

Abstract
Human parainfluenza virus type 3 (HPIV3) is a major respiratory pathogen in humans. Failure to induce immunological memory associated with HPIV3 infection has been attributed to inhibition of lymphocyte proliferation. We demonstrate that the inability of mixed lymphocytes to respond to virally infected antigen-presenting cells is due to an interleukin-2-dependent, nonapoptotic mechanism involving natural killer (NK) cells and their influence is exerted in a contact-dependent manner. These results suggest a novel regulatory mechanism for NK cells during HPIV3 infection, offering an explanation for viral persistence and poor memory responses.
Human parainfluenza virus type 3 (HPIV3) is a major respiratory pathogen responsible for pneumonia, croup, and bronchiolitis in infants and children (7, 9, 13). Reinfection is a common occurrence (13, 15). This clinical characteristic is distinct from infection with most viral infections, such as measles, mumps (15), and subtype-specific influenza (6), which, in most cases, result in lifelong immunity against reinfection with the same strain. Even in the presence of circulating neutralizing antibodies, individuals have been shown to be susceptible to reinfection (3). This feature of HPIV3 has resulted in failure to design an effective vaccine against this major pathogen. It has been suggested that HPIV3 can reinfect and persist in a human host due to its ability to inhibit T-cell proliferation (12, 15). However, mechanisms underlying this reduced proliferating capacity remain poorly defined (12, 15, 16). In this study, we investigate the role of NK cells in T-cell proliferation during HPIV3 infection and describe a novel control mechanism involving NK cells, a population largely overlooked in the milieu of human immune regulation.
It has been shown that HPIV3 inhibits proliferation in anti-CD3 activated peripheral blood mononuclear cell (PBMC) cultures (15). We examined the influence of the cell subsets in the PBMCs on the ability of HPIV3 to inhibit T-cell proliferation. To explore the role of antigen-presenting cells (APCs), we investigated proliferation of mixed lymphocytes or purified CD3+ T lymphocytes to allogeneic HPIV3-infected APCs.
CD14+ monocytes were purified from PBMCs (obtained from buffy coats of healthy donors from the Irish Blood Transfusion Service) to >90% using an antibody/magnetic cell sorter (MACS) magnetic bead separation protocol. These cells (2 × 105/ml) were cultured in RPMI (with 10% fetal calf serum, 1% l-glutamine, and 1% penicillin/streptomycin) and were untreated (medium), treated with tumor necrosis factor alpha (TNF-α) (as a positive stimulus), infected with HPIV3 (50% tissue culture infective dose [TCID50]/ml of 6) or influenza virus (a respiratory virus associated with acute rather than persistent infection [TCID50/ml of 7]) for 2 h at 37°C. Cells were washed and irradiated. To ensure cells incubated with virus were indeed infected, we identified expression of HPIV3/influenza virus nucleoprotein (NP) through reverse transcriptase PCR in the relevant cells (data not shown). CD14− mixed lymphocytes or CD3+ T cells were isolated from a different donor using appropriate MACS separation columns. Mixed lymphocytes or CD3+ T cells (2 × 106/ml) were added to the infected or treated CD14+ cells described above and incubated at 37°C for 5 days. Additional mixed-lymphocyte or T-cell-APC cocultures were activated with phorbol myristate acetate (PMA) and anti-CD3 as a positive control. Proliferation was evaluated using a [3H]thymidine assay, by adding 2.5 μCi/well [3H]thymidine to the cocultures for the last 5 h of incubation. Cells were harvested onto filter mats and analyzed on a scintillation counter. Human interleukin-10 (IL-10), IL-2, and gamma interferon (IFN-γ) enzyme-linked immunosorbent assay kits (R&D) were used to quantify cytokine levels in supernatants.
We have previously demonstrated strong maturation of APCs to IFN-like dendritic cells (DCs), following HPIV infection, as indicated by the presence of CD83, CD40, CD80, CD86, and major histocompatibility complex class II markers (10). In keeping with an earlier study (15), we found HPIV3-infected APCs failed to induce allogeneic lymphocyte proliferation compared with influenza virus-infected, TNF-α-treated, or unstimulated CD14+ cells. Strikingly, proliferation of CD3+ purified T cells cultured with HPIV3-infected APCs was not lower than those observed with influenza virus-infected or TNF-α-treated samples (Fig. 1A), indicating that a cell subtype in the CD14− mixed lymphocytes had a role in HPIV3 inhibition of T-cell proliferation.
FIG. 1.

Mixed-lymphocyte and purified CD3 T-cell proliferation and IL-10 cytokine profiles. Mixed lymphocytes were cultured with medium alone, with allogeneic APCs alone, with APCs plus TNF-α, with APCs infected with HPIV3, with APCs infected with influenza virus (Flu), or with PMA and anti-CD3 (aCD3) alone. (A) Proliferation profiles of mixed lymphocytes or purified T cells cocultured with APCs. *, P < 0.05. (B) IL-10 levels in supernatants from mixed lymphocytes or purified T cells cocultured with APCs. *, P < 0.05.
In contrast to previous studies (15, 16), IL-10 was not significantly induced in lymphocytes stimulated by HPIV3-infected APCs compared to influenza virus infection or TNF-α treatment (Fig. 1B). These results suggest that failure of lymphocytes to proliferate to HPIV3-infected DCs is not due to IL-10 but also points toward the presence of a regulatory group of cells in the mixed-lymphocyte population. To investigate this, we isolated CD3− CD14− cells and restored them to the CD3+ cocultures. Addition of these cells significantly reduced CD3+ T-cell proliferation in HPIV3-infected cells (Fig. 2A). This was not observed in TNF-α-treated or influenza virus-infected cells (data not shown). To establish if this interaction was contact dependent, we repeated the experiments using transwells. CD14+ monocytes were cultured with allogeneic CD14− CD3− cells in the upper chamber of an eight-well strip insert with 0.2-μm pores on a 96-well plate. In the lower chamber, 1 × 105 CD3+ T cells were cultured with 1 × 104 monocytes. Cultures were incubated for 5 days and analyzed for proliferation levels as described previously.
FIG. 2.

Inhibition of lymphocyte proliferation is mediated by CD14− CD3− cells in a contact-dependent manner. (A) Purified T cells were cocultured with medium alone, with allogeneic APCs infected with HPIV, with APCs infected with HPIV, and with CD14− CD3− cells. Results are representative of three experiments. aCD3, anti-CD3; FLU, influenza virus. *, P < 0.05. (B) In the same set of experiments using the same donor cell population as for panel A, T cells were cocultured with APCs and CD14− CD3− cells in transwells alone, with TNF-α, with HPIV3, or with influenza virus. Results are representative of three experiments.
Separation of the cells restored T-cell proliferation (Fig. 2B). These results suggest inhibition of T-cell proliferation is not due to regulatory cytokines but to a contact-dependent mechanism by the CD3− CD14− cell population in the lymphocyte culture.
Earlier studies suggested HPIV3-induced apoptosis of APCs may have a role in poor memory response to HPIV3 (12). However, in a recent study we observed considerably lower levels of apoptotic cells induced by HPIV3 infection of CD14+ monocytes compared with HPIV3 infection of IL-4-generated APCs (10). Consistent with this, allogeneic mixed lymphocytes cocultured with HPIV3-infected CD14+ cells did not demonstrate significant apoptosis compared to influenza virus-infected cultures (Fig. 3A). One reason for the contrast between our findings and those of a previous study (12) is that we used directly infected monocytes, while they used artificially generated IL-4-producing DCs. We have recently shown that artificially generated DCs skew immune responses to infections. Furthermore, we have demonstrated that artificially generated IL-4 DCs were more susceptible to apoptosis than directly infected monocytes or IFN-α DCs (10).
FIG. 3.

Inhibition of lymphocyte proliferation is not mediated by apoptosis but is dependent on IL-2. (A) Apoptosis of mixed lymphocytes was measured by annexin V-propidium iodide staining 5 days post-coculture with allogeneic APCs with medium alone, with TNF-α, with HPIV3, or with influenza virus. Lymphocyte populations were gated, and percentages of early and late apoptotic cells are shown in the lower right quadrant and upper right quadrant, respectively. Results are representative of three experiments. (B) IL-2 levels in the supernatants from cocultures described above. FLU, influenza virus. *, P < 0.05. (C) Lymphocyte proliferation with APCs with medium alone, with APCs plus TNF-α, with APCs plus HPIV3 (PIV), or with HPIV3-infected APCs with IL-2. *, P < 0.05.
As IL-2 is a crucial growth factor for T cells (5), levels of this cytokine in supernatants of HPIV3-infected DC-allogeneic mixed-lymphocyte cocultures were examined. We found lymphocyte IL-2 production was severely impaired in HPIV3-infected cocultures compared to influenza virus or TNF-α samples (Fig. 3B). Moreover, addition of IL-2 to these cultures restored the proliferating capacity of these cells (Fig. 3C), emphasizing the essential role of IL-2 in T-cell proliferation during HPIV3 infections.
The CD14− CD3− populations consist of a heterogenous group with CD19+ (B cells) and CD56+ (NK cells) predominating. We examined surface markers from uninfected and HPIV3-infected CD3− CD14− cells to assess variations in the phenotypic profiles from different cell populations. The only difference observed between uninfected and infected cells was an upregulation of the NK lineage marker, CD56, from the HPIV3-infected cells (data not shown).
In humans, several autoimmune disorders such as multiple sclerosis, rheumatoid arthritis, and systemic lupus erythematosus are associated with decreased NK activity (4, 8, 14). Other reports have demonstrated that NK cells, particularly those high in CD56, play a role in the regulation of T cells (2, 17). Therefore, we investigated the potential of NK cells to inhibit T-cell proliferation from the HPIV3-infected cocultures. Addition of these CD56+ NK cells to purified CD3+ T-cell cocultures significantly reduced T-cell proliferation from HPIV3-infected cocultures compared to influenza virus-infected, TNF-α-treated, or untreated cocultures (Fig. 4). This effect was not observed following addition of purified CD19+ cells (data not shown). These results strongly suggest that NK cells from this CD3− CD14− population play a role in T-cell inhibition from HPIV3-infected mixed-lymphocyte cocultures. To further investigate this, we examined the cell cycling of T cells by propidium iodide staining. This identified that T cells are retained in the G0/G1 phase of the cell cycle in HPIV3-infected cultures compared to influenza virus-infected, TNF-α-treated, or untreated cells. Normal cell cycle status is restored to these cultures following NK depletion (data not shown).
FIG. 4.
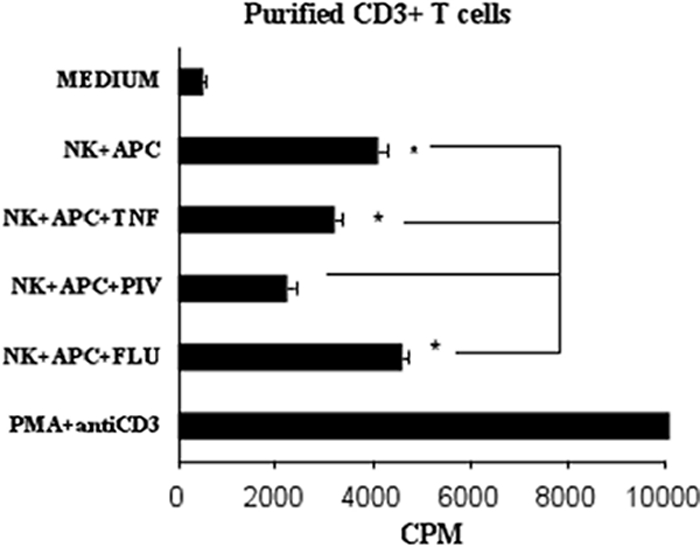
NK cells reduce proliferation of purified CD3+ T cells from HPIV3-infected cocultures. Purified T cells were cocultured with medium alone, with APCs and NK cells alone or with TNF-α, with HPIV3 (PIV), or with influenza virus (FLU), or T cells were treated with PMA and anti-CD3. Results are representative of two experiments. *, P < 0.05.
This study has identified that the failure of HPIV3 to induce T-cell proliferation in allogeneic cocultures can be restored by purification of the CD3+ T cells from the mixed-lymphocyte population. We have also demonstrated that the inability of mixed lymphocytes to respond to virally infected DCs was dependent on IL-2 levels and not due to the induction of apoptosis. Finally, we show that the NK cells in the CD14− CD3− component of the mixed lymphocytes are responsible for the inhibition of T-cell proliferation and their influence is exerted in a contact-dependent manner. Current thinking on immune regulation suggests that, in addition to the control of responses to self, regulatory mechanisms exist which limit collateral damage during infection and that this response often comes with inappropriate immune activation, resulting in chronic or persistent infections (1, 11). We hypothesize that NK cells regulate proliferation of overactive T cells. This prevents bystander immune activation and limits immune damage, the ultimate price being viral persistence and poor memory responses. These findings demonstrate immune regulation by NK cells during viral infection, revealing the complex human immune response to danger. Furthermore, these findings suggest that this mechanism may be more widely employed by other immunosuppressive viruses, as a means of subverting immune responses.
Acknowledgments
This work was financially supported by the European Union 5th Framework Program (EU contract no. QLK2-CT-2002-01722) and Dublin City University.
We also acknowledge the assistance of Grazia Cusi, University of Siena, Italy, and Wolfgang Neubert, Max Plank Institute, Munich, Germany.
Footnotes
Published ahead of print on 9 July 2008.
REFERENCES
- 1.Accapezzato, D., V. Francavilla, M. Paroli, M. Casciaro, L. V. Chircu, A. Cividini, S. Abrignani, M. U. Mondelli, and V. Barnaba. 2004. Hepatic expansion of a virus-specific regulatory CD8(+) T cell population in chronic hepatitis C virus infection. J. Clin. Investig. 113963-972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bielekova, B., M. Catalfamo, S. Reichert-Scrivner, A. Packer, M. Cerna, T. Waldmann, H. McFarland, P. Henkart, and R. Martin. 2006. Regulatory CD56bright natural killer cells mediate immunomodulatory effects of IL-2Rα-targeted therapy (daclizumab) in multiple sclerosis. Proc. Natl. Acad. Sci. USA 1035941-5946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bloom, H., K. Johnson, R. Jacobsen, and R. Chanocj. 1961. Recovery of parainfluenza viruses from adults with upper respiratory illness. Am. J. Hyg. 7450-59. [Google Scholar]
- 4.Combe, B., R. Pope, B. Darnell, and N. Talal. 1984. Modulation of natural killer cell activity in the rheumatoid joint and peripheral blood. Scand. J. Immunol. 20551-558. [DOI] [PubMed] [Google Scholar]
- 5.Gaffen, S. L. and K. D Liu. 2004. Overview of interleukin-2 function, production and clinical applications. Cytokine 28109-123. [DOI] [PubMed] [Google Scholar]
- 6.Gorse, G., T. O'Connor, F. Newman, M. Mandava, P. Mendelman, J. Wittes, and P. Peduzzi. 2004. Immunity to influenza in older adults with chronic obstructive pulmonary disease. J. Infect. Dis. 19011-19. [DOI] [PubMed] [Google Scholar]
- 7.Henrickson, K. J. 2003. Parainfluenza viruses. Clin. Microbiol. Rev. 16242-264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kastrukoff, L. F., A. Lau, R. Wee, D. Zecchini, R. White, and D. W. Paty. 2003. Clinical relapses of multiple sclerosis are associated with ‘novel’ valleys in natural killer cell functional activity. J. Neuroimmunol. 145103-114. [DOI] [PubMed] [Google Scholar]
- 9.Lee, M., D. Greenberg, S. Yeh, R. Yogev, K. Reisinger, J. Ward, M. Blatter, I. Cho, S. Holmes, J. Cordova, M. August, W. Chen, H. Mehta, K. Coelingh, and P. Mendelman. 2001. Antibody responses to bovine parainfluenza virus type 3 (PIV3) vaccination and human PIV3 infection in young infants. J. Infect. Dis. 184909-913. [DOI] [PubMed] [Google Scholar]
- 10.Noone, C., E. Manahan, R. Newman, and P. Johnson. 2007. Artificially generated dendritic cells misdirect antiviral immune responses. J. Leukoc. Biol. 81952-956. [DOI] [PubMed] [Google Scholar]
- 11.O'Garra, A., P. L. Vieira, P. Vieira, and A. E. Goldfeld. 2004. IL-10-producing and naturally occurring CD4+ Tregs: limiting collateral damage. J. Clin. Investig. 1141372-1378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Plotnicky-Gilquin, H., D. Cyblat, J. P. Aubry, Y. Delneste, A. Blaecke, J. Y. Bonnefoy, N. Corvaia, and P. Jeannin. 2001. Differential effects of parainfluenza virus type 3 on human monocytes and dendritic cells. Virology 28582-90. [DOI] [PubMed] [Google Scholar]
- 13.Schmidt, A. C., R. B. Couch, G. J. Galasso, F. G. Hayden, J. Mills, B. R. Murphy, and R. M. Chanock. 2001. Current research on respiratory viral infections: Third International Symposium. Antiviral Res. 50157-196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sibbitt, W. L., Jr., P. M. Mathews, and A. D. Bankhurst. 1983. Natural killer cell in systemic lupus erythematosus. Defects in effector lytic activity and response to interferon and interferon inducers. J. Clin. Investig. 711230-1239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sieg, S., C. Muro-Cacho, S. Robertson, Y. Huang, and D. Kaplan. 1994. Infection and immunoregulation of T lymphocytes by parainfluenza virus type 3. Proc. Natl. Acad. Sci. USA 916293-6297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sieg, S., C. King, Y. Huang, and D. Kaplan. 1996. The role of interleukin-10 in the inhibition of T-cell proliferation and apoptosis mediated by parainfluenza virus type 3. J. Virol. 704845-4848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Trivedi, P. P., P. C. Roberts, N. A. Wolf, and R. H. Swanborg. 2005. NK cells inhibit T cell proliferation via p21-mediated cell cycle arrest. J. Immunol. 1744590-4597. [DOI] [PubMed] [Google Scholar]