

Abstract
CD4+ T-cell depletion is the hallmark of AIDS pathogenesis. Multiple mechanisms may contribute to the death of productively infected CD4+ T cells and innocent-bystander cells. In this study, we characterize a novel mechanism in which human immunodeficiency virus type 1 (HIV-1) infection preferentially depletes peripheral memory CD4+ T cells before the completion of reverse transcription. Using a recombinant HIV-1 carrying the green fluorescent protein reporter gene, we demonstrate that memory CD4+ T cells were susceptible to infection-induced cell death at a low multiplicity of infection. Infected memory CD4+ T cells underwent rapid necrotic cell death. Killing of host cells was dependent on X4 envelope-mediated viral fusion, but not on virion-associated Vpr or Nef. In contrast to peripheral resting CD4+ T cells, CD4+ T cells stimulated by mitogen or certain cytokines were resistant to HIV-1-induced early cell death. These results demonstrate that early steps in HIV-1 infection have a detrimental effect on certain subsets of CD4+ T cells. The early cell death may serve as a selective disadvantage for X4-tropic HIV-1 in acute infection but may play a role in accelerated disease progression, which is associated with the emergence of X4-tropic HIV-1 in the late stage of AIDS.
Human immunodeficiency virus type 1 (HIV-1) is the causative agent of AIDS. CD4+ T cells are the main source of virus production and endure progressive depletion over the course of infection (21). While the CD4+ T-cell count serves as one of the major clinical predictors of disease progression, the mechanisms involved in CD4+ T-cell depletion are not clearly understood. HIV-1 infection of activated CD4+ T cells in vitro results in cell death. Expression of several different HIV-1 proteins, including protease, Vif, Vpr, Vpu, Env, Tat, and Nef, has been reported to induce apoptosis or enhance host cell response to apoptotic signals (19). However, most CD4+ T cells destined to die in the lymph nodes of patients with chronic HIV-1 infection and in simian immunodeficiency virus (SIV)-infected rhesus macaques are not productively infected (17). Even in acute HIV-1 and SIV infection, when more than 50% of CD4+ T cells in gastrointestinal lamina propria are depleted (20, 38, 41, 43), only 7% of gastrointestinal CD4+ T cells are found to express HIV-1 RNA at levels detectable by in situ hybridization (38), although a larger fraction have detectable HIV-1 DNA (41). Therefore, CD4+ T-cell death can be dissociated from productive HIV-1 infection and subsequent viral-gene expression.
Multiple indirect killing mechanisms have been proposed to result in HIV-1-induced CD4+ T-cell depletion. Increased T-cell turnover and chronic immune activation play important roles in a host's failure to maintain CD4+ T-cell homeostasis (13, 33, 42, 45, 60). Upregulation of tumor necrosis factor alpha, Fas ligand, and tumor necrosis factor-related apoptosis-inducing ligand (TRAIL), as well as increased sensitivity of CD4+ T cells to death ligand-mediated apoptosis, contributes to the demise of uninfected CD4+ T cells (3, 12, 27, 73). In addition, HIV-1 Tat and Vpr have also been proposed to act in trans to affect the viability of uninfected CD4+ T cells (29, 37). Extensive in vitro studies have revealed that extracellular HIV-1 Env triggers cell death through a variety of mechanisms (1, 55), including signaling through CD4 and coreceptor (4, 72), syncytium formation (39, 54), and induction of autophagy of uninfected CD4+ T cells (14).
While many studies aimed at understanding CD4+ T-cell depletion in AIDS have focused on uninfected CD4+ T cells, less is known about the fate of HIV-1-infected CD4+ T cells that do not express viral genes. Several groups have addressed the question in the setting of nonproductive infection with defective virions, which are estimated to constitute 99.9% of HIV-1 particles in plasma (56, 61). Esser et al. demonstrated that aldrithiol 2-inactivated HIV-1 particles, but not recombinant HIV-1 gp120, trigger CD4+ T-cell death when added to peripheral blood mononuclear cells (PBMCs) (15). It has been proposed that aldrithiol 2 HIV-1-treated plasmacytoid dendritic cells produce type I interferon (IFN) and stimulate CD4+ T cells to express TRAIL (26). An alternative hypothesis is that noninfectious viral particles may directly kill infected CD4+ T cells. However, studies that tested this hypothesis in activated CD4+ T cells with UV-inactivated HIV-1 have yielded inconsistent results (28, 35). Interpretation of these data is complicated by the differences between experimental systems, particularly the presence of multiple immune cell types and a prolonged culture period, which increases the susceptibility of primary CD4+ T cells to cell death. How HIV-1 infection affects host CD4+ T cells in the absence of viral replication remains a subject of controversy.
HIV-1 infection of resting CD4+ T cells from peripheral blood does not lead to viral-gene expression due to blocks at multiple steps prior to integration (64, 75). In chronically infected individuals, the predominant form of HIV-1 in CD4+ T cells is unintegrated virus, which provides an inducible latent reservoir (7, 11, 63). In previous studies, we demonstrated that preintegration latency reflects a complex set of competing processes, including slow reverse transcription in resting cells, the decay of rescuable virus before and after the completion of reverse transcription, and the virus-induced death of host cells (58, 76). We reported that resting peripheral blood CD4+ T cells infected by recombinant HIV-1 die more quickly than mock-infected CD4+ T cells (76). In this study, we extend those findings and demonstrate that resting CD4+ T cells, particularly memory CD4+ T cells, are susceptible to HIV-1-induced cytolysis in the absence of viral-gene expression, while CD4+ T cells stimulated by mitogen are not. We further demonstrate that rapid memory CD4+ T-cell death depends on X4 envelope-mediated HIV-1 fusion. Furthermore, cytokines modulate the susceptibility of memory CD4+ T cells to HIV-1-induced death. We propose a new mechanism of X4 HIV-1-induced T-cell death that may play a role in AIDS pathogenesis.
MATERIALS AND METHODS
Isolation and culture of primary human CD4+ T cells.
Human PBMCs were isolated from the blood of healthy donors by Ficoll-Paque (Amersham Pharmacia) density centrifugation. Memory and naïve CD4+ T cells were further isolated from fresh PBMCs with negative-selection kits for memory and naïve CD4+ T cells, respectively (Miltenyi Biotec) and cultured at 106 cells/ml in RPMI supplemented with 10% heat-inactivated fetal bovine serum (Gemini) (culture medium). In some experiments, the following cytokines (R&D) were added to the culture medium of memory CD4+ T cells 48 h before the cells were infected: interleukin 2 (IL-2) (20 ng/ml), IL-7 (20 ng/ml), IL-15 (20 ng/ml), IFN-α (1,000 U/ml), IFN-β (1000 U/ml), and MIP-1α (20 ng/ml). CD4+ T-cell activation was achieved though stimulation of PBMCs with 0.5 μg/ml phytohemagglutinin (PHA) for 3 days. Activated CD4+ T cells were isolated using a negative-selection kit for CD4+ T cells (Miltenyi Biotec) and cultured in RPMI supplemented with 10% heat-inactivated fetal bovine serum, 100 U/ml recombinant human IL-2, and 2% supernatant from activated PBMCs.
Generation of retroviral vectors.
The HIV-1-based reporter construct NL4-3-green fluorescent protein (GFP) was used to generate recombinant virus for infection (76). In this construct, the GFP coding sequence is inserted in frame into the Env coding region, along with an endoplasmic reticulum retention signal to retain the Env-GFP fusion protein in the endoplasmic reticulum. A protease mutant reporter virus, D25N, was constructed by introducing a GAT→AAT mutation in the protease active site using a Quickchange II site-directed mutagenesis kit (Stratagene) with the following PCR primers: sense, 5′-GGCAATTAAAGGAAGCTCTATTAAATACAGGAGCAGATGATACAG-3′; antisense, 5′-CTGTATCATCTGCTCCTGTATTTAATAGAGCTTCCTTTAATTGCC-3′. To generate a Nef− Vpr− mutant vector, a deletion of a 204-nucleotide fragment including the first 72 nucleotides of nef was achieved by the ligation of the HapI/BlpI-restricted NL4-3-GFP backbone after it had been treated with mung bean nuclease at 37°C for 20 s. An A→G mutation at the start codon of Vpr was introduced by PCR-mediated mutagenesis with two sets of primers: (i) 5303R, 5′-GGAGTCTCCATAGAATGGAGGAAA-3′, and ATG mutant L, 5′-CTGGGGCTTGTTCCACCTGTCCTCTGTCAGTTTCCTAACA-3′; (ii) ATG mutant R, 5′-GACAGGTGGAACAAGCCCCAGAAGACCAA-3′, and 5744L, 5′-GCAGAATTCTTATTATGGCTTCCACT-3′. All point mutations were verified by sequencing.
Reporter virus particles coated with HIV-1 envelope protein were generated by transfecting 293T cells with an appropriate proviral vector and X4 HIV-1 envelope expression vector, as previously described (76). Supernatant containing viral particles was collected 60 h after transfection. Cell debris was removed from the supernatant by centrifugation at 470 × g for 5 min and subsequent filtration through a 0.22-μm filter. Viral particles in the supernatant were pelleted through a 20% sucrose cushion by ultracentrifugation at 112,000 × g at 4°C for 1.75 h. The virus pellet was resuspended in the culture medium and stored at −80°C. The virus titer was determined by infection of 2 × 106 activated CD4+ T cells with serial dilutions of a virus stock and analysis of GFP-expressing cells at 72 h postinfection.
Infection of primary CD4+ T cells.
Infection of CD4+ T cells was carried out by 2 h of spinoculation, as previously described (51), followed by 0.5 h of incubation at 37°C. Then, the cells were washed once with phosphate-buffered saline (PBS) supplemented with 2% fetal calf serum and cultured in culture medium at 106 cells/ml in 96-well flat-bottom plates. In some experiments, CD4+ T cells were treated with a 10 μM concentration of the reverse transcriptase inhibitor lamivudine (3TC) (obtained from the NIH AIDS Research and Reference Reagent Bank) for 12 h prior to infection. In some experiments, the fusion inhibitor T1249 was added to the medium at the beginning of infection at a concentration of 1 μg/ml and kept in the culture in order to block viral entry (76).
Phenotypic analysis by flow cytometry.
To quantify the percentage of dead cells, CD4+ T cells were incubated in the culture medium containing 200 nM Mitotracker Red CMXRos (Invitrogen) at 37°C for 30 min before flow cytometry analysis. Dead cells were readily identified by reduced inner-mitochondrial-membrane transmembrane potential (ΔΨm). Flow cytometry data were collected on a FACSCalibur (Becton Dickinson) and analyzed with CellQuest software. To detect active caspase 3, cells were fixed and permeabilized with Cytofix/Cytoperm (BD Biosciences) and stained with rabbit anti-active caspase 3 polyclonal antibody (CM1; BD PharMingen). Carboxyfluorescein diacetate, succinimidyl ester (CFSE) labeling was carried out by incubation of 106 CD4+ T cells in 20 μl PBS containing 5 μm CFSE (Invitrogen) for 3 min at room temperature. The labeling reaction was stopped by adding an equivalent volume of fetal bovine serum to the cells. The cells were washed three times before being used in coculture experiments.
Western blotting.
Electrophoresis of proteins was performed using the NuPage system (Invitrogen) according to the manufacturer's protocol. In brief, CD4+ T cells were washed once in ice-cold PBS and incubated in lysis buffer (1% sodium dodecyl sulfate, 60 mM Tris, pH 7.6, and a Complete mini-protease inhibitor cocktail tablet [Roche]) on ice for 15 min. The cell lysate was mixed with 2-mercaptoethanol and 4× loading buffer (Invitrogen) and boiled at 100°C for 5 min before being loaded onto a NuPage 4 to 12% Bis-Tris gel. Following electrophoresis, the proteins were transferred to a polyvinylidene difluoride membrane (Immobilon; Millipore). Western blots were stained with monospecific NuMA serum, monospecific U1-70K serum, and monospecific PARP serum (gifts of Antony Rosen, Johns Hopkins School of Medicine) in blotting buffer (10 mM Tris, pH 7.6, 0.5% NP-40 [Sigma], 0.15 M NaCl). Western Lighting Chemiluminescence Reagent Plus (Perkin-Elmer Life Science) was used to develop the blot. Images were captured using Fluorchem 5500 (Alpha Innotech). Membranes were reprobed by treating them with Restore Western blot stripping buffer (Pierce) and were stained with antibody recognizing β-actin (Sigma). To analyze the amount of virion-associated gp120, viral lysate was prepared in the same way as cell lysate. The resulting Western blot was stained with HIV-1 gp120 monoclonal antibody 902 (NIH AIDS Research and Reference Reagent Program). To confirm that the Nef− Vpr− mutant construct did not express either Nef or Vpr, lysates of 293T cells transfected with the wild-type or mutant construct were prepared as described above. The Western blot was stained with antibody recognizing Vpr (a gift of Xiaofang Yu, Johns Hopkins School of Public Health) and HIV-1 IIIB Nef monoclonal antibody AE6 (NIH AIDS Research and Reference Reagent Program).
Electron microscopy.
Cells were processed for transmission electron microscopy as previously described (25). Briefly, the cells were fixed for 1 h at room temperature in a solution containing 3.0% formaldehyde, 1.5% glutaraldehyde, and 2.5% sucrose in 100 mM cacodylate (pH 7.4). The cells were gently pelleted, washed in 100 nM cacodylate, and subsequently embedded in 2% ultra-low-temperature gelling agarose. After being trimmed into 1-mm3 pieces, samples were stained with OsO4 (1 h at 4°C), washed once in double-distilled H2O, and incubated overnight in Kellenberger's uranyl acetate. The pellet was then dehydrated through a graded series of ethanol and embedded in Embed-812. Sections were cut on a Leica Ultracut UCT ultramicrotome, collected onto 400-mesh nickel grids, poststained in uranyl acetate and lead citrate, and observed at 100 kV in an FEI Tecnai 12 transmission electron microscope equipped with a Soft Imaging System MegaView III digital camera and analysis software.
RESULTS
Preferential depletion of memory CD4+ T cells by HIV-1.
To study CD4+ T-cell death in nonproductive HIV-1 infection, we infected freshly isolated, enriched peripheral CD4+ T cells with X4-tropic recombinant HIV-1 virions in the presence of 3TC, a reverse transcriptase inhibitor. The 3TC blocked the formation of a complete reverse-transcription product and therefore allowed us to examine the cytopathicity caused by early events in the HIV-1 life cycle. A significant percentage of CD4+ T cells in infected cultures displayed the characteristic phenotype of dead cells with reduced cell size, loss of ΔΨm, and positive staining for annexin V and 7-aminoactinomycin D (Fig. 1A). If the HIV-1 fusion inhibitor T1249 was added at the beginning of infection, HIV-1-induced cell death was inhibited, as judged by normal cell size, unchanged ΔΨm, and several other criteria, including the electron microscopic morphology (Fig. 2B to E). Interestingly, exposure to HIV-1 caused an increase in the percentage of annexin V+ 7-aminoactinomycin D− CD4+ T cells that was not blocked by 3TC or T1249. This is likely due to initial binding of gp120 to CD4+ T cells, as the subset was not observed in CD4+ T cells infected by vesicular stomatitis virus G (VSV-G)-pseudotyped HIV-1 (see Fig. S1 in the supplemental material). Our results clearly establish that early events in the HIV-1 life cycle trigger CD4+ T-cell death before the completion of reverse transcription.
FIG. 1.

Preferential depletion of memory CD4+ T cells by HIV-1. (A) HIV-1 infection induced CD4+ T-cell death. Peripheral CD4+ T cells were pretreated with the reverse transcriptase inhibitor 3TC before infection with recombinant HIV-1 in the presence or absence of the entry inhibitor T1249. The cell phenotype was characterized at 4 h postinfection. 7AAD, 7-aminoactinomycin D. (B) Preferential depletion of memory CD4+ T cells. Phenotypes of live CD4+ T cells in the uninfected and infected cultures were analyzed at 4 h postinfection. (C) Differential susceptibilities of naïve, memory, and activated CD4+ T cells to HIV-1-induced cell death. Naïve and memory CD4+ T cells were isolated from the same PBMC sample. Activated CD4+ T cells were isolated from PBMCs that were stimulated with PHA for 3 days. All cells were pretreated with 3TC before infection with recombinant HIV-1 at an MOI of 1. The percentage of dead cells was determined at 4 h postinfection by monitoring ΔΨm dissipation. The data represent the average result of independent experiments with different donors. Naïve and memory CD4+ T cells, n = 4; PHA-stimulated CD4+ T cells, n = 7. The P value was determined by paired Student t tests. The error bars represent standard errors. w/, with; w/o, without.
FIG. 2.

Infection with X4 HIV-1 induces rapid necrotic death of resting memory CD4+ T cells. (A) HIV-1-induced cell death increased with the amount of input virus. Memory CD4+ T cells were pretreated with 3TC and infected with recombinant HIV-1 at MOI of 0.1, 0.2, 0.4, 0.8, and 1.6. Virus input is indicated as the amount of p24 antigen. Cell death was analyzed at 4 h postinfection by monitoring ΔΨm dissipation. The data represent the average of four independent experiments with different donors. The error bars represent standard errors. w/, with; w/o, without. (B) Kinetics of memory CD4+ T-cell depletion. 3TC-treated memory CD4+ T cells were mock infected or infected with recombinant HIV-1 at an MOI of 1 in the presence of T1249. Percentages of viable cells were calculated based on the numbers of cells that excluded trypan blue. The data presented are averages of duplicate wells and are representative of three independent experiments. (C) Caspase-independent cell death. Apoptosis was induced in memory CD4+ T cells by staurosporin as a control. Lane 1, uninfected cells; lane 2, cells treated with 2 μM staurosporin; lane 3, cells infected in the presence of T1249; lane 4, cells infected in the absence of T1249. Cell viability was determined at 4 h postinfection by monitoring ΔΨm dissipation. Cell lysates were analyzed by Western blotting for the cleavage of NuMa (205 kDa→185 kDa), PARP (113 kDa→89 kDa), and U1-70K (70 kDa→40 kDa). (D) Absence of active caspase 3 in infection-induced cell death. Uninfected cells (lane 1), staurosporin-treated cells (lane 2), and cells infected in the presence (lane 3) or the absence (lane 4) of the entry inhibitor were analyzed by flow cytometry for ΔΨm and active caspase 3. (E) X4 HIV-1-induced cell death has the morphological hallmarks of necrosis. Shown are electron microscopic images of uninfected CD4+ T cells (a), CD4+ T cells infected in the presence of T1249 (b and c), staurosporin-treated memory CD4+ T cells (d), and CD4+ T cells infected in the absence of T1249 (e and f). n, nucleus; m, mitochondria. Bars = 5.0 μm (a, b, d, and e) and 0.5 μm (c and f). Virions are indicated by arrows.
We next investigated whether subsets of CD4+ T cells have differential susceptibilities to HIV-1-induced early cell death. We examined viable CD4+ T cells for the expression of CD62L and CD45RO at 4 hours postinfection. Compared to the uninfected culture, the percentage of CD45ROhigh memory CD4+ T cells decreased in the infected culture while the percentage of CD45ROlow CD62Lhigh naïve CD4+ T cells increased (Fig. 1B). The change in the proportion of CD4+ T-cell subsets was probably a result of memory cells being preferentially depleted by viral infection. We further tested the hypothesis with naïve and memory CD4+ T cells isolated from PBMCs. More infected cells than uninfected cells died in both naïve and memory CD4+ T-cell subsets. However, the percentage of dead cells was about twice as high in the infected memory CD4+ T cells as in the infected naïve CD4+ T cells (Fig. 1C). Activated CD4+ T cells isolated from PHA-stimulated PBMCs were also infected with an equivalent amount of virus in the presence of 3TC. In contrast to unstimulated CD4+ T cells, similar percentages of dying cells were found in infected and uninfected activated CD4+ T cells. This likely reflects activation-induced cell death (Fig. 1C). Thus, for activated CD4+ T cells, activation-induced cell death is much more prominent, and the cells do not appear to be susceptible to the same kind of HIV-1-induced cell killing observed in quiescent CD4+ T cells.
HIV induces rapid necrotic death of memory CD4+ T cells.
We sought to further characterize the loss of memory CD4+ T cells due to nonproductive infection with HIV-1. CD45ROhigh CD4+ T cells of 99% purity (see Fig. S2 in the supplemental material) were isolated from PBMCs of HIV-1-negative donors and treated with 3TC for 12 h. Infection was carried out with recombinant HIV-1 at multiplicities of infection (MOI) of 0.1, 0.2, 0.4, 0.8, and 1.6. With increasing amounts of virus, the percentages of memory CD4+ T cells undergoing infection-induced cell death also increased (Fig. 2A). Notably, even with low MOI, such as 0.1 and 0.2, infection-induced cell death was consistently observed with memory CD4+ T cells from different donors, albeit only in small fractions of the infected cultures (see Fig. S3 in the supplemental material). The death of memory CD4+ T cells occurred very rapidly, within 6 hours postinfection (Fig. 2B). To address whether the cells died from caspase-mediated apoptosis, we performed Western blots to look for known markers of caspase activity. The nuclear proteins NuMA, PARP, and U1-70K were cleaved by caspase 3 during apoptosis induced by staurosporin (9, 68). However, in cultures of infected memory CD4+ T cells in which a large fraction of cells were killed by nonproductive HIV-1 infection, no cleavage of NuMA, PARP, or U1-70K was observed (Fig. 2C). The percentage of cells harboring active caspase 3 remained the same in cultures undergoing infection-induced cell death as in uninfected cultures (Fig. 2D). Syncytium formation was also not observed in infected cultures (data not shown). We conclude that rapid death induced by nonproductive infection in resting memory CD4+ T cells is independent of caspase activity.
Morphological analysis by electron microscopy has been used to distinguish necrosis from apoptosis (18, 77). CD4+ T cells infected by recombinant HIV-1 in the presence of entry inhibitor had the same morphology as uninfected cells. Intact mitochondria were found in both cell populations (Fig. 2E, a, b, and c). Apoptotic features, such as chromatin condensation (nuclear pyknosis) and an intact cytoplasm, were readily detected in staurosporin-induced cell death (Fig. 2E, d). However, this was not observed in resting CD4+ T cells infected by recombinant HIV-1. Instead, dead cells in the infected culture were swollen. The cytoplasm appeared electron lucent (Fig. 2E, e). We also observed swollen, deformed mitochondria in infected memory CD4+ T cells (Fig. 2E, f). Necrosis is defined as a type of cell death that involves plasma membrane permeabilization without hallmarks of apoptosis or massive autophagic vacuolization (16, 18, 77). Our data suggest that HIV-1 induces memory CD4+ T cells to undergo necrotic cell death at an early stage of infection.
HIV-1 cytolysis depends on X4 envelope-mediated viral entry.
HIV-1 can induce “innocent-bystander” killing of uninfected CD4+ T cells through several mechanisms. To determine whether bystander killing plays a role in rapid virus-induced death of memory cells, we mock infected CFSE-labeled memory CD4+ T cells at the same time that we infected autologous memory CD4+ T cells with recombinant HIV-1. At the end of the spinoculation, the two cell populations were mixed and cocultured. If entry inhibitor T1249 was added to the culture, only spontaneous cell death was observed in both cell populations. In the absence of T1249, the percentage of dying cells was found to increase in infected cells, but not in mock-infected cells (Fig. 3A). The results of this experiment indicate that CD4+ T-cell death is the result of direct infection, rather than bystander killing.
FIG. 3.
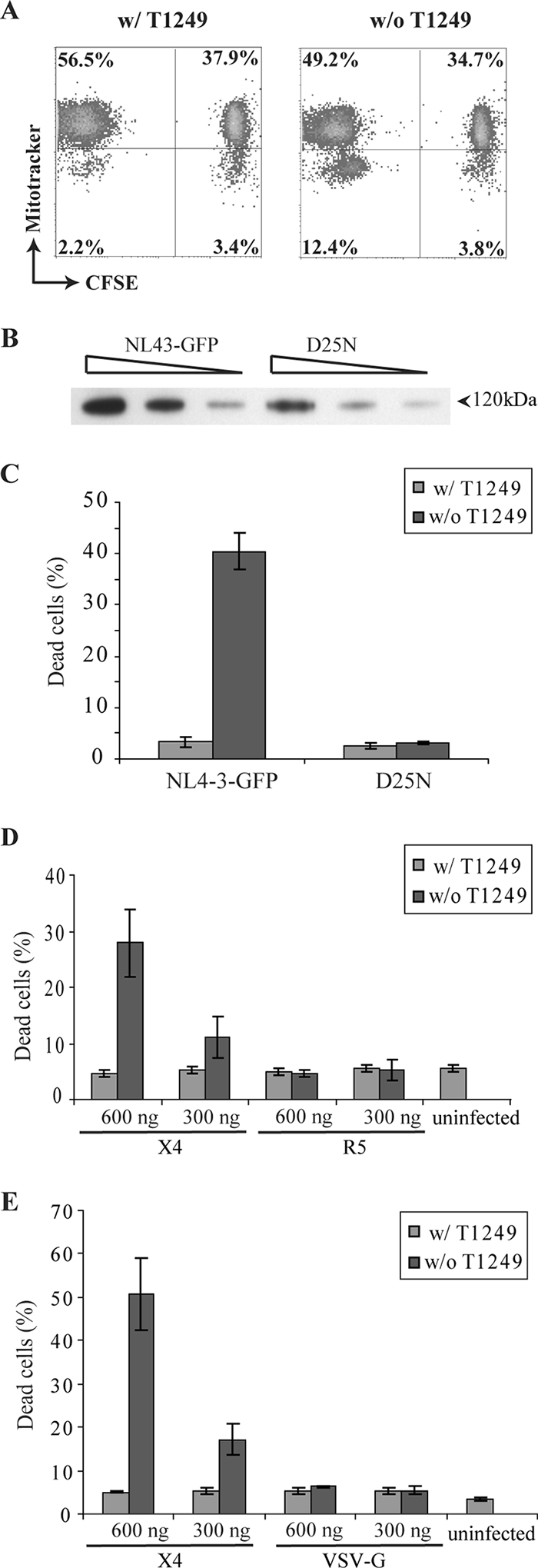
HIV cytolysis depends on X4 envelope-mediated viral fusion. (A) Direct killing of infected cells. CFSE-labeled memory CD4+ T cells were mock infected and mixed with HIV-1-infected autologous memory CD4+ T cells after spinoculation. Cell death was analyzed at 4 h postinfection. Increased cell death was noted only in the directly infected (CFSE-negative) cells. (B) Standardization of virion preparation based on virion-associated gp120. A series of twofold dilutions of recombinant HIV-1 virions and the protease mutant virus D25N were analyzed by Western blotting. (C) Protease mutant virus did not induce memory CD4+ T-cell death. Memory CD4+ T cells were infected by recombinant HIV-1 at an MOI of 1 or by the protease mutant virus D25N that was normalized at the level of virion-associated gp120. Cell death was analyzed at 4 h postinfection by monitoring ΔΨm dissipation. The data presented are averages of duplicate wells and are representative of three independent experiments. The error bars represent standard deviations. w/, with; w/o, without. (D) R5 HIV-1 recombinant virus did not induce memory CD4+ T-cell death. R5 virus was normalized to X4 virus based on p24 antigen. The percentage of dead cells was determined at 4 h postinfection by monitoring ΔΨm dissipation. The data represent the average of three independent experiments with different donors. The error bars represent standard errors. (E) VSV-G-mediated viral entry did not induce memory CD4+ T-cell death. VSV-G-pseudotyped virus was normalized to X4 virus based on p24 antigen. Cell death was analyzed at 24 h postinfection by monitoring ΔΨm dissipation. The data represent the average of three independent experiments with different donors. The error bars represent standard errors.
The results presented above suggest that viral entry into memory CD4+ T cells can induce cell death. To investigate the role of virion-associated envelope protein in triggering memory CD4+ T-cell death, we exposed cells to viruses carrying equivalent amounts of envelope protein but differing in their capacities to enter cells. We generated a protease mutant virus that carried a mutation, D25N, in the enzyme active site (31). Previous studies have shown that the protease mutant virus is defective in fusion with the host cell plasma membrane due to an immature viral core (47, 74). Unprocessed Gag protein drives the assembly of mutant virions, which are released into the supernatant. To remove free gp120 in the viral stock, we pelleted viral particles through a sucrose cushion. Protease mutant virus was normalized to recombinant HIV-1 based on the amount of gp120 incorporated into virions (Fig. 3B). In contrast to infection with recombinant HIV-1, infection with the protease mutant virus did not induce cell death in memory CD4+ T cells (Fig. 3C). These data suggest that HIV-1 Env binding to receptors is not enough to induce memory CD4+ T-cell death. Instead, postbinding events, such as fusion, are required.
Both X4- and R5-tropic HIV-1 envelope proteins mediate viral fusion at the plasma membrane. We compared the cytopathicities of HIV-1 virions coated with different HIV-1 envelope proteins. Memory CD4+ T cells were infected with equivalent amounts of X4 or R5 pseudovirus based on p24 antigen. No cell death was observed in R5 HIV-1-infected memory CD4+ T cells (Fig. 3D). The lack of cytopathicity probably reflects inefficient R5 HIV-1 entry due to a low surface expression level of CCR5 on unstimulated memory CD4+ T cells (57). In contrast to HIV-1 envelope protein, VSV-G glycoprotein allows HIV-1 pseudovirus to enter CD4+ T cells through endocytosis. VSV-G-mediated pH-dependent fusion is triggered in the late endosome (2). We tested whether VSV-G-pseudotyped HIV-1 has the same cytopathic effect on memory CD4+ T cells as the recombinant virus that utilizes CXCR4. Memory CD4+ T cells were infected with X4-utilizing virus and VSV-G-pseudotyped virions normalized by the amount of p24 antigen. Cell death was observed only in X4 HIV-1-infected memory CD4+ T cells (Fig. 3E). Memory CD4+ T cells infected with VSV-G-pseudotyped virions displayed the same viability at 24 h postinfection as uninfected cells. Therefore, HIV-1-induced early cell death depends on X4 Env-mediated viral fusion.
HIV-1 early cytolysis is independent of virion-associated Nef and Vpr.
We next tested whether virion accessory proteins play any role in the observed early cell death. HIV-1 packages approximately 10 to 100 Nef molecules per virion. It has been suggested that Nef enhances HIV-1 infectivity in part by facilitating the penetration of the viral core through the cytoskeleton (8, 62). Vpr is an accessory protein that is packaged into virions in large amounts. Virion-associated Vpr has been reported to arrest activated CD4+ T cells at the G2 phase of the cell cycle. This arrest occurs even in the presence of reverse transcriptase inhibitor and may lead to cell death (59). To determine whether Nef and Vpr participate in the killing of memory CD4+ T cells, we introduced a silent mutation at the initiating ATG of vpr and deleted the first 72 nucleotides at the 5′ end of the nef coding region in the provirus NL4-3-GFP (Fig. 4A). Nef− Vpr− HIV-1 caused as much memory CD4+ T-cell death as the recombinant HIV-1 with intact Vpr and Nef (Fig. 4B), indicating that neither virion-associated Nef nor Vpr is involved in HIV-1-induced early memory CD4+ T-cell death.
FIG. 4.

Neither virion-associated Nef nor Vpr is necessary for early memory CD4+ T-cell death. (A) The absence of Nef and Vpr expression in 293T cells transfected by a Nef− Vpr− mutant construct. 293T cell lysates were analyzed by Western blotting with anti-Nef and anti-Vpr antibodies and anti-HIV-1 human serum. w/, with; w/o, without. (B) Nef− Vpr− HIV-1 induces early memory CD4+ T-cell death. The Nef− Vpr− virus concentration was normalized to wild-type virus based on p24 antigen. The percentage of dead cells was determined at 4 h postinfection by monitoring ΔΨm dissipation. The data presented are averages of duplicate wells and are representative of three independent experiments. The error bars represent standard errors.
Cytokines modulate memory CD4+ T-cell susceptibility to HIV-1 killing.
Unlike peripheral CD4+ T cells, memory CD4+ T cells in lymphoid tissue support productive HIV-1 infection, probably under the influence of cytokines in the microenvironment (38, 71). To investigate whether cytokines modulate the susceptibility of memory CD4+ T cells to HIV-1-induced early cell death, we cultured memory CD4+ T cells in medium containing various cytokines for 2 days before infection. At 4 h postinfection, we compared the viability of cytokine-treated cells to that of cells that had not been treated (Fig. 5A and B). Consistent with their important roles in T-cell survival and proliferation (6), the cytokines IL-2, IL-7, and IL-15 improved the viability of both uninfected memory CD4+ T cells and infected cells. Type I IFN has been suggested to induce CD4+ T-cell death by upregulating TRAIL expression (26). In our culture system, type I IFN treatment had minimal effect on the viability of uninfected memory CD4+ T cells. However, type I IFN rendered memory CD4+ T cells resistant to HIV-1-induced early cell death. It has been reported that the chemokine MIP-1α secreted by macrophages induces chemotaxis and renders resting CD4+ T cells permissive to productive HIV-1 infection (67). We observed that MIP-1α-treated memory CD4+ T cells were less susceptible to HIV-1-induced cell death.
FIG. 5.

Cytokines modulate memory CD4+ T-cell susceptibility to HIV-1 killing. (A and B) Memory CD4+ T cells were treated with cytokines for 48 h before infection with X4 recombinant HIV-1. Cell death was detected by flow cytometric analysis at 4 h postinfection. The data represent the average of duplicate wells. Each panel shows the results from an independent experiment with different donors. w/, with; w/o, without. (C) Expression levels of CXCR4 on cytokine-stimulated memory CD4+ T cells. The mean fluorescence intensity (MFI) for CXCR4 staining is indicated. The error bars represent standard errors.
Since we demonstrated that HIV-1 cytolysis depends on X4-mediated viral entry, one possible explanation for the reduced susceptibility to HIV-1-induced cell death could be low expression levels of the HIV-1 coreceptor CXCR4 on memory CD4+ T-cell surfaces upon cytokine stimulation. We found that type I IFN-treated memory CD4+ T cells expressed less CXCR4 on the cell surface than untreated cells (Fig. 5C). However, under other cytokine treatments, the surface expression level of CXCR4 was unchanged or increased compared to untreated memory CD4+ T cells. Therefore, our data suggest that several cytokines modulate memory CD4+ T-cell susceptibility to HIV-1-induced early cell death through mechanisms other than preventing viral entry.
DISCUSSION
HIV-1 infection of CD4+ T cells is initiated when the virus binds to CD4 and a coreceptor on the cell surface. Conformational changes in HIV-1 gp120 triggered by CD4 binding facilitate coreceptor binding, leading ultimately to the exposure of a fusion peptide, which mediates fusion of the viral envelope with the plasma membrane. After entry and uncoating, reverse transcription occurs within a ribosome-sized preintegration complex that is composed of viral proteins and host factors. Subsequent nuclear import of the preintegration complex and integration of the viral genome into the host chromosome are essential for the virus to establish productive infection. Recent discoveries of APOBEC3G and TRIM5a highlight host restrictions imposed on the virus prior to integration (23, 65). In this study, we monitored the effects of early events in the HIV-1 life cycle on the viability of the host CD4+ T cells. We demonstrated that X4-tropic HIV-1 triggers the death of quiescent peripheral CD4+ T cells, particularly memory CD4+ T cells, even in the presence of reverse transcriptase inhibitors. In contrast, CD4+ T cells stimulated by mitogen or cytokines are resistant to this mechanism of killing. We have provided the first evidence that early steps of HIV-1 infection have detrimental effects on certain subsets of CD4+ T cells.
Focusing on isolated memory CD4+ T cells, we demonstrated that HIV-1 directly kills infected resting memory cells, leaving bystander cells viable. We investigated the molecular mechanism involved in HIV-1-induced early memory CD4+ T-cell death. A large body of literature describes HIV-1 envelope-induced apoptosis of uninfected cells through cross-ligation of CD4, stimulation of CXCR4 or CCR5, or cell-cell fusion (1, 55). However, we found that HIV-1 cytopathicity in memory CD4+ T cells depends on not only HIV-1 envelope binding, but also viral fusion at the plasma membrane. First, the fusion inhibitor T1249 efficiently blocked infection-induced CD4+ T-cell death, indicating that cell death occurs upon or after fusion. Second, a protease mutant virus that is inefficient in fusion did not induce cell death, although it carries the same amount of gp120 as the recombinant HIV-1 with wild-type protease. Finally, substitution of VSV-G envelope protein for HIV-1 envelope protein abolished the cytopathicity of HIV-1 viral particles, as the pseudotyped virus released viral core into the host cell cytoplasm through fusion in the endocytic compartment. Deletion of both Nef and Vpr, two virion-associated proteins involved in early events in HIV-1 infection, did not affect the cytopathicity of HIV-1 for CD4+ T cells. It remains possible that other viral proteins, such as protease, may play roles in early cytolysis. Fusion-induced thymocyte depletion has been reported in HIV-1-infected human fetal thymus organ cultures (44). Our results provide evidence that CXCR4-tropic HIV-1 depletes peripheral memory CD4+ T cells through a novel mechanism that also depends on fusion.
Consistent with the notion that early steps of HIV-1 infection trigger host cell death, memory CD4+ T-cell death occurs within 6 h postinfection. We characterized infection-induced cell death both biochemically and morphologically. Annexin V-positive staining and ΔΨm dissipation have been observed in both apoptotic and necrotic cells (34, 46, 70). The lack of caspase activation, chromatin condensation, and autophagic vacuoles leads us to conclude that infected CD4+ T cells undergo necrotic cell death. Although necrosis has been considered an uncontrolled form of cell death under nonphysiological stress, some studies suggest that necrosis may be another form of induced cell death involved in the pathogenesis of certain viral infections, such as Sindbis virus infection of motor neurons (10, 24, 48) and severe acute respiratory syndrome coronovirus (69). Several groups have demonstrated that the death of productively infected CD4+ T cells or T-cell lines is necrotic rather than apoptotic (32, 36, 53). Whether CD4+ T cells die from necrosis, apoptosis, or autophagy in HIV-1-infected individuals might depend on the nature of the triggering signal and the CD4+ T-cell subsets involved. One concern with the culture model is the large amount of virus that is used to overcome the inefficiency of in vitro infection. While we did not know how many virions per cell are required to trigger necrosis of memory CD4+ T cells, we observed rapid cell death in memory CD4+ T cells infected at an MOI equivalent to 0.1. In other words, the same amount of reporter virus could productively infect only 10% of activated CD4+ T cells. Therefore, necrosis of infected cells is not due to an extremely high MOI.
In contrast to memory CD4+ T cells, we found that naïve CD4+ T cells were less susceptible to fusion-dependent HIV-1 cytopathicity and that activated CD4+ T cells were resistant. The mechanism underlying this differential susceptibility is not understood, since CXCR4 is expressed at sufficiently high levels on all CD4+ T-cell subsets. Interestingly, we found that several cytokines reduced memory CD4+ T-cell susceptibility to HIV-1-induced early cell death, even though some cytokines upregulated CXCR4 expression on the cell surface. It is possible that different survival mechanisms that normally control the homeostasis of naïve, memory, and activated CD4+ T cells determine whether host cells die upon viral entry. Selective depletion of subsets of CD4+ T cells has also been reported in rhesus macaques infected with CXCR4-utilizing simian-human immunodeficiency virus (SHIV). Within 2 weeks after intravenous inoculation, X4-tropic SHIV causes profound loss of peripheral CD4+ T cells, but not of the CD4+ T cells within the intestinal mucosa (22). Another study reported that both peripheral naïve CD4+ T cells and central memory CD4+ T cells are rapidly depleted by X4-tropic SHIV in infected macaques (50). While 32 to 88% of circulating naïve CD4+ T cells produce virus at day 10 postinoculation, only 0.8 to 4.2% of memory CD4+ T cells in the blood release infectious virus (49). These data are in agreement with our finding that infected memory CD4+ T cells die at the early stage of viral infection.
Preferential depletion of memory CD4+ T cells may contribute to the pathogenesis of CXCR4-utilizing HIV-1. X4-tropic HIV-1 is rarely associated with acute infection, even in cases of intravenous transmission. Cytotoxic T lymphocytes and humoral control have been proposed as postmucosal selective forces against X4-tropic virus (40). However, in the late stage of disease, X4-tropic HIV-1 emerges in about 50% of infected individuals and is associated with accelerated loss of CD4+ T cells and faster disease progression. Previous studies have attributed the profound cytopathicity of X4 HIV-1 to syncytium formation, infection, and destruction of a broad range of host cells, including naïve CD4+ T cells and thymocytes (5, 30). Based on our data, we propose that in acute infection when the majority of peripheral CD4+ T cells are quiescent, the death of host cells without producing progeny virus represents a selective disadvantage for X4-tropic HIV-1. However, in the late stage of the disease, more CD4+ T cells are activated, and they provide the major source for X4 virus production. The nonproductive infection of memory CD4+ T cells has little effect on virus proliferation. Instead, the direct killing of memory CD4+ T cells damages the host's immune response to the virus, as the central role of memory CD4+ T cells has been demonstrated in rhesus macaques infected with SIV (52, 66). Furthermore, necrotic cells release intracellular molecules through disrupted plasma membranes and elicit proinflammatory responses. Necrosis of HIV-1-infected memory CD4+ T cells may fuel chronic immune activation that affects not only CD4+ T cells, but many immune cell types, and eventually leads to the onset of AIDS. Further understanding of how HIV-1 induces early cell death and what factors determine the susceptibility of CD4+ T cells will shine new light on the host-virus interaction and provide new targets for therapy.
Supplementary Material
Footnotes
Published ahead of print on 2 July 2008.
Supplemental material for this article may be found at http://jvi.asm.org/.
REFERENCES
- 1.Ahr, B., V. Robert-Hebmann, C. Devaux, and M. Biard-Piechaczyk. 2004. Apoptosis of uninfected cells induced by HIV envelope glycoproteins. Retrovirology 112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Aiken, C. 1997. Pseudotyping human immunodeficiency virus type 1 (HIV-1) by the glycoprotein of vesicular stomatitis virus targets HIV-1 entry to an endocytic pathway and suppresses both the requirement for Nef and the sensitivity to cyclosporin A. J. Virol. 715871-5877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Badley, A. D., D. H. Dockrell, A. Algeciras, S. Ziesmer, A. Landay, M. M. Lederman, E. Connick, H. Kessler, D. Kuritzkes, D. H. Lynch, P. Roche, H. Yagita, and C. V. Paya. 1998. In vivo analysis of Fas/FasL interactions in HIV-infected patients. J. Clin. Investig. 10279-87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Berndt, C., B. Mopps, S. Angermuller, P. Gierschik, and P. H. Krammer. 1998. CXCR4 and CD4 mediate a rapid CD95-independent cell death in CD4+ T cells. Proc. Natl. Acad. Sci. USA 9512556-12561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Blaak, H., A. B. van't Wout, M. Brouwer, B. Hooibrink, E. Hovenkamp, and H. Schuitemaker. 2000. In vivo HIV-1 infection of CD45RA+CD4+ T cells is established primarily by syncytium-inducing variants and correlates with the rate of CD4+ T cell decline. Proc. Natl. Acad. Sci. USA 971269-1274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Boyman, O., J. F. Purton, C. D. Surh, and J. Sprent. 2007. Cytokines and T-cell homeostasis. Curr. Opin. Immunol. 19320-326. [DOI] [PubMed] [Google Scholar]
- 7.Bukrinsky, M. I., T. L. Stanwick, M. P. Dempsey, and M. Stevenson. 1991. Quiescent T lymphocytes as an inducible virus reservoir in HIV-1 infection. Science 254423-427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Campbell, E. M., R. Nunez, and T. J. Hope. 2004. Disruption of the actin cytoskeleton can complement the ability of Nef to enhance human immunodeficiency virus type 1 infectivity. J. Virol. 785745-5755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Casciola-Rosen, L. A., D. K. Miller, G. J. Anhalt, and A. Rosen. 1994. Specific cleavage of the 70-kDa protein component of the U1 small nuclear ribonucleoprotein is a characteristic biochemical feature of apoptotic cell death. J. Biol. Chem. 26930757-30760. [PubMed] [Google Scholar]
- 10.Chen, Y. B., S. Y. Seo, D. G. Kirsch, T. T. Sheu, W. C. Cheng, and J. M. Hardwick. 2006. Alternate functions of viral regulators of cell death. Cell Death Differ. 131318-1324. [DOI] [PubMed] [Google Scholar]
- 11.Chun, T. W., L. Carruth, D. Finzi, X. Shen, J. A. DiGiuseppe, H. Taylor, M. Hermankova, K. Chadwick, J. Margolick, T. C. Quinn, Y. H. Kuo, R. Brookmeyer, M. A. Zeiger, P. Barditch-Crovo, and R. F. Siliciano. 1997. Quantification of latent tissue reservoirs and total body viral load in HIV-1 infection. Nature 387183-188. [DOI] [PubMed] [Google Scholar]
- 12.de Oliveira Pinto, L. M., S. Garcia, H. Lecoeur, C. Rapp, and M. L. Gougeon. 2002. Increased sensitivity of T lymphocytes to tumor necrosis factor receptor 1 (TNFR1)- and TNFR2-mediated apoptosis in HIV infection: relation to expression of Bcl-2 and active caspase-8 and caspase-3. Blood 991666-1675. [DOI] [PubMed] [Google Scholar]
- 13.Douek, D. C., L. J. Picker, and R. A. Koup. 2003. T cell dynamics in HIV-1 infection. Annu. Rev. Immunol. 21265-304. [DOI] [PubMed] [Google Scholar]
- 14.Espert, L., M. Denizot, M. Grimaldi, V. Robert-Hebmann, B. Gay, M. Varbanov, P. Codogno, and M. Biard-Piechaczyk. 2006. Autophagy is involved in T cell death after binding of HIV-1 envelope proteins to CXCR4. J. Clin. Investig. 1162161-2172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Esser, M. T., J. W. Bess, Jr., K. Suryanarayana, E. Chertova, D. Marti, M. Carrington, L. O. Arthur, and J. D. Lifson. 2001. Partial activation and induction of apoptosis in CD4+ and CD8+ T lymphocytes by conformationally authentic noninfectious human immunodeficiency virus type 1. J. Virol. 751152-1164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Festjens, N., T. Vanden Berghe, and P. Vandenabeele. 2006. Necrosis, a well-orchestrated form of cell demise: signalling cascades, important mediators and concomitant immune response. Biochim. Biophys. Acta 17571371-1387. [DOI] [PubMed] [Google Scholar]
- 17.Finkel, T. H., G. Tudor-Williams, N. K. Banda, M. F. Cotton, T. Curiel, C. Monks, T. W. Baba, R. M. Ruprecht, and A. Kupfer. 1995. Apoptosis occurs predominantly in bystander cells and not in productively infected cells of HIV- and SIV-infected lymph nodes. Nat. Med. 1129-134. [DOI] [PubMed] [Google Scholar]
- 18.Galluzzi, L., M. C. Maiuri, I. Vitale, H. Zischka, M. Castedo, L. Zitvogel, and G. Kroemer. 2007. Cell death modalities: classification and pathophysiological implications. Cell Death Differ. 141237-1243. [DOI] [PubMed] [Google Scholar]
- 19.Gougeon, M. L. 2003. Apoptosis as an HIV strategy to escape immune attack. Nat. Rev. Immunol. 3392-404. [DOI] [PubMed] [Google Scholar]
- 20.Guadalupe, M., E. Reay, S. Sankaran, T. Prindiville, J. Flamm, A. McNeil, and S. Dandekar. 2003. Severe CD4+ T-cell depletion in gut lymphoid tissue during primary human immunodeficiency virus type 1 infection and substantial delay in restoration following highly active antiretroviral therapy. J. Virol. 7711708-11717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Haase, A. T. 1999. Population biology of HIV-1 infection: viral and CD4+ T cell demographics and dynamics in lymphatic tissues. Annu. Rev. Immunol. 17625-656. [DOI] [PubMed] [Google Scholar]
- 22.Harouse, J. M., A. Gettie, R. C. Tan, J. Blanchard, and C. Cheng-Mayer. 1999. Distinct pathogenic sequela in rhesus macaques infected with CCR5 or CXCR4 utilizing SHIVs. Science 284816-819. [DOI] [PubMed] [Google Scholar]
- 23.Harris, R. S., K. N. Bishop, A. M. Sheehy, H. M. Craig, S. K. Petersen-Mahrt, I. N. Watt, M. S. Neuberger, and M. H. Malim. 2003. DNA deamination mediates innate immunity to retroviral infection. Cell 113803-809. [DOI] [PubMed] [Google Scholar]
- 24.Havert, M. B., B. Schofield, D. E. Griffin, and D. N. Irani. 2000. Activation of divergent neuronal cell death pathways in different target cell populations during neuroadapted Sindbis virus infection of mice. J. Virol. 745352-5356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hendricks, L. C., M. McCaffery, G. E. Palade, and M. G. Farquhar. 1993. Disruption of endoplasmic reticulum to Golgi transport leads to the accumulation of large aggregates containing beta-COP in pancreatic acinar cells. Mol. Biol. Cell 4413-424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Herbeuval, J. P., A. W. Hardy, A. Boasso, S. A. Anderson, M. J. Dolan, M. Dy, and G. M. Shearer. 2005. Regulation of TNF-related apoptosis-inducing ligand on primary CD4+ T cells by HIV-1: role of type I IFN-producing plasmacytoid dendritic cells. Proc. Natl. Acad. Sci. USA 10213974-13979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Herbeuval, J. P., J. Nilsson, A. Boasso, A. W. Hardy, M. J. Kruhlak, S. A. Anderson, M. J. Dolan, M. Dy, J. Andersson, and G. M. Shearer. 2006. Differential expression of IFN-alpha and TRAIL/DR5 in lymphoid tissue of progressor versus nonprogressor HIV-1-infected patients. Proc. Natl. Acad. Sci. USA 1037000-7005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Holm, G. H., and D. Gabuzda. 2005. Distinct mechanisms of CD4+ and CD8+ T-cell activation and bystander apoptosis induced by human immunodeficiency virus type 1 virions. J. Virol. 796299-6311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jacotot, E., L. Ravagnan, M. Loeffler, K. F. Ferri, H. L. Vieira, N. Zamzami, P. Costantini, S. Druillennec, J. Hoebeke, J. P. Briand, T. Irinopoulou, E. Daugas, S. A. Susin, D. Cointe, Z. H. Xie, J. C. Reed, B. P. Roques, and G. Kroemer. 2000. The HIV-1 viral protein R induces apoptosis via a direct effect on the mitochondrial permeability transition pore. J. Exp. Med. 19133-46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kitchen, S. G., and J. A. Zack. 1997. CXCR4 expression during lymphopoiesis: implications for human immunodeficiency virus type 1 infection of the thymus. J. Virol. 716928-6934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kohl, N. E., E. A. Emini, W. A. Schleif, L. J. Davis, J. C. Heimbach, R. A. Dixon, E. M. Scolnick, and I. S. Sigal. 1988. Active human immunodeficiency virus protease is required for viral infectivity. Proc. Natl. Acad. Sci. USA 854686-4690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kolesnitchenko, V., L. King, A. Riva, Y. Tani, S. J. Korsmeyer, and D. I. Cohen. 1997. A major human immunodeficiency virus type 1-initiated killing pathway distinct from apoptosis. J. Virol. 719753-9763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kovacs, J. A., R. A. Lempicki, I. A. Sidorov, J. W. Adelsberger, B. Herpin, J. A. Metcalf, I. Sereti, M. A. Polis, R. T. Davey, J. Tavel, J. Falloon, R. Stevens, L. Lambert, R. Dewar, D. J. Schwartzentruber, M. R. Anver, M. W. Baseler, H. Masur, D. S. Dimitrov, and H. C. Lane. 2001. Identification of dynamically distinct subpopulations of T lymphocytes that are differentially affected by HIV. J. Exp. Med. 1941731-1741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kroemer, G., B. Dallaporta, and M. Resche-Rigon. 1998. The mitochondrial death/life regulator in apoptosis and necrosis. Annu. Rev. Physiol. 60619-642. [DOI] [PubMed] [Google Scholar]
- 35.LaBonte, J. A., N. Madani, and J. Sodroski. 2003. Cytolysis by CCR5-using human immunodeficiency virus type 1 envelope glycoproteins is dependent on membrane fusion and can be inhibited by high levels of CD4 expression. J. Virol. 776645-6659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lenardo, M. J., S. B. Angleman, V. Bounkeua, J. Dimas, M. G. Duvall, M. B. Graubard, F. Hornung, M. C. Selkirk, C. K. Speirs, C. Trageser, J. O. Orenstein, and D. L. Bolton. 2002. Cytopathic killing of peripheral blood CD4+ T lymphocytes by human immunodeficiency virus type 1 appears necrotic rather than apoptotic and does not require env. J. Virol. 765082-5093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Li, C. J., D. J. Friedman, C. Wang, V. Metelev, and A. B. Pardee. 1995. Induction of apoptosis in uninfected lymphocytes by HIV-1 Tat protein. Science 268429-431. [DOI] [PubMed] [Google Scholar]
- 38.Li, Q., L. Duan, J. D. Estes, Z. M. Ma, T. Rourke, Y. Wang, C. Reilly, J. Carlis, C. J. Miller, and A. T. Haase. 2005. Peak SIV replication in resting memory CD4+ T cells depletes gut lamina propria CD4+ T cells. Nature 4341148-1152. [DOI] [PubMed] [Google Scholar]
- 39.Lifson, J. D., G. R. Reyes, M. S. McGrath, B. S. Stein, and E. G. Engleman. 1986. AIDS retrovirus induced cytopathology: giant cell formation and involvement of CD4 antigen. Science 2321123-1127. [DOI] [PubMed] [Google Scholar]
- 40.Margolis, L., and R. Shattock. 2006. Selective transmission of CCR5-utilizing HIV-1: the ‘gatekeeper’ problem resolved? Nat. Rev. Microbiol. 4312-317. [DOI] [PubMed] [Google Scholar]
- 41.Mattapallil, J. J., D. C. Douek, B. Hill, Y. Nishimura, M. Martin, and M. Roederer. 2005. Massive infection and loss of memory CD4+ T cells in multiple tissues during acute SIV infection. Nature 4341093-1097. [DOI] [PubMed] [Google Scholar]
- 42.McCune, J. M. 2001. The dynamics of CD4+ T-cell depletion in HIV disease. Nature 410974-979. [DOI] [PubMed] [Google Scholar]
- 43.Mehandru, S., M. A. Poles, K. Tenner-Racz, V. Manuelli, P. Jean-Pierre, P. Lopez, A. Shet, A. Low, H. Mohri, D. Boden, P. Racz, and M. Markowitz. 2007. Mechanisms of gastrointestinal CD4+ T-cell depletion during acute and early human immunodeficiency virus type 1 infection. J. Virol. 81599-612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Meissner, E. G., L. Zhang, S. Jiang, and L. Su. 2006. Fusion-induced apoptosis contributes to thymocyte depletion by a pathogenic human immunodeficiency virus type 1 envelope in the human thymus. J. Virol. 8011019-11030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mohri, H., S. Bonhoeffer, S. Monard, A. S. Perelson, and D. D. Ho. 1998. Rapid turnover of T lymphocytes in SIV-infected rhesus macaques. Science 2791223-1227. [DOI] [PubMed] [Google Scholar]
- 46.Munoz, L. E., B. Frey, F. Pausch, W. Baum, R. B. Mueller, B. Brachvogel, E. Poschl, F. Rodel, K. von der Mark, M. Herrmann, and U. S. Gaipl. 2007. The role of annexin A5 in the modulation of the immune response against dying and dead cells. Curr. Med. Chem. 14271-277. [DOI] [PubMed] [Google Scholar]
- 47.Murakami, T., S. Ablan, E. O. Freed, and Y. Tanaka. 2004. Regulation of human immunodeficiency virus type 1 Env-mediated membrane fusion by viral protease activity. J. Virol. 781026-1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Nargi-Aizenman, J. L., and D. E. Griffin. 2001. Sindbis virus-induced neuronal death is both necrotic and apoptotic and is ameliorated by N-methyl-d-aspartate receptor antagonists. J. Virol. 757114-7121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Nishimura, Y., C. R. Brown, J. J. Mattapallil, T. Igarashi, A. Buckler-White, B. A. Lafont, V. M. Hirsch, M. Roederer, and M. A. Martin. 2005. Resting naive CD4+ T cells are massively infected and eliminated by X4-tropic simian-human immunodeficiency viruses in macaques. Proc. Natl. Acad. Sci. USA 1028000-8005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Nishimura, Y., T. Igarashi, A. Buckler-White, C. Buckler, H. Imamichi, R. M. Goeken, W. R. Lee, B. A. Lafont, R. Byrum, H. C. Lane, V. M. Hirsch, and M. A. Martin. 2007. Loss of naive cells accompanies memory CD4+ T-cell depletion during long-term progression to AIDS in simian immunodeficiency virus-infected macaques. J. Virol. 81893-902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.O'Doherty, U., W. J. Swiggard, and M. H. Malim. 2000. Human immunodeficiency virus type 1 spinoculation enhances infection through virus binding. J. Virol. 7410074-10080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Okoye, A., M. Meier-Schellersheim, J. M. Brenchley, S. I. Hagen, J. M. Walker, M. Rohankhedkar, R. Lum, J. B. Edgar, S. L. Planer, A. Legasse, A. W. Sylwester, M. Piatak, Jr., J. D. Lifson, V. C. Maino, D. L. Sodora, D. C. Douek, M. K. Axthelm, Z. Grossman, and L. J. Picker. 2007. Progressive CD4+ central memory T cell decline results in CD4+ effector memory insufficiency and overt disease in chronic SIV infection. J. Exp. Med. 2042171-2185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Park, I. W., E. Kondo, L. Bergeron, J. Park, and J. Sodroski. 1996. Effects of human immunodeficiency virus type 1 infection on programmed cell death in the presence or absence of Bcl-2. J. Acquir. Immune Defic. Syndr. Hum. Retrovirol. 12321-328. [DOI] [PubMed] [Google Scholar]
- 54.Perfettini, J. L., M. Castedo, R. Nardacci, F. Ciccosanti, P. Boya, T. Roumier, N. Larochette, M. Piacentini, and G. Kroemer. 2005. Essential role of p53 phosphorylation by p38 MAPK in apoptosis induction by the HIV-1 envelope. J. Exp. Med. 201279-289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Perfettini, J. L., M. Castedo, T. Roumier, K. Andreau, R. Nardacci, M. Piacentini, and G. Kroemer. 2005. Mechanisms of apoptosis induction by the HIV-1 envelope. Cell Death Differ. 12(Suppl. 1)916-923. [DOI] [PubMed] [Google Scholar]
- 56.Piatak, M., Jr., M. S. Saag, L. C. Yang, S. J. Clark, J. C. Kappes, K. C. Luk, B. H. Hahn, G. M. Shaw, and J. D. Lifson. 1993. High levels of HIV-1 in plasma during all stages of infection determined by competitive PCR. Science 2591749-1754. [DOI] [PubMed] [Google Scholar]
- 57.Pierson, T., T. L. Hoffman, J. Blankson, D. Finzi, K. Chadwick, J. B. Margolick, C. Buck, J. D. Siliciano, R. W. Doms, and R. F. Siliciano. 2000. Characterization of chemokine receptor utilization of viruses in the latent reservoir for human immunodeficiency virus type 1. J. Virol. 747824-7833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Pierson, T. C., Y. Zhou, T. L. Kieffer, C. T. Ruff, C. Buck, and R. F. Siliciano. 2002. Molecular characterization of preintegration latency in human immunodeficiency virus type 1 infection. J. Virol. 768518-8531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Poon, B., K. Grovit-Ferbas, S. A. Stewart, and I. S. Chen. 1998. Cell cycle arrest by Vpr in HIV-1 virions and insensitivity to antiretroviral agents. Science 281266-269. [DOI] [PubMed] [Google Scholar]
- 60.Rosenzweig, M., M. A. DeMaria, D. M. Harper, S. Friedrich, R. K. Jain, and R. P. Johnson. 1998. Increased rates of CD4+ and CD8+ T lymphocyte turnover in simian immunodeficiency virus-infected macaques. Proc. Natl. Acad. Sci. USA 956388-6393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Rusert, P., M. Fischer, B. Joos, C. Leemann, H. Kuster, M. Flepp, S. Bonhoeffer, H. F. Gunthard, and A. Trkola. 2004. Quantification of infectious HIV-1 plasma viral load using a boosted in vitro infection protocol. Virology 326113-129. [DOI] [PubMed] [Google Scholar]
- 62.Schaeffer, E., R. Geleziunas, and W. C. Greene. 2001. Human immunodeficiency virus type 1 Nef functions at the level of virus entry by enhancing cytoplasmic delivery of virions. J. Virol. 752993-3000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Spina, C. A., J. C. Guatelli, and D. D. Richman. 1995. Establishment of a stable, inducible form of human immunodeficiency virus type 1 DNA in quiescent CD4 lymphocytes in vitro. J. Virol. 692977-2988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Stevenson, M., T. L. Stanwick, M. P. Dempsey, and C. A. Lamonica. 1990. HIV-1 replication is controlled at the level of T cell activation and proviral integration. EMBO J. 91551-1560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Stremlau, M., C. M. Owens, M. J. Perron, M. Kiessling, P. Autissier, and J. Sodroski. 2004. The cytoplasmic body component TRIM5α restricts HIV-1 infection in Old World monkeys. Nature 427848-853. [DOI] [PubMed] [Google Scholar]
- 66.Sun, Y., S. R. Permar, A. P. Buzby, and N. L. Letvin. 2007. Memory CD4+ T-lymphocyte loss and dysfunction during primary simian immunodeficiency virus infection. J. Virol. 818009-8015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Swingler, S., B. Brichacek, J. M. Jacque, C. Ulich, J. Zhou, and M. Stevenson. 2003. HIV-1 Nef intersects the macrophage CD40L signalling pathway to promote resting-cell infection. Nature 424213-219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Taimen, P., and M. Kallajoki. 2003. NuMA and nuclear lamins behave differently in Fas-mediated apoptosis. J. Cell Sci. 116571-583. [DOI] [PubMed] [Google Scholar]
- 69.Tan, Y. J., S. G. Lim, and W. Hong. 2007. Regulation of cell death during infection by the severe acute respiratory syndrome coronavirus and other coronaviruses. Cell Microbiol. 92552-2561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Tsujimoto, Y., and S. Shimizu. 2007. Role of the mitochondrial membrane permeability transition in cell death. Apoptosis 12835-840. [DOI] [PubMed] [Google Scholar]
- 71.Unutmaz, D., V. N. KewalRamani, S. Marmon, and D. R. Littman. 1999. Cytokine signals are sufficient for HIV-1 infection of resting human T lymphocytes. J. Exp. Med. 1891735-1746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Vlahakis, S. R., A. Algeciras-Schimnich, G. Bou, C. J. Heppelmann, A. Villasis-Keever, R. C. Collman, and C. V. Paya. 2001. Chemokine-receptor activation by env determines the mechanism of death in HIV-infected and uninfected T lymphocytes. J. Clin. Investig. 107207-215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Westendorp, M. O., R. Frank, C. Ochsenbauer, K. Stricker, J. Dhein, H. Walczak, K. M. Debatin, and P. H. Krammer. 1995. Sensitization of T cells to CD95-mediated apoptosis by HIV-1 Tat and gp120. Nature 375497-500. [DOI] [PubMed] [Google Scholar]
- 74.Wyma, D. J., J. Jiang, J. Shi, J. Zhou, J. E. Lineberger, M. D. Miller, and C. Aiken. 2004. Coupling of human immunodeficiency virus type 1 fusion to virion maturation: a novel role of the gp41 cytoplasmic tail. J. Virol. 783429-3435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Zack, J. A., S. J. Arrigo, S. R. Weitsman, A. S. Go, A. Haislip, and I. S. Chen. 1990. HIV-1 entry into quiescent primary lymphocytes: molecular analysis reveals a labile, latent viral structure. Cell 61213-222. [DOI] [PubMed] [Google Scholar]
- 76.Zhou, Y., H. Zhang, J. D. Siliciano, and R. F. Siliciano. 2005. Kinetics of human immunodeficiency virus type 1 decay following entry into resting CD4+ T cells. J. Virol. 792199-2210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Zong, W. X., and C. B. Thompson. 2006. Necrotic death as a cell fate. Genes Dev. 201-15. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.