purdue University). Vesicular stomatitis virus (VSV) was the Indiana strain used in our previous studies (43).
Virus propagation.
The alphavirus field isolates were propagated to make working stocks by one passage at low multiplicity on BHK-21 cells in MEM containing 0.2% bovine serum albumin and 10 mM HEPES, pH 7.5. Titers were determined by plaque assay on BHK-21 cells. Serial passaging of the indicated alphaviruses was performed on BHK-21 cells by five cycles of infection at low multiplicity. Flavivirus isolates were propagated to make working stocks by one passage at low multiplicity on C6/36 cells in DMEM containing 2% delipidated heat-inactivated FCS. Ten mM HEPES, pH 8.0, was added to the medium after harvesting and before freezing at −80°C. Stocks were titered by infectious center assay on C6/36 cells as described below.
Infectious center assay.
To evaluate the cholesterol dependence of infection, control and cholesterol-depleted C6/36 mosquito cells were infected with serial dilutions of virus for 2 h at 28°C and cultured overnight (alphaviruses) or for 2 to 4 days at 28°C (flaviviruses) in medium containing 20 mM NH4Cl to prevent secondary infection. Infected cells were quantitated by indirect immunofluorescence using polyclonal antibodies to the SFV or SIN envelope proteins (35, 57), monoclonal antibody (MAb) I1 to the VSV G protein (29) (obtained from Doug Lyles), and mouse polyclonal hyperimmune ascitic fluid against DV2 (obtained from Robert Tesh) or against YFV (American Type Culture Collection). For ease of comparison of virus infection of control and depleted cells, in each assay, the titers on control cells were normalized to a value that permitted representation of all of the virus titers on a log scale (33).
Fusion infection assay.
To evaluate the cholesterol dependence of the virus-membrane fusion, serial dilutions of virus were incubated with control and cholesterol-depleted C6/36 cells for 90 min on ice (33). Virus fusion with the plasma membrane was induced by treatment for 1 min at pH 5.7 at 28°C. The cells were then incubated overnight (alphaviruses) or for 2 days (DV) at 28°C in medium containing 20 mM NH4Cl to prevent secondary infection, and the cells infected due to low-pH-triggered fusion were quantitated by immunofluorescence as described above. Previous experiments demonstrated that infection after treatment at neutral pH was negligible compared with that obtained after low-pH treatment (32, 33). For ease of comparison of virus fusion with control and depleted cells, in each assay, the titers on control cells were normalized to a value that permitted representation of all of the virus titers on a log scale (33).
Production of SFV E1′ and DV2 E′ in Drosophila S2 cells.
Truncated forms of the SFV E1 and DV2 E proteins (see Fig. 4A) were obtained by inducible expression in Drosophila S2 cells similar to previous studies of DV proteins (18). The DV2 prM-E sequences (nucleotides 439 to 2121 in the DV2-NGC infectious clone JES18) (44) were PCR amplified and cloned into the BglII/XhoI sites of pMT/Bip/V5-His A (Invitrogen). The resultant truncated E sequence, termed E′, ends at residue E G395 followed by the V5 epitope and a six-histidine tag from the vector. The SFV p62-E1 sequences (nucleotides 8242 to 11022 in the pSP6-SFV4 infectious clone) (34) were PCR amplified and cloned into the KpnI/XbaI sites of the pMT/V5-His A vector (Invitrogen). The resultant truncated E1 sequence, termed E1′, ends at residue E1 P383, followed by the V5 epitope and a six-histidine tag from the vector. The virus protein-encoding sequences of both constructs were confirmed. The constructs were cotransfected with the pCo-BLAST selection vector into Drosophila S2 cells using calcium phosphate, and stable cell populations were selected using the recommended methods (Invitrogen).
Proteins were expressed by growth of the stable cells in Drosophila serum-free medium (Invitrogen, CA, or HyClone, UT) for 7 to 10 days at 27°C in a shaking flask at 115 rpm in the presence of 750 μM copper sulfate to induce expression. The secreted E′ and E1′ proteins were purified from the culture medium by affinity chromatography on a chelating Sepharose column that interacts with the copper bound to the His tag (30). Proteins were further purified by size exclusion chromatography using a Superdex 200 column. Protein concentrations were estimated by a Bio-Rad assay versus an immunoglobulin G standard or by quantitative Western blot analysis.
Liposomes.
Liposomes were prepared by freeze-thawing and extrusion (7), using TAN buffer (20 mM triethanolamine, pH 8.0, and 130 mM NaCl), a 1:1:1:3 molar ratio of 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine, 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphoethanolamine, porcine brain sphingomyelin (Avanti Polar Lipids, Alabaster, AL) and cholesterol (Steraloids, Inc., Wilton, NH), and [3H]cholesterol or [3H]photocholesterol as indicated. The resultant liposome preparations contained approximately 6 mM lipid and 1 μCi [3H]cholesterol (Amersham) per ml or 50 μCi [3H]photocholesterol per ml. [3H]photocholesterol (6-azi-5α-cholestan-3β-ol) was synthesized by Merck and Co. exactly as described previously (54) and was a kind gift of David Silver of the Albert Einstein College of Medicine (59). Cholesterol-deficient liposomes were prepared by maintaining the 1:1:1 molar ratio of phospholipids described above but without cholesterol. Liposomes were stored at 4°C under N2 conditions and used within 2 weeks of preparation.
Photoaffinity labeling of SFV E1′ and DV2 E′.
In a 150-μl reaction mixture, SFV E1′ or DV2 E′ was mixed with [3H]photocholesterol liposomes at final concentrations of 50 to 100 μg protein/ml and 1 mM lipid. The samples were incubated at 25°C for 5 min, adjusted to pH 5.75 by adding a precalibrated volume of 0.5 N acetic acid or 0.3 M MES (morpholinoethanesulfonic acid), further incubated at 25°C for 30 min, and then adjusted to pH 8.0 by the addition of 0.5 N NaOH or 0.3 M TEA (triethanolamine) buffer. A parallel control reaction mixture was maintained at pH 8.0 throughout. Aliquots were removed for analysis by liposome coflotation and sucrose density gradient sedimentation as described below. The remaining reaction mixture was split into two aliquots each containing 2.5 to 5.0 μg protein in 55 μl. Samples were placed in a tissue culture grade 96-well plate that had been pretreated with 75 μl of 50 μg/ml bovine serum albumin for 30 min at room temperature. Half of the samples were collected prior to UV treatment, the corresponding wells were washed three times with 55 μl TN buffer (50 mM Tris-HCl, pH 7.4, 100 mM NaCl), and the sample and washes were pooled. The remaining samples were treated with UV light for 20 min on ice using an inverted UV transilluminator (Fotodyne 3-3500; four 15-W, 312-nm UV bulbs) placed 1.5 to 2.0 cm above the samples and then collected as above. A total of 5 μl of each sample was analyzed by quantitative Western blot analysis to calculate protein recovery after UV treatment. The remaining samples were precipitated for 2 h on ice with 10% trichloroacetic acid in the presence of 15 μg of carrier DNA. The samples were centrifuged for 15 min at 14,000 rpm at 4°C, and the pellets were washed three times with 1 ml of ice-cold acetone. The pellets were air dried for 10 min, resuspended in sodium dodecyl sulfate (SDS) sample buffer, and analyzed on 11% polyacrylamide gels. Gels were fixed in 25% isopropanol-10% acetic acid for 30 min, incubated with Amplify (GE Healthcare) for 30 min, dried, and exposed to Hyperfilm (GE Healthcare) with intensifying screens on either side at −80°C. For quantitation, the films were scanned using an Epson Perfection Photoscanner and Silverfast Ai software and analyzed using Quantity One software.
The bacterial toxin perfringolysin O (PFO) was expressed and purified as previously described (50) and was a kind gift of Rodney Tweten of the University of Oklahoma Health Sciences Center. PFO (50 μg/ml) was incubated with 1 mM photocholesterol liposomes at neutral pH for 10 min at 37°C and then exposed to UV light and analyzed as described above.
Liposome coflotation analysis.
The protein-liposome samples described above were adjusted to a final volume of 0.4 ml containing 20% sucrose (wt/wt) in TAN buffer, pH 8.0, and layered on top of a 0.3-ml 60% sucrose cushion in TLS-55 ultracentrifuge tubes. The samples were overlaid with 1.2 ml of 15% sucrose and 0.2 ml 5% sucrose (wt/wt), all in TAN buffer, pH 8.0. Gradients were centrifuged for 3 h at 54,000 rpm at 4°C and seven 0.3-ml fractions were collected from the top. The top three and the bottom four fractions were pooled separately, TCA precipitated, and analyzed by quantitative Western blotting using SFV E1-3 (21) and DV 4G2 MAbs (11). Proteins were quantitated using Odyssey Licor software, and the amount floating with the liposomes in the top of the gradient was calculated.
Sucrose density gradient sedimentation analysis.
Protein-liposome samples described above were solubilized with 1.5% octyl-β-d-glucopyranoside (β-OG) for 1 h at room temperature. Fifty-microliter samples were layered onto step gradients consisting of 200 μl 5%, 400 μl 10%, 400 μl 20%, and 200 μl 40% sucrose (wt/wt) in TAN buffer, pH 8.0, containing 1 M NaCl and 1% β-OG. Gradients were centrifuged at 4°C for 5 h at 54,000 rpm in a TLS-55 rotor, fractionated, and analyzed by SDS-polyacrylamide gel electrophoresis (SDS-PAGE) and quantitative Western blotting using MAbs SFV E1-3 (21) and DV 4G2 (17).
Reconstitution of SFV envelope proteins into photocholesterol liposomes.
The SFV envelope proteins were reconstituted into photocholesterol liposomes by detergent dialysis (1). In brief, 0.6 μmol of the lipid mixture described above plus four times this mass of octylglucoside and 5 μCi [3H]photocholesterol were dried on a rotary evaporator, lyophilized for 2 h, and resuspended in 100 μl of TN buffer containing 25 mM octylglucoside and purified SFV (10 μg of virus protein). The clear solution obtained was dialyzed for a total of 36 h using 3× 250 ml 20 mM MES-130 mM NaCl, pH 7.0. The proteoliposomes, thus prepared, were treated at the indicated pH and analyzed for photolabeling and gradient flotation as described above. Control liposomes without virus were prepared in parallel and mixed with SFV E1′, and the samples were tested for photolabeling and gradient flotation.
MβCD treatment of proteoliposomes.
SFV-E1′ or DV2 E′ was mixed with 1 mM liposomes labeled with trace amounts of [3H]cholesterol and incubated at neutral or low pH as described above. The samples were then treated with 20 mM MβCD (Sigma Chemical Co., MO) for 30 min at 37°C as previously described (1) and analyzed for gradient flotation of the [3H]cholesterol marker and the viral proteins as described above.
RESULTS
Cholesterol requirements for alphavirus infection and fusion.
In order to test the cholesterol requirements of alphaviruses that had not been passaged in tissue culture, we compared infection by our wt SFV laboratory strain and the relatively cholesterol-independent srf-3 mutant with infection by one SIN strain and two SFV strains that were originally isolated in Africa and passaged in mice. As described previously (43, 57), the wt SFV infection of cholesterol-depleted C6/36 mosquito cells was decreased by more than 1,000-fold compared to that of control cells (Fig. 1A). In contrast, infection of cholesterol-deficient cells by the srf-3 mutant was more efficient, although it still decreased by about 40-fold compared to infection of control cells. Importantly, the cholesterol-independent rhabdovirus VSV infected both cell types equivalently, confirming that the differences observed with SFV were not due to cytotoxicity or inhibition of the endocytic entry pathway (Fig. 1A) (9, 43). The African field isolates SFVA, SFVB, and SIN were all strongly cholesterol dependent, with the difference between infection in depleted versus control cells ranging from 2 to 4 logs. The cholesterol dependence of virus fusion was then tested by evaluating the low-pH-triggered fusion of prebound virus with the plasma membrane of control or sterol-depleted cells (Fig. 1B) (57). Fusion by the wt SFV was strongly cholesterol dependent, showing a decrease of more than 5 logs on cholesterol-depleted cells, while fusion of the srf-3 mutant showed a decrease of greater than 2 logs on depleted cells. The membrane fusion reactions of SFVA, SFVB, and SIN were all markedly cholesterol dependent, with decreases ranging from ∼4 to 6 logs on sterol-depleted cells.
FIG. 1.
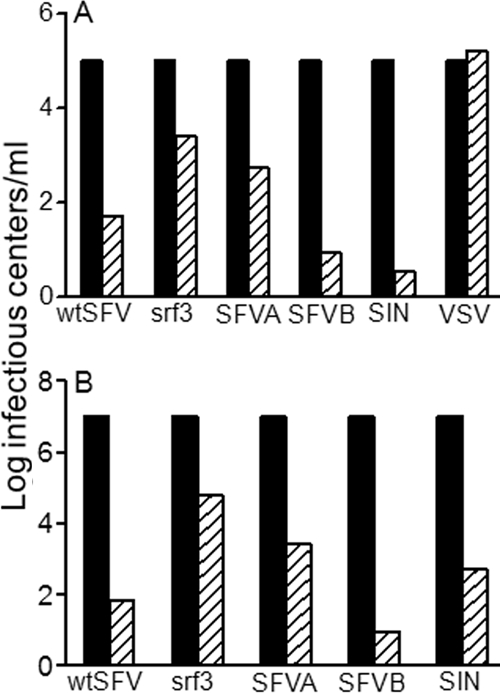
Infection and fusion by SFV and SIN are naturally cholesterol dependent. (A) Infection. Control (black bars) and cholesterol-depleted (hatched bars) C6/36 mosquito cells were infected with serial dilutions of wt SFV, the srf-3 mutant, VSV, or field isolates of SIN and SFV (SFVA and SFVB). Primary virus infection was quantitated by immunofluorescence and normalized to 105 infectious centers/ml on control cells. (B) Fusion. Serial dilutions of viruses (indicated as for panel A) were bound to control (black bars) and cholesterol-depleted (hatched bars) C6/36 cells on ice, and fusion was induced by treatment at pH 5.7 for 1 min at 28°C. The resultant infection was quantitated as for panel A and normalized to 107 infectious centers/ml on control cells. Panels A and B represent the average of two to three independent experiments.
These assays were performed with stocks of the field isolates that had been expanded only once on tissue culture cells. Notably, virus adaptation to binding cell surface heparan sulfate was found to occur after two to three passages of SIN on BHK cells (26). To address the possible role of tissue culture passage in the alphavirus cholesterol-dependent phenotype, we serially passaged wt SFV, the srf-3 mutant, SFVA, and SFVB five times in BHK cells and then tested their cholesterol dependence (Fig. 2). The requirement for cholesterol during infection and fusion was unaltered by multiple passages of any of the isolates, including the relatively cholesterol-independent srf-3 mutant and the highly cholesterol-dependent SFVB. Thus, these data suggest that the cholesterol dependence of virus fusion is an intrinsic property of the alphavirus infection pathway.
FIG. 2.
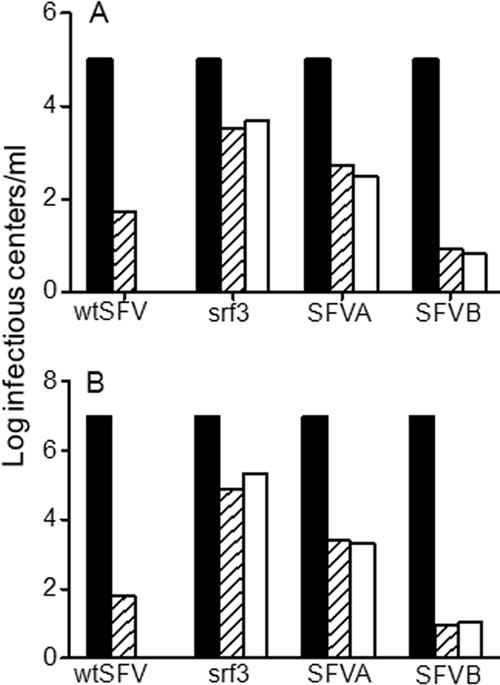
The cholesterol dependence of SFV isolates is stable. (A) Infection. The srf-3 mutant and SFV field isolates (SFVA and SFVB) were serially passaged five times on cholesterol-containing BHK cells, and the cholesterol dependence of infection was measured as for Fig. 1A. The black bars indicate infection of control cells by each virus (normalized to 105 infectious centers/ml), and the hatched and white bars indicate infection of cholesterol-depleted cells by the original and the passaged virus stocks, respectively. (B) Fusion. The cholesterol dependence of fusion was determined for the indicated viruses (with labeling as for Fig. 2A), with the titer on control cells (black bars) normalized for each virus to 107 infectious centers/ml. Panels A and B represent the average of two independent experiments.
Cholesterol requirements for flavivirus infection and fusion.
We used a variety of laboratory and clinical isolates of DV to test the requirements for cholesterol in virus infection and fusion. The strains included the New Guinea C strain of DV2, the Western Pacific strain of DV1, the clinical isolates D1ci and D2ci, the 16681 strain of DV2 isolated from a patient with dengue hemorrhagic fever, and the laboratory reference strains D1rs and D2rs. In addition, we tested the 17D vaccine strain of YFV. All of the DV strains and YFV showed comparable levels of infection and fusion on control and cholesterol-depleted cells, while in the same experiments, SFV was strongly cholesterol dependent (Fig. 3A and B). Coinfection experiments showed that the sterol dependence of SFV was unaffected by the presence of DV, confirming that the DV stocks did not somehow modify the cholesterol-depleted cells (data not shown). Thus, the class II fusion proteins of alphaviruses and flaviviruses, although highly related structurally, clearly differ in their requirement for cholesterol during membrane fusion.
FIG. 3.
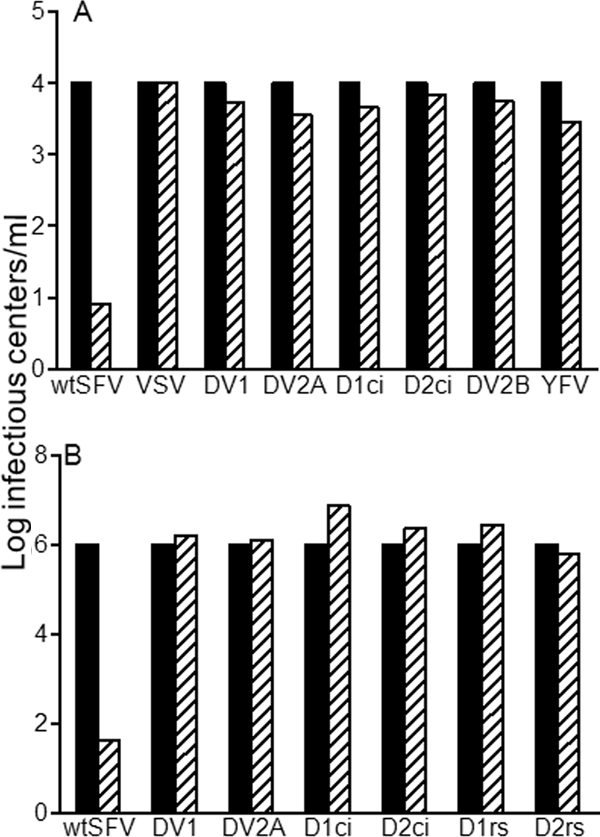
DV infection and fusion are not cholesterol dependent. (A) Infection. The cholesterol dependence of infection by wt SFV, VSV, YFV, and various strains of DV were measured as for Fig. 1A, with the titer on control cells (black bars) normalized to 104 infectious centers/ml. DV2A refers to DV2 New Guinea C, DV2B refers to DV2 16681, and D1ci and D2ci refer to DV1 and DV2 clinical isolates (see Materials and Methods for details). (B) Fusion. The cholesterol dependence of fusion by the indicated viruses was measured as for Fig. 1B, with the titer on control cells (black bars) normalized to 106 infectious centers/ml. D1rs and D2rs refer to DV1 and DV2 reference strains (see Materials and Methods for details). Panels A and B represent the average of two independent experiments.
Interaction of the DV and SFV fusion protein ectodomains with cholesterol.
Previous studies have shown that the 3β-hydroxyl group of cholesterol is critical in promoting SFV E1-membrane insertion and virus fusion (23, 25, 43) and that following low-pH-induced membrane insertion, the SFV E1* ectodomain interacts with cholesterol-enriched membrane domains (1). These findings suggested that there might be a specific and direct interaction of the SFV E1 protein with cholesterol in the target membrane. To assess this, we produced soluble ectodomains of the SFV and DV fusion proteins and tested them for interaction with [3H]photocholesterol.
Ectodomains of the wt SFV E1 and DV2 NGC E proteins were expressed in Drosophila S2 cells using a strategy similar to that previously described for DV E protein (see Materials and Methods and references 18 and 40). In each case, the fusion protein was coexpressed with the companion protein p62 (SFV) or prM (DV) to facilitate correct folding and transport (19, 41). Fusion proteins were truncated at the end of domain III prior to the stem and TM domains (Fig. 4A) and will be referred to as SFV E1′ and DV2 E′, respectively. Both E1′ and E′ were efficiently secreted into the S2 culture medium upon induction. The secreted SFV E1′ appeared to be monomeric and without detectable E2 protein (Fig. 4C) (data not shown), while the secreted DV E′ was a homodimer, in keeping with previous reports (40).
The purified ectodomains were tested for liposome binding and homotrimer formation. Proteins were mixed with liposomes containing 50 mol% cholesterol, a concentration optimal for SFV E1* insertion and trimerization (1, 25). The liposomes also contained 50 μCi [3H]photocholesterol/ml. The protein-liposome mixtures were incubated at low or neutral pH, and the efficiency of protein-membrane insertion was quantitated by a liposome coflotation assay (Fig. 4B). Both SFV E1′ and DV E′ efficiently associated with liposomes at low pH but not at neutral pH. Analysis of the samples by sucrose density gradient sedimentation demonstrated that both E1′ and E′ efficiently converted to the homotrimer form at low pH (Fig. 4C). Under the conditions of the assay, comparable molar amounts of the SFV and DV proteins were membrane associated and migrated as trimers (Fig. 4B and C).
We then tested the interaction of the E1′ and E′ proteins with [3H]photocholesterol. The protein-liposome mixtures were exposed to UV light to activate photocholesterol cross-linking, and labeling was evaluated by SDS-PAGE and fluorography (Fig. 4D). We consistently observed the labeling of SFV E1′ (n = 7). Labeling was dependent on both prior low pH incubation and on exposure to UV light. A small amount of E1′ labeling was observed for the pH 8.0 sample, but this labeling was consistently less than that observed for the low pH E1′. This may represent labeling of the small amount of E1′ that is stably liposome associated at pH 8.0 (Fig. 4B), or it could reflect a transient membrane interaction of the E1′ monomer at neutral pH. We also observed significant labeling of PFO, a member of the cholesterol-dependent cytolysins. These bacterial pore-forming toxins have been shown to require cholesterol for membrane interaction and pore formation (50, 56), and thus, we predicted that PFO should act as a positive control in this experiment. In contrast, the membrane-inserted DV E′ protein, although present at similar concentrations as those of the SFV protein, did not show efficient labeling with [3H]photocholesterol (Fig. 4D). Quantitation of duplicate parallel labeling experiments showed that the label associated with low-pH-treated, UV-cross-linked SFV E1′ was 13- to 16-fold higher than the background labeling of this sample in the absence of UV cross-linking. In comparison, labeling of low-pH-treated, UV-cross-linked DV E′ was only 1.4-fold higher than this background level.
Specificity of SFV E1′ photocholesterol labeling.
Given the very short half-life of the active carbene that is generated by UV irradiation of the diazirine ring (5), labeling of a protein by [3H]photocholesterol has been taken to reflect its specific cholesterol binding (54). Our data thus suggest that SFV E1′ has a direct physical interaction with cholesterol in the target membrane. However, it is possible that the SFV E1 trimer contains hydrophobic surfaces that nonspecifically attract cholesterol, resulting in labeling that is not reflective of the membrane-inserted region(s) of the protein. Alternatively, in our liposome system, photocholesterol may nonspecifically label any regions of proteins that lie within the liposome membrane rather than selectively labeling cholesterol-interacting membrane domains. Although the lack of labeling of the membrane-inserted DV protein argues against the latter explanation, we addressed the issue of specificity in detail by comparing the SFV E1′ ectodomain with the full-length E1 protein reconstituted into liposomes via its TM domain. Viral E1 and E2 were solubilized and reconstituted into liposomes containing [3H]photocholesterol using detergent dialysis as previously described (1). The liposomes containing reconstituted E1 were incubated at either neutral or low pH. Under the latter condition, the reconstituted E1 will convert to the trimer form. In a parallel reaction, protein-free liposomes containing [3H]photocholesterol were prepared by detergent dialysis and then mixed with SFV E1′ and incubated at neutral pH or at low pH to trigger protein-membrane insertion. Liposome coflotation experiments confirmed comparable levels of protein-membrane association for reconstituted full-length E1 and low-pH-treated E1′ (Fig. 5A). These proteoliposome samples were tested for photocholesterol labeling. As observed with the previous liposome system, E1′ was labeled by [3H]photocholesterol in a low-pH- and UV-dependent reaction (Fig. 5B). In contrast, full-length E1 and E2 reconstituted into membranes via their TM domains were not labeled with [3H]photocholesterol, even when the samples had been treated at low pH to trigger E1 HT formation. Reconstituted E1 can mediate liposome fusion, but our previous data suggest that due to the speed and geometry of the fusion reaction, only a small proportion of the E1 fusion loops insert into target membranes (1). The lack of labeling could further suggest that the reconstituted E1 protein does not efficiently insert into its “own” membrane, but further studies are required to determine this.
FIG. 5.
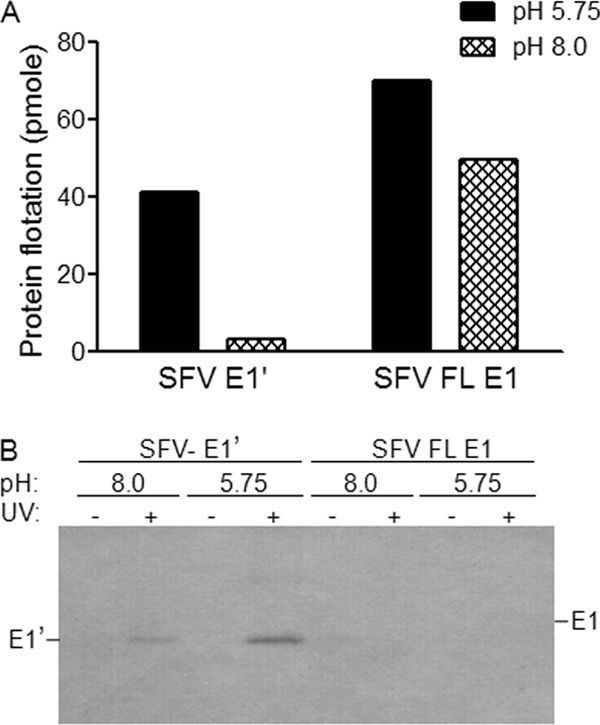
The TM domain of SFV E1 does not bind [3H]photocholesterol. The SFV envelope proteins E1 and E2 were reconstituted by detergent dialysis into liposomes containing [3H]photocholesterol. Protein-free [3H]photocholesterol-labeled liposomes were prepared in parallel by detergent dialysis and then mixed with E1′. The protein-liposome mixtures were treated at the indicated pH for 30 min at 25°C. (A) Liposome coflotation of SFV E1′ versus reconstituted full-length (FL) E1. Aliquots of the samples were tested for liposome association as described for Fig. 4B. The increase in FL E1 in the top of the gradient after low pH treatment may reflect fusion between proteoliposomes leading to more efficient flotation. (B) Photoaffinity labeling of SFV E1′ versus reconstituted full-length E1. Aliquots of the samples were exposed to UV light and analyzed for photocholesterol labeling as for Fig. 4D. The positions of SFV E1′ and SFV E1 are indicated based on parallel Western blot analysis. Panels A and B are representative examples of two independent experiments.
Together, our results thus indicate that photocholesterol labeling of the SFV E1 protein is dependent on the efficient interaction of the protein with the target membrane through the fusion loop rather than reflecting bulk labeling of protein regions in the bilayer or nonspecific interactions of cholesterol with the E1 homotrimer.
Differential partitioning of membrane-inserted SFV E1′ and DV2 E′.
A role for cholesterol in the membrane association of the class II fusion protein ectodomains could reflect a general requirement for a platform to orient and cluster the soluble monomers, thereby promoting lateral contacts and trimerization, or it could indicate a more specific lipid-protein interaction. Liposome flotation studies showed that cholesterol was required for the stable membrane association of both SFV E1′ and DV E′, while the association of both proteins with cholesterol-deficient liposomes was at background levels (Fig. 6A). We then compared the proteins' physical interaction with cholesterol-enriched membrane domains by taking advantage of our previous finding that the treatment of liposomes with MβCD specifically extracted both cholesterol and the membrane-inserted SFV E1* ectodomain (1). We triggered the insertion of SFV E1′ and DV2 E′ into cholesterol-containing liposomes at low pH, treated the membranes with MβCD, and quantitated the flotation of both the [3H]cholesterol marker and the viral protein. Treatment with MβCD efficiently extracted most of the cholesterol from proteoliposomes containing either E1′ or E′ proteins (Fig. 6B). As observed previously with SFV E1*, the SFV E1′ protein was quantitatively released from the membrane by MβCD treatment (Fig. 6A). In contrast, the majority of the membrane-inserted DV E′ protein remained associated with the membranes and floated in the gradient after MβCD extraction (Fig. 6A). Thus, although cholesterol promoted the initial membrane insertion of DV E′, the protein did not strongly associate with cholesterol-enriched membrane domains.
FIG. 6.
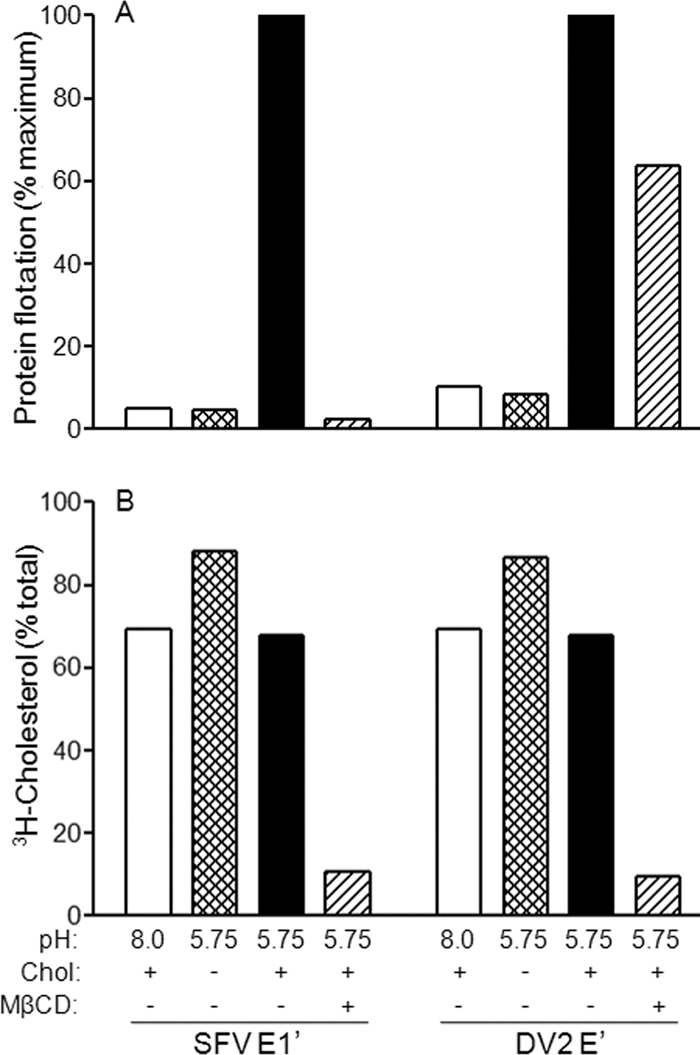
Membrane-inserted SFV fusion protein but not DV2 fusion protein associates with cholesterol-rich membrane microdomains. (A) Protein-liposome binding. SFV E1′ or DV2 E′ was mixed as indicated with liposomes containing 50 mol% cholesterol or cholesterol-deficient liposomes. Both liposome types contained trace amounts of [3H]cholesterol as a marker. The samples were treated at pH 8.0 or pH 5.75 for 30 min at 25°C, and samples were then extracted as indicated with 20 mM MβCD for 30 min at 37°C. All samples were then analyzed for protein-liposome binding by gradient flotation as for Fig. 4B. (B) Flotation of [3H]cholesterol. The percentage of [3H]cholesterol recovered in the top of the gradient was determined for each of the samples in Fig. 6A by liquid scintillation counting. Panels A and B represent the average of two independent experiments.
DISCUSSION
Differential cholesterol requirements of the flaviviruses and alphaviruses.
Taken together, our results reveal striking differences between the fusion reactions of members of the alphaviruses and flaviviruses. SFV and SIN fusions were strongly promoted by cholesterol, a property that was observed with both field isolates and viruses that had been extensively passaged in tissue culture. The membrane-inserted SFV fusion protein reacted with photocholesterol and was specifically released from membranes by cholesterol extraction. In contrast, membrane fusion and infection by a variety of DV isolates and YFV 17D were cholesterol independent. The membrane-inserted DV fusion protein did not react with photocholesterol and did not coextract with cholesterol. Thus, in spite of their structural similarities, our data reveal important mechanistic differences between these alphavirus and flavivirus fusion proteins. The specific cholesterol interaction properties of these proteins appear to be intrinsic features rather than the product of virus selection in tissue culture.
Virus-cell fusion experiments test the complete fusion reaction rather than the initial lipid mixing step. These experiments definitively demonstrated that DV does not require cholesterol in the target membrane for efficient membrane fusion, in keeping with the lack of E′ labeling by photocholesterol and with the resistance of the membrane-inserted E′ to extraction by MβCD. However, liposome coflotation assays showed that cholesterol strongly promoted the initial interaction of DV E′ with target liposomes (Fig. 6A). Previous liposome binding studies with the fusion protein ectodomain of TBE also demonstrated a cholesterol requirement (52). We propose that stable E′-membrane binding reflects formation of the homotrimer conformation, which would be anchored in the liposome membrane by the relatively shallow interactions of three fusion loops. In contrast, a single fusion loop would not be sufficient to mediate stable membrane interaction, a hypothesis that is supported by studies of monomeric TBE fusion proteins at pH 10 (53). In this model, cholesterol would promote the trimerization of E′ proteins simply by promoting their lateral clustering and concentration in the membrane, thus leading to increased trimerization and stable membrane interaction. In contrast, the E proteins on virus particles are already highly organized and concentrated (41), allowing the efficient interaction of monomers to form stable membrane-inserted trimers. Thus, protein clustering by cholesterol would not promote more efficient flavivirus-membrane fusion, in keeping with our flavivirus fusion results.
Our results clearly demonstrate efficient membrane fusion by DV in a cholesterol-depleted insect cell system, as scored by envelope protein production during primary virus infection. In contrast, the removal of cholesterol from mammalian cells by MβCD treatment had been reported to inhibit DV production (46). The mechanism of inhibition was recently addressed by two reports that came to different conclusions. Medigeshi et al. reported that pretreatment with MβCD inhibits endocytic uptake of WNV but that virus replication is unaffected by the postinfection depletion of cholesterol (38). However, studies of DV infection showed that cholesterol depletion by MβCD treatment causes only a mild inhibition of entry but a more severe inhibition of intracellular replication (28). These differences with our results could reflect differences in the properties of endocytosis between mammalian and insect cells, or differential requirements for cholesterol during virus replication in the two systems. It is also possible that the results reflect the general difficulties of cholesterol depletion in mammalian cells. Clearly, however, our studies with insect cells demonstrate that cholesterol is not required for entry and membrane fusion by DV and YFV.
Role of alphavirus-cholesterol interactions in vivo.
Our previous data on the srf-3 mutant showed that its increased cholesterol independence is due to a single point mutation of E1 proline 226 to serine (57). Selection for growth of SFV on cholesterol-depleted mosquito cells strongly favors the P226S mutation, which was independently isolated multiple times (6). Studies of SIN virus demonstrate that conversion of the SIN ij loop sequence to that of the srf-3 mutant also produces increased cholesterol independence for SIN (35). Other replacements, such as A226, also function in this context (35). E1 residue 226 is on the ij loop, adjacent to the fusion loop (47), and thus, these results suggest that the ij loop could act by regulating a cholesterol-dependent interaction of the fusion loop with the target membrane. These tissue culture studies are especially interesting in light of recent findings with CHIKV. During the CHIKV epidemic in Réunion between 2005 and 2006, virus strains with a point mutation of E1 alanine 226 to valine (A226V) were preferentially isolated (48). The predominant CHIKV vector during the epidemic was Aedes albopictus, the Asian tiger mosquito, and the A226V virus is better able to reproduce in this vector and to be transmitted to mammalian hosts (55, 58). The A226V mutation also makes the virus more cholesterol dependent (55). While the connection between vector adaptation and cholesterol dependence is not yet clear, these studies indicate that selection pressures in the wild can affect both of these alphavirus traits and produce important medical and epidemiological consequences.
Photocholesterol as a probe for membrane protein-cholesterol interactions.
The use of [3H]photocholesterol was pioneered in whole-cell studies and provided the first evidence that the synaptic vesicle membrane protein synaptophysin interacts with cholesterol (54). While a number of cellular membrane proteins are labeled with both photocholesterol and photoactivatable phospholipids, synaptophysin is preferentially labeled by photocholesterol, in agreement with the observed functional effects of cholesterol depletion on synaptophysin (54). Photocholesterol also preferentially labels a number of other cellular membrane proteins, including the known cholesterol-interacting protein caveolin (54); the Niemann-Pick type C1 protein, a membrane protein that is involved in cholesterol transport and contains a sterol sensing domain (42); and syntaxin, a SNARE protein involved in membrane fusion (27). The physical properties of photocholesterol in liposomes are similar to those of cholesterol (39), suggesting its potential for labeling studies in model membranes.
Here, we demonstrated photocholesterol labeling of membrane-inserted SFV E1 ectodomains using a liposome system. We also observed efficient labeling of PFO, a bacterial toxin whose membrane insertion is strongly cholesterol dependent (50, 56). In validation of this liposome system, we demonstrated that the DV E′ protein did not show significant labeling, even though the structure of this membrane-inserted fusion protein is very similar to that of SFV E1′. Perhaps most importantly, however, we showed that the hydrophobic TM domains of SFV E1 and E2 were not susceptible to labeling when reconstituted into liposomes containing photocholesterol. Thus, in this liposome system, we did not observe generalized photocholesterol labeling of protein regions located within the membrane. Similar to the photolabeling studies in cells, in our system, photoactivatable cholesterol represents only a small fraction of the total cholesterol in the liposome membrane. Labeling is thus observed in the presence of a large excess of unlabeled cholesterol, and the stoichiometry of protein-sterol interaction is at present unknown.
E1 cholesterol interaction site.
How might cholesterol affect the alphavirus E1 protein and the virus membrane fusion reaction? Studies of the stages of membrane fusion suggest that the primary role of cholesterol is to promote stable E1-membrane interaction (reviewed in reference 19). This could occur through a direct role of cholesterol in the initial insertion of the fusion loop into the target membrane or through subsequent stabilization of the fusion loop after its non-cholesterol-dependent insertion. We cannot currently differentiate between these two possible mechanisms. Cholesterol is clearly required for stable virus-membrane binding through the fusion loop (23), and the presence of cholesterol-containing membranes promotes low-pH-dependent conformational changes in E1 (7). The labeling of E1′ with photocholesterol and the efficient release of membrane-bound E1′ by extraction of membrane cholesterol strongly suggest that cholesterol directly binds to E1. Such binding could occur through the fusion loop, which is known to insert into the target membrane; through the ij loop, which is known to modulate virus cholesterol dependence; or through some combination of these E1 regions. Characterization of the binding site and its properties would help to define the mechanism of the cholesterol effect in alphavirus fusion and its possible differences in various virus strains in nature. Our studies suggest that protein-cholesterol interaction sites may be identified through the use of photocholesterol cross-linking.
Acknowledgments
We are grateful to David Silver for his helpful advice and input on the photocholesterol labeling and for providing us with photocholesterol. We thank Rod Tweten for providing purified PFO and for helpful advice; Aravinda de Silva, Richard Kinney, Richard Kuhn, John Roehrig, Richard Stockert, and Robert Tesh for providing virus strains; and Douglas Lyles, Milton Schlesinger, and Robert Tesh for providing antibodies. We also thank the members of our laboratory for many helpful discussions and suggestions, Anuja Ogirala for technical assistance, and Rod Tweten and the members of our laboratory for critical reading of the manuscript.
This work was supported by a grant to M.K. from the Public Health Service (R01 GM57454) and by Cancer Center Core Support Grant NIH/NCI P30-CA13330.
Footnotes
Published ahead of print on 16 July 2008.
REFERENCES
- 1.Ahn, A., D. L. Gibbons, and M. Kielian. 2002. The fusion peptide of Semliki Forest virus associates with sterol-rich membrane domains. J. Virol. 763267-3275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Allison, S. L., J. Schalich, K. Stiasny, C. W. Mandl, and F. X. Heinz. 2001. Mutational evidence for an internal fusion peptide in flavivirus envelope protein E. J. Virol. 754268-4275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Barrett, A. D., and S. Higgs. 2007. Yellow fever: a disease that has yet to be conquered. Annu. Rev. Entomol. 52209-229. [DOI] [PubMed] [Google Scholar]
- 4.Bressanelli, S., K. Stiasny, S. L. Allison, E. A. Stura, S. Duquerroy, J. Lescar, F. X. Heinz, and F. A. Rey. 2004. Structure of a flavivirus envelope glycoprotein in its low-pH-induced membrane fusion conformation. EMBO J. 23728-738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Brunner, J. 1993. New photolabeling and crosslinking methods. Annu. Rev. Biochem. 62483-514. [DOI] [PubMed] [Google Scholar]
- 6.Chatterjee, P. K., C. H. Eng, and M. Kielian. 2002. Novel mutations that control the sphingolipid and cholesterol dependence of the Semliki Forest virus fusion protein. J. Virol. 7612712-12722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chatterjee, P. K., M. Vashishtha, and M. Kielian. 2000. Biochemical consequences of a mutation that controls the cholesterol dependence of Semliki Forest virus fusion. J. Virol. 741623-1631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Clarke, T. 2002. Dengue virus: break-bone fever. Nature 416672-674. [DOI] [PubMed] [Google Scholar]
- 9.Cleverley, D. Z., H. M. Geller, and J. Lenard. 1997. Characterization of cholesterol-free insect cells infectible by baculoviruses: effects of cholesterol on VSV fusion and infectivity and on cytotoxicity induced by influenza M2 protein. Exp. Cell Res. 233288-296. [DOI] [PubMed] [Google Scholar]
- 10.Corver, J., A. Ortiz, S. L. Allison, J. Schalich, F. X. Heinz, and J. Wilschut. 2000. Membrane fusion activity of tick-borne encephalitis virus and recombinant subviral particles in a liposomal model system. Virology 26937-46. [DOI] [PubMed] [Google Scholar]
- 11.Crill, W. D., and G. J. Chang. 2004. Localization and characterization of flavivirus envelope glycoprotein cross-reactive epitopes. J. Virol. 7813975-13986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Enserink, M. 2007. Infectious diseases. Chikungunya: no longer a third world disease. Science 3181860-1861. [DOI] [PubMed] [Google Scholar]
- 13.Gibbons, D. L., M.-C. Vaney, A. Roussel, A. Vigouroux, B. Reilly, J. Lepault, M. Kielian, and F. A. Rey. 2004. Conformational change and protein-protein interactions of the fusion protein of Semliki Forest virus. Nature 427320-325. [DOI] [PubMed] [Google Scholar]
- 14.Gollins, S. W., and J. S. Porterfield. 1986. pH-dependent fusion between the flavivirus West Nile and liposomal model membranes. J. Gen. Virol. 67157-166. [DOI] [PubMed] [Google Scholar]
- 15.Gubler, D. J. 2002. Epidemic dengue/dengue hemorrhagic fever as a public health, social and economic problem in the 21st century. Trends Microbiol. 10100-103. [DOI] [PubMed] [Google Scholar]
- 16.Heinz, F. X., and S. L. Allison. 2000. Structures and mechanisms in flavivirus fusion. Adv. Virus Res. 55231-269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hiramatsu, K., M. Tadano, R. Men, and C. J. Lai. 1996. Mutational analysis of a neutralization epitope on the dengue type 2 virus (DEN2) envelope protein: monoclonal antibody resistant DEN2/DEN4 chimeras exhibit reduced mouse neurovirulence. Virology 224437-445. [DOI] [PubMed] [Google Scholar]
- 18.Ivy, J., E. Nakano, and D. Clements. December 2000. Subunit immunogenic composition against dengue infection. U.S. patent 6,165,477.
- 19.Kielian, M. 2006. Class II virus membrane fusion proteins. Virology 34438-47. [DOI] [PubMed] [Google Scholar]
- 20.Kielian, M. 1995. Membrane fusion and the alphavirus life cycle. Adv. Virus Res. 45113-151. [DOI] [PubMed] [Google Scholar]
- 21.Kielian, M., S. Jungerwirth, K. U. Sayad, and S. DeCandido. 1990. Biosynthesis, maturation, and acid-activation of the Semliki Forest virus fusion protein. J. Virol. 644614-4624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kielian, M., M. R. Klimjack, S. Ghosh, and W. A. Duffus. 1996. Mechanisms of mutations inhibiting fusion and infection by Semliki Forest virus. J. Cell Biol. 134863-872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kielian, M. C., and A. Helenius. 1984. The role of cholesterol in the fusion of Semliki Forest virus with membranes. J. Virol. 52281-283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kinney, R. M., S. Butrapet, G. J. Chang, K. R. Tsuchiya, J. T. Roehrig, N. Bhamarapravati, and D. J. Gubler. 1997. Construction of infectious cDNA clones for dengue 2 virus: strain 16681 and its attenuated vaccine derivative, strain PDK-53. Virology 230300-308. [DOI] [PubMed] [Google Scholar]
- 25.Klimjack, M. R., S. Jeffrey, and M. Kielian. 1994. Membrane and protein interactions of a soluble form of the Semliki Forest virus fusion protein. J. Virol. 686940-6946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Klimstra, W. B., K. D. Ryman, and R. E. Johnston. 1998. Adaptation of Sindbis virus to BHK cells selects for use of heparan sulfate as an attachment receptor. J. Virol. 727357-7366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lang, T., D. Bruns, D. Wenzel, D. Riedel, P. Holroyd, C. Thiele, and R. Jahn. 2001. SNAREs are concentrated in cholesterol-dependent clusters that define docking and fusion sites for exocytosis. EMBO J. 202202-2213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lee, C. J., H. R. Lin, C. L. Liao, and Y. L. Lin. 2008. Cholesterol effectively blocks entry of flavivirus. J. Virol. 826470-6480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lefrancois, L., and D. S. Lyles. 1982. The interaction of antibody with the major surface glycoprotein of vesicular stomatitis virus. I. Analysis of neutralizing epitopes with monoclonal antibodies. Virology 121157-167. [PubMed] [Google Scholar]
- 30.Lehr, R. V., L. C. Elefante, K. K. Kikly, S. P. O'Brien, and R. B. Kirkpatrick. 2000. A modified metal-ion affinity chromatography procedure for the purification of histidine-tagged recombinant proteins expressed in Drosophila S2 cells. Protein Expr. Purif. 19362-368. [DOI] [PubMed] [Google Scholar]
- 31.Lescar, J., A. Roussel, M. W. Wien, J. Navaza, S. D. Fuller, G. Wengler, and F. A. Rey. 2001. The fusion glycoprotein shell of Semliki Forest virus: an icosahedral assembly primed for fusogenic activation at endosomal pH. Cell 105137-148. [DOI] [PubMed] [Google Scholar]
- 32.Liao, M., and M. Kielian. 2005. The conserved glycine residues in the transmembrane domain of the Semliki Forest virus fusion protein are not required for assembly and fusion. Virology 332430-437. [DOI] [PubMed] [Google Scholar]
- 33.Liao, M., and M. Kielian. 2005. Domain III from class II fusion proteins functions as a dominant-negative inhibitor of virus-membrane fusion. J. Cell Biol. 171111-120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Liljeström, P., S. Lusa, D. Huylebroeck, and H. Garoff. 1991. In vitro mutagenesis of a full-length cDNA clone of Semliki Forest virus: the small 6,000-molecular-weight membrane protein modulates virus release. J. Virol. 654107-4113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lu, Y. E., T. Cassese, and M. Kielian. 1999. The cholesterol requirement for Sindbis virus entry and exit and characterization of a spike protein region involved in cholesterol dependence. J. Virol. 734272-4278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mandl, C. W., H. Kroschewski, S. L. Allison, R. Kofler, H. Holzmann, T. Meixner, and F. X. Heinz. 2001. Adaptation of tick-borne encephalitis virus to BHK-21 cells results in the formation of multiple heparan sulfate binding sites in the envelope protein and attenuation in vivo. J. Virol. 755627-5637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Marquardt, M. T., and M. Kielian. 1996. Cholesterol-depleted cells that are relatively permissive for Semliki Forest virus infection. Virology 224198-205. [DOI] [PubMed] [Google Scholar]
- 38.Medigeshi, G. R., A. J. Hirsch, D. N. Streblow, J. Nikolich-Zugich, and J. A. Nelson. 2008. West Nile virus entry requires cholesterol-rich membrane microdomains and is independent of αvβ3 integrin. J. Virol. 825212-5219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mintzer, E. A., B. L. Waarts, J. Wilschut, and R. Bittman. 2002. Behavior of a photoactivatable analog of cholesterol, 6-photocholesterol, in model membranes. FEBS Lett. 510181-184. [DOI] [PubMed] [Google Scholar]
- 40.Modis, Y., S. Ogata, D. Clements, and S. C. Harrison. 2003. A ligand-binding pocket in the dengue virus envelope glycoprotein. Proc. Natl. Acad. Sci. USA 1006986-6991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mukhopadhyay, S., R. J. Kuhn, and M. G. Rossmann. 2005. A structural perspective of the flavivirus life cycle. Nat. Rev. Microbiol. 313-22. [DOI] [PubMed] [Google Scholar]
- 42.Ohgami, N., D. C. Ko, M. Thomas, M. P. Scott, C. C. Chang, and T. Y. Chang. 2004. Binding between the Niemann-Pick C1 protein and a photoactivatable cholesterol analog requires a functional sterol-sensing domain. Proc. Natl. Acad. Sci. USA 10112473-12478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Phalen, T., and M. Kielian. 1991. Cholesterol is required for infection by Semliki Forest virus. J. Cell Biol. 112615-623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Polo, S., G. Ketner, R. Levis, and B. Falgout. 1997. Infectious RNA transcripts from full-length dengue virus type 2 cDNA clones made in yeast. J. Virol. 715366-5374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rey, F. A., F. X. Heinz, C. Mandl, C. Kunz, and S. C. Harrison. 1995. The envelope glycoprotein from tick-borne encephalitis virus at 2 Å resolution. Nature 375291-298. [DOI] [PubMed] [Google Scholar]
- 46.Reyes-Del Valle, J., S. Chavez-Salinas, F. Medina, and R. M. Del Angel. 2005. Heat shock protein 90 and heat shock protein 70 are components of dengue virus receptor complex in human cells. J. Virol. 794557-4567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Roussel, A., J. Lescar, M.-C. Vaney, G. Wengler, G. Wengler, and F. A. Rey. 2006. Structure and interactions at the viral surface of the envelope protein E1 of Semliki Forest virus. Structure 1475-86. [DOI] [PubMed] [Google Scholar]
- 48.Schuffenecker, I., I. Iteman, A. Michault, S. Murri, L. Frangeul, M. C. Vaney, R. Lavenir, N. Pardigon, J. M. Reynes, F. Pettinelli, L. Biscornet, L. Diancourt, S. Michel, S. Duquerroy, G. Guigon, M. P. Frenkiel, A. C. Brehin, N. Cubito, P. Despres, F. Kunst, F. A. Rey, H. Zeller, and S. Brisse. 2006. Genome microevolution of chikungunya viruses causing the Indian Ocean outbreak. PLoS Med. 3e263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Smit, J. M., R. Bittman, and J. Wilschut. 1999. Low-pH-dependent fusion of Sindbis virus with receptor-free cholesterol- and sphingolipid-containing liposomes. J. Virol. 738476-8484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Soltani, C. E., E. M. Hotze, A. E. Johnson, and R. K. Tweten. 2007. Structural elements of the cholesterol-dependent cytolysins that are responsible for their cholesterol-sensitive membrane interactions. Proc. Natl. Acad. Sci. USA 10420226-20231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Stiasny, K., S. Brandler, C. Kossl, and F. X. Heinz. 2007. Probing the flavivirus membrane fusion mechanism by using monoclonal antibodies. J. Virol. 8111526-11531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Stiasny, K., C. Koessl, and F. X. Heinz. 2003. Involvement of lipids in different steps of the flavivirus fusion mechanism. J. Virol. 777856-7862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Stiasny, K., C. Kossl, J. Lepault, F. A. Rey, and F. X. Heinz. 2007. Characterization of a structural intermediate of flavivirus membrane fusion. PLoS Pathog. 3e20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Thiele, C., M. J. Hannah, F. Fahrenholz, and W. B. Huttner. 2000. Cholesterol binds to synaptophysin and is required for biogenesis of synaptic vesicles. Nat. Cell Biol. 242-49. [DOI] [PubMed] [Google Scholar]
- 55.Tsetsarkin, K. A., D. L. Vanlandingham, C. E. McGee, and S. Higgs. 2007. A single mutation in chikungunya virus affects vector specificity and epidemic potential. PLoS Pathog. 3e201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Tweten, R. K. 2005. Cholesterol-dependent cytolysins, a family of versatile pore-forming toxins. Infect. Immun. 736199-6209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Vashishtha, M., T. Phalen, M. T. Marquardt, J. S. Ryu, A. C. Ng, and M. Kielian. 1998. A single point mutation controls the cholesterol dependence of Semliki Forest virus entry and exit. J. Cell Biol. 14091-99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Vazeille, M., S. Moutailler, D. Coudrier, C. Rousseaux, H. Khun, M. Huerre, J. Thiria, J. S. Dehecq, D. Fontenille, I. Schuffenecker, P. Despres, and A. B. Failloux. 2007. Two Chikungunya isolates from the outbreak of La Reunion (Indian Ocean) exhibit different patterns of infection in the mosquito, Aedes albopictus. PLoS ONE 2e1168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wang, N., D. L. Silver, C. Thiele, and A. R. Tall. 2001. ATP-binding cassette transporter A1 (ABCA1) functions as a cholesterol efflux regulatory protein. J. Biol. Chem. 27623742-23747. [DOI] [PubMed] [Google Scholar]
- 60.White, J., and A. Helenius. 1980. pH-dependent fusion between the Semliki Forest virus membrane and liposomes. Proc. Natl. Acad. Sci. USA 773273-3277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zhang, Y., W. Zhang, S. Ogata, D. Clements, J. H. Strauss, T. S. Baker, R. J. Kuhn, and M. G. Rossmann. 2004. Conformational changes of the flavivirus E glycoprotein. Structure (Cambridge) 121607-1618. [DOI] [PMC free article] [PubMed] [Google Scholar]